

Abstract

Humins are carbonaceous, polymeric byproducts formed during the acid-catalyzed condensed phase transformation of biomass-derived moieties and are responsible for significant carbon loss and catalyst deactivation. There exists very limited knowledge about their formation chemistry and composition. Infrared spectra of humins formed during the dehydration of glucose/fructose to 5-HMF show that the furan ring and the hydroxy methyl group of 5-HMF are present in humins, but the carbonyl group is not. Based on this, aldol addition and condensation between 5-HMF and other derived species are proposed as the main reactions that initiate humin formation. Hence, in this work, density functional theory (DFT)-based calculations are performed to compute the reaction pathways, activation barriers, and reaction free energies associated with all elementary reaction steps in the 5HMF-initiated, acid-catalyzed reactions leading to humin formation. The humin formation is initiated with the rehydration of HMF to form 2,5-dioxo-6-hydroxy-hexanal or DHH (key promoter of humin formation), followed by its keto–enol tautomerization and aldol addition and condensation with HMF. The rate-determining step in this pathway is the aldol-addition reaction between the DHH-derived enols with 5-HMF. Within the implicit solvation approximation, the formation of the 5-HMF-DHH dimer is slightly endergonic, whereas the 5-HMF rehydration leading to DHH is thermodynamically downhill. This mechanistic understanding of initiation reactions for humins could pave the way to screen and design solvent and catalyst systems to deter their formation.
1. Introduction
In the recent past, second-generation lignocellulosic biomass has attracted much attention worldwide as a renewable source of carbon to produce chemicals.1 It is the only sustainable source of carbon that can be transformed into chemical feedstock.2−4 Lignocellulosic biomass consists of three main structural units: hemicellulose, lignin, and cellulose.5 Cellulose is a polymer of glucose units connected by glycoside linkages that accounts for 40–50% of a typical lignocellulosic feedstock.6 The selective transformation of biomass feedstocks to chemicals is carried out mainly in the condensed phase at moderate conditions (temperature below 400 K) and in the presence of homogeneous and heterogeneous catalysts.7 Biomass molecules undergo a wide variety of reactions, such as isomerization, hydrolysis, hydrogenation, dehydration, and aldol addition, to name a few, to obtain valuable platform chemicals such as 5-hydroxymethylfurfural (5-HMF), furfural, tetrahydro furfural (THF), glyceraldehyde (GHA), and levulinic acid (LA).8 Among these, 5-HMF is considered a key building block for the polymer industry and for producing biofuels.9,10 The acid-catalyzed dehydration of cellulose-derived carbohydrates to furans has been extensively studied in the literature.11−13 However, the dehydration of biomass species to 5-HMF also leads to the formation of humins as a byproduct.14 Humins are polymeric, carbonaceous species that are formed during catalytic deconstruction and conversion reactions of cellulosic biomass. Their formation lowers the selectivity toward the desired platform chemicals, causes significant carbon loss, deactivates catalysts, and may cause severe processing and separation problems.15 Humin formation also drastically decreases the selectivity toward 5-HMF in sugar dehydration, since 10–50 wt % of the initial substrate is converted to humins.16 Deposition of humins may also damage the reactor and its components.17−21 The process economics of biorefineries can be significantly improved by inhibiting the formation of humins and this requires a thorough understanding of their formation process and molecular structure. However, the majority of the focus in the biomass community has been on improving the selectivity of the desired products, with little attention to the chemistry of the formation of the undesired product, humins. The addition of aprotic solvents to the aqueous reaction phase during the dehydration of sugars has been shown to improve the selectivity toward 5-HMF though.22−25
Investigating humin formation is challenging due to their complex and unknown molecular structure. Likewise, during the condensed phase catalytic conversion of biomass, several reactions occur simultaneously, making it difficult to isolate humin formation reactions. Since humins are polymeric and insoluble oligomers, it is challenging to determine their chemical structure and identify the intermediates that can potentially lead to their formation. It has been proposed that humins formed during the catalytic dehydration reactions of cellulose-derived biomass molecules consist mainly of furan rings and aliphatic linkers and are rich in functional groups such as carboxyl, carbonyl, and hydroxyl.26 Moreover, Cheng et al.27 reported that a certain fraction of humins is a conglomerate of oligomers interacting weakly rather than being large macromolecules. Zandvoort et al.28 characterized humins derived from glucose, fructose, and xylose in aqueous sulfuric acid. Based on Fourier transform infrared (FTIR) spectroscopy, it was suggested that humin formation during the transformation of hexoses occurs via 5-HMF, which further undergoes polycondensation processes by electrophilic substitution with the formation of ether or acetal bonds between the furan rings. However, Dee and Bell29 performed kinetic studies during the acid-catalyzed hydrolysis of cellulose, where glucose is the main intermediate species undergoing dehydration to 5-HMF. A corollary of that work was that the reduction of humin formation was attributed to the decrease of 5-HMF formation, suggesting that humins are formed due to the polymerization and condensation of 5-HMF and glucose. Patil and Lund30,31 based on the IR spectra of acid-catalyzed humins formed from 5-HMF, fructose, and glucose showed that the furan ring and the hydroxyl methyl group of 5-HMF were present in humins but the carbonyl group was not. Thus, the absence of the carbonyl group in humins suggested that 5-HMF is polymerized through its carbonyl group. Additionally, it was observed that humins were derived from 5-HMF and not from levulinic acid (LA) or formic acid (FA), which are rehydration products of 5-HMF. Their findings are consistent with a mechanism proposed by Horvat32 for identifying 2,5-dioxo-6-hydroxy-hexanal (DHH) as a promoter of humin formation, which, in turn, is formed from the hydration of 5-HMF. According to this, aldol addition and condensation are proposed as the main reaction pathways between DHH and 5-HMF or any carbohydrate molecule present in the reacting system to initiate humin formation. An overall pathway for the conversion of cellulose-derived glucose to 5-HMF and subsequent degradation of 5-HMF to humins and other products is shown in Scheme 1.
Scheme 1. Glucose Conversion Pathway to 5-HMF and the Rehydration of 5-HMF to LA and DHH Leading to the Formation of Humins.

