

Abstract
Exposure of mitochondrial DNA (mtDNA) to the cytosol activates innate immune responses. But the mechanisms by which mtDNA crosses the inner mitochondrial membrane are unknown. Here, we found that the inner mitochondrial membrane protein prohibitin 1 (PHB1) plays a critical role in mtDNA release by regulating permeability across the mitochondrial inner membrane. Loss of PHB1 results in alterations in mitochondrial integrity and function. PHB1‐deficient macrophages, serum from myeloid‐specific PHB1 KO (Phb1MyeKO) mice, and peripheral blood mononuclear cells from neonatal sepsis patients show increased interleukin‐1β (IL‐1β) levels. PHB1 KO mice are also intolerant of lipopolysaccharide shock. Phb1‐depleted macrophages show increased cytoplasmic release of mtDNA and inflammatory responses. This process is suppressed by cyclosporine A and VBIT‐4, which inhibit the mitochondrial permeability transition pore (mPTP) and VDAC oligomerization. Inflammatory stresses downregulate PHB1 expression levels in macrophages. Under normal physiological conditions, the inner mitochondrial membrane proteins, AFG3L2 and SPG7, are tethered to PHB1 to inhibit mPTP opening. Downregulation of PHB1 results in enhanced interaction between AFG3L2 and SPG7, mPTP opening, mtDNA release, and downstream inflammatory responses.
Keywords: AFG3L2, MIMP, mtDNA, PHB, SPG7
Subject Categories: Immunology, Organelles
Loss of the inner mitochondrial membrane protein PHB1 promotes innate immune responses by altering mitochondrial membrane integrity and mtDNA cytoplasmic release.

Introduction
The mitochondrion plays an important role in immunity, and it harbors contents serving as effective agonists of inflammation, the most notable of which is mtDNA (West & Shadel, 2017). Previous studies have reported that the release of mtDNA into the cytoplasm triggers innate immunity and inflammation (Kim et al, 2019; Wang et al, 2021). In response to cytosolic exposure of mtDNA, two major DNA‐sensing signaling pathways will be activated: one is the cyclic GMP‐AMP synthase‐Stimulator of interferon genes protein (cGAS‐STING) pathway, and the other is the inflammasome pathway (Kim et al, 2019; Wang et al, 2021). In the cGAS‐STING pathway, double‐stranded DNA exposed to the cytosol will bind to cGAS, which in turn activates STING and subsequently causes phosphorylation of TANK‐binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3), resulting in the induction of type I interferon (Wu et al, 2013). In the inflammasome pathway, several pattern recognition receptors, such as NOD, LRR, and Pyrin domain‐containing protein 3 (NLRP3) and absent in melanoma 2 (AIM2), oligomerize and bind to the adaptor molecule ASC and the effector molecule pro‐caspase‐1 under the stimulation of pathogen‐associated molecular patterns (PAMPs) or damage‐associated molecular pattern (DAMPs) (Schroder & Tschopp, 2010). Consequently, pro‐caspase‐1 is cleaved to the mature form of caspase‐1, which then cleaves pro‐interleukin‐1β (IL‐1β) and Gasdermin D to promote the pro‐inflammatory process and pyroptosis (Swanson et al, 2019). The activation of inflammasomes is associated with several diseases, including sepsis, atherosclerosis, diabetes, and neurodegeneration (Swanson et al, 2019). Although it is widely known that mtDNA release is required for inflammatory responses (Shimada et al, 2012), the exact mechanism of how mtDNA is released from mitochondria into the cytoplasm remains enigmatic.
Perturbations in mitochondrial outer and inner membrane permeability (MOMP and MIMP) are the main causes of mtDNA release (Riley & Tait, 2020). Cumulative evidence has revealed that mtDNA effluxes through the outer mitochondrial membrane via mechanisms involving voltage‐dependent anion channel (VDAC) oligomers and BCL2 antagonist/killer and BCL2 associated X, apoptosis regulator (BAK/BAX) macropores (McArthur et al, 2018; Kim et al, 2019). Upon MOMP transition, short mtDNA fragments are released into the cytosol through VDAC oligomers in cells lacking mitochondrial endonuclease G (Kim et al, 2019). During cellular apoptosis, mtDNA is released into the cytosol through BAK/BAX macropores (McArthur et al, 2018; Riley et al, 2018). Unfortunately, the clear mechanism of MIMP releasing mtDNA remains to be elucidated. On the inner mitochondrial membrane, several proteins contribute to the formation and opening of mPTP (Izzo et al, 2016), which participates in mtDNA release that triggers the inflammatory response (Yu et al, 2020; Wang et al, 2021). However, the specific proteins that regulate the opening of mPTP for mtDNA release remain unclear.
Several proteins localized in the mitochondrial inner membrane were reported to be components of mPTP and regulate MIMP, including adenine nucleotide translocase (ANT) (Woodfield et al, 1998), the F1Fo ATP synthase (Bonora et al, 2013; Giorgio et al, 2013), and spastic paraplegia 7 protein (SPG7) (Shanmughapriya et al, 2015). These factors are thought to be required for the opening of mPTP to mediate ion transport between the cytosol and the mitochondrial matrix (Bonora et al, 2021). However, it remains unclear how these factors are coordinated to regulate MIMP and to promote mtDNA release through the inner mitochondrial membrane.
Prohibitin (PHB) 1 is localized to the inner mitochondrial membrane and interacts with PHB2. Approximately 12 to 16 heterodimers form a ring‐shaped complex that maintains mitochondrial structure and function (Artal‐Sanz & Tavernarakis, 2009). Kasashima et al (2008) reported that loss of PHB1 impaired the organization of mitochondrial nucleoids (mtDNA/protein complexes) and reduced the level of the core nucleoid component, mitochondrial transcription factor A (TFAM). These disorders in mitochondrial nucleoids result in a decrease in the copy number of mtDNA (Kasashima et al, 2008). Furthermore, He et al (2012) isolated mitochondrial nucleoids and found PHB1 in the nucleoid fraction. Thus, PHB1 was proposed to be a possible component of mitochondrial nucleoids. In addition, PHB1 deficiency has been reported to promote inflammation and increase sensitivity to liver injury (Ko et al, 2010). Mice with intestinal epithelial cell deletion of Phb1 exhibit spontaneous ileal inflammation (Jackson et al, 2020). However, the mechanistic link between PHB1 perturbation, mtDNA release, and inflammation has not been revealed.
The m‐AAA protease complex is assembled in a heterohexamer form with AFG3‐like protein 2 (AFG3L2) and SPG7 or in a homohexamer form with only AFG3L2 (Levytskyy et al, 2017). Knockdown of SPG7, the potential regulator of mPTP, impairs ion efflux from mitochondria to cytosol (Shanmughapriya et al, 2015). In addition, SPG7 has been demonstrated to interact with cyclophilin D (CYPD), an essential component of mPTP (Crompton et al, 1998; Shanmughapriya et al, 2015). PHB1 may negatively regulate the proteolysis activity of the m‐AAA protease complex to mediate the degradation of mitochondrial proteins (Steglich et al, 1999). This raised the question of whether PHB1 promotes mtDNA release by regulating SPG7‐CYPD‐dependent opening of mPTP. Here, we propose that PHB1 may be a potential factor that localizes in the mitochondrial inner membrane to regulate mtDNA release in response to inflammatory stresses.
Results
Loss of Phb1 induces inflammation in vivo and in vitro
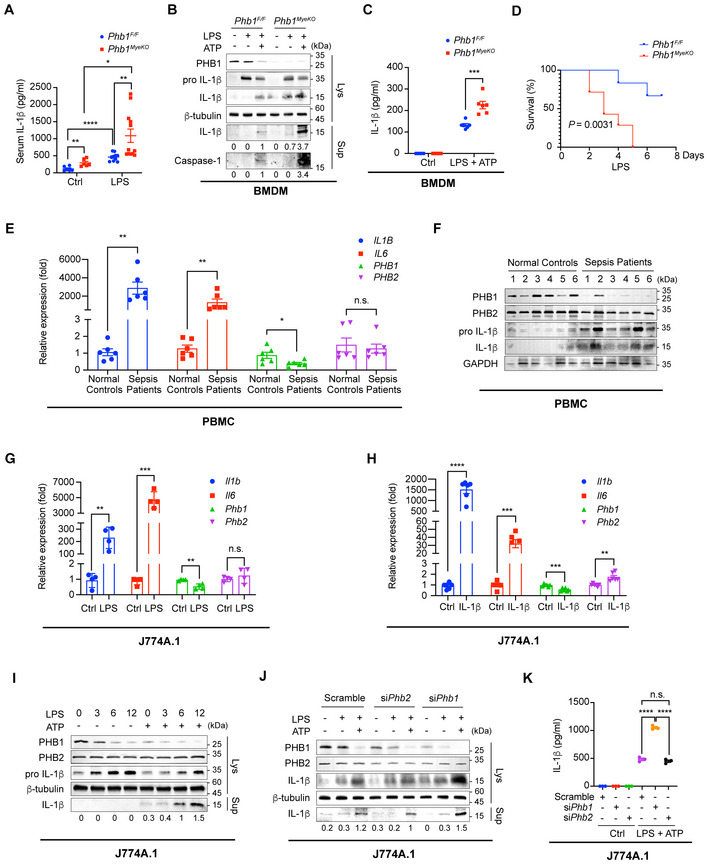
Phb1 −/− mice were not obtained from heterozygous intercrosses at the expected Mendelian ratio (Appendix Fig S1A). Therefore, we crossed Phb1 F/F and LysM‐Cre mice to generate Phb1 F/F : LysM‐Cre mice (hereafter called Phb1 MyeKO ) that lack Phb1 in mature myeloid cells, including macrophages and neutrophils (Appendix Fig S1B). Myeloid cells were extracted from the bone marrow of Phb1 MyeKO mice and differentiated into bone marrow–derived macrophages (BMDMs). Phb1 was not expressed in these cells (Appendix Fig S1C and D). Surprisingly, when we performed phenotypic screening on the genetically manipulated mice, we found that the serum level of IL‐1β is spontaneously increased in Phb1 MyeKO mice relative to Phb1 F/F mice (Fig 1A). After LPS administration, serological IL‐1β is increased in both Phb1 F/F mice and Phb1 MyeKO mice, and the increase is more pronounced in Phb1 MyeKO mice (Fig 1A). Accordingly, BMDMs extracted from Phb1 MyeKO mice have a higher level of cleaved IL‐1β relative to BMDMs from Phb1 F/F mice (Fig 1B). When lipopolysaccharide (LPS)‐primed BMDMs are extracted from Phb1 MyeKO mice and activated by addition of ATP, they secrete more cleaved Caspase‐1 and mature IL‐1β into the culture medium (Fig 1B and C). Subsequently, we examined the survival rate of mice treated with a high dose of LPS to induce septic shock. We found that the survival rate of Phb1 MyeKO mice is much lower than Phb1 F/F mice (Fig 1D).
Figure 1. Loss of Phb1 induces inflammation in vivo and in vitro .
-
AAfter being randomized, 8‐ to 12‐week‐old and sex‐matched Phb1 F/F mice and Phb1 MyeKO mice were administered with PBS or LPS (30 mg/kg) for 12 h via intraperitoneal injection. Serum was collected and ELISA assays were used to detect the level of IL‐1β (n = 6, 10, 6, and 12 mice for each group, respectively). Data presented as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 (Student's t‐test).
-
BBMDMs were extracted from Phb1 F/F or Phb1 MyeKO mice and stimulated by 20% culture supernatant of L929 cells for 7 days. Differentiated BMDMs were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min, the cells were lysed and the culture medium was collected. Western blotting (WB) assays were used to detect protein levels in whole cell lysates (Lys) and culture medium (Sup).
