

Abstract

Synthesis of a new series of 20 compounds bearing the thieno[2,3-d]pyrimidine-4-one scaffold was achieved. The inhibitory activity of these compounds was performed over 60 cell lines of human tumor at single and five dose concentrations. Compounds 20, 22, and 23 exhibited potent growth inhibitions toward the majority of the tested NCI 60 cell lines. Compounds 20 and 23 were the most active compounds with (MG-MID) TGI, GI50, and LC50 values of 16.2, 3.3, 50.1 and 67.7, 6.6, 100, respectively. Also, both compounds showed 7- and 4-fold better activity, respectively, than the standard antitumor agent 5-fluorouracil. Therefore, compounds 20 and 23 were selected to measure their ability to inhibit the dihydrofolate reductase enzyme (DHFR) in comparison to methotrexate (MTX) as a reference drug. Compound 20 was a more potent inhibitor of DHFR (IC50 = 0.20 μM) than MTX (IC50 = 0.22 μM). Molecular modeling studies were performed in the DHFR active site, and it showed compatibility with the results obtained from biological studies. Finally, the results showed that compound 20 is a strong antitumor agent and potent inhibitor of DHFR. In addition, this compound induced cell-cycle arrest in SNB-75 cells in the G2/M phase and the apoptosis process in the Pre-G phase. Compound 20 also increased the level of both caspases-3 and 9 by 11.8- and 50.3-fold, respectively.
1. Introduction
Currently, cancer is the leading cause of fatality worldwide. The dihydrofolate reductase (DHFR) enzymatic pathway has been a significant goal in cancer therapy because DHFR is essential for reducing dihydrofolate into tetrahydrofolate1 to afford one-carbon moieties for the synthesis of new thymidylate and purine nucleotides. Therefore, the inhibition of the DHFR enzyme leads to cancer cell death.2
Many thieno[2,3-d]pyrimidines have been reported as potent nonclassical lipophilic DHFR inhibitors.3,4 Binding of thieno[2,3-d]pyrimidines to the DHFR enzyme is compared to folic acid binding; it is in the “flipped” mode where the sulfur of the thiophene pharmacophore was found to be able to mimic the 4-amino of methotrexate (MTX).3,5,6
Thieno[2,3-d]pyrimidine moieties are characterized by selectively targeting the tumor cells instead of the host normal cells because they target folate receptors (FRs), which are highly expressed in cancer cells, rather than target reduced folate carriers (RFCs), which are expressed in normal cells (Figure 1).3,7
Figure 1.

Tumor-targeting technique of thieno[2,3-d]pyrimidines.
As shown in Figure 2, reported structural features of thieno[2,3-d]pyrimidines as DHFR inhibitors showed that the nitrogen atom at position 1 and the NH2 group at position 2 are essential for the formation of a salt bridge with the key amino acid Glu30.8 In addition, the oxygen atom at position 4 is crucial for forming a hydrogen bond with Val115, Ile7, and Tyr121 amino acids.3 Also, elongation of R2 at position 6 by replacing the methyl group with an ethyl group increases the potency against DHFR through increasing the lipophilicity of these inhibitors, which can facilitate passive diffusion.6 Moreover, the presence of tricyclic benzo[4,5]thieno[2,3-d]pyrimidine-4-one was found to be more selective for human DHFR (hDHFR) in comparison to MTX. This selectivity is due to the presence of the sulfur atom at position 7, which is superimposed onto the MTX 4-NH2 group.3 Besides, it forms intermolecular H-bonds with Ile7 and Val115 amino acids through its carbonyl group and Tyr121 amino acid through its hydroxyl group.8 Since the size of the sulfur atom is smaller than the size of carbon atoms and larger than the size of the nitrogen atom, the thiophene moiety, present in benzo[4,5]thieno[2,3-d]pyrimidines, is used to mimic the activity of the larger quinazoline ring and the smaller pyrrole ring of MTX and pemetrexed (PMX), respectively.6
Figure 2.

Design of new thieno[2,3-d]pyrimidine analogues as DHFR inhibitors.
The aim of this work is to design new thieno[2,3-d]pyrimidine analogues accompanied by manipulation in their structures to evaluate their inhibitory effects toward DHFR activity. Moreover, in vitro cytotoxic activity against complete NCI 60 cell-line panels was evaluated, and the highest active compounds were selected to test their hDHFR inhibitory effect. Apoptotic and cell-cycle arrest analyses against the SNB-75 cell line were performed as well as the role of caspase-3 and caspase-9 in the signaling pathway of DHFR inhibitor-induced apoptosis was determined.
2. Results and Discussion
2.1. Chemistry
Target compounds’ synthesis was achieved through two schemes, Scheme 1 (for synthesis of compounds 7–16) and Scheme 2 (for synthesis of compounds 17–24). In Scheme 1, compound 2 was synthesized according to the Gewald reaction9 through the interaction of cycloheptanone (1), ethyl cyanoacetate, and elemental sulfur in absolute ethanol using triethylamine (TEA) as a base catalyst. Reaction of ethyl carboxylate 2 with aryl isothiocyanates (R = H or NO2) at room temperature using pyridine or THF as the solvent, respectively, gave thiourea derivatives 3 and 4. Confirmation of structure 4 was done using 1H NMR, in which two additional NH signals at 8.07 and 12.58 ppm appeared. Moreover, the presence of the aromatic ring was detected by the proton signals at 7.54–8.07 ppm. In the IR spectra, there was a (C=S) band at 853 and 1206 cm–1, an (NO2) band at 1342–1509 cm–1, and a band at 3190 cm–1 characteristic to (NH), while the characteristic bands for the (NH2) group at 3322 and 3465 cm–1 disappeared.
Scheme 1. Reagents and Conditions: (a) Ethyl 2-Cyanoacetate, Sulfur, Et3N, rt; (b) Ar-SCN, Pyridine or THF, rt; (c) KOH/H2O, 100 °C; (d) R–X, Acetone, K2CO3, 50 °C or rt; (e) R–X, DMF, K2CO3, rt.

Scheme 2. Reagents and Conditions: (a) NH2NH2, Ethanol, rt; (b) Ar-CHO, EtOH, 90 °C; (c) Acetylacetone, EtOH, 90 °C.

The synthesis of pyrimidin-4-ones 5 and 6 was performed through the cyclization of carbothioamides 3 and 4 in an aqueous potassium hydroxide solution.10 The structure of the target compounds was confirmed in 1H NMR spectra by the disappearance of the NH protons and the appearance of a new (SH) proton at 1.24–1.27 ppm.11
Two forms of tautomers, thiol and thione, could be expected from the cyclization of compounds 3 and 4, under basic conditions.12,13 From the NMR spectra, only the thiol tautomer of 5 and 6 was detected due to the presence of the (SH) proton at 1.27 ppm and the (C–SH) carbon at 147.8 ppm (Figure 3).
Figure 3.

Possibilities of thiol-thione tautomerism.
S-Alkylation of pyrimidin-4-one 5 was carried out through the reaction with several alkyl halides in acetone using K2CO3 as a base under reflux to prepare 2-(substituted thio)pyrimidin-4-ones 7–10.14 The formation of the ester derivative 11 was found to be temperature dependent as it was obtained at room temperature instead of under a refluxing condition. The 1H NMR spectrum of compound 7 confirms its structure by the appearance of two benzylic protons at 4.37 ppm and the aromatic protons at 7.29–7.52 ppm. Also, 1H NMR spectra of compounds 8–10 showed the presence of the signals of aliphatic protons at 0.84–3.02 ppm. Compound 11 showed triplet signals in its 1H NMR spectrum at 1.22 ppm, singlet signals at 3.91 ppm, and quartet signals at 4.12 ppm attributed to −OCH2CH3, −SCH2, and −OCH2CH3 protons, respectively. The signals of these groups in the 13C NMR spectrum were detected at 14.65, 34.83, and 61.52 ppm, respectively.
