

Abstract

Biofilm formation is an adaptive resistance mechanism that pathogens employ to survive in the presence of antimicrobials. Pseudomonas aeruginosa is an infectious Gram-negative bacterium whose biofilm allows it to withstand antimicrobial attack and threaten human health. Chronic wound healing is often impeded by P. aeruginosa infections and the associated biofilms. Previous findings demonstrate that 600 Da branched polyethylenimine (BPEI) can restore β-lactam potency against P. aeruginosa and disrupt its biofilms. Toxicity concerns of 600 Da BPEI are mitigated by covalent linkage with low-molecular-weight polyethylene glycol (PEG), and, in this study, PEGylated BPEI (PEG350-BPEI) was found exhibit superior antibiofilm activity against P. aeruginosa. The antibiofilm activity of both 600 Da BPEI and its PEG derivative was characterized with fluorescence studies and microscopy imaging. We also describe a variation of the colony biofilm model that was employed to evaluate the biofilm disruption activity of BPEI and PEG-BPEI.
Introduction
Chronic wound infections present a significant burden to patients, clinicians, and the entire health care system. In the United States, total Medicare spending on chronic wound care is estimated to be around $32 billion annually,1 in part because biofilms stifle and complicate healing processes. Pseudomonas aeruginosa (P. aeruginosa) is a dangerous pathogen commonly found in wound infections. Wound infections aggravate the spread of pathogens and antimicrobial resistance genes because patients reside in and are transported among, out-patient clinical facilities, in-patient hospitals, long-term care facilities, long-term acute care facilities, and skilled nursing home facilities.2−4 Ineffective treatment of infected wounds can lead to tissue necrosis, bacteremia, and septicemia, allowing the spread of P. aeruginosa infections to major organs and the central nervous system.5
The danger to human health increases with multidrug-resistant (MDR) P. aeruginosa infections, which are listed as a serious threat by the Centers for Disease Control and Prevention 2019 Annual Report. The threat is heightened by the ability of P. aeruginosa to aggregate and form biofilms. Microbial biofilms are communities of microorganisms enclosed in a protective extracellular polymeric substance (EPS) matrix.6 Biofilm formation is an adaptive survival mechanism that pathogens activate as a physical protection from external stresses (i.e., antibiotics and antimicrobials, nutrient deprivation, sheer forces). Bacteria within biofilms have stronger resilience compared to free-floating planktonic bacteria and thus are harder to eradicate.7 Biofilm formation by MDR strains of P. aeruginosa continue to pose a threat to human health, and hence new antimicrobial and antibiofilm agents are required.8
We have previously identified low molecular weight 600 Da branched polyethylenimine (600 Da BPEI) as a broad-spectrum antibiotic potentiator that can overcome antibiotic resistance and restore the susceptibility of MDR strains of Gram-positive and Gram-negative bacteria to existing antibiotics.9−13 We have also reported its antibiofilm properties and suggested that the cationic nature of BPEI enables electrostatic interactions with anionic targets in biofilms. However, the primary amines of 600 Da BPEI create cytotoxicity concerns that cannot be overlooked. As a result, we previously reported a modification by covalent attachment of a low molecular weight (350 MW) polyethylene glycol (PEG) group to 600 Da BPEI (PEG350-BPEI) led to a reduction in acute toxicity in a mouse model.14 We have also reported its antibiofilm properties and suggested that the cationic nature of BPEI enables electrostatic interactions with anionic targets in biofilms. However, the primary amines of 600 Da BPEI create cytotoxicity concerns that cannot be overlooked. In the same report, we used nuclear magnetic resonance (NMR) spectroscopy to follow the PEGylation of 600 Da BPEI and verified that the reaction eached completion.14 Additionally, PEGylation of BPEI improved the dispersal of methicillin-resistant Staphylococcus epidermidis (MRSE) biofilms while retaining β-lactam potentiation activity.14
In this work, we continue to investigate 600 Da BPEI and its less toxic derivative, PEG350-BPEI, in the eradication of P. aeruginosa biofilms, including biofilms of a MDR clinical isolate using more complex infection models. The colony biofilm model was used to produce static biofilms, and this model was a particularly useful method to observe the effect(s) of antimicrobials on static biofilms that are known to form in wound infections.15 This model produces more robust biofilms compared to a minimum biofilm eradication concentration (MBEC) assay.16 We also applied an ex vivo porcine skin model where we evaluated the effectiveness of our compounds in an infected tissue. Treatments with 600 Da BPEI and PEG350-BPEI resulted in an overall reduction of bacterial colony forming units (CFU). We also demonstrate a reduction in biofilm biomass through a modified crystal violet assay and imaging. Thermodynamic interactions between 600 Da BPEI with DNA, a major stabilizing component of the biofilm architecture, were measured. Additional insights regarding the action of biofilm disruption are provided through analysis of electron microscopy images.
Experimental Procedures
Materials
P. aeruginosa bacterial stocks were purchased from American Type Culture Collection (ATCC BAA-47, a PAO1 strain, and ATCC 27853), and the multidrug-resistant strain (OU1) was obtained from the University of Oklahoma Health Sciences Center using appropriate Institutional Review Boards (IRB) protocols and procedures. Chemicals and antibiotic were purchased from Sigma-Aldrich (erythromycin, growth media, and microscopy fixatives). 600 Da BPEI was purchased from Polysciences, Inc., and PEG350-BPEI is synthesized and characterized as previously described.14
Biofilm Biomass Disruption Assay
This assay is as adapted from a protocol previously described by Lam et al.11 with slight modification. A subculture of P. aeruginosa cells was grown from a cryogenic stock overnight (final OD600 = 1.0) at 37 °C. A presterilized 96-well plate was inoculated with 100 μL of M63 media/well (22 mM KH2PO4, 40 mM K2HPO4, 15 mM (NH4)2SO4, 1 mM MgSO4, 0.4% arginine)15 and 1 μL of 1 colony/mL in M63 media. The plate was incubated for 24 h at 37 °C to form established biofilms. After incubation, the planktonic bacteria were carefully removed by turning the plate over and removing excess liquid with gentle shaking. The plate was then washed by gently submerging in a small tub of water with shaking. To stain the biomass, 125 μL of 0.1% crystal violet was added to each well and incubated at room temperature for 10–15 min. After incubation, the crystal violet was removed, and the excess dye was washed 3–4 times. After washing, the plate was allowed to dry for 1 h.
To treat the biofilms, different treatment concentrations were added to the biofilm plate for a total volume of 100 μL (in Tris-HCl 0.01 M pH = 7). The negative control was treated with Tris buffer alone. The positive control was treated with 100 μL of 30% acetic acid for 2 min. After 3 h of incubation at 37 °C, the dissolved solutions were carefully transferred into a separate flat-bottom 96-well plate for OD550 measurements. Statistical analysis among treated samples was preformed using t test, p < 0.01.
Biofilm Killing Assay
Biofilm Growth
A subculture of P. aeruginosa cells was grown from a cryogenic stock overnight at 37 °C (OD600 = 1.0). To grow biofilms, sterilized 13 mm polycarbonate membranes (Isopore, Merck Millipore Ltd.) were placed on (tryptic soy agar) TSA plates and inoculated with 1 μL of cell culture from 1 colony/mL in tryptic soy broth (TSB). One colony/mL of P. aeruginosa is approximately 5 × 105 CFU/mL,9 thus a 1 μL inoculation is about 5 × 102 CFU. The plates were incubated at 37 °C for 24 h. After incubation, the biofilms were subjected to immersion treatment or cellulose filtration disc treatment.
Immersion Treatment
The membranes with mature biofilms were transferred into solutions of 500 μL of Tris buffer with or without antibiofilm agents. The samples were incubated in the treatment solutions overnight at 37 °C. After incubation, the membranes, along with the treatment culture, were transferred into vials and were subjected to sonication for 10 min. Cell viability was identified via agar plating with enumeration of colony formation units or by measuring solution turbidity via optical density with visible light spectroscopy at 600 nm (OD600).