-
CBMDMs were extracted from Phb1 F/F or Phb1 MyeKO mice and stimulated with 20% culture supernatant of L929 cells for 7 days. Differentiated BMDMs were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. ELISA assays were used to detect protein levels in the culture medium. Data of BMDMs from the separate mice (n = 6 mice for each group) are presented as mean ± SEM. ***P < 0.001 (Student's t‐test).
-
DAfter randomized, 4 weeks old and sex‐matched Phb1 F/F mice and Phb1 MyeKO mice were administered with PBS or LPS (300 mg/kg) to induce septic shock via intraperitoneal injection. The life span of Phb1 F/F mice (n = 6) and Phb1 MyeKO mice (n = 7) was recorded.
-
EPBMCs were extracted from normal controls and patients with sepsis (n = 6 individuals). Intracellular mRNA levels were detected by real‐time quantitative PCR (RT–qPCR) assays (mean ± SEM). n.s., no significance, *P < 0.05, **P < 0.01 (Student's t‐test).
-
FPBMCs were extracted from normal controls and infant patients with sepsis. Protein levels in whole cell lysates were detected by WB assays.
-
G, HJ774A.1 cells were stimulated with LPS (200 ng/ml) for 6 h or recombinant IL‐1β (100 pg/ml) for 24 h. Intracellular mRNA levels were detected by RT–qPCR assays. Data are from four independent experiments for (G) and six independent experiments for (H) (mean ± SEM). n.s., no significance, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student's t‐test).
-
IAfter J774A.1 cells were stimulated with LPS (200 ng/ml) for 0, 3, 6, 12 h and ATP (4 mM) for 45 min, the cells were lysed, and the culture medium was collected. Protein levels in whole cell lysates (Lys) and culture medium (Sup) were detected by WB assays.
-
JAfter J774A.1 cells with Phb1 or Phb2 knockdown were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min, the cells were lysed, and the culture medium was collected. Protein levels in whole cell lysates (Lys) and culture medium (Sup) were detected by WB assays.
-
KAfter J774A.1 cells with Phb1 or Phb2 knockdown were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min, the culture medium was collected. ELISA assays were used to detect IL‐1β levels in the culture medium. Data are from three independent experiments (mean ± SEM). n.s., no significance, ****P < 0.0001 (Student's t‐test).
Source data are available online for this figure.
Sepsis is a clinically common disease typically characterized by a systemic inflammatory response of the human body (Angus & van der Poll, 2013). Guo et al (2020) found that PHB1 mRNA levels in peripheral blood mononuclear cells (PBMCs) were lower in patients with sepsis than in healthy controls. To confirm the connection between PHB1 and this severe inflammatory disease, we extracted PBMCs from infant patients with sepsis and found that the transcription levels of IL1B and IL6 are upregulated in these cells (Fig 1E). The transcription and protein levels of PHB1 are decreased in PBMCs from infant sepsis patients, while the PHB2 levels are relatively stable (Fig 1F).
Based on the observations in vivo, we used LPS and ATP stimuli to generate a cellular inflammatory model to further investigate whether PHB1 participates in innate immune responses. Similar to the data collected from PBMCs in infant patients with sepsis, synergistic stimulation of cells from the macrophage cell line J774A.1 with LPS enhances the transcription of Il1b and Il6 (Fig 1G). The mRNA and protein levels of PHB1 but not PHB2 are decreased in the same batch of cell lysate presplit into two parts (Fig 1G). Consistently, like LPS, exogenous IL‐1β also triggers MyD88‐signaling to increase transcription of Il1b and Il6, with a similar effect of LPS on PHB1 mRNA levels in J774A.1 (Fig 1G and H). Of note, the direct stimulation of IL‐1β mildly increases the PHB2 mRNA levels (Fig 1H). In addition, significantly elevated secretion of IL‐1β into the supernatant is detected after co‐stimulation with LPS and ATP (Fig 1I). We found that small interfering RNA (siRNA) knockdown of Phb1 in LPS + ATP‐stimulated J774A.1 macrophages also increases the secretion of IL‐1β into the supernatant, whereas knockdown of Phb2 does not promote the production of cleaved IL‐1β (Fig 1J and K).
Loss of PHB1 disturbs mitochondrial homeostasis and induces membrane permeability
PHB1 has been reported to maintain multiple mitochondrial functions, including m‐AAA protease activity (Steglich et al, 1999), oxidative phosphorylation (Bourges et al, 2004; Jian et al, 2017), cristae shape (Kong et al, 2014), and the copy number of mitochondrial DNA (Kasashima et al, 2008). Consistent with previous studies, we confirmed the major localization of PHB1 to mitochondria in immune and nonimmune cells (Appendix Fig S2A). We generated a HeLa cell line with stable knockdown (KD) of Phb1. Single cell‐expressing shRNA was selected with GFP+, and reduction in PHB1 expression was confirmed by Western blotting (Appendix Fig S2B). In cells with Phb1 deficiency, we observed the disorders in the mitochondrial network and excessive fragmentation of mitochondria indicated by increased circular‐ or dot‐shaped mitochondrial staining patterns (Fig 2A).
Figure 2. PHB1 is required to maintain mitochondrial homeostasis and membrane permeability.

-
A, BGFP‐expressing HeLa cells with stable Phb1 knockdown were fixed and subjected to immunofluorescence (IF) analysis to detect mitochondria (MitoTracker, red; A) or ΔΨm (TMRM, red; B). Images shown are representative of at least three independent experiments. White boxed regions in the panels in (A) are enlarged. Scale bar: 10 μm.
-
CFlow cytometric analysis of ΔΨm (TMRM). HeLa cells with stable Phb1 knockdown were treated with CsA (2 μM) for 30 min or FCCP (4 μM) for 5 min.
-
DThe mitochondrial respiratory capacity of J774A.1 cells with or without Phb1 knockdown was measured with a Seahorse XFe96 analyzer. Data are from at least three independent experiments (mean ± SEM). n.s., no significance. **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student's t‐test).
-
EHeLa cells with stable PHB1 ablation were incubated with H2O2 (2 μM) and CsA (2 μM) for 30 min. Flow cytometric analysis of intracellular ROS levels (DHE).
-
F, GConfocal microscopy analyses of mitochondrial Ca2+ in time‐series mode. HeLa cells stably expressing 4mt‐RCaMPh with or without Phb1 knockdown were treated with CsA (2 μM) for 30 min. During the acquisition of fluorescence images (F), cells were treated with histamine (200 μM). Traces of mitochondrial Ca2+ (4mt‐RCaMPh) dynamics are shown in (G) (mean ± SEM). n = 16, 16, 15, 16 (from the top to the bottom). Scale bar: 10 μm. *P < 0.05 relative to Scramble group, #P < 0.05 relative to shPHB1 group (Student's t‐test).
Source data are available online for this figure.
Next, we detected mitochondrial membrane potential (ΔΨm) in the Phb1 KD HeLa cells with tetramethylrhodamine methyl ester (TMRM), a fluorescent dye that usually accumulates in the mitochondrial matrix of intact mitochondria (Perry et al, 2011). We found that ΔΨm is lost upon knockdown of Phb1 (Fig 2B and C). Given that the opening of mPTP may be attributed to the loss of ΔΨm (Perry et al, 2011), we blocked the opening of mPTP by the inhibitor of CYPD, cyclosporin A (CsA). We found that the loss of ΔΨm caused by Phb1 deficiency was inhibited by treatment with CsA (Fig 2B and C). As a control, ΔΨm is lost when cells are treated with Carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (FCCP), which impairs the mitochondrial electron transport chain (ETC) and proton flux (Fig 2C). This indicates that PHB1 is required for maintenance of ΔΨm, and PHB1 maintains ΔΨm by inhibiting the opening of mPTP and MIMP (Fig 2C).
To investigate whether PHB1 participates in the functions of ETC, we examined mitochondrial respiration. Knockdown of PHB1 decreases the basal oxygen consumption rate, the respiratory capacity, and the ATP production of mitochondria in J774.1 cells (Fig 2D). Additionally, knockdown of Phb1 increases the production of reactive oxygen species (ROS) under physiological or H2O2 stress conditions, while the treatment of CsA restores this excessive production (Fig 2E).
Given that the opening of mPTP impacts mitochondrial Ca2+ transient, we further investigated whether PHB1 regulates mitochondrial Ca2+ uptake. We examined the mitochondrial Ca2+ transient in the Phb1 KD HeLa cell line stably expressing a Ca2+ indicator, 4mt‐RCaMPh. As described previously (Zhao et al, 2019), the fluorescence intensity of 4mt‐RCaMPh reflects the free Ca2+ level in mitochondria. Histamine triggers rapid Ca2+ influx from the cytoplasm into mitochondria (Zhao et al, 2019). We found that Phb1 knockdown impairs the uptake of Ca2+ in mitochondria, and this impairment is ameliorated by the mPTP inhibitor CsA (Fig 2F and G, Movies [Link], [Link]). CsA was not considered that impacts mitochondrial Ca2+ uptake (Yun et al, 2014), so this amelioration may be attributed to preventing the effects of Phb1 deficiency on other events and to transiently increasing mitochondrial Ca2+ level.
Of note, Phb1 knockdown does not cause excessive cell death under physiological or oxidative stress conditions compared with the scramble control (Appendix Fig S2C).
PHB1 deficiency promotes mtDNA release via CYPD‐ and VDAC‐associated mPTP opening
Given that the loss of PHB1 promotes the opening of mPTP, we wanted to know whether mtDNA escapes from mitochondria under the same conditions. Using SG‐ALK (Han et al, 2020) and an anti‐DNA antibody (Yu et al, 2020) to indicate DNA, we found that knockdown of Phb1 but not Phb2 decreases the co‐localization between mtDNA and mitochondria in J774.1 cells (Fig 3A and B; Appendix Fig S3A). As described in a previous study (Yu et al, 2020), we detected mtDNA levels in the cytosol with qPCR assays and found that PHB1 deficiency enhances the abundance of cytoplasmic mtDNA in J774.1 cells (Fig 3C; Appendix Fig S3B).
Figure 3. PHB1 knockdown promotes mPTP‐dependent mtDNA release.

-
AThe co‐localization between mtDNA (SG‐ALK, green) and mitochondria (MitoTracker, red) was examined in J774A.1 cells with or without knockdown of Phb1 or Phb2. White boxed regions in the panels are enlarged. The white arrows indicate mtDNA outside mitochondria. Scale bar: 10 μm.
-
BQuantitative analysis of data from (A). Data are from 10, 8, and 7 images from three independent experiments (left to right, respectively) (mean ± SEM). n.s., no significance, ***P < 0.001 (Student's t‐test).
-
CJ774A.1 cells with or without Phb1 knockdown were lysed, and DNA was extracted. qPCR assays were used to detect the mtDNA levels (D‐loop) in the cytosol relative to nuclear DNA levels (Tert) in the whole cell lysates. Data are from six independent experiments (mean ± SEM). ****P < 0.0001 (Student's t‐test).
-
DBMDMs isolated from Phb1 F/F mice and Phb1 MyeKO mice were fixed and subjected to IF analysis to detect the co‐localization between mitochondria (MitoTracker, red) and mtDNA (SG‐ALK, green). White boxed regions in the panels are enlarged. The white arrows indicate mtDNA outside mitochondria. Scale bar: 10 μm.
-
EQuantitative analysis of data from (D). Data are from 10 and 8 images from three independent experiments (mean ± SEM). **P < 0.01 (Student's t‐test).
-
FPBMCs were extracted from Phb1 F/F mice and Phb1 MyeKO mice. Cells were lysed, and DNA was extracted. qPCR assays were used to detect the levels of mtDNA (D‐loop) in the cytosol relative to the levels of nuclear DNA levels (Tert) in the whole cell lysates. Data of PBMC are from seven Phb1 F/F mice and Phb1 MyeKO mice, respectively (mean ± SEM). **P < 0.01 (Student's t‐test).