To prepare 2-(substituted thio)pyrimidin-4-ones 12-1, S-alkylation of pyrimidin-4-one 6 was performed by reacting with several alkyl halides in DMF using K2CO3 as the base, stirring at room temperature.15,16 Confirmation of structure 12 was done using the 1H NMR spectrum by the presence of the two benzylic protons at 4.39 ppm and the aromatic protons at 7.50–8.39 ppm. The structures of compounds 13 and 14 were detected in 1H NMR spectra by additional singlet protons at 3.85 ppm and multiplet protons in the range of 1.45–1.89 ppm, attributed to methyl and cyclopentyl groups, respectively. For compound 15, ester group signals were observed in the 1H NMR spectrum at 1.22 (−OCH2CH3) and 4.12 (−OCH2CH3) ppm, in addition to singlet (−SCH2) protons at 3.96 ppm. The IR spectra also showed the characteristic carbonyl ester band at 1733 cm–1. Singlet signals at 3.22 and 3.95 ppm attributed to the propargaylic proton and −SCH2 protons, respectively, were observed in the 1H NMR spectrum of compound 16. The same groups’ signals were recorded in the 13C NMR spectrum at 74.79, 79.35, and 21.13 ppm.
As shown in Scheme 2, acetohydrazide 17 was prepared by stirring the ester derivative 11 with hydrazine hydrate in absolute ethanol at room temperature for 48 h.17 The 1H NMR spectrum of acetohydrazide 17 showed new signals appeared at 4.26 and 9.29 ppm, respectively, attributed to the protons of NH2 and NH of the hydrazide structure along with the disappearance of the signals of the ethoxy group. Also, the IR spectrum showed the appearance of the carbonyl hydrazide band at 1645 cm–1 and the disappearance of the carbonyl easter band at 1736 cm–1.
Schiff bases 18–23 were synthesized through the reaction of different aromatic aldehydes with acetohydrazide 17 using ethanol as a solvent.16 The 1H NMR spectra of these compounds showed additional signals in the range from 6.72 to 8.28 ppm owing to the aromatic ring and singlet signals in the range from 7.87 to 8.53 ppm owing to the ylidenic proton HC=N.
It was observed that two signal sets of −SCH2, −NH, and −N=CH groups of these compounds were observed in their 1H NMR spectra as trans and cis conformers. In the cis conformer, −NH and −N=CH protons’ lines appeared upfield, while in the trans conformer, the lines of the protons of the same groups appeared downfield (Figure 4).18
Figure 4.

Geometrical isomers and conformers of Schiff bases 18–23.
The Knorr pyrazole reaction was utilized for the synthesis of pyrazole 24 through the interaction of acetohydrazide 17 with acetylacetone in absolute ethanol.19 No signals belonging to NH or NH2 protons were displayed for this compound in its 1H NMR spectrum; instead, new singlet signals appeared at 6.39, 1.7, and 1.99 ppm, attributed to CH–pyrazole and the two CH3 groups, respectively.
2.2. Biology
2.2.1. Cytotoxicity Screening
For screening the antitumor activity, a screening panel assay for in vitro disease-oriented human cells was run at the National Cancer Institute (NCI). It was dependent on the usage of a single dose of tested compounds (10 μM) in the complete NCI 60 cell-line panel assay that contains cancer subpanels, namely, breast, renal, prostate, melanoma, colon, CNS, non-small-cell lung, ovarian, and leukemia cancer cells.20,21 The capability of the tested compounds to produce inhibition of cancer growth was represented by the percentage growth inhibition (GI %) (Table 1).
Table 1. (GI %) Percentage Growth Inhibition Resulting from Compounds’ Concentration of 10 μM (4, 12–23) of Subpanel Tumor Cell Linesa.
| subpanel tumor cell lines (sample conc. = 10 μM) | % growth
inhibition (GI %) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 13 | 14 | 15 | 16 | 18 | 19 | 20 | 22 | 23 | |
| leukemia | |||||||||||
| CCRF-CEM | 36 | 7 | 10 | 7 | 11 | 12 | 30 | 22 | 54 | 24 | 30 |
| HL-60(TB) | 30 | 3 | 8 | 13 | 12 | 9 | – | 22 | – | – | 14 |
| RPMI-8226 | 53 | 7 | 2 | – | 9 | 16 | 7 | 9 | 34 | 44 | 34 |
| SR | 38 | 15 | 22 | 11 | 24 | 12 | 16 | 17 | 89 | 62 | 78 |
| non-small-cell lung cancer | |||||||||||
| HOP-62 | – | 27 | 18 | – | 53 | 7 | – | – | 80 | 47 | 79 |
| HOP-92 | 100 | 6 | 8 | 18 | 20 | 13 | – | – | 18 | 27 | 50 |
| A549/ATCC | 15 | 19 | – | 8 | 34 | 6 | 9 | 2 | 91 | 73 | 94 |
| EKVX | 21 | 15 | 19 | 18 | 13 | 13 | 21 | 20 | 30 | 53 | |
| NCI-H460 | 31 | 26 | 15 | 10 | 52 | 6 | 4 | – | 90 | 81 | 99 |
| NCI-H522 | 42 | 19 | 20 | 15 | 32 | 20 | 20 | 24 | 56 | 47 | 79 |
| NCI-H322M | 30 | 12 | 15 | 10 | 23 | 11 | – | 3 | 33 | 21 | 56 |
| NCI-H23 | 31 | 46 | 1 | 1 | 26 | 5 | 3 | 2 | 63 | 46 | 71 |
| colon cancer | |||||||||||
| HCC-2998 | 3 | – | – | 6 | 18 | – | – | – | – | 3 | 61 |
| HCT-116 | 30 | 16 | - | 9 | 49 | 23 | 3 | 3 | 126 | 96 | 93 |
| COLO 205 | – | 1 | – | 2 | 18 | – | – | – | 62 | 11 | 67 |
| HCT-15 | 33 | 6 | 3 | 6 | 15 | 8 | 3 | 15 | 52 | 47 | 57 |
| HT29 | – | 8 | 2 | – | 22 | 2 | – | – | 74 | 68 | 55 |
| KM12 | 28 | – | 6 | 13 | 11 | 9 | – | – | 58 | 53 | 62 |
| SW-620 | 1 | 5 | 9 | 1 | 19 | 6 | – | – | 47 | 48 | 73 |
| CNS cancer | |||||||||||
| SF-268 | 11 | 2 | 12 | 4 | 29 | 10 | – | – | 92 | 56 | 63 |
| SF-295 | 13 | 32 | 6 | 2 | 23 | 11 | 7 | 7 | 84 | 24 | 81 |
| SF-539 | 8 | 5 | 2 | – | 25 | – | – | – | 139 | 99 | 66 |
| SNB-75 | 14 | 15 | 18 | 3 | 89 | 7 | – | – | 165 | 94 | 102 |
| U251 | 23 | 16 | 19 | 18 | 47 | 6 | – | 5 | 93 | 51 | 95 |
| melanoma | |||||||||||
| LOX IMVI | 16 | 6 | 8 | 3 | 21 | 21 | 40 | 16 | 63 | 24 | 63 |
| MALME-3 | 28 | 9 | 1 | 4 | 23 | 5 | – | 28 | 89 | 26 | 65 |
| M14 | 7 | 6 | 4 | 6 | 25 | 15 | – | 7 | 49 | 29 | 54 |
| MDA-MB-435 | 12 | 3 | 1 | – | 23 | 10 | – | 12 | 40 | 28 | 67 |
| SK-MEL-2 | 34 | – | – | – | 10 | 3 | 3 | 34 | 9 | 10 | 22 |
| SK-MEL-28 | 15 | – | – | – | 16 | – | – | 15 | 52 | 26 | 53 |
| SK-MEL-5 | 57 | 4 | 3 | 5 | 12 | 9 | 9 | – | 50 | 33 | 54 |
| UACC-257 | 30 | 9 | 6 | 10 | 11 | 3 | – | 30 | 26 | 34 | 36 |
| UACC-62 | 52 | 13 | 15 | 8 | 26 | 8 | 45 | 52 | 17 | 35 | 92 |
| ovarian cancer | |||||||||||
| 786-0 | 7 | 5 | 7 | 7 | 29 | – | – | – | 36 | 20 | 55 |
| A498 | 50 | 20 | 9 | 6 | 15 | 1 | 45 | 11 | 17 | 65 | |
| ACHN | 15 | 33 | 16 | 15 | 48 | 10 | 3 | 17 | 74 | 20 | 75 |
| CAKI-1 | 21 | 29 | 8 | – | 49 | 32 | 14 | 24 | 113 | 41 | 91 |
| RXF 393 | 16 | 16 | 27 | 13 | 34 | 6 | – | 95 | 39 | 106 | |
| SN12C | 45 | 12 | 34 | 28 | 16 | 12 | 23 | 26 | 84 | 34 | 44 |
| prostate cancer | |||||||||||
| PC-3 | 57 | 55 | 17 | 10 | 27 | 25 | 6 | 14 | 55 | 62 | 123 |
| DU-145 | 9 | 1 | 5 | 4 | 61 | 2 | – | – | 113 | 101 | 77 |
| breast cancer | |||||||||||
| MCF7 | 30 | 14 | 11 | 15 | 27 | 21 | 1 | 17 | 74 | 55 | 65 |
| MDA-MB-231/ATCC | 21 | 25 | 10 | 7 | 50 | 25 | 9 | 24 | 126 | 49 | 57 |
| HS 578T | 9 | 18 | – | – | 61 | 2 | – | – | 113 | 101 | 77 |
| BT-549 | 6 | – | – | – | 3 | – | 2 | 11 | 52 | 62 | 29 |
| T-47D | 12 | 61 | 15 | 12 | 24 | 19 | 17 | 17 | 84 | 36 | 39 |
| MDA-MB-468 | 29 | 17 | 3 | – | 44 | – | – | – | 103 | 81 | 74 |
| MG-MID | 25 | 11 | 8 | 6 | 28 | 9 | 2 | 5 | 65 | 44 | 62 |
– represents GI ≤ 10%. Bold numbers represent the high % GI. Underlined numbers represent GI ≥100% “Lethal activity”. MG-MID represents full-panel mean-graph midpoint.