Cellulose Filtration Disc Treatment
Membranes with mature biofilms were transferred to a fresh TSA plate, and sterilized 7 mm cellulose filtration discs were carefully placed on top of the biofilm. A micropipette was used to deliver 40 μL of treatment (600 Da BPEI or PEG350-BPEI in Tris buffer) or control (Tris buffer only). The biofilms were incubated at 37 °C for 8 h. After treatment, the membranes and cellulose filtration discs were transferred into vials with 10 mL of Tris buffer and subjected to sonication for 15 min to detach biofilms. After vortex mixing to create homogeneous solutions, the amount of viable bacterial cells was determined via enumeration of colonies on agar plates using a spot plate technique.17 Spot plating is an alternative to the more common method of spreading bacterial suspensions onto agar. Briefly, homogeneous cell suspensions in 10 mL Tris buffer were serially diluted up to nine times. Then, 10 μL aliquots of each dilution were spotted onto TSA plates and incubated overnight at 37 °C. Mean viable cell counts were converted to log10 units, and standard deviations were calculated (n = 5 per treatment condition).
Porcine Explant Model
The ex vivo model on porcine explants used in this study consisted of 8 mm biopsied explants (3 mm thick) taken from the dorsal area without fat and subcutaneous tissue and shaved to remove hair (Pel-Freez, LLC). The explants were exposed to povodone iodine prep solution (10% povodone iodine) for approximately 30 s, placed on soft TSA agar, inoculated with 1 μL P. aeruginosa BAA-47 (PAO1) cell culture (from 1 colony/mL in TSB), and incubated for 24 h at 37 °C. The povodone iodine prep solution step was added to mimic skin preparation prior to surgeries. These “uninfected explants” were sampled and identified to have ∼108Staphylococcal cells using Mannitol red agar. After incubation, the explants were transferred into a 24-well plate containing treatment solutions (1000 μL) and controls (buffer only). The treatment plate was incubated for another 24 h at 37 °C. After treatment, each explant was aseptically placed into a 15 mL sterile tube with 10 mL Tris buffer. The explants were homogenized using a tissue homogenizer, sonicated for 15 min, and cell viabilities were enumerated via spot plating as previously described. Mean viable cell counts were converted to log10 units, and standard deviations were calculated (n = 5 per treatment condition).
Scanning Electron Microscopy
P. aeruginosa ATCC BAA-47 (PAO1) biofilms grown on sterilized polycarbonate membranes for 24 h were submerged in primary fixative (5% glutaraldehyde in 0.1 M cacodylate buffer) and incubated at 4 °C for 2 days. The membranes were removed from fixing solution and air-dried for at least 72 h at 25 °C. After drying, the membranes were mounted on aluminum stubs with carbon tape and sputter-coated with AuPd. Scanning electron microscopy images were taken using a Zeiss NEON at 5 kV accelerating voltage. For the treated samples, 24 h biofilms were treated with hydrated 7 mm cellulose filtration discs. The biofilms were treated with 40 μL of Tris buffer or 1024 μg/mL of 600 Da BPEI at time = 0, followed by a second dose/rehydration at time = 4 h. After a total of 8 h incubation at 37 °C, the 7 mm cellulose filtration discs were carefully removed, and the biomass remaining on the polycarbonate membrane was fixed and imaged as described above.
Isothermal Titration Calorimetry
To identify any binding interactions between 600 Da BPEI and DNA, isothermal titration calorimetry (ITC; MicroCal PEAQ-ITC, Malvern Inc., Malvern, U.K.) was used. Briefly, 600 Da BPEI (3.2 mg/mL in Tris-HCl buffer) was titrated into a DNA solution (DNA Sodium Salt, Fish Sperm Ampresco; 1.0 mg/mL in Tris-HCl buffer) in 2 μL lasting 4 s and separated by 150 s time intervals at 25 °C. Controls were performed, and the experiment was done in duplicate.
Calgary Device Fluorescence Experiment
A subculture of the P. aeruginosa BAA47 (PAO1) was grown from a cryogenic stock overnight at 37 °C as described above. To produce biofilms, the Calgary device18 plate was inoculated with 100 μL M63 media plus 1 μL of a stock culture made from 1 colony/mL of P. aeruginosa BAA47 in M63 media. The plates were incubated for overnight at 37 °C. After incubation, the Calgary device lid containing the prongs was transferred into a challenge plate containing increasing concentrations of 600-Da BPEI plus controls. The resulting challenge plate with lid was left to incubate overnight at 37 °C. The next day, the Calgary device lid was transferred to a fresh 96-well plate with fixative agent (5% glutaraldehyde) and left overnight at 4 °C. The next day, the prong was detached from the lid, and set on a cover glass for imaging. CLSM experiments were performed using Nikon AX confocal laser scanning microscope (λEX = 405 nm and λEM = 460 nm). Image analysis was performed using Nikon Image Software (NIS-Elements Viewer).
Results and Discussion
The pseudomonal biofilm matrix is composed of bacterial cells, extracellular polysaccharides, rhamnolipids, biofilm matrix-related proteins, nucleic acids, secondary metabolites, and water.19 These components can assemble into a matrix that protects the bacterial population within the biofilm. Some interactions between the biofilm components also play a significant role in maintaining the structural integrity of the biofilm matrix. For example, extracellular DNA (eDNA) can interact with proteins and exopolysaccharides via charge–charge interactions.7 Hence, cationic compounds, like 600 Da BPEI and its PEGylated derivatives, can bind to the biofilm matrix and destabilize inter- and intramolecular interactions within the matrix. This creates an environment that increases bacterial susceptibility to external agents, such as antimicrobial compounds. We have previously shown the ability of 600 Da BPEI to disrupt bacterial EPS of Gram-positive staphylococcal biofilms and Gram-negative P. aeruginosa using the MBEC model.11,14,16,20
The colony biofilm model is a useful method to test antimicrobial solutions. Figure 1 shows an overview of the growth and treatment conditions employed in our study. Biofilms grown via the biofilm colony model for 24 h show a visible biomass deposited on the surface of the polycarbonate membrane. To confirm the presence of bacteria and EPS after 24 h incubation, scanning electron microscopy (SEM) was utilized to visualize the bacteria (Figure 2). The biofilm matrix of P. aeruginosa is composed of three major exopolysaccharides: alginate, Psl and Pel. Alginate is a polymer of (1,4)-linked β-d-mannuronate and α-l-guluronate, Psl is repeating pentamer consisting of d-mannose, l-rhamnose, and d-glucose residues, and Pel is a N-acetyl glucosamine (GlcNAc)- and N-acetyl galactosamine (GalNAc)-rich polysaccharide.21,22 These exopolysaccharides are not only crucial in maintaining the biofilm matrix structure but also play a role in increasing antimicrobial resistance of P. aeruginosa cells within the biofilms.23,24 These exopolysaccharides, along with other biofilm components (i.e., proteins and eDNA), create a three-dimensional matrix.
Figure 1.