-
GHeLa cells expressing TFAM‐FLAG with or without Phb1 knockdown were incubated with CsA (2 μM) for 30 min. Cells were fixed and subjected to IF analysis to detect the co‐localization between mitochondria (anti‐TOMM20, red) and TFAM‐FLAG (anti‐FLAG, green). White boxed regions in the panels are enlarged. The white arrows indicate mtDNA outside mitochondria. The yellow arrows indicate mtDNA inside mitochondria. Scale bar: 10 μm.
-
HQuantitative analysis of data from (G). Data from 30, 27, 25, and 24 images from three independent experiments (mean ± SEM). ****P < 0.0001 (Student's t‐test).
-
IHeLa cells with or without Phb1 knockdown were incubated with H2O2 (5 mM) for 20 min and CsA (4 μM) for 12 h. Cells were lysed, and DNA was extracted. qPCR assays were used to detect mtDNA levels (ND1) in the cytosol and nuclear DNA levels (18S) in the whole cell lysates. Data are from four independent experiments (mean ± SEM). **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student's t‐test).
-
J, KHeLa cells with or without Phb1 knockdown were incubated with VBIT‐4 (10 μM) for 1 h and subsequently lysed, and DNA was extracted. qPCR assays were used to detect mtDNA levels (ND1 in HeLa cells or D‐loop in J774A.1 cells) in the cytosol relative to nuclear DNA levels (TERT in HeLa cells or Tert in J774A.1 cells) in the whole cell lysates. Data are from three independent experiments (mean ± SEM). n.s., no significance, *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t‐test).
-
LJ774A.1 cells with or without Phb1 knockdown were lysed. The whole cell lysates were immunoprecipitated by IgG or anti‐ASC antibody and the resulting proteins were detected by WB assay.
-
MBMDMs isolated from Phb1 F/F mice and Phb1 MyeKO mice were incubated with MCC950 (50 nM) for 30 min and were subsequently stimulated by LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. Protein levels in whole cell lysates and culture medium were detected by WB assay.
Source data are available online for this figure.
To further evaluate mtDNA release upon PHB1 knockout, we extracted the myeloid cells from the bone marrow of Phb1 MyeKO mice and induced them to differentiate into BMDMs. Consistent with the observations in J774A.1 cells, the co‐localization between mtDNA and mitochondria is impaired in BMDMs (Fig 3D and E; Appendix Fig S3C). In addition, PBMCs from Phb1 MyeKO mice show an increased abundance of cytoplasmic mtDNA (Fig 3F). In nonimmune HeLa cells, Phb1 knockdown also impairs the co‐localization between mtDNA and mitochondria, and enhances the cytoplasmic mtDNA level in nonimmune HeLa cells (Fig 3G and H; Appendix Fig S3D–H). The effects of Phb1 knockdown are also inhibited by CsA, as evidenced by multiple probes to indicate DNA, including TFAM, SG‐ALK, and anti‐DNA antibody (Fig 3G and H; Appendix Fig S3D and E).
H2O2 is an established activator of mPTP opening. It also induces the oligomerization of VDAC (Keinan et al, 2010; Kim et al, 2019). When incubated with H2O2, HeLa cells showed an increased cytoplasmic mtDNA level (Fig 3I). When the cells are treated with H2O2 and CsA, the level of mtDNA within mitochondria is mostly restored (Fig 3I). Accordingly, confocal microscopy shows that the co‐localization between mtDNA and mitochondria is reduced in response to H2O2, whereas CsA blocked the H2O2‐induced efflux of mtDNA (Appendix Fig S4A). By monitoring Ca2+ levels in the mitochondrial matrix, we confirmed that H2O2 stimulation, comparable with Phb1 silencing, impairs Ca2+ transient in mitochondria (Appendix Fig S4B and C).
Importantly, VBIT‐4, an inhibitor of the mitochondrial outer membrane protein VDAC, also suppresses the release of mtDNA caused by Phb1 knockdown both in immune cells and nonimmune cells (Fig 3J and K).
Phb1 deficiency promotes the assembly of inflammasomes
Previous studies reported that the activation of NLRP3‐inflammasomes contributes to the progression of sepsis (Jin et al, 2017; Ren et al, 2021). Thus, we asked whether PHB1 deficiency promotes the activation of NLRP3‐inflammasomes in sepsis. In J774A.1 cells, Phb1 knockdown enhances the interaction between NLRP3 and ASC, two key components required for inflammasome assembly but does not alter the expression of either of them (Fig 3L).
To investigate whether the formation of the NLRP3‐inflammasome is the major reason for IL‐1β cleavage promoted by loss of PHB1, we blocked the activity of NLRP3 activity using its specific inhibitor, MCC950. We found that in MCC950‐treated BMDMs from Phb1 MyeKO mice, Phb1 knockout fails to increase the levels of cleaved IL‐1β in both cytosol and medium (Fig 3M).
Since AIM2, contributing to the activation of AIM2‐inflammasome, serves as a double‐strand DNA sensor, we also investigated its role in Phb1 deficiency‐relative inflammasome activity. Firstly, we did not observe the alteration in AIM2 levels upon the stimulation of poly (dA:dT). Under the Phb1 deficiency condition, the stimulation of poly (dA:dT) promoted increased production of IL‐1β (Appendix Fig S5A). Subsequently, we asked whether AIM2‐inflammasomes are involved in the production of IL‐1β upon inflammatory stresses. We noticed that AIM2 ablation slightly decreased the production of IL‐1β in Phb1‐depleted cells (Appendix Fig S5B).
The cytosolic mtDNA and ROS promoted by Phb1‐depletion contribute to the inflammatory responses
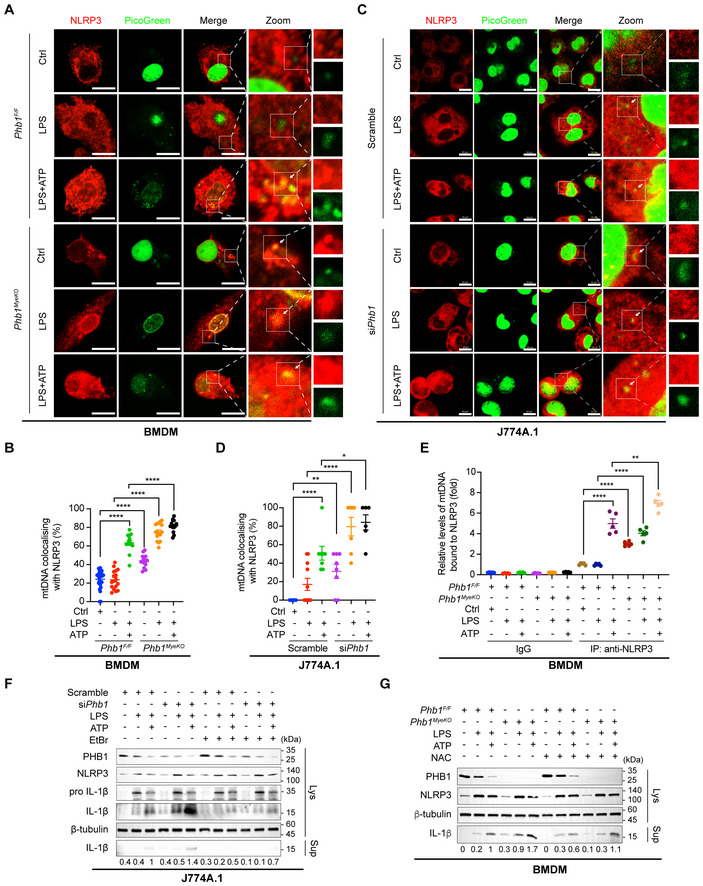
Previous studies reported that the cytosolic mtDNA can bind to NLRP3 and is involved in the activation of NLRP3‐inflammasomes (Shimada et al, 2012; Pan et al, 2018; Li et al, 2020; Wang et al, 2021). To investigate whether the effluxed mtDNA by Phb1 deficiency is sensed by NLRP3, we firstly examined the co‐localization between mtDNA and NLRP3. In physiological conditions, the co‐localization between mtDNA and NLRP3 is rarely observed in BMDMs derived from Phb1 F/F mice or in cultured J774A.1 cells (Fig 4A–D). Upon combined stimulation with LPS and ATP, more mtDNA co‐localizes with NLRP3 (Fig 4A–D). The co‐localization occurs in Phb1 knockdown J774A.1 cells or BMDMs from Phb1 MyeKO mice even in the absence of LPS and is more obvious with LPS alone or combined treatment with LPS and ATP (Fig 4A–D). In addition, we precipitated NLRP3 in BMDMs with an anti‐NLRP3 antibody and evaluated the abundance of mtDNA that bound to NLRP3. We found that knockout of Phb1 increases the enrichment of mtDNA on NLRP3, regardless of physiological conditions or being stimulated by inflammatory stresses of LPS and ATP (Fig 4E).
Figure 4. Cytosolic mtDNA and ROS promoted by Phb1 depletion contribute to the inflammatory responses.
-
ABMDMs isolated from Phb1 F/F mice and Phb1 MyeKO mice were stimulated with LPS (200 ng/ml) for 6 h, and ATP (4 mM) for 45 min. Cells were fixed and subjected to IF analysis to detect the co‐localization between NLRP3 (anti‐NLRP3, red) and mtDNA (PicoGreen, green). White boxed regions in the panels are enlarged. The white arrows indicate mtDNA co‐localized with NLRP3. Scale bar: 10 μm.
-
BQuantitative analysis of data from (A). Data are from at least 14 images from three independent experiments (mean ± SEM). ****P < 0.0001 (Student's t‐test).
-
CJ774A.1 cells with or without Phb1 knockdown were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. Cells were fixed and subjected to IF analysis to detect the co‐localization between NLRP3 (anti‐NLRP3, red) and mtDNA (PicoGreen, green). White boxed regions in the panels are enlarged. The white arrows indicate mtDNA co‐localized with NLRP3. Scale bar: 10 μm.
-
DQuantitative analysis of data from (C). Data are from at least six images from three independent experiments (mean ± SEM). *P < 0.05, **P < 0.01, ****P < 0.0001 (Student's t‐test).
-
EJ774A.1 cells with or without Phb1 knockdown were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. After cells were lysed, NLRP3 was immunoprecipitated by anti‐NLRP3 antibody. mtDNA levels (D‐loop) from immunoprecipitants were detected by qPCR in J774A.1 cells with or without Phb1 knockdown. Data are from five independent experiments (mean ± SEM). **P < 0.01, ****P < 0.0001 (Student's t‐test).
-
FJ774A.1 cells with Phb1 knockdown were incubated with EtBr (150 ng/ml) for 3 days, followed by LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. WB was used to detect the protein levels in whole cell lysates and culture medium.
-
GBMDMs isolated from Phb1 F/F mice and Phb1 MyeKO mice were treated with NAC (2 mM) for 8 h, LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. WB assay was used to detect the protein levels in whole cell lysates and in the culture medium.
Source data are available online for this figure.
Also, we investigated whether the other DNA conjugate AIM2 recognizes mtDNA and promotes the production of IL‐1β in Phb1‐deficient cells. We observed that Phb1 deficiency fails to increase the co‐localization between mtDNA and AIM2, regardless of in physiological conditions or upon inflammatory stimulation of LPS and ATP (Appendix Fig S5C and D). Additionally, we precipitated AIM2 in J774A.1 cells with an anti‐AIM2 antibody and detected the abundance of mtDNA binding to AIM2. Although poly (dA:dT) increases the binding between mtDNA and AIM2, Phb1 deficiency did not promote more enrichment of mtDNA on AIM2 (Appendix Fig S5E).