In the current investigation, the antitumor activity of a broad spectrum was observed by the majority of the tested compounds 4, 12–23. It was found that different antitumor activities were related to different structural characteristics. Generation of different substituted thio derivatives was aimed to discover their structure–activity relationship (SAR). The substituted thio derivatives 7–11 showed low potency versus tumor cell lines and were regarded as the least effective participants. However, introduction of an electron-withdrawing nitro group as in derivatives 12–16 increased the antitumor activities. Incorporation of unsaturation as in derivatives 12 and 16 showed potent antitumor activities compared to saturated derivatives 13–15. Different derivatives of Schiff bases were prepared to discover their SAR. The introduction of a hydroxyl group produced Schiff base derivatives with a low antitumor activity such as 18 and 19, suggesting that a hydrophilic group is not preferable. However, exchanging the hydrophilic group with a lipophilic one such as a dimethyl amino group and a fluorine atom resulted in potent compounds 23 and 20, respectively.
NCI assay results revealed that many compounds showed high selectivity with potent growth inhibition. Compounds with high selectivity include compound 4, which is obviously selective with GI % of about 100% against the cancer cell line of the non-small-cell lung, compounds 13 and 16 with GI % ranging from 32 to 34% against the renal cancer cell line, compound 18 showed selectivity against the melanoma cell line with GI % ranging from 40 to 45%, and compound 19 also showed GI % ranging from 30 to 34% against leukemia and melanoma cell lines. It is worthy to mention that compounds 20, 22, and 23 possessed the highest potency and also showed lethal activity against certain cancer cell lines. All of the tested cell lines were highly sensitive to compound 22 with very high GI % values.
Compounds 20 and 23 proved to be the best among all tested compounds, with GI % mean values of 65 and 62%, respectively. Therefore, both compounds passed the primary anticancer test at 10 μM concentration and were selected by NCI for conducting further assays at five concentrations (5-log dose range), which could better capture the potency of these compounds. Monitoring of GI50, TGI, and LC50 as the three response parameters for each cell line was done (Figures 5–7).
Figure 5.

Dose–response curve of compound 20 in all cancer lines.
Figure 7.
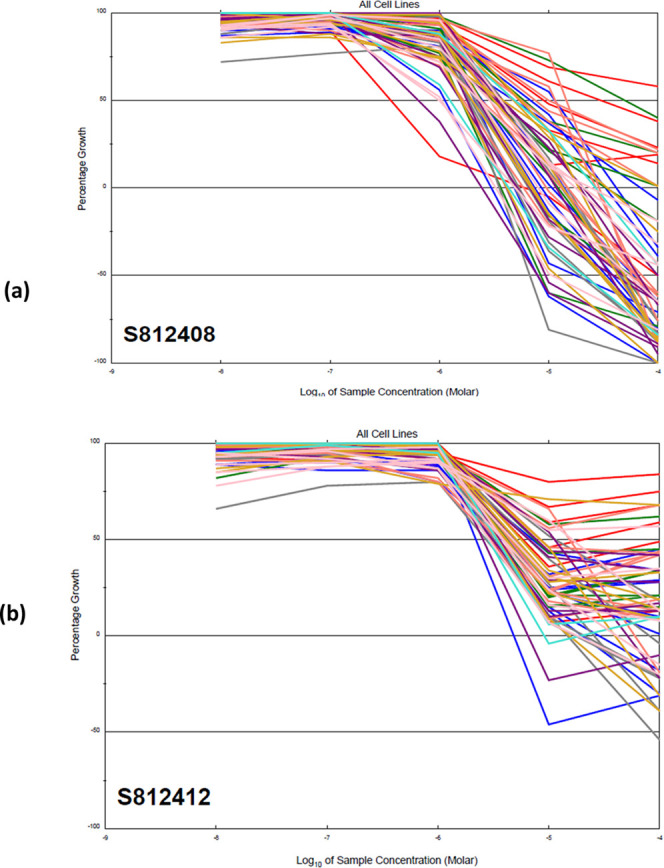
Dose–response graphs of the NCI 60 cell-line panel exposed to (a) compound 20 (NSC 812408) and (b) compound 23 (NSC 812412). The graph is coded with the color of the tissue origin: pink for breast cancer, turquoise for prostate cancer, gold for renal cancer, coral for the melanoma cell line, purple for ovarian cancer, green for colon cancer, gray for central nervous system cancer, blue for lung cancer, and red for the leukemia cell line.
Figure 6.

Dose–response curve of compound 23 in all cancer lines.
As shown in Table 2, compound 20 was 7-fold more active than the standard 5-FU, with full-panel mean-graph midpoint (MG-MID) TGI, GI50, and LC50 values of 16.2, 3.3, and 50.1, respectively. However, compound 23 was 4-fold more active than the standard drug, with MG-MID TGI, GI50, and LC50 values of 67.7, 6.6, and 100, respectively.
Table 2. Mean Sensitivity of Compounds 20 and 23 across All Cell Lines.
| full-panel mean-graph midpoint MG-MID (mM) |
|||
|---|---|---|---|
| compound | GI50 | TGI | LC50 |
| 20 | 3.3 | 16.2 | 50.1 |
| 23 | 6.6 | 67.7 | 100 |
| 5-flourouracil (5-FU) | 22.6 | >100 | >100 |
2.2.2. Human Dihydrofolate Reductase (hDHFR) Inhibition Assay
Regarding the NCI results, compounds 20 and 23 were selected for the assay of hDHFR inhibition using MTX as a reference drug. It was observable that these compounds exhibited high inhibitory activity (IC50 = 0.20 and 0.28 μM, respectively), which was comparable to the inhibitory activity of MTX (IC50 = 0.22 μM). The results suggested that hDHFR inhibition might be responsible for the cytotoxic activity of the tested compounds (Table 3).
Table 3. hDHFR Inhibitory Activity of Compounds 20 and 23.
| compound | hDHFR inhibition IC50 (μM) |
|---|---|
| 20 | 0.20 |
| 23 | 0.28 |
| MTX | 0.22 |
2.2.3. Analysis of Cell-Cycle Arrest
Previous assays showed that compound 20 has broad-spectrum activity that was remarkable against many tested cell lines of human cancer, especially against the SNB-75 cell line. Therefore, it was more investigated, relative to MTX, to explore its mechanism of action. Thus, measurement of DNA contents of the SNB-75 cell line was carried out depending on the flow cytometry technique in which the cell line was treated first with compound 20 for 24 h and propidium iodide was used for staining before measuring the DNA contents (Table 4 and Figure 8).