To summarize, there is sufficient experimental evidence to conclude that 5-HMF is a precursor of humin formation in the sugar dehydration reaction and to suggest that rehydration and addition–condensation reactions between 5-HMF and its derivatives initiate humin formation. Hence, to investigate the humin formation chemistry in detail and to explore the kinetic and thermodynamic feasibility of the reaction pathways, density functional theory (DFT)-based calculations are performed in this paper. Individual elementary reaction steps are simulated and associated activation free energy barriers and reaction free energies are computed. We aim to investigate aldol addition/condensation as a primary humin growth reaction, preceded by the hydration of 5-HMF and its keto–enol tautomerization to DHH. DHH is suggested to be a key intermediate leading to humin formation. We also endeavor to get an insight into the effect of the solvation environment on the energetics of humin formation, using implicit water and dimethyl sulfoxide (DMSO) solvent models.
2. Methodology
DFT calculations are performed using Gaussian 09 code.33 The hybrid functional RM06-2X is used in combination with the 6-311++G (2d, p) basis set. This theory level is deemed sufficient to study the chemistry of cellulose-derived molecules.34,35 For the geometry optimization and transition-state (TS) calculations, no constraints are applied to the atoms. The TS search is performed using the Berny algorithm, as implemented in the Gaussian code. Geometry optimization is accompanied by frequency calculations at the same level to distinguish the optimized structures as the TS and the local minima, indicated by the presence and absence of an imaginary frequency (which corresponds to the reaction coordinate), respectively. Intrinsic reaction coordinate (IRC) calculations are performed to confirm that the obtained TS is linked with the anticipated reactant and product on the potential energy surface. The polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM) is used during all geometry optimization calculations and TS search. Implicit solvation in the continuum model is implemented using the dielectric constant (ε) of DMSO and water. The reaction free energies in the gas phase and in the implicit solvent phase are reported at 433 K. Self-consistent field (SCF) selected for finding electronic structure configuration was very close equivalent to 1.00D-06 in convergence in energy change.
The Zundel cation,36 which is an important and simplified structure for the solvated proton H5O2+, was chosen as the acid catalyst.37 A conformational analysis was carried out to determine the ground state of the initial system formed by a DHH molecule interacting with a Zundel structure. All energies are reported in kJ/mol.
3. Results and Discussion
The complete reaction network investigated in this work, starting from 5-HMF, is shown in Scheme 2. The reaction steps involved are (i) the rehydration of 5-HMF to DHH; (ii) the keto–enol tautomerization of DHH, resulting in the formation of four enols 1,2-enol, 2,3-enol, 4,5-enol, and 5,6-enol; (iii) subsequent reaction of each enol with the protonated 5-HMF (5-HMF+) via aldol addition; and (iv) further two-step condensation reaction of the rest of the dimers except for those derived from the aldol-addition reaction between 1,2-enol and 5-HMF+. All of the reaction steps and intermediate species are numbered in Scheme 2 and referenced throughout the manuscript.
Scheme 2. Schematic Illustrating Reaction Pathways for Humin Formation through 5-HMF Rehydration to DHH, DHH Keto–Enol Tautomerization, and Further Dimerization Reaction with 5-HMF.