Subsequently, we generated ρ0 J774A.1 cells by depleting mtDNA in J774A.1 cells with the treatment of EtBr for 3 days as described previously (Nakahira et al, 2011; Rongvaux et al, 2014; Appendix Fig S6A and B). We found that in ρ0 cells, mtDNA depletion decreased the production of IL‐1β with the stimulation of LPS and ATP. Upon mtDNA depletion, knockdown of Phb1 fails to elevate the level of cleaved IL‐1β, as compared to normal J774A.1 cells (Fig 4F). These results suggest that the maturation of IL‐1β induced by PHB1 deficit is dependent on mtDNA. Increased intracellular ROS also triggers the activation of NLRP3‐inflammasomes (Zhong et al, 2016). We found that NAC can decrease the ROS levels in J774A.1 cells (Appendix Fig S6C) and thus inhibit the increased production of IL‐1β induced by Phb1 deficiency, seemly with less impact than mtDNA (Fig 4F and G).
The previous studies reported that cytoplasmic exposure of mtDNA also activates the cGAS‐STING pathway, which promotes the expression of interferon (IFN) and IFN‐stimulated genes (ISGs) (Riley & Tait, 2020). We used BMDMs from Phb1 MyeKO mice to examine the downstream genes of the cGAS‐STING pathway and found that Phb1 knockout activates the transcription of two ISGs, Isg15 and Cxcl10, in both physiological conditions and the stimulation of H2O2, which is considered to induce mtDNA release (Appendix Fig S7A and B).
PHB1 regulates the formation of mPTP by modulating the interaction between SPG7 and AFG3L2
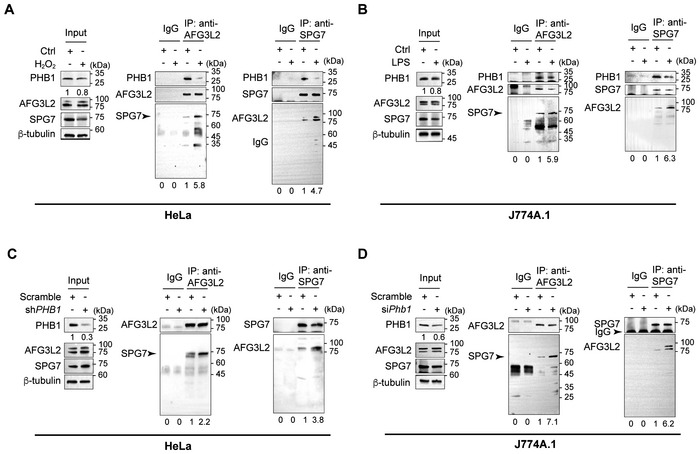
SPG7 binds the mPTP core component CYPD in the mitochondrial matrix and VDAC in the outer mitochondrial membrane. The interaction between SPG7 and AFG3L2 indicates the existence of mPTP (Shanmughapriya et al, 2015). Oligomycin Sensitivity‐Conferring Protein Subunit of ATP synthase (OSCP) has also been reported to bind CYPD (Giorgio et al, 2013). Therefore, we first wanted to know whether PHB1 can interact with OSCP, VDAC, SPG7, or AFG3L2. As shown in Appendix Fig S6A, overexpressed PHB1‐FLAG precipitates AFG3L2 and SPG7 but not VDAC or OSCP (Appendix Fig S8A). Therefore, we focused on how PHB1 regulates SPG7‐ and AFG3L2‐mediated mPTP opening.
We found that PHB1 strongly interacts with SPG7 and AFG3L2 under normal conditions in HeLa cells (Fig 5A). However, in H2O2‐stimulated HeLa cells, the PHB1 level is reduced and more SPG7 associates with AFG3L2 endogenously (Fig 5A). Accordingly, more endogenous AFG3L2 can be also precipitated by anti‐SPG7 antibody (Fig 5A). Consistent with the stimulatory effect of H2O2 in nonimmune HeLa cells, LPS treatment also reduces the PHB1 level and enhances the endogenous interaction between SPG7 and AFG3L2 in J774A.1 cells (Fig 5B).
Figure 5. PHB1 regulates the formation of mPTP by modulating the interaction between SPG7 and AFG3L2.
-
AHeLa cells were incubated with H2O2 (5 mM) for 20 min and then lysed. The whole cell lysate was immunoprecipitated by IgG, anti‐AFG3L2 antibody, or anti‐SPG7 antibody. WB was used to detect the SPG7 or AFG3L2 levels, respectively, in the precipitated products.
-
BJ774A.1 cells were stimulated with LPS (200 ng/ml) for 6 h and lysed. The whole cell lysate was immunoprecipitated by IgG, anti‐AFG3L2 antibody, or anti‐SPG7 antibody. The respective levels of precipitated SPG7 or AFG3L2 were examined by WB.
-
C, DHeLa cells or J774A.1 cells with or without Phb1 knockdown were lysed. The whole cell lysate was immunoprecipitated by IgG, anti‐AFG3L2 antibody, or anti‐SPG7 antibody. The respective levels of precipitated SPG7 or AFG3L2 were examined by WB.
Source data are available online for this figure.
Similar to the effect of H2O2 or LPS, both of which open mPTP, the knockdown of Phb1 itself significantly enhances the interaction between endogenous SPG7 and AFG3L2 in both HeLa cells and J774A.1 cells (Fig 5C and D).
SPG7 and AFG3L2 are both required for mtDNA release and inflammation activation mediated by PHB1 deficiency
Above, we showed that mPTP opening and mtDNA release induced by PHB1 deficiency is VDAC‐ and CYPD‐dependent. Shanmughapriya et al revealed that SPG7, as a core component of mPTP, interacts with VDAC and CYPD to promote the opening of mPTP. Additionally, SPG7 binds AFG3L2 to form the m‐AAA protease complex, which is regulated by PHB1 (Shanmughapriya et al, 2015). Thus, we asked whether SPG7 and AFG3L2 are involved in mtDNA release and inflammation activation induced by oxidative stress, LPS, or Phb1 knockdown.
Upon treatment of Phb1 KD HeLa cells with H2O2, the levels of SPG7 and AFG3L2 remain stable (Fig 5A; Appendix Fig S9A). This indicates that SPG7 and AFG3L2 do not depend on their altered expression to participate in the opening of mPTP. Additionally, in Phb1 KD HeLa cells, the disorder in mitochondrial Ca2+ uptake is recovered by additional knockdown of SPG7 or AFG3L2 (Appendix Fig S9B–E). We found that the co‐localization between mtDNA and mitochondria is reduced in Phb1‐depleted J774A.1 cells or HeLa cells, whereas additional knockdown of SPG7 or AFG3L2‐ restores the co‐localization (Fig 6A and B; Appendix Fig S9F). Subsequently, we verified that synergistically knocking down PHB1/SPG7 or PHB1/AGF3L2 suppresses the cytoplasmic mtDNA level, which is increased by knockdown of PHB1 alone (Fig 6C). Additionally, the increased production of cleaved IL‐1β induced by PHB1 knockdown is repressed with additional knockdown of SPG7 or AFG3L2 (Fig 6D–F).
Figure 6. SPG7 and AFG3L2 are both required for mtDNA release and inflammatory responses mediated by Phb1 knockdown.
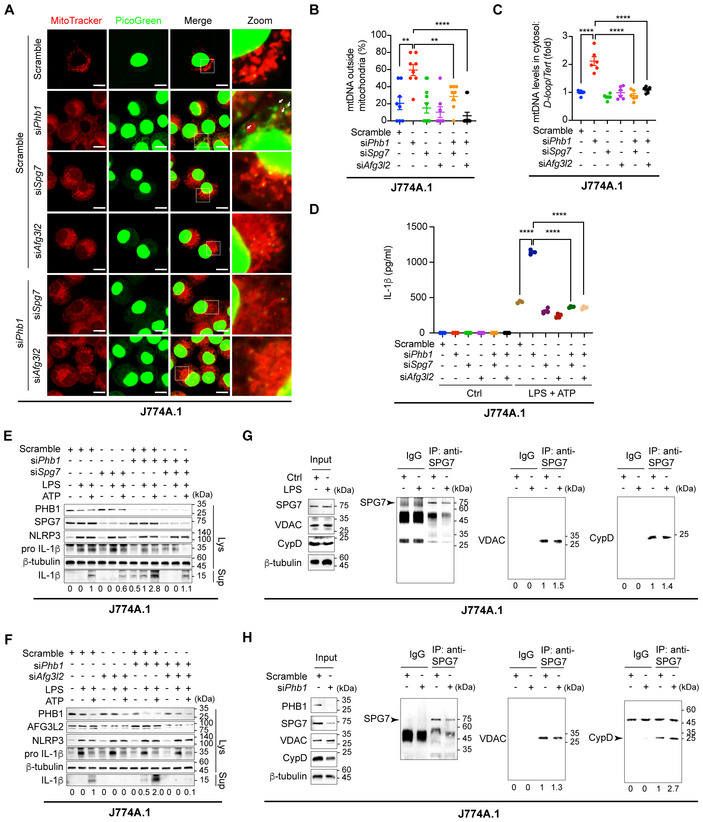
- J774A.1 cells with knockdown of Phb1 alone, Afg3l2 alone, Spg7 alone, Phb1 + Afg3l2, or Phb1 + Spg7 were fixed and subjected to IF analysis to assess the co‐localization between mitochondria (MitoTracker, red) and mtDNA (PicoGreen, green). White boxed regions in the panels are enlarged. The white arrows indicate mtDNA outside mitochondria. Scale bar: 10 μm.
- Quantitative analysis of the data from (A). Data are from at least 12 images from three independent experiments (mean ± SEM). **P < 0.01, ****P < 0.0001 (Student's t‐test).
- J774A.1 cells with knockdown of Phb1 alone, Afg3l2 alone, Spg7 alone, Phb1 + Afg3l2, or Phb1 + Spg7 were lysed, and DNA was extracted. qPCR was used to detect the mtDNA levels (D‐loop) in the cytosol relative to the nuclear DNA levels (Tert) in the whole cell lysate. Data are from six independent experiments (mean ± SEM). ****P < 0.0001 (Student's t‐test).
- J774A.1 cells with knockdown of Phb1 alone, Afg3l2 alone, Spg7 alone, Phb1 + Afg3l2, or Phb1 + Spg7 were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. The culture medium was collected and ELISA assays were used to detect IL‐1β levels in the culture medium. Data are from four independent experiments (mean ± SEM). ****P < 0.0001 (Student's t‐test).
- J774A.1 cells with Phb1 knockdown, Spg7 knockdown, or combined Phb1 and Spg7 knockdowns were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. WB assay was used to detect the protein levels in whole cell lysates and culture medium.
- J774A.1 cells with Phb1 knockdown, Afg3l2 knockdown, and combined Phb1 and Afg3l2 knockdowns were stimulated with LPS (200 ng/ml) for 6 h and ATP (4 mM) for 45 min. WB assay was used to detect the protein levels in whole cell lysates and culture medium.
- J774A.1 cells were stimulated with LPS (200 ng/ml) for 6 h and lysed. The whole cell lysate was immunoprecipitated by IgG or anti‐SPG7 antibody. WB was used to detect the CYPD and VDAC levels in the precipitated products.
- J774A.1 cells with or without Phb1 knockdown were lysed. The whole cell lysates were immunoprecipitated by IgG or anti‐SPG7 antibody. WB was used to detect the CYPD and VDAC levels in the precipitated products.
Source data are available online for this figure.
The interaction of SPG7 with VDAC or with CYPD is not influenced by PHB knockdown
SPG7/VDAC or SPG7/CYPD interaction mediates the opening of mPTP as evidenced by a previous study (Shanmughapriya et al, 2015). Therefore, we examined whether the interaction between SPG7/VDAC and SPG7/CYPD will be altered under conditions of inflammation or Phb1 knockdown. Under each condition, we failed to see an altered interaction between SPG7/VDAC and SPG7/CYPD in J774A.1 cells (Fig 6F–H). These results suggest that the opening of mPTP is not determined by alterations in the interactions between SPG7, VDAC, and CYPD, or by changes in the levels of these proteins.