Table 4. Analysis of Compound 20 Cell-Cycle Arrest Using Flow Cytometry Relative to MTX.
| analysis
of cell-cycle arrest (%) |
||||
|---|---|---|---|---|
| G0/G1 | S | G2/M | pre-G1 | |
| 20/SNB-75 | 33.76 | 29.72 | 36.52 | 24.11 |
| MTX/SNB-75 | 29.36 | 26.81 | 43.83 | 27.38 |
| control/SNB-75 | 58.24 | 36.28 | 5.48 | 1.38 |
Figure 8.

Cell-cycle distribution of the SNB-75 cell line by compound 20.
The cell proportion in the S phase after treating SNB-75 cells with compound 20 decreased to 29.72% when compared to the control cell (36.28%). In addition, there was an increase in the cell proportion up to 36.52% in the G2/M phase due to compound 20 in comparison to the control cell (5.48%). This indicates that the G2/M phase is the one in which cells were arrested. On the other hand, treatment with compound 20 leads to the pre-G1 population (24.11%), while that of the controlled cell is 1.38%, which indicates that compound 20 had a great influence on cell-cycle distribution and induced cytotoxicity as a result of cell-cycle arrest
2.2.4. Apoptotic Assay
Cells were treated with compound 20 and suspended in Annexin V-FITC and PI for 24 h. Annexin V-FITC and PI were used for detecting apoptosis and necrosis, respectively. The apoptotic cell population was increased in SNB-75-treated cells in comparison to the untreated cells. These results indicated that compound 20 induced apoptosis by 24.11% (Table 5 and Figure 9).
Table 5. Apoptotic Activities of Compound 20.
| apoptosis |
||||
|---|---|---|---|---|
| compounds | early | late | necrosis | total |
| 20/SNB-75 | 8.49 | 13.43 | 2.19 | 24.11 |
| MTX/SNB-75 | 6.78 | 18.08 | 2.52 | 27.38 |
| control/SNB-75 | 0.73 | 0.28 | 0.37 | 1.38 |
Figure 9.

(A) SNB-75 cell line’s cell-cycle distribution by compound 20. (B) Apoptotic effect of compound 20 on the human SNB-75 cell line.
2.2.5. Detection of Protein Levels of Caspases-3 and 9
Determination of caspases-3 and caspase-9 levels is regarded as an essential method to detect the pathway involved in the process of apoptosis. The level of caspase-3 and caspase-9 in SNB-75 cells was measured after 24 h treatment of these cells with compound 20. It was found that our compound increased the level of caspase-3 and caspase-9 by 11.8- and 50.3-fold, respectively, which is compatible with cell-cycle arrest analysis data (Table 6).
Table 6. Caspases-3 and Caspase-9 (ng/mL) Detection Assay.
| compound | caspase-3 | caspase-9 | caspase-3 fold | caspase-9 fold |
|---|---|---|---|---|
| 20 | 590.5 | 33.2 | 11.8 | 50.3 |
| control/MCF7 | 50 | 0.66 |
2.2.6. In Silico Study
According to the significant results of hDHFR inhibition activity, it was exciting to further study the docking of compounds 20, 22, and 23, which had the highest DHFR inhibitory activity, into the binding site of hDHFR and explore the interaction of these compounds in the hDHFR pocket with its amino acids. Docking was done using molecular operating environment (MOE) software version 2014.09. The crystal structure of the DHFR complex with MTX (PDB code 1U72) was important to identify the 2D ligand interaction of DHFR and MTX. The pattern of binding between the DHFR receptor active site and MTX revealed the key amino acids of the hDHFR pocket through the trifurcated H-bonds that were built with the “catalytic triad” Ile7, Tyr121 and Val115 residues, and the pocket residues Glu30 and Asn64 form stable H-bonds22 (Figure 10).
Figure 10.

2D ligand interaction of DHFR and MTX.
The 2D binding configuration shows that skeletons of our compounds are deeply inhumed inside the pocket, and a tortuous “L” structure was formed by those compounds through the bridge region, which enables them to interact with most of the key amino acids. They can interact with Asn64 through their amide linkers, with Val115 through their thiophene moieties, with Arg70 through their pyridine rings, and with Phe31 through their aromatic rings. Although they have the same structures, they have slightly different enzymatic activities as there is a specific pattern of configuration for each compound inside the pocket. Therefore, compound 20 has the ability to bind to the key pocket residue Tyr121 (Figure 11A), while compound 23 does not. The activity of 23 is reduced due to this binding, where the thiophene moiety is positioned away from Tyr121 (Figure 11B). For compound 22, its nitro group at the meta position enables it to interact with most of the key amino acids such as Arg70 and Asn64. It can also bind to Phe31 through its aromatic ring. Its different configuration inside the pocket directs its thiophene ring away from both key amino acids yr121 and Val115. This binding shows why compound 22 has reduced activity (Figure 11C).
Figure 11.

(A) 2D binding configuration of DHFR and compound 20. (B) 2D binding configuration of DHFR and compound 22. (C) 2D binding configuration of DHFR and compound 23.
3. Conclusions
In this work, new thieno[2,3-d]pyrimidines 4–24 were synthetized and evaluated for their cytotoxic activity against complete NCI 60 cell-line panels. The target compounds 20, 22, and 23 are characterized by having the highest DHFR inhibitory activity. The docking studies that were carried out proved that these compounds are highly potent as DHFR inhibitors. Moreover, compound 20 induced Pre-G apoptosis and G2/M cell-cycle arrest in SNB-75 cells.
4. Experimental Section
4.1. Chemistry
The Stuart apparatus was used for recording of the melting points, and they were uncorrected. NMR spectra (1H and 13C) were recorded on Bruker 400 MHz and Joel 500M FT NMR spectrometers with tetramethyl silane (TMS) as the standard. (DMSO-d6) and (CDCl3) were the used solvents, and chemical shifts were obtained in δ (ppm) at the Unite of Nuclear Magnetic Resonance, Faculties of Science and Pharmacy, Mansoura University. Recording of mass spectra (MS) was done by the Hewlett Packard 5988 spectrometer (EI) in Al-Azhar University, Cairo, Egypt, and the spectrometer of Advion compact mass (ESI) in the Scientific laboratory of Nawah, Cairo, Egypt. IR spectra (KBr) (υ in cm–1) were recorded at the Unit of Central Laboratory in the Pharmacy Faculty of Mansoura University using the IR spectrometer of Nicolet iS10FT. TLC was used for recording the reaction time using silica gel plates (60 F245E). Merk. Spot visualization was done using UV (366 nm). All of the chemicals used were from Aldrich, and they needed no further purification. Compounds 2,233,24 and 5(25) have been described elsewhere.
4.1.1. Synthesis of Ethyl 2-[3-(4′-Nitrophenyl)carbothioamido]-5,6,7,8-tetrahydro-3H-cyclohepta[b]thiophene-3-carboxylate (4)
p-Nitrophenyl isothiocyanate (0.72 g, 4 mmol) was added to compound 2 solution (0.96 g, 4 mmol) in THF (10 mL). This mixture was stirred for 24 h at room temperature. After that, it was poured on crushed ice, followed by separation of the precipitated solid by filtration, washing with water, and recrystallization from EtOH, giving an orange solid; yield = 95%, melting point = 171–173 °C. IR (KBr, υ, cm–1): 1342, 1509 (NO2), 1546 (C=S), 1662 (C=O), 3190 (NH). 1H NMR (400 MHz, CDCl3): δ 1.31 (t, J = 7.1 Hz, 3H, CH2CH3), 1.61 (m, 4H, 2CH2), 1.81 (m, 2H, CH2), 2.71 (m, 2H, CH2), 2.99 (m, 2H, CH2) 4.24 (q, J = 7.1 Hz, 2H, CH2CH3), 7.54 (d, J = 9.0 Hz, 2H, Ar-H), 8.07 (s, 1H, NH; D2O exchangeable), 8.25 (d, J = 9.0 Hz, 2H, Ar-H), 12.58 (s, 1H, NH; D2O exchangeable).