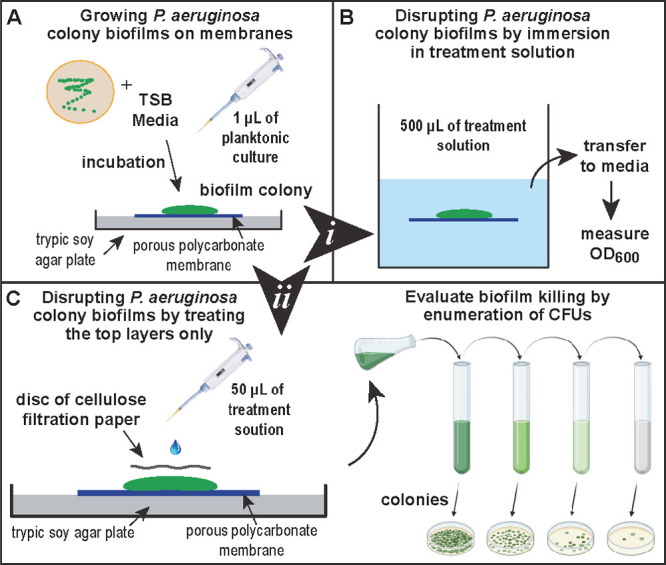
Illustration of the modified colony biofilm model. (A) Side profile of the biofilm colony grown on top of a porous membrane resting on TSA agar. (B) Treatment option (i) in a bulk solution of treatment agent. (C) Treatment option (ii) used cellulose filtration discs to deliver treatment to the biofilm surface and prevent the treatment solution from evaporating, followed by CFU counting.
Figure 2.

Scanning electron micrographs of ATCC BAA-47 (PAO1) P. aeruginosa grown via colony biofilm model. Thick layers of P. aeruginosa cells are stacked on top of each other after 24 h (A). Closer inspection reveals a thick EPS surrounding the bacterial cells (B), encasing the bacterial cells. The SEM image in (A) shows a thick layer of rod-shaped P. aeruginosa cells embedded within the biomass. The biofilm cross-section shows layers of cells and EPS the comprise the biofilm (B). These SEM images confirm that P. aeruginosa cells are coated with EPS before treatments were added.
Treatment solutions were applied via immersion (method i in Figure 1) and cellulose filtration discs (method ii in Figure 1). Starting from a 5 × 102 CFU inoculation, 24 h of growth increases the biomass to 5 × 109 CFU/mL for ATCC BAA-47 (PAO1), 10 × 106 CFU/mL for ATCC 27853, and 2 × 107 CFU/mL for the MDR strain OU1. P. aeruginosa BAA-47 is a PAO1 strain isolated from a wound in 1954,25,26 and ATCC 27853 was isolated from a hospital blood specimen in 1971.27 The MDR clinical isolate OU1 exhibits resistance against aztreonam, ceftazidime, ciprofloxacin, meropenem, and piperacillin/tazobactam.9 We have previously identified the ability of 600 Da BPEI to resensitize β-lactams against MDR P. aeruginosa using checkerboard and MBEC assays.9
After growing the P. aeruginosa biofilms using the modified colony biofilm model (Figure 1A), we subjected the biofilms to biofilm killing assays. The first assay used a biofilm killing treatment model where the whole membrane is immersed into a treatment solution. A volume of 500 μL Tris buffer was prepared with varying concentrations of 600 Da BPEI. Then, the established biofilms were exposed to treatments overnight. Using this treatment model, complete bacterial eradication (CFU/mL = 0) was observed when 512 μg/mL 600 Da BPEI was applied to BAA-47 (PAO1) biofilms (∼5 × 109 CFU/mL). This treatment delivery provides about 420 nmol of 600 Da BPEI. However, with this treatment model, we observed a passive dispersal (i.e., erosion) of the biofilm when the membrane is immersed into a liquid solution. This could be caused by environmentally induced dispersion because of the shift from solid agar interface to liquid treatments.28 Dispersion of the biofilm may turn sessile biofilm bacteria into planktonic cells, which are more susceptible to antimicrobials, including 600 Da BPEI and PEG350-BPEI.9,12,14,16 Although we observed efficacy using the immersion treatment model, it is also possible that the 600 Da BPEI molecules accessed the biofilm via the pores in the polycarbonate membrane. Hence, 600 Da BPEI could have bypassed the biofilm outer layers and attacked the biofilm at a weaker location. Thus, a follow-up study was designed to treat the top layers of the biofilm only. A sterile cellulose filtration paper disc was applied to the biofilm surface, and then treatments were applied to the paper disc (Figure 1C). This ensures the treatment solutions stay on top of the biofilm, reduces evaporation, and resembles clinical wound treatments.29 The application of the 600 Da BPEI and PEG350-BPEI kills P. aeruginosa cells within the biofilm. This activity is quantified by measuring the viable cell concentration in colony forming units (CFU/mL) after treatment with Tris buffer only, 600 Da BPEI, or PEG350-BPEI. The difference in CFU/mL between Tris and 600 Da BPEI (or PEG350-BPEI) treated samples is used to calculate log reduction values (LRVs). Dose-dependent LRVs are shown in Figure 3.
Figure 3.
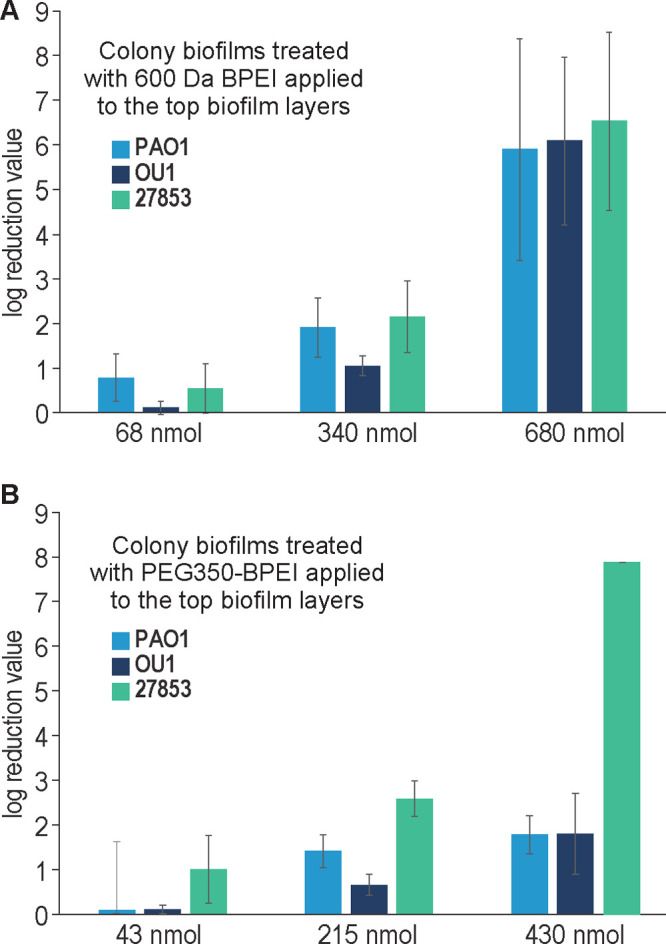
LRVs of 600 Da BPEI (A) and PEG350-BPEI (B). Error bars denote standard deviation (n = 5). Increasing nmol amounts of 600 Da BPEI delivered to the biofilms increases LRVs across different P. aeruginosa strains. LRVs of the highest concentration 600 Da BPEI are better than of PEG350-BPEI except for one strain: ATCC 27853.