Discussion
Recently, evidence has accumulated that mtDNA is important for the initiation of sterile innate immune responses (Riley & Tait, 2020). The channels that allow mtDNA to cross through the outer mitochondrial membrane have been extensively studied (McArthur et al, 2018; Kim et al, 2019). For instance, it has been reported that BAX and BAK are recruited to the outer mitochondrial membrane where they form macropores, followed by cytochrome C release and inner mitochondrial membrane herniation prior to the release of mtDNA and activation of the cGAS‐STING pathway (McArthur et al, 2018; Riley et al, 2018). The authors claimed that, during this extreme event, the manner in which mtDNA passes through the inner mitochondrial membrane is independent of the opening of mPTP.
Evidence from another group has demonstrated that during oxidative stress, mtDNA is released through macropores formed by oligomerization of VDACs and thus encounters the DNA sensor cGAS, which leads to TBK1 phosphorylation and IFN production (Kim et al, 2019). The authors proposed that the opening of mPTP induces mitochondrial inner membrane instability and mtDNA release (Kim et al, 2019). Yu et al (2020) showed that mtDNA passes through both mitochondrial membranes via mPTP in amyotrophic lateral sclerosis (ALS). Although the authors provided different mechanisms by which mtDNA escapes from the outer mitochondrial membrane, these studies failed to clarify how mtDNA passes through the inner mitochondrial membrane, nor did they identify which factors play key roles in MIMP by regulating mPTP opening.
More notably, although mPTP is involved in VDAC‐dependent mtDNA release induced by oxidative stress and pro‐inflammatory conditions (Kim et al, 2019; Yu et al, 2020), it does not appear to be involved in BAX/BAK‐dependent mtDNA release induced by proapoptotic conditions (McArthur et al, 2018; Riley et al, 2018). Also, VDAC‐mPTP channels can only release mtDNA fragments of about 110 bp, whereas the opening of BAX/BAK macropores can cause inner mitochondrial membrane herniation and nucleoid release into the cytoplasm (McArthur et al, 2018; Kim et al, 2019). Therefore, further work is required to determine whether different stress conditions can induce different molecularly mediated MIMPs and whether various forms of MIMPs can mediate the efflux of different sizes of mtDNA.
The current evidence showed there were different insights in the specific sensor to recognize mtDNA and mediate the downstream pathways. Although NLRP3, recognizing PAMPs and DAMPs, seems to sense oxidated mtDNA and lead to the activation of NLRP3‐inflammasomes (Shimada et al, 2012; Pan et al, 2018; Li et al, 2020; Wang et al, 2021), whether mtDNA directly triggers this event remains controversial (Holley & Schroder, 2020; Riley & Tait, 2020). Nevertheless, other factors involving defects in mitochondrial function (including loss of membrane potential, increased ROS, or perturbations in mitochondrial electron transport chain) are also associated with inflammasome activation (Zhou et al, 2011; Zhong et al, 2016; Billingham et al, 2022). Additionally, other sensors represented by cGAS and AIM2 can directly recognize mtDNA with their specific DNA‐binding domain and activate IFN‐relative pathways (Kim et al, 2019; Yu et al, 2020) and AIM2‐inflammasomes (Wang et al, 2021), respectively. Recently, Xian et al (2022) found that upon the stimulations of NLRP3 activators, ox‐mtDNA reaches the cytosol via mPTP‐ and VDAC‐ dependent channels and activates NLRP3‐inflammasomes and cGAS‐STING pathways. However, the results from our laboratory (not shown) and other laboratories show the limitations of using 8‐OH‐dG to stain ox‐mtDNA. They are not very specific and the fluorescence of ox‐mtDNA shows a diffuse distribution, which could be related to the principle of the method itself, in which all oxidized DNA in the cell is stained, causing a large amount of background fluorescence. A more convincing method to solve this difficulty remains to be developed in this field (Zhong et al, 2018; Shu et al, 2021). In the present study, we did confirm that mtDNA releases and is involved in these pathways above and found that Phb1 deficiency‐dependent mtDNA release may be associated with the activation of both NLRP3‐inflammasomes and cGAS‐STING pathways. Meanwhile, we also found that Phb1 deficiency‐dependent high ROS levels get involved in NLRP3‐inflammasomes activation. Thus, PHB1 may regulate inflammatory responses via multiple effects on mitochondrial functions.
EtBr is widely used to generate ρ0 cell lines with low mtDNA levels for investigation of the roles of mtDNA in inflammasomes and cGAS‐STING axis (Yu et al, 2020; Wang et al, 2021). EtBr treatment also has profound effects on cellular homeostasis, including increased intracellular ROS level, glycolysis, and reduced autophagic level (Nacarelli et al, 2014; Warren et al, 2017). Of note, increased ROS level and glycolysis as well as reduced autophagic level promote the activation of NLRP3‐inflammasome (Zhou et al, 2011; Xie et al, 2016; Zhong et al, 2016), whereas lower mtDNA level blocks NLRP3‐inflammasomes activation (Zhong et al, 2016). Consistent with the previous studies (Zhong et al, 2016; Wang et al, 2021), we also generated the ρ0 J774A.1 cells and found that mtDNA depletion blocks the activation of inflammasomes. Thus, we believe that methodology should be further improved for investigating the roles of mtDNA in the activation of inflammasomes and cGAS‐STING pathways.
Under these circumstances, we paid more attention to the mechanisms of MIMP during inflammatory responses and investigated what factors regulate MIMP, leading to mtDNA crossing the inner mitochondrial membrane into the cytosol in the present study. We propose that the release of mtDNA from the mitochondrial matrix firstly requires contact with inner mitochondrial membrane proteins or lipids and then transport across the inner and outer membranes. PHB1 emerged in our search for mtDNA‐interacting proteins located in the inner mitochondrial membrane. We think it could be a perfect candidate because it can regulate GEP1 and USP1 to maintain the levels of cardiolipin and phosphatidylethanolamine (PE) in the inner mitochondrial membrane (Osman et al, 2009). PHB1 assists its binding partner MDM33 to maintain cardiolipin and PE levels in the inner mitochondrial membrane (Klecker et al, 2015). Thus, PHB1 has been considered to be an essential scaffold in the mitochondrial inner membrane. More importantly, the association between PHB1 and mtDNA was found several years ago. Kasashima et al (2008) identified that PHB1 is a mitochondrial nucleoid component and maintains the copy number of mtDNA. Therefore, PHB1 is very likely to be a critical protein for mtDNA efflux.
There are vigorous debates regarding the composition of mPTP. Shanmughapriya et al demonstrated that SPG7 is a core component of mPTP and interacts with VDAC1 and CYPD. They also showed that SPG7 knockdown impairs the mitochondrial calcium retention capacity (Shanmughapriya et al, 2015). By contrast, Bernardi and Forte (2015, 2016) proposed that SPG7 is an indirect regulator of mPTP opening and MIMP rather than a core component of mPTP. In the last decade, some evidence suggests that mPTP is formed by the rearrangement of ATP synthase (Giorgio et al, 2013). Therefore, the exact mechanism underlying mPTP formation is still controversial, and it is possible that the composition of mPTP may differ under diverse stimulation conditions.
Our study identified that PHB1, a core factor in the inner mitochondrial membrane, controls mPTP formation and MIMP by regulating the interactions between SPG7 and AFG3L2. Specifically, under normal conditions, two inner membrane resident proteins, AFG3L2 and SPG7, are tethered to PHB1. However, under oxidative stress‐ or LPS‐induced mPTP opening conditions, or in cells with Phb1 knockdown, the binding between AFG3L2 and SPG7 is significantly reinforced, thus promoting the formation of mPTP. Although genetic deficiency of CYPD seems to have no effect on inflammasome responses (Allam et al, 2014), the loss of another potential mPTP regulator, SPG7, did block the activation of NLRP3‐inflammasomes in our study. In this way, mtDNA is exposed to the cytosol and activates downstream inflammatory responses (Fig 7).
Figure 7. Proposed working model of the role of PHB1 under normal and inflammatory stress conditions.
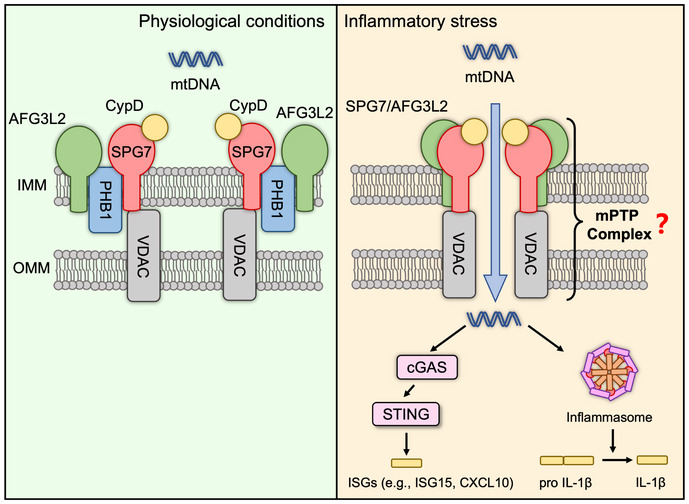
Under normal physiological conditions, two inner membrane resident proteins, AFG3L2 and SPG7, are tethered to PHB1. The mitochondrial permeability transition pore (mPTP) is inactive and mtDNA is retained in the matrix. In cells under inflammatory stress (e.g., induced by LPS), or in cells with PHB1 knockdown, the interaction between AFG3L2 and SPG7 is significantly enhanced, which promotes the formation of mPTP. This leads to mtDNA release and downstream inflammatory responses.
The present study also provides evidence of an anti‐inflammatory role for PHB1 in sepsis, consistent with the previous study (Guo et al, 2020). During T‐cell activation, PHB1 expression at the plasma membrane is upregulated (Yurugi et al, 2012; Buehler et al, 2018). In LPS‐induced acute lung injury, the PHB1 level is also increased in alveolar epithelial cells (Zhang et al, 2018). By contrast, more studies have shown that LPS stimulation downregulates the PHB1 level in epithelial cells and that overexpression of PHB1 alleviates inflammation and restores body weight (Theiss et al, 2009, 2011). Unfortunately, these studies failed to reveal the mechanism by which PHB1 regulates inflammation, whereas our study found that the downregulation of PHB1 in response to LPS stimulation promotes the formation of mtDNA‐dependent inflammasomes, which may lead to the progression of sepsis. We also noticed that IL‐1β levels were around 100 pg/ml in Phb1 F/F mice without LPS treatment, which may lead to inflammatory status, but the previous studies also showed similar levels in the control group (Zhong et al, 2016; Chen et al, 2020). Although some individuals had higher basal inflammation, any of their behavioral manifestations were normal. Collectively, our results reveal novel mechanistic roles of PHB1 not only in the initiation of innate immunity but also in the downstream responses.
Materials and Methods
Mice
LysM‐Cre mice were obtained from Yiming Xu's Lab, Guangzhou Medical University, China. Phb1 F/F mice were generated by Biocytogen Pharmaceuticals Co., Ltd (Beijing, China). LysM‐Cre mice were crossed with Phb1 F/F mice to generate Phb1 MyeKO mice. Phb1 F/F littermates were used as controls in all assays in this study. Whole body Phb1 +/− mice were generated by Biocytogen Pharmaceuticals Co., Ltd (Beijing, China). The drug administration of animals was according to the blind strategy. All mice were bred in the eligible animal housing facility at Guangzhou Medical University according to the guidelines of the Institutional Animal Care and Use Committee of UCSD (ID: GY2019‐017).