4.1.2. Synthesis of 3-(4-Nitrophenyl)-2-sulfanyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (6)
A solution of compound 4 (4.19 g, 10 mmol) in aqueous KOH (6%, 60 mL) was refluxed for 2 h and then concentrated. The concentrate was dissolved in water and neutralized with HCl (1 M). The precipitated solid was filtered, washed with water, dried, and recrystallized from EtOH to give a yellow solid; yield = 88%, melting point = 223–225 °C, IR (KBr, υ, cm–1): 1345, 1520 (NO2), 1648 (C=O), 3407 (NH). 1H NMR (400 MHz, CDCl3): δ 1.22 (s, 1H, SH), 1.67 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.73 (m, 2H, CH2), 3.13 (m, 2H, CH2), 7.42 (d, J = 7.4 Hz, 2H, Ar-H). 8.37 (d, J = 7.4 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 27.25, 27.57, 27.70, 29.39, 32.25, 117.69, 124.57, 124.99, 130.17, 130.23, 138.28, 147.80.
4.1.3. General Procedure for the Synthesis of 2-(Alkylthio)-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-ones (7–11)
The appropriate alkyl halide (1.5 mmol) was added to a mixture of K2CO3 (0.18 g, 1 mmol) and compound 5 (0.32 g, 1 mmol) in acetone (5 mL). It was refluxed at 60 °C for 8 h and was TLC-monitored precisely. After reaction accomplishment, acetone was evaporated under a vacuum, and the obtained residue was recrystallized from EtOH.
4.1.3.1. 2-(Benzylthio)-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (7)
Using benzyl chloride (0.19 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 74%, melting point = 237–239 °C. 1H NMR (400 MHz, CDCl3): δ 1.66 (m, 4H, 2CH2), 1.89 (m, 2H, CH2), 2.87 (m, 2H, CH2), 3.30 (m, 2H, CH2), 4.37 (s, 2H, CH2–S), 7.29 (m, 5H, Ar-H), 7.38 (d, J = 7.8 Hz, 2H, Ar-H), 7.52 (d, J = 7.8 Hz, 3H, Ar-H).
4.1.3.2. 2-(Methylthio)-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (8)
Using methyl iodide (0.21 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 70%, melting point = 187–189 °C, IR (KBr, υ, cm–1): 1509 (olefinic C=C), 1668 (C=O). 1H NMR (400 MHz, DMSO-d6): δ 1.60 (m, 4H, 2CH2), 1.85 (m, 2H, CH2), 2.42 (s, 3H, CH3), 2.84 (m, 2H, CH2), 3.18 (m, 2H, CH2), 7.38 (d, J = 6.7 Hz, 2H, Ar-H). 7.52 (d, J = 6.7 Hz, 3H, Ar-H).
4.1.3.3. 2-(Ethylthio)-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (9)
Using ethyl iodide (0.22 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 72%, melting point = 157–159 °C, IR (KBr, υ, cm–1): 1509 (C=C), 1683 (C=O). 1H NMR (400 MHz, CDCl3): δ 1.23 (t, J = 7.4 Hz, 3H, S-CH2CH3), 1.61 (m, 4H, 2CH2), 1.80 (m, 2H, CH2), 2.76 (m, 2H, CH2), 3.02 (2H, q, J = 7.4 Hz, S–CH2CH3), 3.2 (m, 2H, CH2), 7.24 (d, J = 6.7 Hz, 2H, Ar-H). 7.50 (d, J = 6.7 Hz, 3H, Ar-H).
4.1.3.4. 2-(Butylthio)-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (10)
Using butyl iodide (0.28 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 76%, melting point = 170–172 °C. 1H NMR (400 MHz, CDCl3): δ 0.84 (t, J = 7.3 Hz, 3H, CH2CH2CH2CH3), 1.32 (dq, J = 7.3–14.5 Hz, 2H, CH2CH2CH2CH3), 1.53 (m, J = 7.3 Hz, 2H, CH2CH2CH2CH3), 1.62 (m, 4H, 2CH2), 1.80 (m, 2H, CH2), 2.76 (m, 2H, CH2), 3.02 (m, J = 7.3 Hz, 2H, CH2CH2CH2CH3), 3.2 (m, 2H, CH2), 7.24 (d, J = 6.3 Hz, 2H, Ar-H), 7.50 (d, J = 6.3 Hz, 3H, Ar-H).
4.1.3.5. Ethyl 2-[(4-Oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-5H-2-yl)thio]acetate (11)
Using ethyl chloroacetate (0.18 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 88%, melting point = 188–190 °C, IR (KBr, υ, cm–1): 1517 (C=C), 1680 (C=O amide), 1736 (CO, ester).1H NMR (400 MHz, DMSO-d6): δ 1.22 (t, J = 7.1 Hz, 3H, COOCH2CH3), 1.59 (m, 4H, 2CH2), 1.80 (m, 2H, CH2), 2.83 (m, 2H, CH2), 3.17 (m, 2H, CH2), 3.91 (s, 2H, CH2–S), 4.12 (q, J = 7.1 Hz, 2H, COOCH2CH3), 7.43 (m, 2H, Ar-H), 7.59 (m, 3H, Ar-H). 13C NMR (100 MHz, DMSO-d6): 14.65, 27.25, 27.47, 27.83, 29.38, 32.42, 34.83, 61.52, 119.58, 129.77, 130.49, 136.99, 158.78, 158.25, 160.09, 168.67.
4.1.4. General Procedure for the Synthesis of 2-Alkylthio-3-(4-nitrophenyl)-6,7,8,9-hexahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-ones (12–16)
The appropriate alkyl halide (1.5 mmol) and a mixture of K2CO3 (0.18 g, 1 mmol) and compound 6 (0.37g, 1 mmol) in DMF (5 mL) were stirred for about 24 h at room temperature. The reaction mixture was poured onto iced water, the formed solid was filtrated and washed with water, and crystallization from ethanol was done.
4.1.4.1. 2-Benzylthio-3-(4-nitrophenyl)-6,7,8,9-hexahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (12)
Using benzyl chloride (0.19 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 74%, melting point = 214–216 °C, IR (KBr, υ, cm–1): 1350, 1513 (NO2), 1686 (C=O), 1613 (C=C).1H NMR (400 MHz, CDCl3): δ 1.68 (m, 4H, 2CH2), 1.89 (m, 2H, CH2), 2.88 (m, 2H, CH2), 3.30 (m, 2H, CH2), 4.37 (s, 2H, S-CH2), 7.31 (m, 5H, Ar-H), 7.50 (d, 2H, Ar-H), 8.39 (d, 2H, Ar-H).
4.1.4.2. 2-Methylthio-3-(4-nitrophenyl)-6,7,8,9-hexahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (13)
Using methyl iodide (0.21 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 77%, melting point = 223–225 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.60 (m, 4H, 2CH2), 1.85 (m, 2H, CH2), 2.82 (m, 2H, CH2), 3.17 (m, 2H, CH2), 3.85 (s, 3H, CH3), 7.73 (d, J = 7.1 Hz, 2H, Ar-H). 8.36 (d, J = 7.1 Hz, 2H, Ar-H).
4.1.4.3. 2-Cyclopentythio-3-(4-nitrophenyl)-6,7,8,9-hexahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (14)
Using cyclopentyl bromide (0.15 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 71%, melting point = 199–201 °C. 1H NMR (400 MHz, CDCl3): δ 1.45–2.12 (m, 14H, 7CH2), 2.85–3.18 (m, 4H, 2CH2), 3.93 (p, 1H, CH), 7.78 (d, J = 8.0 Hz, 2H, Ar-H), 8.40 (d, J = 8.0 Hz, 2H, Ar-H). 13C NMR (100 MHz, DMSO-d6): 21.13, 27.26, 27.52, 27.85, 29.40, 32.39, 79.35, 74.79, 119.77, 125.27, 131.82, 136.28, 137.06, 141.88, 148.79, 154.78, 158.09, 160.20.
4.1.4.4. Ethyl 2-[3-(4-Nitrophenyl)-4-oxo-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl]thioacetate (15)
Using ethyl chloroacetate (0.18 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 88%, melting point = 213–215 °C, IR (KBr, υ, cm–1): 1350, 1519 (NO2), 1679 (C=O, amide), 1733 (CO, ester). 1H NMR (400 MHz, DMSO-d6): δ 1.22 (t, J = 7.1 Hz, 3H, CH2CH3), 1.60 (m, 4H, 2CH2), 1.81 (m, 2H, CH2), 2.83 (m, 2H, CH2), 3.17 (m, 2H, CH2), 3.96 (s, 2H, CH2-S), 4.13 (q, J = 7.1 Hz, 2H, CH2CH3), 7.83 (d, J = 8.8 Hz, 2H, Ar-H), 8.44 (d, J = 8.8 Hz, 2H, Ar-H).