To deliver effective quantities of 600 Da BPEI or PEG350-BPEI while also keeping the liquid on the biofilm surface, low volumes of high concentration solutions were used. The 40 μL volume of a 1024 μg/mL solution of 600 Da BPEI delivers 68 nmol to the biofilm surface. This results in a LRV <1 (Figure 3A). However, 40 μL of a 1024 μg/mL solution of PEG350-BPEI delivers 43 nmol and produces a LRV close to 1 for P. aeruginosa 27853 (Figure 3B). For P. aeruginosa BAA-47 (PAO1) and MDR-PA OU1, LRVs show that PEG350-BPEI is less effective than 600 Da BPEI. However, higher amounts of 600 Da BPEI or PEG350-BPEI produce larger LRVs for all strains (Figure 3). Both 600 Da BPEI and PEG350-BPEI were effective at killing P. aeruginosa biofilms, including biofilms of MDR-PA OU1. It appears that PEGylation may reduce the eradication activity of 600 Da BPEI, but this effect is strain dependent. For P. aeruginosa ATCC 27853, the LRVs for PEG350-BPEI are higher than 600 Da BPEI even though less of the compound was used. In a comparative genomic study between P. aeruginosa ATCC 27853 and PAO1, results suggest that ATCC 27853 expresses Psl genes at a much higher rate compared to PAO1 in the development of colony biofilms.27 The Psl locus contains the PslA-O gene that encodes for the mannose and galactose components of the matrix. In the same comparative study, it was reported that both PAO1 and ATCC 27853 expressed low pel and alginate biosynthesis genes.27 High levels of the polysaccharides mannose and galactose in ATCC 27853 biofilms, and low levels of the anionic pel and alginate components, may leave the bacterial cells more susceptible to the action of 600 Da BPEI and PEG350-BPEI compared to the BAA-47 (PAO1) and MDR strains. High amounts ion anionic EPS components would attract and bind greater fractions of the 600 Da BPEI or PEG350-BPEI dose. This action would leave a smaller fraction of BPEI remaining to kill the bacteria via attack on its outer membrane. Finally, the data in Figure 3 demonstrate that PEG350-BPEI can be used to kill P. aeruginosa biofilms. These data do not demonstrate the amount required for in vivo studies, where it is not our intention to eradicate P. aeruginosa biofilms with a single high-concentration dose. Instead, we envision that low-concentration doses of PEG350-BPEI will be applied to wounds as a topical agent administered as a monotherapy or in combination with IV/oral antibiotics. Topical use lowers toxicity concerns, whereas the risk is greater if PEG-BPEI was intended for internal use. PEG-BPEI may be a useful therapeutic against wound infections by simultaneously providing anti-inflammation and infection prevention, in addition to antibiofilm properties that assist with localized wound decolonization.20 This will reduce inflammation and tissue damage and accordingly will shorten the inflammatory phase of wound healing, lowering the chance that wound infections lead to bacteremia, sepsis, and death. PEG-BPEI will complement other wound treatment interventions, such as treatment with antibiotics and wound debridement.
While the colony biofilm model provides a good high-throughput model for testing antimicrobials against biofilms, a limitation of this model is that the biofilm is still grown on an artificial substrate. Hence, we used a porcine explant model to address such an issue to evaluate the efficacy of 600 Da BPEI and PEG350 BPEI against infections grown on tissues. This method offers additional challenges to treatment because the skin is a rich substrate that supports bacterial growth and poses a barrier to the penetration of antimicrobials.30
The ex vivo porcine skin infection model consisted of 8 mm discs created with a biopsy punch. The explants were infected with ∼102 CFU of a P. aeruginosa inoculum. After 24 h incubation, the P. aeruginosa bacterial count increased to ∼108, as determined by counting colonies on a Pseudomonas-selective centrimide agar (referred to as t = 0 in Figure 4). Figure 4 shows that treatment of the explants with a single dose of 1024 μg/mL 600-Da BPEI and PEG350-BPEI; both resulted in an overall total bacterial decrease in CFU recovered. These concentrations are equivalent to 1706 nmol and 1077 nmol, respectively, almost 4× the amount of moles needed to eradicate the biofilms formed using the colony biofilm method (Figure 1B). This shows that the infection model on ex vivo skin hinders the ability of antimicrobials to eliminate infections. Developing and testing potentiator compounds in an ex vivo model can provide a more realistic environment because the bacteria can penetrate skin spore and hair follicles to form robust biofilms. Adding an antibiotic to this treatment increased the eradication of the bacterial biofilm, as discussed below.
Figure 4.

Log CFU per mL of PAO1 suspension that survived after a single dose of each treatment in the ex vivo porcine skin model. Data set shows that before treatments, at time point zero (t = 0), there were ∼108 cells in the porcine skin. Buffer (Tris)-treated samples continued to grow more cells (∼109), and treatments with 600 Da BPEI and PEG350-BPEI caused a CFU reduction as observed in the colony biofilm model (n = 5; errors denote standard deviation. Data analysis indicates a significant difference between each of treatments, p-value <0.01).
We previously reported erythromycin to interact synergistically with 600 Da BPEI against both planktonic Gram-positive and Gram-negative pathogens.20 Erythromycin is a bacteriostatic antibiotic that falls under the macrolide class.31 In our previous report, we attributed the synergy between 600 Da BPEI and erythromycin to increased drug influx. Typically, macrolides such as erythromycin diffuse across the outer membrane to get to the bacterial cell interior. However, the lipopolysaccharides (LPSs) of the outer membrane hinder macrolides from passing through easily.32 We suggest that 600 Da BPEI can reduce the drug diffusion barriers by interacting with LPS, thus allowing the antibiotic into the cell and perform its mechanism of action.9Figure 5 shows that immersing the porcine punches in 1024 μg/mL of erythromycin only causes a 1.68 log CFU/mL reduction compared to buffer (Tris)-treated samples.
Figure 5.
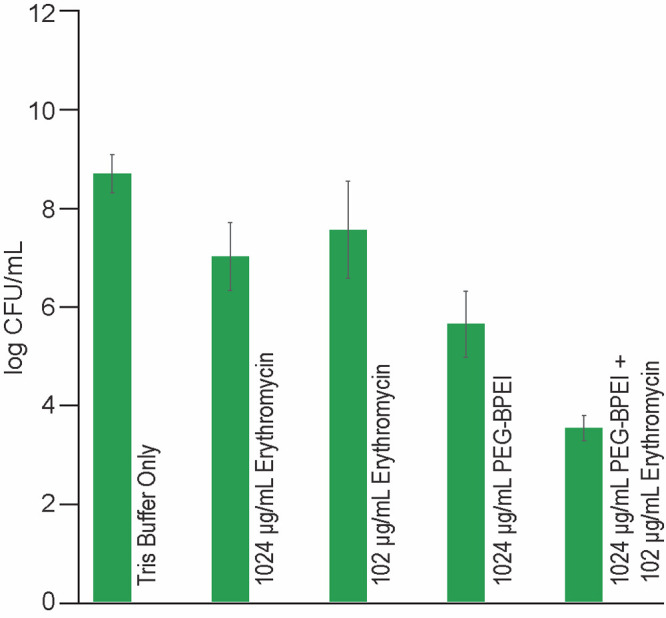
Log CFU per mL of PAO1 suspension that survived after a single dose of each type of treatment in the ex vivo porcine skin model. Data set shows that even up to 1024 μg/mL of erythromycin only causes modest CFU/mL reduction (1.68 log CFU/mL) compared to buffer (Tris)-treated punches. Treatments with 102 μg/mL erythromycin alone or 1024 μg/mL PEG350-BPEI alone both caused 1.13 and 3.05 log CFU/mL, respectively. However, a combination of 1024 μg/mL PEG350-BPEI and 102 μg/mL erythromycin produced a 5.17 log CFU/mL reduction (n = 5; errors denote standard deviation. Data analysis indicates a significant difference between each of treatments, p-value <0.01)
Single-dose treatments of 1024 μg/mL PEG350-BPEI alone and 102 μg/mL erythromycin alone reduced the number of viable bacteria (1.13 and 3.05 LRV, respectively). However, combining 1024 μg/mL PEG350-BPEI and 102 μg/mL erythromycin produced a 5.17 LRV. The increased killing ability of the combination treatment demonstrates the benefit of the combination treatment because adding the LRV for each compound alone (1.13 + 3.05 = 4.18 LRV) was less effective than the combination (5.17 LRV). Immersing the porcine samples in treatment solution can be envisioned as an ex vivo model of wound cleaning where the standard of care procedures involve irrigating wounds with saline or wound cleansing agents. The goal of wound irrigation is to cleanse the wound (i.e., remove bacterial contamination, exudates).33 Understandably, this ex vivo treatment model does not account for inflammatory and other host-mediated responses. These limitations of this model will be addressed in future in vivo animal models.