Reagents and antibodies
LPS (E. coli O111:B4) were from Sigma‐Aldrich (L2630). ATP was from Sigma‐Aldrich (A7699). Histamine was from Solarbio (H8240). Cyclosporin A was from Solarbio (C8780). FCCP was from Meilunbio (MB3642). VBIT‐4 was from Selleck Chemicals (S3544). Oligomycin was from Meilunbio (MB5431). Antimycin A was from Enzo Life Sciences (ALX‐380‐075). Rotenone was from Sigma‐Aldrich (R8875). Ethidium Bromide was Sigma‐Aldrich (E7637). Puromycin was from Sigma‐Aldrich (P8833). TMRM was from Anaspec (AS‐88065). DHE was from US EVERBRIGH (D1008). MCC950 was from MCE (HY‐12815A). H2O2 was from Huankai Microbial (029161). DMSO was from Solarbio (D8371). SG‐ALK was obtained from Shoufa Han's Lab, Xiamen University. MitoTracker™ Red CMXRos was from Invitrogen (M7512). Quant‐iT™ PicoGreen™ dsDNA Reagent was from Invitrogen (P11495). Transfection was performed with Hieff Trans™ Liposomal Transfection Reagent (YEASEN, 40802ES02) or Lipofectamine™ 2000 Transfection Reagent (Invitrogen, 11668019). ELISA assays were performed with Mouse IL‐1 beta ELISA Kit (Proteintech Group, KE10003). Antibodies for co‐immunoprecipitation assays, Western blotting assays or immunofluorescence analysis were: anti‐Prohibitin/PHB1 (ab75766, Abcam; MAB10296, Abnova; 10787, Proteintech Group), anti‐PHB2 (14085, Cell Signaling Technology), anti‐NLRP3 (15101, Cell Signaling Technology, AG‐20B‐0014, Adipogen), anti‐ASC (67824, Cell Signaling Technology), anti‐IL‐1β (12426/12242, Cell Signaling Technology), anti‐AFG3L2 (14631‐1‐AP, Proteintech Group), anti‐paraplegin/SPG7 (sc‐514393, Santa‐Cruz), anti‐VDAC (4661S, Cell Signaling Technology), anti‐CypD (18466‐1‐AP, Proteintech Group), anti‐OSCP (sc‐365162, Santa‐Cruz), anti‐Caspase‐1 (24232S, Cell Signaling Technology), anti‐AIM2 (66902‐1‐IG, Proteintech Group), anti‐TOMM20 (11802‐1‐AP, Proteintech Group), anti‐FLAG (66008‐3‐Ig/80010‐1‐RR, Proteintech Group), anti‐HA (3724S, Cell Signaling Technology), anti‐GAPDH (60004‐1‐Ig, Proteintech Group), anti‐β‐tubulin (10068‐1‐AP, Proteintech Group), anti‐DNA (AC‐30‐10, Progen), HRP‐conjugated Affinipure Goat Anti‐Mouse IgG(H + L) (SA00001‐1, Proteintech Group), HRP‐conjugated Affinipure Goat Anti‐Rabbit IgG(H + L) (SA00001‐2, Proteintech Group), Alexa Fluor 488 goat anti‐Rabbit (A11008, Invitrogen), Alexa Fluor 594 goat anti‐Rabbit (A11037, Invitrogen), Alexa Fluor 488 goat anti‐Mouse (A32723, Invitrogen), Alexa Fluor 594 donkey anti‐Mouse (A21203, Invitrogen), Mouse IgG (3420, Cell signaling technology), Rabbit IgG (2729, Cell signaling technology). RIPA lysis was from SAB (SA7951). The protease inhibitor cocktail was from Beyotime (P1006); PMSF (Phenylmethanesulfonyl fluoride) was from Beyotime (ST505).
Cell culture and infections
HeLa cell line and 293T‐cell line were obtained from the National Collection of Authenticated Cell Cultures. J774A.1 cell line was obtained from Kunming Cell Bank. CAS. Primary bone marrow–derived macrophages (BMDM) were obtained from gene‐modified mice. As the previous study described (Hornung et al, 2008), after being extracted from the bone marrow, marrow cells were stimulated by 20% vol/vol L929 cultural medium for 7 days to differentiate into macrophages. HeLa cells and 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher, 12800‐017) supplemented with 10% (v:v) fetal bovine serum (FS301‐02, TRANS) at 37°C with 5% (v:v) CO2. J774A.1 cells and BMDM were cultured in DMEM with 15% (v:v) fetal bovine serum (FS301‐02, TRANS) at 37°C with 5% (v:v) CO2. ShPHB1, 4mt‐RCaMPh, or scrambled sequences in lentiviral PLKO.1‐puro plasmid were transfected in 293T cells, and the lentivirus was packed. HeLa cells were infected by collected lentivirus containing target sequences. Monoclonal cell lines were selected and maintained by 3 μg/ml puromycin. Cell lines were recently authenticated by STR profiling and tested for mycoplasma contamination.
Isolation of peripheral blood mononuclear cells (PBMCs)
Peripheral venous blood of patients with neonatal septicemia and an equal number of normal controls were collected from The Second Affiliation Hospital of Guangzhou Medical University (n = 6, respectively) were collected from PBMCs were extracted with Human PBMCs separation kit (P8670, Solarbio). Institutional review board approval and ethics committee approval were granted for this study and informed consent was signed by the statutory guardian of patients before samples were collected. The collection of peripheral venous blood and extraction of PBMCs were according to the blind strategy. According to the manufacturer's instructions, 5 ml of peripheral venous blood was obtained in tubes with EDTA. Blood was slowly layered on 5 ml Ficoll–Hypaque, followed by the centrifuge at 800 g for 35 min at room temperature (RT) with slow acceleration and deceleration. The buffy coat in gradient was collected and mixed with PBS to 8 ml, followed by the centrifuge at 400 g for 10 min at RT. The pellet was suspended with Red blood cell lysis buffer and incubated for 10 min at 4°C. Washed with PBS, cells were collected in 1.5 ml EP centrifuge tubes for further assays.
Plasmid constructs and transfection
4mt‐RcaMPh plasmid was a gift from Xin Pan's Lab, National Center of Biomedical Analysis, China. According to our previous study (Hu et al, 2021), For other plasmids, target cDNA with the homologous sequence was amplified with Golden Star T6 PCR Super Mix (TSE101, Tsingke). Expression plasmids were digested by restriction enzymes (Takara). Target cDNA was cloned into a digested vector with homologous recombination by Hieff Clone® Plus One Step Cloning Kit (10911ES20, YEASEN). ASC gene was cloned into pCMV expression vectors with HA tag. ASC gene, NLRP3 gene, TFAM gene, and PHB1 gene were cloned into pCMV expression vector with FLAG tag.
The primers were provided as follows:
ASC: 5′‐CGCTCTAGCCCGGGCGGATCCATGGGGCGCGCGCGCGACGCCAT‐3′ (Forward), 5′‐GTATGGGTATCTAGACTCGAGGCTCCGCTCCAGGTCCTCCACCA‐3′ (Reverse)
NLRP3: 5′‐GGGAATTCGTCGACTGGATCCATGAAGATGGCAAGCACCCGCTGCAAG‐3′ (Forward), 5′‐TGAGATGAGTTTCTGCTCGAGCCAAGAAGGCTCAAAGACGACGGTCAGC‐3′ (Reverse)
TFAM: 5′‐GGGAATTCGTCGACTGGATCCATGGCGTTTCTCCGAAGCATGT‐3′ (Forward), 5′‐TGAGATGAGTTTCTGCTCGAGACACTCCTCAGCACCATATTTTCG‐3′ (Reverse)
PHB1: 5′‐TACCGAGGAGATCTGCCGCCGCGATCGCCATGGCTGCCAA‐3′ (Forward); 5′‐CCGCGTACGCGTCTGGGGCAGCTGGAGGAGCACGGACTGC‐3′ (Reverse)
PHB1 shRNAs, SPG7 shRNAs, and scramble control shRNAs were inserted into GV248 vectors (Shanghai GeneChem Co., Ltd; Tsingke Biotechnology Co., Ltd.). The shRNA target sequences were provided as follows:
Scramble: 5′‐TTCTCCGAACGTGTCACGT‐3′
PHB1: 5′‐AGTGTTTGAGTCCATTGGCAA‐3′
SPG7: 5′‐AATGCCTACGTTAAGCTATAC ‐3′
AFG3L2: 5′‐GCTAGAGTCCGAGACTTATTT‐3′
Expression plasmids and shRNA were transfected into cells for 36–48 h according to the manufacturer's instructions of transfection reagents, followed by harvest for Western blotting, RT–qPCR, and immunofluorescence analysis.
Transient transfection with siRNA
PHB1, PHB2, SPG7, or AFG3L2 were knocked down by siRNA that was designed and synthesized by Kidan Biosciences co., Ltd (Guangzhou, China).
The sequences were provided as follows:
Scramble: 5′‐UUCUCCGAACGUGUCACGUTT‐3′ (sense), 5′‐ACGUGACACGUUCGGAGAATT‐3′ (antisense)
Phb1: 5′‐CGUCAAUAUCACACUGCGAAUTT‐3′ (sense), 5′‐AUUCGCAGUGUGAUAUUGACGTT‐3′ (antisense)
Phb2: 5′‐GCCUCAUUAAGGGUAAGAAAUTT‐3′ (sense), 5′‐AUUUCUUACCCUUAAUGAGGCTT‐3′ (antisense)
Spg7: 5′‐GCAUGUCUUCAUUGAUCUUTT‐3′ (sense), 5′‐AAGAUCAAUGAAGACAUGCCT‐3′ (antisense)
Afg3l2: 5′‐UGUUGCCCACCGUACUCAUUATT‐3′ (sense), 5′‐UAAUGAGUACGGUGGGCAACATT‐3′ (antisense)
Aim2: 5′‐GGAAGCCAUCAGAGAAGAUTT‐3′ (sense), 5′‐AUCUUCUCUGAUGGCUUCCTG‐3′ (antisense)
Transfection was performed with Hieff Trans™ Liposomal Transfection Reagent or Lipofectamine™ 2000 Transfection Reagent according to the manufacturer's instructions. The efficiency of knockdown was verified by RT–qPCR and Western blotting assays.
Enzyme‐linked immunosorbent assay (ELISA)
ELISA kits were used to detect mouse IL‐1β levels in mice serum (dilution, 1:200) and culture medium according to the manufacturer's instructions.
Western blotting
According to our previous study, we performed the WB assays (Hu et al, 2021). Cells were harvested and lysed by RIPA lysis buffer (RIPA lysis: cocktail: PMSF = 100:1:1). Following centrifuge at 16,215 g for 20 min at 4°C, the supernatant was collected, and the concentration of total proteins was detected by bicinchoninic acid (BCA) kit (23227, ThermoFisher Scientific). All samples were normalized and boiled at 100°C for 10 min. Samples and Protein Marker (20352ES76, YEASEN) were separated by SDS–PAGE with Genshare CFAS any KD PAGE (Genshare Biological), then transferred to PVDF membrane. Membranes carrying samples proteins were blocked by 5% BSA (Bovine Serum Albumin, 4240GR100, BioFroxx) or fat‐free milk for 1 h at RT and then were incubated with primary antibodies overnight at 4°C. After being washed by TBST buffer 4 times for 10 min, membranes were incubated with secondary antibodies for 1 h at RT. Subsequently, following washing by TBST buffer 4 times for 10 min, the blots were detected with High‐sig ECL Western Blotting Substrate (180‐5001, Tanon) and GelView 6000 Pro (Biolight, China).