4.1.4.5. 3-(4-Nitrophenyl)-2-(prop-2-yn-1-ylthio)-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-(5H)-one (16)
Using propargyl bromide (0.17 g, 1.5 mmol) as the alkyl halide yielded a white solid; yield = 86%, melting point = 204–206 °C, IR (KBr, υ, cm–1): 1350, 1519 (NO2), 1682 (C=O), 3287 (CH SP). 1H NMR (400 MHz, DMSO-d6): δ 1.61(m, 4H, 2CH2), 1.81(m, 2H, CH2), 2.86 (m, 2H, CH2), 3.18 (m, 2H, CH2), 3.22 (s, 1H, −CHS−), 3.95 (s, 1H, −CHS−), 7.82 (d, J = 8.7 Hz, 2H, Ar-H). 8.42 (d, J = 8.6 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): 24.90, 27.27, 27.56, 27.87, 29.40, 32.41, 32.94, 45.60, 74.79, 119.51, 125.15, 131.77, 135.73, 137.01, 142.46, 148.59, 156.83, 158.21, 160.66.
4.1.5. Synthesis of 2-(4-Oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thioacetohydrazide (17)
A mixture of thioacetate 11 (0.41 g, 1 mmol) and hydrazine hydrate (0.10 g, 2 mmol) in absolute ethanol (4 mL) was stirred for 12 h at room temperature. When the TLC showed that the reaction was complete, filtration and purification using column chromatography (ethyl acetate/petroleum ether) gave a white solid, yield = 58%, melting point = 240–242 °C, IR (KBr, υ, cm–1): 1514 (olefinic C=C), 1645 (CO, hydrazide), 1687 (C=O, amide), 3263, 3323 (NH2). 1H NMR (400 MHz, DMSO-d6): δ 1.60 (m, 4H, 2CH2), 1.83 (m, 2H, CH2), 2.84 (m, 2H, CH2), 3.19 (m, 2H, CH2), 3.78 (s, 2H, CH2-S), 4.26 (s, 2H, NH2; exchangeable with D2O), 7.42 (m, 2H, Ar-H), 7.57 (m, 3H, Ar-H), 9.29 (s, 1H, NH; exchangeable with D2O).
4.1.6. General Procedure for the Synthesis of N′-Arylidene-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazide (18–23)
A hydrazide 17 suspension (0.43 g, 1 mmol) in absolute ethanol (3 mL) was added to the appropriate aromatic aldehyde (1 mmol). It was heated at 70 °C for 24 h. When the TLC showed that the reaction was complete, the separated solid was filtered and purified with silica gel chromatography (ethyl acetate/petroleum ether).
4.1.6.1. Synthesis of N′-(4-Hydroxybenzylidene)-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazid (18)
Using 4-hydroxybenzaldehyde (0.12 g) yielded a white solid; yield = 89%, melting point = 267–269 °C, IR (KBr, υ, cm–1): 1607 (C=N), 1649 (C=O, hydrazide), 1688 (C=O, amide), 3067 (NH), 3261 (OH). 1H NMR (400 MHz, DMSO-d6): δ 1.55 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.82 (m, 2H, CH2), 3.18 (m, 2H, CH2), 3.92 and 4.35 (s, 2H, S-CH2, trans/cis conformers), 6.81 (m, 2H, Ar-H), 7.43 (m, 4H, Ar-H), 7.58 (m, 3H, Ar-H), 7.90 and 8.07 (s, 1H, N=CH, trans/cis conformers), 9.89 (s, 1H, NH, trans/cis conformers), 11.45 and 11.55 (s, 1H, OH). 13C NMR (100 MHz, DMSO-d6): 27.26, 27.49, 27.85, 29.37, 32.42, 34.80, 116.14, 119.50, 125.48, 129.04, 129.35, 129.83, 130.01, 135.67, 136.17, 136.97, 144.29, 157.30, 158.40, 159.70, 160.29, 168.60.
4.1.6.2. Synthesis of N′-(3-Hydroxy-4-methoxybenzylidene)-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]aceto- hydrazide (19)
Using 3-hydroxy-4-methoxybenzaldehyde (0.15 g) yielded a white solid; yield = 87%, melting point = 247–249 °C, IR (KBr, υ, cm–1): 1514 (olefinic C=C), 1585 (C=N), 1677 (C=O-amide), 3099 (NH), 3261 (OH). 1H NMR (400 MHz, DMSO-d6): δ 1.59 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.82 (m, 2H, CH2), 3.14 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 3.91 and 4.35 (s, 2H, S-CH2, trans/cis conformers), 6.98 (d, J = 7.3 Hz, 2H, Ar-H), 7.17 (s, 1H, Ar-H), 7.52 (m, 5H, Ar-H), 7.87 and 8.03 (s, 1H, N=CH, trans/cis conformers), 9.27 and 9.34 (s, 1H, NH, trans/cis conformers), 11.48 and 11.58 (s, 1H, OH, trans/cis conformers). 13C NMR (100 MHz, DMSO-d6): 27.26, 27.49, 27.85, 29.37, 32.42, 34.80, 116.14, 119.50, 125.48, 129.04, 129.35, 129.83, 130.01, 135.67, 136.17, 136.97, 144.29, 157.30, 158.40, 159.70, 160.29, 168.60.
4.1.6.3. Synthesis of N′-(4-Fluorobenzylidene)-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazid (20)
Using 4-flourobenzaldehyde (0.12 g) yielded a white solid; yield = 90%, melting point = 247–249 °C, IR (KBr, υ, cm–1): 1514 (olefinic C=C), 1676 (C=O, amide), 3069 (NH). 1H NMR (400 MHz, DMSO-d6): δ 1.58 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.81 (m, 2H, CH2), 3.18 (m, 2H, CH2), 3.95 and 4.37 (s, 2H, s, 2H, S-CH2, trans/cis conformers), 7.28 (t, J = 5.8 Hz, 2H, Ar-H), 7.43 (t, J = 5.8 Hz, 2H, Ar-H) 7.58 (m, 2H, Ar-H), 7.73 (m, 3H, Ar-H), 8.01 and 8.19 (s, 1H, N=CH, trans/cis conformers), 11.66 and 11.77 (s, 1H, NH, trans/cis conformers).
4.1.6.4. Synthesis of N′-(4-Nitrobenzylidene)-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazid (21)
Using 4-nitrobenzaldehyde (0.15 g) yielded a gray solid; yield = 85%, melting point = 232–234 °C, IR (KBr, υ, cm–1): 1340, 1515 (NO2), 1591 (olefinic C=C), 1680 (C=O, amide), 3079 (NH). 1H NMR (400 MHz, DMSO-d6): δ 1.58 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.81 (m, 2H, CH2), 3.19 (m, 2H, CH2), 3.99 and 4.41 (s, 2H, s, 2H, S-CH2, trans/cis conformers), 7.43 (d, J = 6.4 Hz, 2H, Ar-H) 7.58 (d, J = 6.6 Hz, 3H, Ar-H), 7.95 (m, 2H, Ar-H), 8.13 (s, 1H, N=CH), 8.28 (m, 2H, Ar-H), 11.97 (s, 1H, NH; D2O exchangeable).
4.1.6.5. Synthesis of N′-(3-Nitrobenzylidene)-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazid (22)
Using 4-nitrobenzaldehyde (0.15 g, 1 mmol) yielded a gray solid; yield = 88%, melting point = 250–252 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.58 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.81 (m, 2H, CH2), 3.18 (m, 2H, CH2), 3.92 and 4.43 (s, 2H, s, 2H, S-CH2, trans/cis conformers), 7.45 (m, 3H, Ar-H), 7.58 (d, J = 6.7 Hz, 2H, Ar-H) 7.74 (s, 1H, Ar-H), 8.21 (m, 3H, Ar-H), 8.51 and 8.53 (s, 1H, N=CH, trans/cis conformers), 11.90 and 12.03 (s, 1H, NH, trans/cis conformers). 13C NMR (100 MHz, DMSO-d6): 27.29, 27.47, 27.83, 29.15, 29.36, 32.39, 119.53, 129.80, 130.04, 130.09, 130.94, 133.49, 135.74, 136.98, 136.98, 141.75, 144.68, 157.30, 158.41, 159.11, 160.21, 169.37.