After identifying the ability of 600 Da BPEI and PEG350-BPEI to reduce bacterial biofilm bioburden, we sought to clarify the mode through which these compounds attack biofilms. In our previous study on MRSE and methicillin-resistant Staphylococcus aureus biofilms, we implemented a biofilm disruption assay via crystal violet staining.12,16 Here, we also applied a similar biofilm disruption assay. Growing P. aeruginosa biofilms on microtiter plates presents a technical challenge because growth in TSB media results in poor biofilm adhesion. The biofilms were observed to slide off of the microtiter plates during washing. However, for P. aeruginosa, it is known that an inverse relationship exists between biofilm formation (attachment) and swarming motility (movement),34,35 and by modulating the amino acid concentrations in the growth media, biofilm formation and attachment can be enhanced. Hence, we inoculated a minimal salts media supplemented with 0.4% arginine as described by Bernier et al.34 With this new procedure, biofilm formation is more uniform and robust for ATCC BAA-47 (PAO1) and ATCC 27853 in microtiter plates. This media, however, did not produce a quantifiable biofilm biomass for the MDR clinical isolate OU1 (data not shown). Biofilms of laboratory reference strain ATCC BAA-47 (PAO1) P. aeruginosa were stained with crystal violet and treated with increasing concentrations of 600 Da BPEI and PEG350-BPEI (2–64 μg/mL). The positive control was 30% acetic acid, and the negative control was Tris buffer. The stained biomass samples were incubated with the treatment solutions for 3 h, and then the dissolved stained biomass was carefully transferred into new 96-well plates for OD550 measurement. The amount of dissolved biomass was directly correlated to the intensity of OD550 values. Measured values were compared to the negative control using Student’s t test. Statistical analysis shows that both 600 Da BPEI and PEG350-BPEI dissolved the biomass in 3 h (n = 12, p-value <0.01), and minimal biomass remained in the original biofilm plate (Figure 6A). Increasing the concentration of 600 Da BPEI increases the amount of biofilm biomass dissolved (Figure 6B). A similar trend is observed with PEG350-BPEI (Figure 6C).
Figure 6.
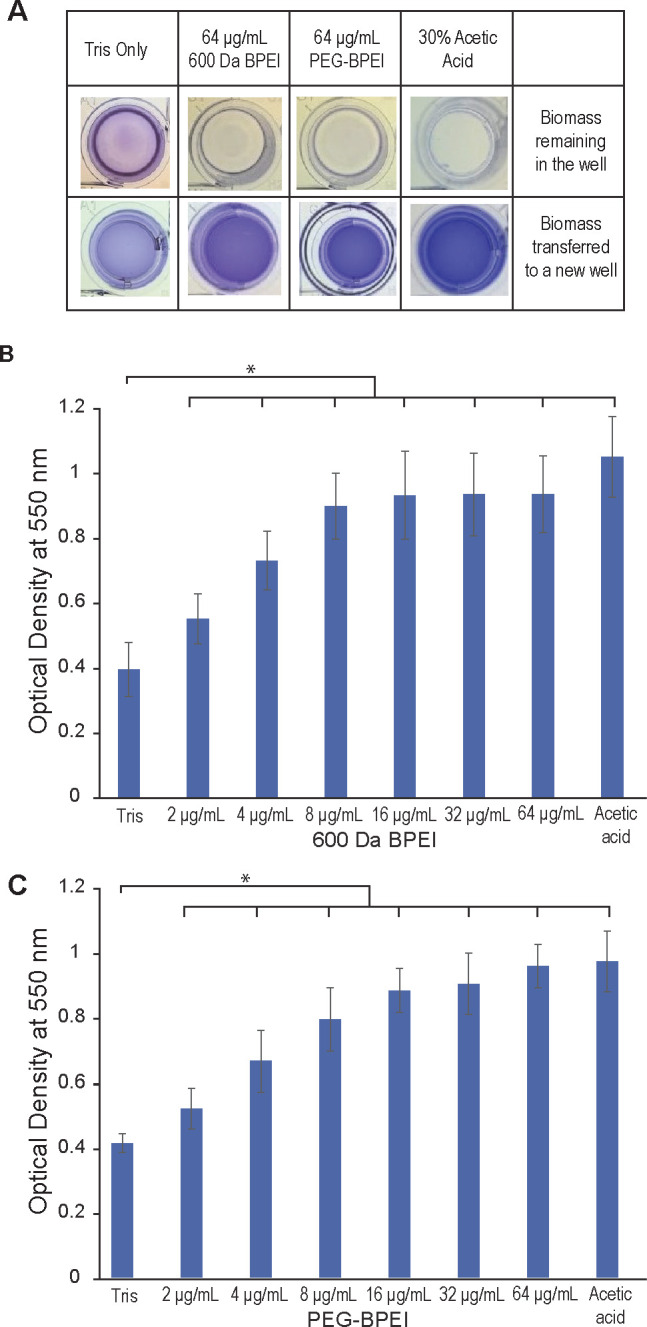
Evaluation of biofilm biomass disruption via crystal violet staining. Biofilms of P. aeruginosa ATCC BAA-47 (PAO1) stained with crystal violet were treated with increasing concentrations of 600 Da BPEI (B) and PEG350-BPEI (C) for 3 h, in addition to negative and positive controls. The dissolved biofilms were transferred into a separate microtiter plate leaving remnants of undissolved biomass in the original microtiter plate (A). The mean OD550 nm of dissolved biofilms was measured and recorded. Error bars denote standard deviation (n = 12). OD550 nm readings show a significant difference between buffer-treated (Tris) control and treatments indicated with an asterisk (t test, p-value <0.01).
Crystal violet assay data comparing the antibiofilm action of 600 Da BPEI and PEG350-BPEI against P. aeruginosa BAA-47 (PAO1) and 27853 are shown in Figure 7. These results show that the amounts of crystal violet dissolved and quantified for the two treatments are indistinguishable (n = 10, p-value >0.01) for these two bacterial strains. Hence, it seems that the apparent ability of 600 Da BPEI to dissolve biomass is unaffected by PEGylation, and thus the differences observed in the LRVs (Figure 3) may be due to differences in bacterial susceptibility to 600 Da BPEI and PEG350-BPEI. Growth inhibition assays of these two compounds, using MDR-PA OU1 and ATCC 27853 strains, show that the minimum inhibitory concentration (MIC) of 600 Da BPEI is 67 μM for both strains. However, the MIC values of PEG350-BPEI are 268 μM and 67 μM against these two strains, respectively.14 This indicates a higher bacterial susceptibility of ATCC 27853 to PEG350-BPEI compared to MDR OU1.
Figure 7.

Biofilm disruption assay via crystal violet staining for P. aeruginosa BAA-47 (PAO1) and 27853 treated with 600 Da BPEI and PEG350-BPEI. The P. aeruginosa biofilms stained with crystal violet were treated with 600 Da BPEI and PEG350-BPEI for 3 h, in addition to negative and positive controls. The mean OD550 nm of dissolved biofilms was measured and recorded. Error bars denote standard deviation (n = 10). OD550 nm values indicate that PEGylated BPEI can disrupt P. aeruginosa with an activity similar to that of 600 Da BPEI (t test, p-value >0.01, denoted by not significant, n.s.). OD550 nm readings show significant difference between buffer-treated (Tris) control and treatments indicated with an asterisk (t test, p-value <0.01).