Carbon atoms are numbered throughout the scheme and each reaction intermediate, and the elementary reaction steps are numbered. Post-DHH formation, the reaction pathways corresponding to 1,2-enol, 2,3-enol, 4,5-enol, and 5,6-enol are color coded using blue, orange, green, and red arrows.
3.1. Rehydration of 5-HMF to form DHH
The hydration of 5-HMF to DHH follows the mechanism described by Markonikov’s electrophilic addition.38,39Scheme 3 shows the detailed reaction mechanism of DHH formation from 5-HMF, corresponding to steps 1–5 in Scheme 2. Figure 1 shows the corresponding free energy profile. Step 1 corresponds to the protonation at C4* of 5-HMF generating an electrophilic site at C5*. The protonation at C4* leads to the formation of intermediate 2. This proton addition at C4* is the rate-determining step for the rehydration of 5-HMF to DHH, with a free energy barrier of ∼93 kJ/mol in both water and DMSO implicit solvation. Further, hydroxylation at C5* leads to the formation of intermediate 3. Subsequently, C3* is protonated to form intermediate 4, promoting furan ring opening. Finally, the −OH group at C5* is deprotonated, leading to a carbonyl group, regeneration of the acid catalyst, and forming DHH, as species 5. The free energy profile reveals that it is an endergonic reaction, with ΔGR slightly more than 30 kJ/mol, in both DMSO and water implicit solvation, as shown in Figure 1.
Scheme 3. Hydration Reaction Mechanism of 5-HMF Leading to DHH Formation.

Blue arrows in the molecular structures indicate electron flow. Transition states of the reactions are depicted using square brackets and atoms not taking part in the reaction are faded. Transition states corresponding to the rehydration of 5-HMF are denoted with the letter R. Indices of the species and carbon atoms numbering correspond to those in Scheme 2.
Figure 1.

Free energy profile for the rehydration of 5-HMF to form DHH. Free energy barriers for each elementary step in water and DMSO are shown in blue and red, respectively. Transition states corresponding to the rehydration of 5-HMF are denoted with the letter R. Free energy values are reported in kJ/mol.
3.2. Keto–Enol Tautomerization of DHH
Keto–enol tautomerization is often involved in the transformation of biomass molecules, such as carboxylic acids and olefins, or the transformation of levoglucosan (LGA) to produce levoglucosenone (LGO), to name a few. DFT calculations have been implemented to investigate these reactions in the past.40−43 The reaction mechanisms for the keto–enol tautomerization of DHH are shown in Scheme 4. The keto–enol tautomerization of DHH takes place via a two-step mechanism. First, DHH is protonated at carbonyl groups at the C2 or C5 carbon, leading to intermediates 6 and 7, respectively. When the carbonyl group at C2 is protonated, intermediate 6 is produced, forming an electrophilic site at C2. Subsequently, the deprotonation at C1 or C3 takes place, leading to the formation of 1,2 or 2,3-enol, respectively. On the other hand, when the carbonyl group at C5 is protonated, intermediate 7 is produced, followed by deprotonation at C4 or C6, leading to the formation of 4,5 or 5,6-enol, respectively. This step restores the Zundel structure, which initially yielded a proton to initiate the keto–enol tautomerization of DHH. The free energy profile for all four possible keto–enol tautomerizations of DHH is shown in Figure 2. The free energy of an isolated 5-HMF is added to allow us to map the complete free energy profile with the subsequent bimolecular reactions. It can be observed that the protonation leading to 4,5-enol and 5,6-enol is kinetically more favorable, leading to intermediate 7 compared to the protonation leading to 1,2- and 2,3-enols through the formation of intermediate 6. Further, it is observed that the TS-3a corresponding to deprotonation at C1 in step 3a leading to 1,2-enol is the least favorable compared to other α-hydrogen transfer of enols studied. This is because the double bond between C1 and C2, just beside the carbonyl group, makes the molecule rigid. Additionally, 1,2-enol shown as intermediate 8 in Figure 2 is the least stable enol compared to other enols formed. This can be a consequence of the formation of a ketene group involving C1 and C2 carbon atoms. This group is more reactive compared to any ketone group formed in the case of 2,3-, 4,5-, or 5,6-enol.
Scheme 4. Reaction Mechanism for the Formation of the Four Possible Enols from DHH.