4.1.6.6. Synthesis of N′-[4-(Dimethylamino)benzylidene]-2-[(4-oxo-3-phenyl-6,7,8,9-tetrahydro-3H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-2-yl)thio]acetohydrazide (23)
Using 4-dimethyl aminobenzaldehyde (0.15 g, 1 mmol) yielded a white solid; yield = 86%, melting point = 229–231 °C, IR (KBr, υ, cm–1): 1513 (olefinic C=C), 1606 (CO– hydrazide), 1677 (C=O, amide), 3083 (NH). 1H NMR (400 MHz, DMSO-d6): δ 1.58 (m, 4H, 2CH2), 1.84 (m, 2H, CH2), 2.81 (m, 2H, CH2), 2.97 (s, 6H, 2CH3), 3.18 (m, 2H, CH2), 3.92–4.34 (s, 2H, S-CH2, trans/cis conformers), 6.72 (m, 2H, Ar-H), 7.43 (m, 3H, Ar-H) 7.55 (m, 4H, Ar-H), 7.87 and 8.03 (s, 1H, N=CH, trans/cis conformers), 11.36 and 11.44(s, 1H, NH, trans/cis conformers). 13C NMR (100 MHz, DMSO-d6): δ 18.65, 27.18, 29.30, 29.37, 32.42, 56.67, 112.18, 128.71, 129.60, 130.17, 136.87, 137.01, 145.14, 147.71, 152.38, 157.28, 159.98, 163.47, 166.41, 168.60.
4.1.7. Synthesis of 2-{[2-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-oxoethyl]thio}-3-phenyl-3,5,6,7,8,9-hexahydro-4H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-one (24)
To hydrazide 17 (1.23 g, 3 mmol) in a minimum volume of hot anhydrous ethanol, acetylacetone (0.30 g, 3 mmol) was added. The reaction mixture was stirred for 3 days at room temperature, followed by evaporation of the solvent at room temperature. The obtained residue was collected and recrystallized from n-hexane to give a white solid; yield = 91%, melting point = 204–206 °C, IR (KBr, υ, cm–1): 1512 (olefinic C=C), 1653 (SCH2CO–N), 1690 (C=O, amide). 1H NMR (400 MHz, DMSO-d6): δ 1.55 (m, 4H, 2CH2), 1.70 (S, 3H, CH3), 1.85 (m, 2H, CH2), 1.99 (S, 3H, CH3), 2.84 (m, 2H, CH2), 3.18 (m, 2H, CH2), 4.14–4.27 (s, 2H, CH2-S), 6.39 (s, 1H, CH, pyrazole), 7.42 (m, 2H, Ar-H), 7.57 (m, 3H, Ar-H). 13C NMR (100 MHz, DMSO-d6): δ 16.35, 26.35, 27.26, 27.49, 27.85, 29.37, 32.41, 37.43, 52.51, 90.94, 119.48, 129.80, 130.03, 130.38, 135.65, 136.29, 136.29, 136.92, 155.95, 157.38, 158.38, 160.35, 164.32.
4.2. Biology
4.2.1. In Vitro Cytotoxicity Screening
Novel synthesized compounds were evaluated for their cytotoxic activities against the 60 cell lines at a single dose of 10 μM. The output from the single dose screening is represented as mean-graph charts of the percentage growth of survived cells and expressed as growth inhibition (GI %). Compounds with significant GI % are prepared at five different concentration levels and evaluated against the 60-cell panel.26
4.2.2. Human Dihydrofolate Reductase (hDHFR) Inhibition Assay
To verify the DHFR activity, an enzyme-linked immunosorbent assay (ELISA) was carried out. In this assay and at room temperature, addition of the standard and tested compounds to the plate wells that were coated with a specific antibody for hDHFR was done. The mean absorbance values of the standard solution and that of the tested compounds were detected. The absorbances and the standard concentrations were represented on the standard curve. In this curve, absorbances were represented on the Y-axis, and the concentrations were represented on the X-axis. The concentration/inhibition curve was utilized for detecting the IC50 values of the tested compounds.27
4.2.3. Analysis of Cell-Cycle Arrest
Measurement of the DNA content was dependent on flow cytometry analysis. The SNB-75 cell was treated with compound 20 in its exponential growth for 24 h. At the end of the incubation period, collection, centrifugation, and fixing of the cell with ice-cold ethanol were performed. The cell was treated with a buffer that contains 0.1% Triton X-100 and was PI-stained. A comparison with the control treated with DMSO was done, in which flow cytometry was used to measure the DNA content.
4.2.4. Apoptotic Assay
In the exponential growth phase, the SNB-75 cell was treated and incubated for 24 h with compound 20. After that, the cells were suspended in a binding buffer, annexin V-FITC, to determine apoptosis and also suspended in PI to determine necrosis. To produce stability of the photo and enhance the signal of fluorescence, the annexin V-FITC conjugate was engineered. Flow cytometry was used to detect and analyze annexin V-FITC binding.28
4.2.5. Detection of Caspase-3 and Caspase-9 Protein Levels
The Invitrogen caspases-3 and -9 human kits (ELISA) were used to detect levels of human active caspases-3 and 9. To lyse the cell, an extraction buffer was added to the cells besides the tested compound. Incubation was carried out at room temperature, and washing of the wells was done 4 times. Incubation again at room temperature for 2 h was performed after active Invitrogen caspase-3 addition. Wells were precisely washed; the chromogen and horseradish peroxidase (HRP) solution (antirabbit IgG) were added and incubated for 30 min at room temperature. At 450 nm, the absorbance was measured.24
4.3. Molecular Modeling Methodology
A molecular docking analysis was performed using MOE software version 2014.09 in accordance with the standard MOE procedure.29 The crystal structure of the DHFR interacting with its inhibitor MTX was obtained from the RSC Protein Data Bank (PDB code 1U72).30,31 Construction of the three DHFR inhibitors (20, 22, and 23) was performed using the library of fragments in the MOE program. The “Amber force field” was used to minimize the energy of the inhibitors. The semiempirical mechanical calculation method “AM1” was adopted for assigning the partial atomic charges. The stochastic conformational search module was used to carry out the stochastic conformational search. All of these methods are performed using MOE program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06078.