A limitation to the crystal violet assay is that the biofilms were stained and dried prior to treatment with the cationic compounds (600 Da BPEI or PEG350-BPEI). Crystal violet binds noncovalently to the biofilm biomass. Hence, false positive conclusions may result if the cationic compounds only replace the bound crystal violet in the biofilm (while not disrupting the matrix). To alleviate these concerns, microscopic analyses were performed. First, fluorescence images were obtained after growing biofilms on a Calgary device18 without drying. It is possible to collect images using the autofluorescence of P. aeruginosa from the fluorescent pigment, pyoverdine (405 nm excitation, 460 nm emission). Pyoverdine is a siderophore responsible for scavenging extracellular iron.36 It is suggested that pyoverdine is regulated by biofilm formation in P. aeruginosa.36 The fluorescence in Figure 8A shows layers P. aeruginosa cells within a thin layer of EPS coating. However, after treatment with 600-Da BPEI, a reduction of both cells and EPS coating is observed (Figure 8B). These data demonstrate that BPEI does indeed disperse the biofilm EPS and embedded bacterial cells. Thus, the release of crystal violet into solution is due to dissolved biomass rather than solubilization of crystal violet while leaving the biomass intact.
Figure 8.

Fluorescence imaging of pyoverdine in P. aeruginosa. Buffer-treated control shows a layer of cells covered in a thin layer of EPS, resulting in a hazy film (A). A reduction of cells and EPS coating is observed when treated with 600-Da BPEI (B).
To further observe the action of 600 Da BPEI against P. aeruginosa biofilms, SEM images were taken to discern the topographical changes within the biofilm environment. Biofilms were grown on polycarbonate membranes and treated with 130 nmol of 600 Da BPEI, or Tris buffer only, using the modified colony biofilm model (Figure 1C). In this method, sterile cellulose filtration paper discs are used to keep the BPEI (or Tris) solution on the biofilm surface. The cellulose paper was carefully removed prior to SEM imaging. A comparison between a buffer-treated control and BPEI-treated biofilm shows that buffer-treated biofilms have a layer of P. aeruginosa cells covered in a layer of EPS (Figure 9 A,C). The noticeable fracture in the biofilm is likely the result of sample drying under the vacuum conditions required for SEM imaging. The SEM images of BPEI-treated biofilms (Figure 9B,D) show a considerable reduction of EPS coating surrounding the bacterial cells. It appears that 600 Da BPEI causes a dissolution of the EPS. This observation is consistent with the results in the biofilm disruption (crystal violet) assay and fluorescence imaging (Figures 6–8). This observation may also be caused by delamination of the biofilm architecture when the cellulose filter paper is removed. Buffer-treated biofilms were handled in a similar manner. Thus, changes in the biofilm EPS are caused by 600 Da BPEI. It is likely that the 600 Da BPEI treatment creates several effects, such as disruption and dissolution of the biofilm EPS, a weaking of the biofilm architecture, and delamination of the biofilm when the cellulose paper is removed. Regardless, the net effect is a reduction in bacterial bioburden on the polycarbonate membrane, and it is conceivable that a similar action would occur in the treatment of wound infections when wound coverings are removed and changed during treatment. Reducing the EPS layer in the biofilms may leave the cells more susceptible to antimicrobial agents and, in this study, allows 600 BPEI greater access to the underlying cells. We previously discussed the mechanism of action of BPEI against P. aeruginosa planktonic cells.9 Those data were interpreted as 600 Da BPEI reducing the diffusion barriers caused by LPS biomolecules in the outer membrane and, at higher concentrations, 600 Da BPEI causing disruption of the outer membrane itself.9 Hence, the dual-effect of dissolving EPS barriers and antimicrobial action at high concentration is a possible explanation for the dose-dependent LRVs shown in Figure 3.
Figure 9.

Scanning electron micrographs of ATCC BAA-47 (PAO1) P. aeruginosa biofilms after treatment via colony biofilm model. Buffer-treated (Tris) biofilms (A, C) are observed to have a film-like layer, whereas the 600 Da BPEI-treated biofilms lack this appearance (B, D). The shape and size of bacteria in 600 Da BPEI-treated biofilms are more resolved compared to the buffer-treated ones. Boxed regions in (A, )B are shown with higher magnification in (C, D).
To dissolve the biofilm matrix, 600 Da BPEI needs to interact with biofilm components, which can destabilize the EPS matrix. One important biofilm component is extracellular DNA (eDNA). Biofilm nucleic acids are important in biofilm formation, cell-to-cell adhesion, cell signaling and maintaining the structural integrity of the biofilm matrix.7 This importance of eDNA allows it to be a possible drug target candidate for antibiofilm treatments. Furthermore, polyethylenimines (PEIs) are widely used transfection agents, hence their interaction with DNA is well-recognized.37,38 To confirm this interaction, ITC data were collected. The thermodynamic profile for titration of 600 Da BPEI into DNA solutions is shown in Figure 10. The binding isotherm illustrates a biphasic response to the increasing concentration of 600 Da BPEI. This response may indicate two binding sites for 600 Da BPEI in the DNA molecules. In a thermodynamic analysis between 600 Da BPEI and DNA, these two types of binding modes were described as (a) binding of the PEI to the DNA groove and (b) external binding of PEI to the phosphate backbone of DNA.38 These possible interactions of the PEI molecules with the eDNA in the matrix can further disorganize its structural integrity, rendering the bacterial cells within the biofilm more susceptible to treatments. In addition, the PEI-DNA interaction may dislocate some of the pre-existing eDNA interactions (i.e., with proteins, exopolysaccharides, metabolites) and inhibit some of its signaling and modulating functions. For example, as the phenazine pyocyanin interacts with eDNA, enhanced electron shuttling within the biofilm is observed as well as increased sputum viscosity.39,40 Inhibiting these interactions may leave the biofilm structure less fit and more vulnerable. Future investigations regarding the delocalization of biofilm components via eDNA-PEI interactions including other biomacromolecular interactions may be addressed in future studies.
Figure 10.

ITC binding isotherm shows that 600 Da BPEI interacts with DNA. As 600 Da BPEI is injected into DNA solutions, initial binding events cause heat to be absorbed and cause DNA conformational reorganization followed by exothermic behavior as additional BPEI molecules binds to the phosphodiester backbone.
Conclusion
PEG-BPEI retains the ability to eradicate P. aeruginosa biofilms in complex model systems. The anionic EPS matrix21,41,42 binds antibiofilm agents, such as Ag+ ions, cationic polyhexamethylene biguanide, and cationic antibiotics, such as tobramycin.43 These agents kill bacteria but do not disrupt the biofilm itself. In contrast, when EPS binds PEG-BPEI, the biofilm disperses.14 Toxicity concerns of 600 Da BPEI can be mitigated by covalent linkage with low molecular weight PEG, and, in this study, PEG350-BPEI was found exhibit superior antibiofilm activity against P. aeruginosa. The antibiofilm activity of both 600 Da BPEI and its PEG derivative was characterized with fluorescence studies and microscopy imaging. These data provide support for development of PEG-BPEI as a wound treatment approach.
PEG-BPEI can be formulated as a gel, cream, or foam for use as a stand-alone or adjunctive therapy that complements other wound treatment interventions. Billions of healthcare dollars are spent on therapeutic devices to alleviate diabetic wounds and pressure ulcers that cause physical pain, disabilities, and poor quality of life.44−46 Delayed healing is caused by bacterial pathogens and their biomolecules that form biofilms in the wound microenvironment. During the inflammation phase of wound healing, immune cells accumulate in response to biofilms and lead to dysregulation of the immune response and excessive inflammation that impairs wound healing, leading to recurrent infection and tissue necrosis.47,48 Excessive inflammatory tissue microenvironments inhibit the migration of keratinocytes, fibroblasts, and the synthesis of new extracellular matrix in the granulation tissue.48 Modulating the innate immune response to biofilms and biofilm EPS with PEG-BPEI expands the possible utility of PEG-BPEI that counteract antibiotic resistance mechanisms.14 This versatility of PEG-BPEI suggests that it may be a useful therapeutic agent by simultaneously providing anti-inflammation and infection prevention that aids with decolonization by dispersing biofilms. This will speed wound healing while also slowing the emergence and spread of antibiotic resistance genes and antibiotic-resistant pathogens.