Blue arrows in the molecular structures indicate the electron flow. Transition states of the reactions are depicted using square brackets and atoms not taking part in the reaction are faded. Transition states corresponding to the keto–enol tautomerization are denoted with the letter E. Indices of the species and carbon atoms numbering correspond to those in Scheme 2.
Figure 2.

Free energy profile for the keto–enol tautomerization reactions. Activation free energy barriers in kJ/mol are also shown in the legend. Transition states corresponding to the keto–enol tautomerization are denoted with the letter E. The labeling of intermediates and transition states is the same as in Scheme 2.
Figure 3 shows the transition-state configurations for the two possible protonation reactions of DHH leading to two possible enolate ions, intermediates 6 and 7. In Figure 3a for E-TS1, it can be seen that the hydroxyl group close to the protonated carbonyl oxygen attached to C2 stabilizes the water cluster. However, for E-TS2 in Figure 3b, the two water molecules are well stabilized with the carbonyl group at C6. It is suggested that this difference in the intermolecular interaction between the DHH and the Zundel structure causes the difference in the activation free energies between the two possible protonation mechanisms of DHH.
Figure 3.

Transition states corresponding to the protonation reaction of the keto–enol tautomerization of DHH: (a) protonation of the C2 carbonyl group of DHH leading to intermediate 6 and (b) protonation of the C5 carbonyl group of DHH leading to intermediate 7. The blue dotted lines indicate the intermolecular interactions between the Zundel structure and DHH polar groups. Oxygen atoms (red), hydrogen atoms (white), and carbon atoms (gray) are shown.
3.3. Aldol-Addition Reaction between DHH and 5-HMF
5-HMF becomes electron deficient when the carbonyl group is protonated, leading to a nucleophilic addition with any enol available. The aldol-addition reaction between 5-HMF+ and the DHH-derived enols has been found to take place in a concerted manner. Scheme 5 shows 5-HMF protonation and four-aldol-addition reactions for each enol derived from DHH. In the case of 1,2-enol, the −OH bonded to C2 is deprotonated to restore the Zundel structure, while C2 is bonded to C1* to form the dimer. Similarly for 2,3-enol, the same −OH group bonded to C2 is deprotonated. However, in this case, C3 becomes electron deficient, reacting with C1* to form the corresponding dimer shown in Scheme 5c. In the case of 4,5- and 5,6-enols, the −OH bonded to C5 is deprotonated, while C4 and C6 now become nucleophilic carbons reacting with C1* of 5-HMF, respectively. Finally, the dimers are formed with a single carbon–carbon bond and a hydroxyl group present at the β-carbon atom, as shown for all of the reactions.