Percent growth inhibition of selected compounds; 13C NMR charts; and 1H NMR charts (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ng H.-L.; Chen S.; Chew E.-H.; Chui W.-K. Applying the designed multiple ligands approach to inhibit dihydrofolate reductase and thioredoxin reductase for anti-proliferative activity. Eur. J. Med. Chem. 2016, 115, 63–74. 10.1016/j.ejmech.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Raimondi M. V.; Randazzo O.; Franca M. L.; Barone G.; Vignoni E.; Rossi D.; Collina S. DHFR inhibitors: reading the past for discovering novel anticancer agents. Molecules 2019, 24, 1140. 10.3390/molecules24061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhou X.; Kisliuk R. L.; Piraino J.; Cody V.; Gangjee A. Design, synthesis, biological evaluation and X-ray crystal structure of novel classical 6,5,6-tricyclic benzo [4,5] thieno [2,3-d] pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors. Bioorg. Med. Chem. 2011, 19, 3585–3594. 10.1016/j.bmc.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-R.; Peng B.; Cao S. L.; et al. Synthesis, cytotoxic evaluation and target identification of thieno [2,3-d] pyrimidine derivatives with a dithiocarbamate side chain at C2 position. Eur. J. Med. Chem. 2018, 154, 324–340. 10.1016/j.ejmech.2018.05.028. [DOI] [PubMed] [Google Scholar]
- Gangjee A.; Qiu Y.; Li W.; Kisliuk R. L. Potent dual thymidylate synthase and dihydrofolate reductase inhibitors: classical and nonclassical 2-amino-4-oxo-5-arylthio-substituted-6-methylthieno [2,3-d] pyrimidine antifolates. J. Med. Chem. 2008, 51, 5789–5797. 10.1021/jm8006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangjee A.; Li W.; Kisliuk R. L.; Cody V.; Pace J.; Piraino J.; Makin J. Design, synthesis, and X-ray crystal structure of classical and nonclassical 2-amino-4-oxo-5-substituted-6-ethylthieno [2,3-d] pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors and as potential antitumor agents. J. Med. Chem. 2009, 52, 4892–4902. 10.1021/jm900490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharaky M.; Kamel M.; Aziz M. A.; Omran M.; Rageh M. M.; Abouzid K. A. M.; Shoumana S. A. Design, synthesis and biological evaluation of a new thieno [2,3-d] pyrimidine-based urea derivative with potential antitumor activity against tamoxifen sensitive and resistant breast cancer cell lines. J. Enzyme Inhib. Med. Chem. 2020, 35, 1641–1656. 10.1080/14756366.2020.1804383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y.; Zhou X.; Desmoulin S. K.; Wu J.; Cherian Ch.; Hou Z.; Matherly L. H.; Gangjee A. Synthesis and biological activity of a novel series of 6-substituted thieno [2,3-d] pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high transporter for cellular entry. J. Med. Chem. 2009, 52, 2940–2951. 10.1021/jm8011323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puterová Z.; Krutošíková A.; Gewald D. V. Gewald reaction: synthesis, properties and applications of substituted 2-aminothiophenes. Arkivoc 2010, 2010, 209–246. 10.3998/ark.5550190.0011.105. [DOI] [Google Scholar]
- Gineinah M. M.; Nasr M. N. A.; Badr S. M. I.; El-Husseiny W. M. Synthesis and antitumor activity of new pyrido [2,3-d] pyrimidine derivatives. Med. Chem. Res. 2013, 22, 3943–3952. 10.1007/s00044-012-0396-0. [DOI] [Google Scholar]
- Wutticharoenwong K.; Soucek M. D. Influence of the Thiol Structure on the Kinetics of Thiol-ene Photopolymerization with Time-Resolved Infrared Spectroscopy. Macromol. Mater. Eng. 2008, 293, 45–56. 10.1002/mame.200700175. [DOI] [Google Scholar]
- Al-Amiery A. A.; Musa A. Y.; Kadhum A. H.; Mohamad A. The use of umbelliferone in the synthesis of new heterocyclic compounds. Molecules 2011, 16, 6833–6843. 10.3390/molecules16086833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socea L. I.; et al. New Heterocyclic Compounds from 1,2,4-triazoles and 1,3,4-Oxadiazoles Class Containing 5H-dibenzo [a,d][7] Annulene Moiety. Rev. Chem. 2017, 68, 2761–2764. 10.37358/RC.17.12.5972. [DOI] [Google Scholar]
- Al-Abdullah E. S.; Asiri H. H.; Lahsasni S.; Habib E. E.; Ibrahim T. M.; El-Emam A. Synthesis, antimicrobial, and anti-inflammatory activity, of novel S-substituted and N-substituted 5-(1-adamantyl)-1,2,4-triazole-3-thiols. Drug Des. Dev. Ther. 2014, 8, 505–518. 10.2147/DDDT.S62465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana J. M.; Sahoo P. K. Chemoselective alkylation of thiols: a detailed investigation of reactions of thiols with halides. Synth. Commun. 1992, 22, 1691–1702. 10.1080/00397919208020489. [DOI] [Google Scholar]
- Amr A. E.-G. E.; Sabry N. M.; Abdulla M. M. Synthesis, reactions, and anti-inflammatory activity of heterocyclic systems fused to a thiophene moiety using citrazinic acid as synthon. Monatsh. Chem. 2007, 138, 699–707. 10.1007/s00706-007-0651-0. [DOI] [Google Scholar]
- El-Azab A. S.; Abdel-Hamide S. G.; Sayed-Ahmed M. M.; Hassan Gh. S.; El-Hadiyah T. M.; Al-Shabanah O. A.; Al-Deeb O. A.; El-Subbagh H. I. Novel 4 (3H)-quinazolinone analogs: synthesis and anticonvulsant activity. Med. Chem. Res., 2013, 22, 2815–2827. 10.1007/s00044-012-0280-y. [DOI] [Google Scholar]
- Rando D. G.; Sato D. N.; Siqueira L.; Malvezzi A.; Leite C. Q. F.; Amaral A. T. d.; Ferreira E. I.; Tavares L. C. Potential tuberculostatic agents. Topliss application on benzoic acid [(5-nitro-thiophen-2-yl)-methylene]hydrazide series. Bioorg. Med. Chem. 2002, 10, 557–560. 10.1016/S0968-0896(01)00313-3. [DOI] [PubMed] [Google Scholar]
- Zerov A. V.; Krupenya T. S.; Petrov A. A.; Yakimovich S. I. Reaction of trifluoromethyl-containing 1,3-dicarbonyl compounds with bis-hydrazides. Russ. J. Org. Chem. 2016, 52, 312–318. 10.1134/S1070428016030039. [DOI] [Google Scholar]
- Grever M. R.; Schepartz S. A.; Chabner B. A. The National Cancer Institute: cancer drug discovery and development program. Semin. Oncol. 1992, 19, 622–638. [PubMed] [Google Scholar]
- Boyd M. R.; Paull K. D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. 10.1002/ddr.430340203. [DOI] [Google Scholar]
- Al-Rashood S. T.; Aboldahab I. A.; Nagi M. N.; Abouzeid L. A.; Abdel-Aziz A. A. M.; Abdel-Hamide S. G.; Youssef K. M.; Al-Obaid A. M.; El-Subbagh H. I. Synthesis, dihydrofolate reductase inhibition, antitumor testing, and molecular modeling study of some new 4 (3H)-quinazolinone analogs. Bioorg. Med. Chem. 2006, 14, 8608–8621. 10.1016/j.bmc.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Fustero S.; Sánchez M.; Barrio P.; Simón A. A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- Scudiero D. A.; Shoemaker R. H.; Paull K. D.; Monks A.; Tierney S.; Nofziger T. H.; Currens M. J.; Seniff D. M.; Boyd R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [PubMed] [Google Scholar]
- Kathiravan M. K.; Shishoo C. J.; Chitre T. S.; Mahadik K. R.; Jain K. S. Efficient Synthesis of Substituted 2-Amino-3-carbethoxythiophenes. Synth. Commun. 2007, 37, 4273–4279. 10.1080/00397910701575889. [DOI] [Google Scholar]
- Hassan G. S.; El-Messery S. M.; Al-Omary F. A. M.; El-Subbagh H. I. Substituted thiazoles VII. Synthesis and antitumor activity of certain 2-(substituted amino)-4-phenyl-1,3-thiazole analogs. Bioorg. Med. Chem. Lett. 2012, 22, 6318–6323. 10.1016/j.bmcl.2012.08.095. [DOI] [PubMed] [Google Scholar]
- Priola J. J.; Calzadilla N.; Baumann M.; Borth N.; Tate C. G.; Betenbaugh M. J. High-throughput screening and selection of mammalian cells for enhanced protein production. Biotechnol. J. 2016, 11, 853–865. 10.1002/biot.201500579. [DOI] [PubMed] [Google Scholar]
- Ou J.; Yu Z.; Qiu M.; et al. Knockdown of VEGFR2 inhibits proliferation and induces apoptosis in hemangioma-derived endothelial cells. Eur. J. Histochem. 2014, 58, 2263 10.4081/ejh.2014.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T.-S.; Le M.-T.; Tran T.-D.; Tran T.-H.; Thai K.-M. Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches. Molecules 2020, 25, 3644. 10.3390/molecules25163644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelaziz O. A.; El Husseiny W. M.; Selim K. B.; Eisa H. M. Dihydrofolate reductase inhibition effect of 5-substituted pyrido [2,3-d] pyrimidines: Synthesis, antitumor activity and molecular modeling study. Bioorg. Chem. 2019, 90, 103076 10.1016/j.bioorg.2019.103076. [DOI] [PubMed] [Google Scholar]
- Li H.; Chen S.; Chew E.-H.; Chui W.-K.; et al. Inhibitors of dihydrofolate reductase as antitumor agents: design, synthesis and biological evaluation of a series of novel nonclassical 6-substituted pyrido [2,3-d] pyrimidines with a three-to five-carbon bridge. Bioorg. Med. Chem. 2018, 26, 2674–2685. 10.1016/j.bmc.2018.04.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.