Acknowledgments
This work was made possible due to the guidance and contributions of Preston Larson, Ph.D., Fernando Esteban Florez, D.D.S., Ph.D., MS, and Anh K Lam, Ph.D. We would also like to thank Dr. Phil Bourne and the Protein Production Core (PPC) at the University of Oklahoma, Norman. PPC is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103640.
Glossary
Abbreviations:
- TSB
tryticase soy broth
- TSA
tryticase soy agar
- Tris
tris(hydroxymethyl)aminomethane buffer
- BPEI
branched polyethylenimine
- PEG
polyethylene glycol
- PEG350-BPEI
pegylated BPEI
- OD550
optical density at 550 nm
- MBEC
minimum biofilm eradication concentration
- MIC
minimum inhibitory concentration
Funding was provided by the National Institutes of Health (CVR, R03AI142420) and The University of Oklahoma. This study was also sponsored by grant W81XWH2210047 from the Department of Defense/USAMRAA (PRMRP #PR210140).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Defense.
The authors declare no competing financial interest.
References
- Nussbaum S. R.; Carter M. J.; Fife C. E.; DaVanzo J.; Haught R.; Nusgart M.; Cartwright D. An Economic Evaluation of the Impact, Cost, and Medicare Policy Implications of Chronic Nonhealing Wounds. Value Health 2018, 21 (1), 27–32. 10.1016/j.jval.2017.07.007. [DOI] [PubMed] [Google Scholar]
- Bhargava A.; Hayakawa K.; Silverman E.; Haider S.; Alluri K. C.; Datla S.; Diviti S.; Kuchipudi V.; Muppavarapu K. S.; Lephart P. R.; Marchaim D.; Kaye K. S. Risk factors for colonization due to carbapenem-resistant Enterobacteriaceae among patients exposed to long-term acute care and acute care facilities. Infect Control Hosp Epidemiol 2014, 35 (4), 398–405. 10.1086/675614. [DOI] [PubMed] [Google Scholar]
- Marchaim D.; Chopra T.; Bogan C.; Bheemreddy S.; Sengstock D.; Jagarlamudi R.; Malani A.; Lemanek L.; Moshos J.; Lephart P. R.; Ku K.; Hasan A.; Lee J.; Khandker N.; Blunden C.; Geffert S. F.; Moody M.; Hiro R.; Wang Y.; Ahmad F.; Mohammadi T.; Faruque O.; Patel D.; Pogue J. M.; Hayakawa K.; Dhar S.; Kaye K. S. The burden of multidrug-resistant organisms on tertiary hospitals posed by patients with recent stays in long-term acute care facilities. Am. J. Infect Control 2012, 40 (8), 760–5. 10.1016/j.ajic.2011.09.011. [DOI] [PubMed] [Google Scholar]
- Henig O.; Cober E.; Richter S. S.; Perez F.; Salata R. A.; Kalayjian R. C.; Watkins R. R.; Marshall S.; Rudin S. D.; Domitrovic T. N.; Hujer A. M.; Hujer K. M.; Doi Y.; Evans S.; Fowler V. G. Jr.; Bonomo R. A.; van Duin D.; Kaye K. S. Antibacterial Resistance Leadership, G. A Prospective Observational Study of the Epidemiology, Management, and Outcomes of Skin and Soft Tissue Infections Due to Carbapenem-Resistant Enterobacteriaceae. Open Forum Infect Dis 2017, 4 (3), ofx157. 10.1093/ofid/ofx157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffin M.; Brochiero E. Repair Process Impairment by Pseudomonas aeruginosa in Epithelial Tissues: Major Features and Potential Therapeutic Avenues. Front Cell Infect Microbiol 2019, 9 (182), 182. 10.3389/fcimb.2019.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlan R. M. Biofilms: microbial life on surfaces. Emerg Infect Dis 2002, 8 (9), 881–90. 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panlilio H.; Rice C. V. The role of extracellular DNA in the formation, architecture, stability, and treatment of bacterial biofilms. Biotechnol. Bioeng. 2021, 118 (6), 2129–2141. 10.1002/bit.27760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S.; Parrino B.; Carbone D.; Pecoraro C.; Diana P. Novel strategies in the war against antibiotic resistance. Future Med. Chem. 2021, 13 (6), 529–531. 10.4155/fmc-2021-0009. [DOI] [PubMed] [Google Scholar]
- Lam A. K.; Panlilio H.; Pusavat J.; Wouters C. L.; Moen E. L.; Rice C. V. Overcoming Multidrug Resistance and Biofilms of Pseudomonas aeruginosa with a Single Dual-Function Potentiator of beta-Lactams. ACS Infect Dis 2020, 6 (5), 1085–1097. 10.1021/acsinfecdis.9b00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M. A.; Lam A. K.; Reed P.; Harney M. C.; Wilson B. A.; Moen E. L.; Wright S. N.; Pinho M. G.; Rice C. V. BPEI-Induced Delocalization of PBP4 Potentiates beta-Lactams against MRSA. Biochemistry 2019, 58 (36), 3813–3822. 10.1021/acs.biochem.9b00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. K.; Hill M. A.; Moen E. L.; Pusavat J.; Wouters C. L.; Rice C. V. Cationic Branched Polyethylenimine (BPEI) Disables Antibiotic Resistance in Methicillin-Resistant Staphylococcus epidermidis (MRSE). ChemMedChem. 2018, 13 (20), 2240–2248. 10.1002/cmdc.201800433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. K.; Wouters C. L.; Moen E. L.; Pusavat J.; Rice C. V. Antibiofilm Synergy of beta-Lactams and Branched Polyethylenimine against Methicillin-Resistant Staphylococcus epidermidis. Biomacromolecules 2019, 20 (10), 3778–3785. 10.1021/acs.biomac.9b00849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panlilio H.; Lam A. K.; Heydarian N.; Haight T.; Wouters C. L.; Moen E. L.; Rice C. V. Dual-Function Potentiation by PEG-BPEI Restores Activity of Carbapenems and Penicillins against Carbapenem-Resistant Enterobacteriaceae. ACS Infect Dis 2021, 7 (6), 1657–1665. 10.1021/acsinfecdis.0c00863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. K.; Moen E. L.; Pusavat J.; Wouters C. L.; Panlilio H.; Ferrell M. J.; Houck M. B.; Glatzhofer D. T.; Rice C. V. PEGylation of Polyethylenimine Lowers Acute Toxicity while Retaining Anti-Biofilm and beta-Lactam Potentiation Properties against Antibiotic-Resistant Pathogens. ACS Omega 2020, 5 (40), 26262–26270. 10.1021/acsomega.0c04111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J. H.; Kadouri D. E.; O’Toole G. A. Growing and analyzing static biofilms. Curr. Protoc Microbiol 2006, 00, 1B.1.1–1B.1.17. 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. K.; Panlilio H.; Pusavat J.; Wouters C. L.; Moen E. L.; Neel A. J.; Rice C. V. Low-Molecular-Weight Branched Polyethylenimine Potentiates Ampicillin against MRSA Biofilms. ACS Med. Chem. Lett. 2020, 11 (4), 473–478. 10.1021/acsmedchemlett.9b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Woo M.; Yan C. Spot plating assay for the determination of survival and plating efficiency of Escherichia coli in sub-MIC levels of antibiotics. JEMI Methods 2017, 1, 26–29. [Google Scholar]