Scheme 5. Reaction Mechanism of the Aldol-Addition Reactions of (a) 1,2-Enol, (b) 2,3-Enol, (c) 4,5-Enol, and (d) 5,6-Enol with 5-HMF+. The Blue Arrows Indicate Electron Flow during the Reactions. Transition States of the Reactions Are Depicted Using Square Brackets and Atoms Not Taking Part in the Reaction Are Faded. Transition States Corresponding to the Aldol Addition Are Denoted with Letters AA. Indices of the Species and Carbon Atoms Numbering Correspond to Those in Scheme 2.
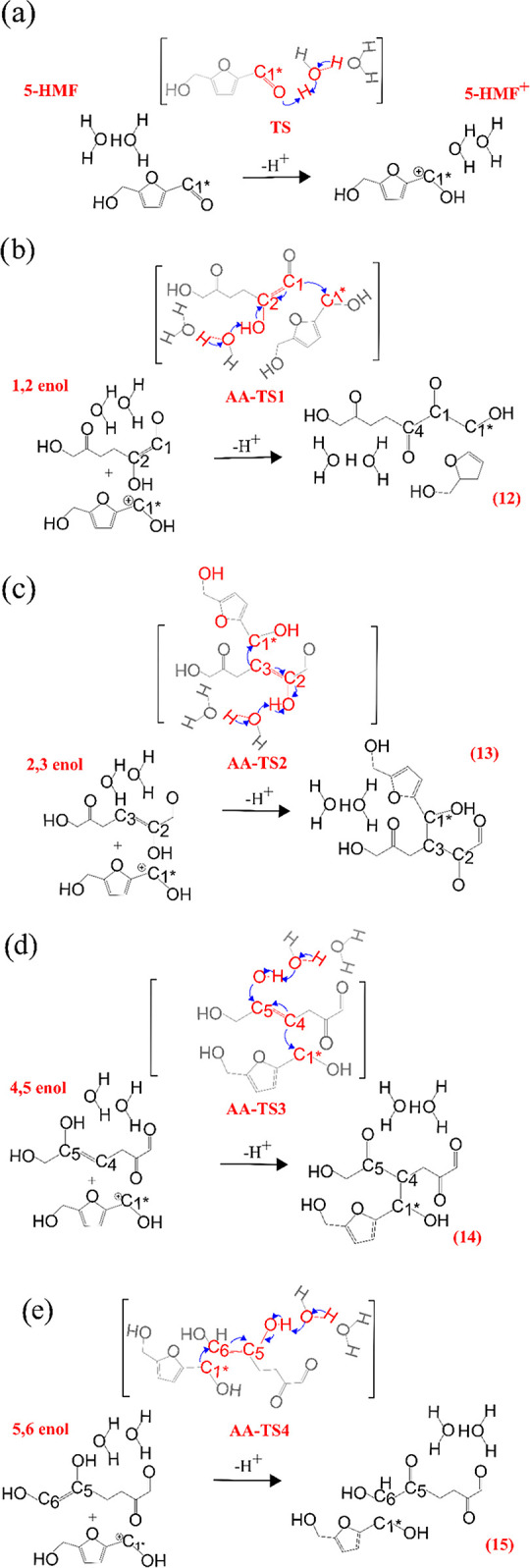
The free-energy barriers for the aldol-addition reactions between 5-HMF+ and the enols are reported in Figure 4. The barriers for the aldol-addition reaction of all enols with the HMF are between 90 and 100 kJ/mol. These values resemble the apparent activation energies of 94 ± 8.8 kJ/mol calculated by Patil et al30 from the kinetic data analysis for the rehydration of 5-HMF at 118–135 °C starting with 0.1 M in an acidic medium. The aldol-addition reaction is identified to be the rate-determining step with the highest energy barriers along the entire reaction pathway. Moreover, no significant differences in the barriers were found between DMSO and water, in the implicit solvent.
Figure 4.
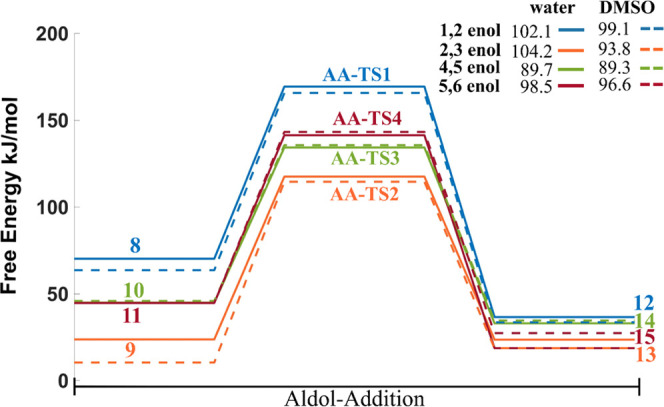
Free-energy profile for aldol-addition reactions. The numbers in the legend indicate the activation free energies. All energy values are reported in kJ.mol–1. Transition states corresponding to the aldol addition are denoted with letters AA. The labeling of intermediates and transition states is the same as in Scheme 2.
3.4. Condensation of Dimers Derived from Enols and 5-HMF Aldol Addition
This reaction proceeds in two steps: loss of hydroxyl group and the subsequent deprotonation of the dimer leading to a C–C double bond, thus producing a water molecule. This reaction only occurs for dimers 13, 14, and 15 due to the presence of α hydrogens (C atom which is α to the newly formed C=O group in the dimer). In the case of the dimer derived from 1,2-enol, the reaction does not take place due to the absence of an α hydrogen.
3.5. Dehydration Intermediate Reaction
Dehydration is the first step for the dimer. Scheme 6 describes the detailed reaction mechanism for the dehydration of the dimers, leading to the formation of humin precursors. The dehydration takes place in a single step involving the protonation of the −OH group attached to C1* and the loss of the hydroxyl group from the dimer, as can be seen in Scheme 6.
Scheme 6. Reaction Mechanism of the Dehydration Reactions for (a) 2,3-Enol-Derived Dimer, (b) 4,5-Enol-Derived Dimer, and (c) 5,6-Enol-Derived Dimer.