Co‐immunoprecipitation
According to our previous study, we performed the co‐immunoprecipitation assays (Hu et al, 2021). For direct co‐IP for intracellular proteins, cells were seeded at 10 cm dishes and were harvested at the confluent degree of 80–90%. Cells were lysed with NP‐40 lysis buffer (P0013F, Beyotime) for 1 h, and the lyses were centrifuged at 16,215 g for 20 min at 4°C. The supernatant was collected, and the concentration of total proteins was detected by BCA kit. Moved 3,000 μg proteins into a fresh 1.5 ml EP centrifuge tube, and then added target antibody into the tube, followed by incubation for 12 h at 4°C. Subsequently, for immunoprecipitation, PureProteome™ Protein A/G Mix Magnetic Beads (LSKMAGAG10, Millipore) or Protein A + G Agaroses (P2055, Beyotime) were prewashed by PBST (PBS with 0.5% Tween) 3 times and were added into this tube and incubated for 3 h (agaroses for 10 h). Beads were washed by RIPA lysis buffer (Tris 10 mM, NaCl 150 mM, NP‐40 1%, SDS 0.1%, deoxycholate 0.1%, pH 7.4) 5 times on ice. All samples were boiled for 10 min.
Immunofluorescence and confocal microscope analysis
According to our previous study, we performed the IF analysis (Hu et al, 2021). For IF analysis, cells were seeded at 12‐well plates that contained glass slides overnight. Subsequently, after were stimulated with LPS 200 ng/ml for 6 h and ATP 4 mM for 45 min, cells were fixed with 4% paraformaldehyde (CF189021, Solarbio) for 10 min, and permeabilized in Immunostaining Permeabilization Buffer with Triton X‐100 (P0096, Beyotime) for 10 min at RT. Following blocking with QuickBlock™ Blocking Buffer (P0260, Beyotime) for 15 min at RT, cells were incubated with primary antibodies diluted in QuickBlock™ Primary Antibody Dilution Buffer (P0262, Beyotime) at 4°C overnight. After being washed by PBS, cells were incubated with Alexa Fluor secondary fluorescent antibodies for 1 h at RT and then were stained by DAPI.
For detection of DNA or mitochondria, cells were seeded at 12‐well plates that contained glass slides overnight. After being stimulated with LPS 200 ng/ml for 6 h and ATP 4 mM for 45 min, cells were incubated with PicoGreen for 1 h, SG‐ALK for 30 min, or MitoTracker for 30 min at 37°C. Subsequently, cells were fixed and stained by DAPI.
Images of cells were acquired by LSM 800 META (Carl Zeiss, Germany) or SP8 confocal microscope (Leica, Germany). The analyses of images were performed by Image J 1.53c (National Institutes of Health, USA).
For time‐series acquisition of images, cells that stably expressed 4mt‐RCaMPh were seeded on dishes with glass walls. Before image acquisition, the culture medium was changed to phenol red‐free DMEM with 10% FBS. Before images were acquired, cells were incubated with CsA 2 μM for 30 min or H2O2 1 mM for 30 min. During a series of image acquisition (interval: 10 s), cells were stimulated with histamine 200 μM.
Images of time series were acquired by LSM 800 META (Carl Zeiss, Germany). The analyses of images were performed by Image J 1.53c (National Institutes of Health, USA). F0 indicated the initial fluorescence intensity, while F indicated the fluorescence intensity at each time point. The plots of mitochondrial Ca2+ levels were indicated with F/F0.
Measurement of mitochondrial membrane potential
For measurement of ΔΨm, following treatment with FCCP 4 μM for 5 min and CsA 2 μM for 30 min, the cells were incubated with 150 nM TMRM for 30 min at 37°C. Subsequently, cells were resuspended with PBS, and ΔΨm was detected by SP8 confocal microscope (Leica, Germany) and CytoFLEX S (Beckman Coulter Life Sciences, USA).
Measurement of reactive oxidative species (ROS)
For measurement of ROS, following treatment with H2O2 500 μM for 30 min, the cells were incubated with 10 μM DHE. Subsequently, cells were resuspended with PBS, and ROS was detected by CytoFLEX S (Beckman Coulter Life Sciences, USA).
Cell death detection assays
The detection of cell death was performed with Annexin V‐FITC Apoptosis Detection Kit (KGA107, KeyGEN BioTECH) and CytoFLEX S (Beckman Coulter Life Sciences, USA).
RNA isolation and quantitative real‐time PCR
According to our previous study, we performed RT–qPCR assays (Hu et al, 2021). Total RNA extraction was performed by HiPure Total RNA Plus Micro Kit (R4122‐02, Magen), and reverse transcription assays were performed by HiScript® II Q RT SuperMix (R223‐01, Vazyme) in ProFlex™ PCR system (ThemoFisher, USA) according to manufacturer's instructions. Real‐time quantitative PCR assays were performed with ChamQ Universal SYBR qPCR Master Mix (Q711‐02, Vazyme) in QuantStudio™ 5 Real‐Time PCR System (ThemoFisher, USA).
The primers sequences were provided as follows:
Human
PHB1: 5′‐ACCACGTAATGTGCCAGTCA‐3′ (Forward), 5′‐TAGT CCTCTCCGATGCTGGT‐3′ (Reverse)
PHB2: 5′‐CGGGCCCAATTCTTGGTAGA‐3′ (Forward), 5′‐TCTGGGCTGCTCGAATCTTG‐3′ (Reverse)
IL1B: 5′‐CCCTGCAGCTGGAGAGTGTGG‐3′ (Forward), 5′‐TGTGCTCTGCTTGAGGTGCT‐3′ (Reverse)
IL6: 5′‐TAGTCCTTCCTACCCCAATTTCC‐3′ (Forward), 5′‐TTGGTCCTTAGCCACTCCTTC‐3′ (Reverse)
GAPDH: 5′‐GAGTCAACGGATTTGGTCGT‐3′ (Forward), 5′‐GACAAGCTTCCCGTTCTCAG‐3′ (Reverse)
Mouse
Phb1: 5′‐TCCCTTGGGTACAGAAACCAATTA‐3′ (Forward), 5′‐TGTGATATTGACGTTCTGCA AGTCT‐3′ (Reverse)
Phb2: 5′‐AGCAGGAACAGCACAGAAGA‐3′ (Forward), 5′‐CGGAGCTTGATATAGCCAGGAT‐3′ (Reverse)
Il1b: 5′‐TGACGGACCCCAAAAGATGA‐3′ (Forward), 5′‐CTGCTGCGAGATTTGAAGCT‐3′ (Reverse)
Il6: 5′‐CCCCAATTTCCAATGCTCTCC‐3′ (Forward), 5′‐CGCACTAGGT TTGCCGAGTA‐3′ (Reverse)
Actb: 5′‐GATTACTGCTCTGGCTCCTAG‐3′ (Forward), 5′‐GACTCATCGTACTCCTGCTTG‐3′ (Reverse)
Isg15: 5′‐CTAGAGCTAGAGCCTGCAG‐3′ (Forward), 5′‐AGTTAGTCACGGACACCAG‐3′ (Reverse)
Cxcl10: 5′‐CCAAGTGCTGCCGTCATTTTC‐3′ (Forward), 5′‐3′GGCTCGCAGGGATGATTTCAA (Reverse)
Cytosolic DNA immunoprecipitation and subcellular fractionation
For the detection of mtDNA binding to NLRP3 or AIM2 in J774A.1 cells, cells were seeded at 6‐wells plates with a density of 10 (Swanson et al, 2019) cells/well. Cells were lysed by NP‐40 lysis buffer and were centrifuged at 16,215 g for 20 min at 4°C. The supernatants were collected, and the concentration of total proteins was detected with BCA kits. Samples with equal protein concentration were moved into fresh 1.5 ml EP centrifuge tubes and were incubated with anti‐NLRP3 antibody or anti‐AIM2 antibody for 12 h at 4°C. Protein A + G Agaroses were prewashed by PBST 3 times and were added into this tube and incubated for 3 h. DNA in immunoprecipitation products was extracted with TIANamp Genomic DNA Kit (DP403, TIANGEN).
For the detection of mtDNA in the cytosol, J774A.1, HeLa cells or PBMCs were seeded at 6‐wells plates with a density of 106 cells/well. Cells were incubated with 1% NP‐40 on ice for 15 min. The supernatants were collected and centrifuged at 16,215 g for 15 min at 4°C. The supernatants were moved into the fresh 1.5 ml EP centrifuge tubes and the DNA was extracted with TIANamp Genomic DNA Kit. The levels of mtDNA were detected by qPCR assays.
The primers sequences were provided as follows:
Human
ND1: 5′‐CACCCAAGAACAGGGTTTGT‐3′ (Forward), 5′‐TGGCCATGGGTATGTTGTTAA‐3′ (Reverse)
D‐LOOP: 5′‐CTATCACCCTATTAACCACTCA‐3′ (Forward), 5′‐TTCGCCTGTAATATTGAACGTA‐3′ (Reverse)
COX3: 5′‐AATCCAAGCCTACGTTTTCACA‐3′ (Forward), 5′‐TGGCCATGGGTATGTTGTTAA‐3′ (Reverse)
18S: 5′‐TAGAGGGACAAGTGGCGTTC‐3′ (Forward), 5′‐CGCTGAGCCAGTCAGTGT‐3′ (Reverse)
TERT: 5′‐TCACGGAGACCACGTTTCAAA‐3′ (Forward), 5′‐TTCAAGTGC TGTCTGATTCCAAT‐3′ (Reverse)
Mouse
D‐loop: 5′‐AATCTACCATCCTCCGTGAAACC‐3′ (Forward), 5′‐TCAGTTTAGCTACCCCCAAGTTTAA‐3′ (Reverse)
Tert: 5′‐CTAGCTCATGTGTCAAGACCCTCTT‐3′ (Forward), 5′‐GCCAGCACGTTTCTCTCGTT‐3′ (Reverse)
Seahorse cell mitochondrial stress test assay
Mitochondrial respiration capacity of J774A.1 cells or HeLa cells with or without PHB1 ablation were detected by Seahorse Cell Mito Stress Test Assay. Before the detection, cells were seeded at XFe96 Cell Culture Microplates (102601‐100, Agilent) with the concentration of 12,000 cells/80 μl per well. Seahorse XFe96 Sensor Cartridge (102601‐100, Agilent) was incubated at 37°C overnight without CO2 for hydration. Cells were washed by XF Cell Mito Stress Test Assay Medium (XF Base Medium [102353‐100, Agilent] with 5.5 mM glucose [G7528, Sigma], 1.0 mM pyruvate [P5280, Sigma], 2 mM L‐glutamine [25030‐081, Life Technologies], adjusted pH to 7.4 at 37°C, and filtered with a 0.2 μm filter [SLGP033RB, Millipore]). Before the detection, cells were incubated with XF Cell Mito Stress Test Assay Medium for 1 h at 37°C without CO2, and assays compounds (port A: oligomycin 1 μM, port B: FCCP 0.5 μM, port C: rotenone 0.5 μM and antimycin A 0.5 μM) were added into incubated Seahorse XFe96 Sensor Cartridge according to manufacturer's instructions. According to the instructions, the assays were performed with Agilent Seahorse XFe96 system (Agilent, USA), and the data were analyzed with Seahorse Wave Desktop and Report Generator software (Agilent, USA).
Septic shock model
For survival analysis of the septic shock model, 3–4 weeks old and sex‐matched mice were then intraperitoneally (i.p.) injected with LPS at a dose of 300 mg/kg body weight. After injection, mice survival was monitored every 24 h. The total duration was 7 days.
For detection of cytokines levels in mice serum, after mice were administrated with LPS for 12 h, the serum was extracted. The levels of cytokines were measured by ELISA kits.