- Ceri H.; Olson M. E.; Stremick C.; Read R. R.; Morck D.; Buret A. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin Microbiol 1999, 37 (6), 1771–6. 10.1128/JCM.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann E. E.; Wozniak D. J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev. 2012, 36 (4), 893–916. 10.1111/j.1574-6976.2011.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. K.; Panlilio H.; Pusavat J.; Wouters C. L.; Moen E. L.; Brennan R. E.; Rice C. V. Expanding the Spectrum of Antibiotics Capable of Killing Multidrug-Resistant Staphylococcus aureus and Pseudomonas aeruginosa. ChemMedChem. 2020, 15 (15), 1421–1428. 10.1002/cmdc.202000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings L. K.; Storek K. M.; Ledvina H. E.; Coulon C.; Marmont L. S.; Sadovskaya I.; Secor P. R.; Tseng B. S.; Scian M.; Filloux A.; Wozniak D. J.; Howell P. L.; Parsek M. R. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. U.S.A. 2015, 112 (36), 11353–11358. 10.1073/pnas.1503058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin M. J.; Nivens D. E.; Weadge J. T.; Howell P. L. Biosynthesis of the Pseudomonas aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front Microbiol 2011, 2, 167. 10.3389/fmicb.2011.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin K. M.; Gordon V. D.; Murakami K.; Borlee B. R.; Wozniak D. J.; Wong G. C.; Parsek M. R. The pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog 2011, 7 (1), e1001264 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings N.; Millan M.; Caldara M.; Rusconi R.; Tarasova Y.; Stocker R.; Ribbeck K. The extracellular matrix Component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog 2013, 9 (8), e1003526 10.1371/journal.ppat.1003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas D.; Holloway B. W.; Schambock A.; Leisinger T. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Molecular & general genetics: MGG 1977, 154 (1), 7–22. 10.1007/BF00265571. [DOI] [PubMed] [Google Scholar]
- Holloway B. W. Genetic recombination in Pseudomonas aeruginosa. J. Gen Microbiol 1955, 13 (3), 572–81. 10.1099/00221287-13-3-572. [DOI] [PubMed] [Google Scholar]
- Cao H.; Lai Y.; Bougouffa S.; Xu Z.; Yan A. Comparative genome and transcriptome analysis reveals distinctive surface characteristics and unique physiological potentials of Pseudomonas aeruginosa ATCC 27853. BMC Genomics 2017, 18 (1), 459. 10.1186/s12864-017-3842-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh K. P.; Sauer K. Biofilm dispersion. Nat. Rev. Microbiol 2020, 18 (10), 571–586. 10.1038/s41579-020-0385-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri G.; Damstetter E.; Phillips T. Choosing a Wound Dressing Based on Common Wound Characteristics. Adv. Wound Care (New Rochelle) 2016, 5 (1), 32–41. 10.1089/wound.2014.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice E. A.; Segre J. A. The skin microbiome. Nat. Rev. Microbiol 2011, 9 (4), 244–53. 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzam K.; Nessel T. A.; Quick J.. Erythromycin. In StatPearls; StatPearls Publishing: Tampa, FL, 2021. [PubMed] [Google Scholar]
- Delcour A. H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794 (5), 808–16. 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K.; Pay J. L.. Wound Irrigation. In StatPearls; StatPearls Publishing: Tampa, FL, 2020. [PubMed] [Google Scholar]
- Bernier S. P.; Ha D. G.; Khan W.; Merritt J. H.; O’Toole G. A. Modulation of Pseudomonas aeruginosa surface-associated group behaviors by individual amino acids through c-di-GMP signaling. Res. Microbiol 2011, 162 (7), 680–8. 10.1016/j.resmic.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole G. A. Microtiter dish biofilm formation assay. J. Vis Exp 2011, (47), 2437. 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D.; Turner K. E.; Kirienko N. V. PqsA Promotes Pyoverdine Production via Biofilm Formation. Pathogens 2018, 7 (1), 3. 10.3390/pathogens7010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A.; Saltik M.; Lehrmann H.; Killisch I.; Mautner V.; Lamm G.; Christofori G.; Cotten M. Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene Ther. 1997, 4 (8), 773–782. 10.1038/sj.gt.3300471. [DOI] [PubMed] [Google Scholar]
- Utsuno K.; Uludag H. Thermodynamics of polyethylenimine-DNA binding and DNA condensation. Biophys. J. 2010, 99 (1), 201–7. 10.1016/j.bpj.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T.; Kutty S. K.; Kumar N.; Manefield M. Pyocyanin facilitates extracellular DNA binding to Pseudomonas aeruginosa influencing cell surface properties and aggregation. PLoS One 2013, 8 (3), e58299 10.1371/journal.pone.0058299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T.; Kutty S. K.; Tavallaie R.; Ibugo A. I.; Panchompoo J.; Sehar S.; Aldous L.; Yeung A. W.; Thomas S. R.; Kumar N.; Gooding J. J.; Manefield M. Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci. Rep 2015, 5, 8398. 10.1038/srep08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloag E. S.; Turnbull L.; Huang A.; Vallotton P.; Wang H.; Nolan L. M.; Mililli L.; Hunt C.; Lu J.; Osvath S. R.; Monahan L. G.; Cavaliere R.; Charles I. G.; Wand M. P.; Gee M. L.; Prabhakar R.; Whitchurch C. B. Self-organization of bacterial biofilms is facilitated by extracellular DNA. P Natl. Acad. Sci. USA 2013, 110 (28), 11541–6. 10.1073/pnas.1218898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaliere R.; Ball J. L.; Turnbull L.; Whitchurch C. B. The biofilm matrix destabilizers, EDTA and DNaseI, enhance the susceptibility of nontypeable Hemophilus influenzae biofilms to treatment with ampicillin and ciprofloxacin. Microbiologyopen 2014, 3 (4), 557–67. 10.1002/mbo3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musken M.; Pawar V.; Schwebs T.; Bahre H.; Felgner S.; Weiss S.; Haussler S. Breaking the Vicious Cycle of Antibiotic Killing and Regrowth of Biofilm-Residing Pseudomonas aeruginosa. Antimicrob Agents Ch 2018, 62 (12), 18. 10.1128/AAC.01635-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. E.; Foster D. S.; Longaker M. T. Management of Chronic Wounds-2018. Jama-J. Am. Med. Assoc 2018, 320 (14), 1481–1482. 10.1001/jama.2018.12426. [DOI] [PubMed] [Google Scholar]
- Lim H. W.; Collins S. A. B.; Resneck J. S. Jr.; Bolognia J. L.; Hodge J. A.; Rohrer T. A.; Van Beek M. J.; Margolis D. J.; Sober A. J.; Weinstock M. A.; Nerenz D. R.; Smith Begolka W.; Moyano J. V. The burden of skin disease in the United States. J. Am. Acad. Dermatol 2017, 76 (5), 958–972. 10.1016/j.jaad.2016.12.043. [DOI] [PubMed] [Google Scholar]
- Gurtner G. C.; Werner S.; Barrandon Y.; Longaker M. T. Wound repair and regeneration. Nature 2008, 453 (7193), 314–21. 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Usui M. L.; Lippman S. I.; James G. A.; Stewart P. S.; Fleckman P.; Olerud J. E. Biofilms and Inflammation in Chronic Wounds. Adv. Wound Care (New Rochelle) 2013, 2 (7), 389–399. 10.1089/wound.2012.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz G.; Bjarnsholt T.; James G. A.; Leaper D. J.; McBain A. J.; Malone M.; Stoodley P.; Swanson T.; Tachi M.; Wolcott R. D. Global Wound Biofilm Expert, P. Consensus guidelines for the identification and treatment of biofilms in chronic nonhealing wounds. Wound Repair Regen 2017, 25 (5), 744–757. 10.1111/wrr.12590. [DOI] [PubMed] [Google Scholar]