Transition states of the reactions are depicted using square brackets and atoms not taking part in the reaction are faded. Transition states corresponding to the dehydration reaction are denoted with the letter D. Indices of the species and carbon atoms numbering correspond to those in Scheme 2.
Energetics of the dehydration reaction is shown in Figure 5. The highest activation barrier is for the dehydration of the dimer derived from 4,5-enol. On the other hand, the kinetically most favorable dimer dehydration is that derived from 5,6-enol. Additionally, the difference in the free energy barriers between water and DMSO implicit solvation, for 4,5- and 5,6-enols, is approximately 20 kJ/mol.
Figure 5.
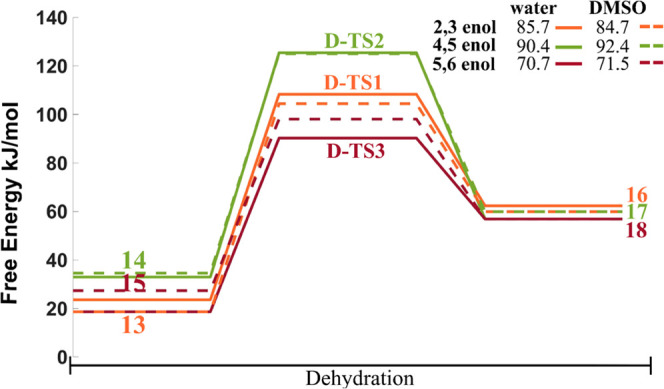
Free energy profile for the dehydration reactions of the dimers derived from 2,3-, 4,5-, and 5,6-enol aldol-addition reaction with 5-HMF. The numbers in the legend indicate the activation free energies. All energy values are reported in kJ.mol–1. Transition states corresponding to the dehydration reaction are denoted with letter D. The labeling of intermediates and transition states is the same as in Scheme 2.
As previously mentioned, the intermolecular interaction between the dimer and the Zundel structure can play a major role in altering the energetics of the reaction. Figure 6 shows the transition states for the three different dehydration reactions. The dehydration of the dimer derived from 5,6-enol has the lowest activation barrier because the −OH groups bonded to C6 and C1 stabilize the Zundel structure in the transition state. This does not occur in the case of the other dimers because of the lack of the −OH group in the immediate vicinity in the transition state. However, for 2,3-enol, the carbonyl group at C2 also stabilizes the water cluster, as shown in Figure 6a.
Figure 6.

Transition states corresponding to the dehydration reaction of (a) 2,3-enol-derived dimers, (b) 4,5-enol-derived dimers, and (c) 5,6-enol-derived dimers. The blue dotted lines indicate the intermolecular interactions between the Zundel structure and the dimer’s polar groups. Oxygen atoms (red), hydrogen atoms (white), and carbon atoms (gray).
3.6. Deprotonation as the Final Reaction Step
The last step in the reaction mechanism is the deprotonation of the dimer to restore the Zundel structure. It is observed in Scheme 7a for the case of dimer 16 (derived from 2,3-enol) that C3 loses the only hydrogen attached to it, forming a double bond with C1*. The same occurs for the rest of the dimers 17 and 18 (derived from 4,5- and 5,6-enols) where C4 and C6 are deprotonated, respectively.
Scheme 7. Reaction Mechanism of Deprotonation Reactions of (a) 2,3-Enol-Derived Dimer, (b) 4,5-Enol-Derived Dimer, and (c) 5,6-Enol-Derived Dimer.

The blue arrows indicate electron flow during the reactions. Transition states of the reactions are depicted using square brackets and atoms not taking part in the reaction are faded. Transition states corresponding to the deprotonation are denoted with letters DP. Indices of the species and carbon atoms numbering correspond to those in Scheme 2.
The free energy barriers corresponding to the deprotonation reaction are shown in Figure 7. A relatively higher energy barrier is required to deprotonate the dimer derived from 4,5-enol. Furthermore, no significant differences are observed between the free energy values of the reaction in water and DMSO implicit solvation.
Figure 7.