Mice Bone marrow cells isolation and differentiation into macrophages
8–12 weeks old and sex‐matched Phb1 F/F mice and Phb1 MyeKO mice were cervically dislocated after anesthetization. Hind legs were dislodged from the acetabulum, and paws and muscles were removed. The femur and the tibia were separated, followed by cutting off both ends of the bones. After being flushed from bones by PBS, the marrow cells were filtered through the 70 μm cell strainer and centrifuged at 250 g for 5 min at 4°C. Cells were incubated with Red blood cells lysis buffer for 10 min at 4°C, followed by resuspended with PBS. Cells were centrifuged at 250 g for 5 min at 4°C. After being resuspended with DMEM (with 15% FBS and 20% L929 cells cultural medium), cells were seeded at 10 cm cell culture dishes at a density of 106/ml and cultured for 7 days to differentiate to macrophages.
For confirming the deletion of Phb1 gene in the genome of bone marrow cells, nucleus DNA was extracted with TIANamp Genomic DNA Kit (DP403, TIANGEN), and was detected by RT–qPCR assays with the primers as follows:
PHB1: 5′‐TCCCTTGGGTACAGAAACCAATTA‐3′ (Forward),
5′‐ TGTGATATTGACGTTCTGCAAGTCT‐3′ (Reverse)
ACTB: 5′‐ GATTACTGCTCTGGCTCCTAG‐3′ (Forward),
5′‐ GACTCATCGTACTCCTGCTTG‐3′ (Reverse)
Statistics
All continuous values were shown as means ± SEM as legends indicated. Values between groups were analyzed by a Student's t‐test or log‐rank test by GraphPad Prism 8.2.1 (GraphPad Software, Inc., USA). A two‐tail P < 0.05 was considered statistically significant.
Author contributions
Du Feng: Conceptualization; resources; supervision; funding acquisition; methodology; writing—original draft; project administration; writing—review and editing. Hao Liu: Formal analysis; investigation; writing—original draft; project administration. Hualin Fan: Formal analysis; investigation; writing—original draft; project administration. Pengcheng He: Formal analysis; funding acquisition; investigation; project administration; writing—review and editing. Haixia Zhuang: Formal analysis; investigation; writing—review and editing. Xiao Liu: Formal analysis; writing—review and editing. Meiting Chen: Formal analysis; writing—review and editing. Wenwei Zhong: Formal analysis; writing—review and editing. Yi Zhang: Formal analysis; writing—review and editing. Cien Zhen: Formal analysis; writing—review and editing. Yanling Li: Formal analysis; writing—review and editing. Huilin Jiang: Formal analysis; writing—review and editing. Tian Meng: Funding acquisition; writing—review and editing. Yiming Xu: Methodology; writing—review and editing. Guojun Zhao: Funding acquisition; investigation; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgment
We gratefully thank Dr. Isabel Hanson for editing the manuscript and Dr. Xin Pan, Institute of Basic Medical Sciences, National Center of Biomedical Analysis for kindly providing the 4mt‐RCaMPh plasmid, and Dr. Shoufa Han, Xiamen University for the DNA dye SG‐ALK. We also acknowledge the core facility of the School of Basic Medical Sciences for instrument and equipment support. This project is supported by the NSFC (No. 32170758, No. 31771531), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (GDUPS, 2018), open research funds from the Sixth Affiliated Hospital of the Guangzhou Medical University, Qingyuan People's Hospital, Guangzhou Ling Nan Ying Cai Project 2018, and Chuang Xin Qiang Xiao project of the Guangzhou Medical University 2019KCXTD015 to Dr. DF, and the National Natural Science Foundation of China (No. 82001205, No. 82170461) to Drs. PH and TM, the Natural Science Foundation of Guangdong (2020A1515010022) and Fundamental and Applied Fundamental Research Project of Guangzhou (202102020016) to Dr. TM.
Data availability
This study includes no data deposited in external repositories.
References
- Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ et al (2014) Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non‐apoptotic caspase‐8 is required for inflammasome priming. EMBO Rep 15: 982–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus DC, van der Poll T (2013) Severe sepsis and septic shock. N Engl J Med 369: 840–851 [DOI] [PubMed] [Google Scholar]
- Artal‐Sanz M, Tavernarakis N (2009) Prohibitin and mitochondrial biology. Trends Endocrinol Metab 20: 394–401 [DOI] [PubMed] [Google Scholar]
- Bernardi P, Forte M (2015) Commentary: SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Front Physiol 6: 320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Forte M (2016) Commentary: the m‐AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Front Physiol 7: 583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, Jacobs HT, Reczek CR, Rashidi A, Zhang P et al (2022) Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol 23: 692–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A et al (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Giorgi C, Pinton P (2021) Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol 23: 266–285 [DOI] [PubMed] [Google Scholar]
- Bourges I, Ramus C, Mousson de Camaret B, Beugnot R, Remacle C, Cardol P, Hofhaus G, Issartel JP (2004) Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria‐encoded subunits and interaction with prohibitin. Biochem J 383: 491–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler U, Schulenburg K, Yurugi H, Šolman M, Abankwa D, Ulges A, Tenzer S , Bopp T, Thiede B, Zipp F et al (2018) Targeting prohibitins at the cell surface prevents Th17‐mediated autoimmunity. EMBO J 37: e99429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xia Y, He F, Fu J, Xin Z, Deng B, He L, Zhou X, Ren W (2020) Serine supports IL‐1beta production in macrophages through mTOR signaling. Front Immunol 11: 1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM (1998) Cyclophilin‐D binds strongly to complexes of the voltage‐dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: 729–735 [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I et al (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110: 5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SD, Yan ST, Li W, Zhou H, Yang JP, Yao Y, Shen MJ, Zhang LW, Zhang HB, Sun LC (2020) HDAC6 promotes sepsis development by impairing PHB1‐mediated mitochondrial respiratory chain function. Aging (Albany NY) 12: 5411–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Zhou X, Shi Y, Inventors; Xiamen University, assignee (2020) SYBR Green derivative and application thereof in mitochondria DNA fluorescent imaging. US patent CN110672572B. 2020‐01‐10
- He J, Cooper HM, Reyes A, di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A et al (2012) Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res 40: 6109–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley CL, Schroder K (2020) The rOX‐stars of inflammation: links between the inflammasome and mitochondrial meltdown. Clin Transl Immunology 9: e01109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang H, Fan H, Meng T, He Z, Huang H et al (2021) The AMPK‐MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17: 1142–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo V, Bravo‐San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26: 655–667 [DOI] [PubMed] [Google Scholar]
- Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, Thompson‐Snipes LA, Dassopoulos T, Feagins LA, Souza RF, Mills JC et al (2020) Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian C, Xu F, Hou T, Sun T, Li J, Cheng H, Wang X (2017) Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J Cell Sci 130: 2620–2630 [DOI] [PubMed] [Google Scholar]
- Jin L, Batra S, Jeyaseelan S (2017) Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol 198: 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasashima K, Sumitani M, Satoh M, Endo H (2008) Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res 314: 988–996 [DOI] [PubMed] [Google Scholar]
- Keinan N, Tyomkin D, Shoshan‐Barmatz V (2010) Oligomerization of the mitochondrial protein voltage‐dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol 30: 5698–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Gupta R, Blanco LP, Yang S, Shteinfer‐Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M et al (2019) VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus‐like disease. Science 366: 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klecker T, Wemmer M, Haag M, Weig A, Böckler S, Langer T, Nunnari J, Westermann B (2015) Interaction of MDM33 with mitochondrial inner membrane homeostasis pathways in yeast. Sci Rep 5: 18344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KS, Tomasi ML, Iglesias‐Ara A, French BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL et al (2010) Liver‐specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology 52: 2096–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Wang Q, Fung E, Xue K, Tsang BK (2014) p53 is required for cisplatin‐induced processing of the mitochondrial fusion protein L‐Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J Biol Chem 289: 27134–27145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levytskyy RM, Bohovych I, Khalimonchuk O (2017) Metalloproteases of the inner mitochondrial membrane. Biochemistry 56: 4737–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li H, Zhang Y‐L, Xin QL, Guan ZQ, Chen X, Zhang XA, Li XK, Xiao GF, Lozach PY et al (2020) SFTSV infection induces BAK/BAX‐dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep 30: 4370–4385.e7 [DOI] [PubMed] [Google Scholar]
- McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359: eaao6047 [DOI] [PubMed] [Google Scholar]
- Nacarelli T, Azar A, Sell C (2014) Inhibition of mTOR prevents ROS production initiated by ethidium bromide‐induced mitochondrial DNA depletion. Front Endocrinol (Lausanne) 5: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brügger B, Westermann B, Langer T (2009) The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol 184: 583–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, He K (2018) Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol 332: 111–120 [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA (2011) Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50: 98–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren GM, Li J, Zhang XC, Wang Y, Xiao Y, Zhang XY, Liu X, Zhang W, Ma WB, Zhang J et al (2021) Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3‐associated inflammatory diseases. Sci Immunol 6: eabe2933 [DOI] [PubMed] [Google Scholar]
- Riley JS, Tait SW (2020) Mitochondrial DNA in inflammation and immunity. EMBO Rep 21: e49799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF et al (2018) Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 37: e99238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY et al (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The Inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S et al (2015) SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell 60: 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L, Hu C, Xu M, Yu J, He H, Lin J, Sha H, Lu B, Engelender S, Guan M et al (2021) ATAD3B is a mitophagy receptor mediating clearance of oxidative stress‐induced damaged mitochondrial DNA. EMBO J 40: e106283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steglich G, Neupert W, Langer T (1999) Prohibitins regulate membrane protein degradation by the m‐AAA protease in mitochondria. Mol Cell Biol 19: 3435–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19: 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss AL, Vijay‐Kumar M, Obertone TS, Jones DP, Hansen JM, Gewirtz AT, Merlin D, Sitaraman SV (2009) Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology 137: 199–208 208.e1‐6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss AL, Laroui H, Obertone TS, Chowdhury I, Thompson WE, Merlin D, Sitaraman SV (2011) Nanoparticle‐based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm Bowel Dis 17: 1163–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LQ, Liu T, Yang S, Sun L, Zhao ZY, Li LY, She YC, Zheng YY, Ye XY, Bao Q et al (2021) Perfluoroalkyl substance pollutants activate the innate immune system through the AIM2 inflammasome. Nat Commun 12: 2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren EB, Aicher AE, Fessel JP, Konradi C (2017) Mitochondrial DNA depletion by ethidium bromide decreases neuronal mitochondrial creatine kinase: implications for striatal energy metabolism. PLoS One 12: e0190456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17: 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP (1998) Direct demonstration of a specific interaction between cyclophilin‐D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 336: 287–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian H, Watari K, Sanchez‐Lopez E, Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS, Karin M (2022) Oxidized DNA fragments exit mitochondria via mPTP‐ and VDAC‐dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55: 1370–1385.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H et al (2016) PKM2‐dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun 7: 13280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C‐H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, de Nardo D et al (2020) TDP‐43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183: 636–649.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun B, Lee H, Ghosh M, Cravatt BF, Hsu KL, Bonventre JV, Ewing H, Gelb MH, Leslie CC (2014) Serine hydrolase inhibitors block necrotic cell death by preventing calcium overload of the mitochondria and permeability transition pore formation. J Biol Chem 289: 1491–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurugi H, Tanida S, Ishida A, Akita K, Toda M, Inoue M, Nakada H (2012) Expression of prohibitins on the surface of activated T cells. Biochem Biophys Res Commun 420: 275–280 [DOI] [PubMed] [Google Scholar]
- Zhang F, Fan D, Mo XN (2018) Prohibitin and the extracellular matrix are upregulated in murine alveolar epithelial cells with LPSinduced acute injury. Mol Med Rep 17: 7769–7773 [DOI] [PubMed] [Google Scholar]
- Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Li T, Chen L, Li L et al (2019) AMPK‐mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol 21: 476–486 [DOI] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez‐Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR et al (2016) NF‐κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164: 896–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez‐Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]