Free energy profile for the deprotonation reactions. The numbers in the legend indicate the activation free energies. All energy values are reported in kJ.mol–1. Transition states corresponding to the deprotonation are denoted with letters DP. The labeling of intermediates and transition states is the same as in Scheme 2.
The transition state structures for the deprotonation of the three dimers are shown in Figure 8. It is noticeable that during the deprotonation of the dimer derived from 4,5-enol, water molecules of the Zundel structure only have one polar group to be stabilized, which is the −OH bonded to C6*. The absence of more polar groups with which the water molecules can interact leads to a higher energy barrier. On the other hand, for cation 16 (dimer derived from 2,3-enol), water molecules are stabilized not only by the −OH bonded to C6* but also by the carbonyl group at C1. In the same way, cation 18 (dimer derived from 5,6-enol) has two −OH groups bonded to C6 and C6* with which the Zundel structure is well stabilized.
Figure 8.

Transition states corresponding to the deprotonation reactions of (a) 2,3-enol-derived dimer, (b) 4,5-enol-derived dimer, and (c) 5,6-enol-derived dimer. The blue dotted line indicates the intermolecular interactions between the Zundel structure and the dimer’s polar groups. Oxygen atoms (red), hydrogen atoms (white), and carbon atoms (gray).
3.7. Complete Free Energy Profile
The free energy profile for the entire reaction pathway presented in Scheme 2 is shown in Figure 9. Aldol-addition reactions have the highest activation free energy barriers in all of the pathways, except for the one in which 1,2-enol is involved. These free energy profiles also give us the thermodynamic perspective of the reactions. In general, reactions corresponding to steps 2, 3, and 5 (protonation, deprotonation, and dehydration) show positive reaction free energies, while the aldol-addition reaction as well as the final deprotonation of the dimers are thermodynamically downhill. However, the overall reaction is slightly endergonic. These results are in agreement with the values reported by Patil et al.30 This could be the reason that humin formation in the condensed phase biomass reactions usually occurs at temperatures above 100 °C.
Figure 9.
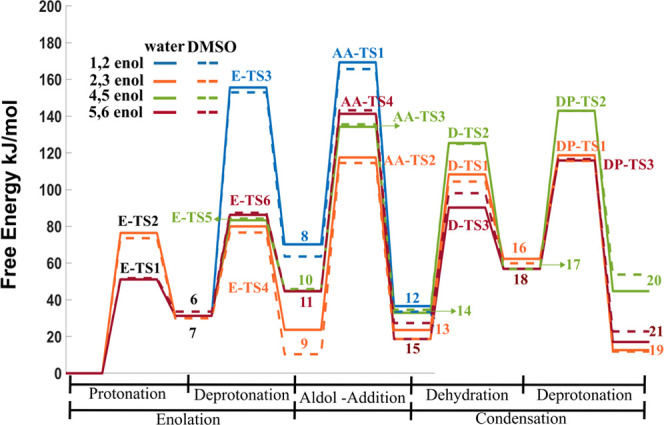
Free energy profile for all of the reactions leading to the formation of the final dimers, as a precursor for humin formation. The activation free energies are calculated with respect to DHH interacting with the Zundel structure and an isolated 5-HMF as the reference. The labeling of intermediates and transition states is the same as in Scheme 2.
Although there is strong experimental evidence that modifying the solvent medium (water vs DMSO–water mixtures) alters the yield of humins, the implicit solvation model does not reveal significant differences in both the kinetics and thermodynamics of the reactions leading to humin formation. Further investigation of the rate-determining aldol-addition reaction step, with explicit solvent molecules in condensed phase (and with finite temperature dynamics), is needed. By identifying the elementary reaction steps and associated activation barriers, the present work laid the foundation for the same.
3.8. Extended Reaction Mechanism for the Formation of Oligomers
The mechanistic understanding and reaction mechanisms developed in this work allow us to predict the subsequent steps for the generation of larger oligomers, as previously pointed out by Patil et al.30Scheme 8 shows the reaction mechanism extended from the dimer derived from 5,6-enol, as the pathway via this enol has the lowest free energy barrier and is thermodynamically most facile. This dimer can further react with 5-HMF molecules until it becomes saturated, forming a pentamer. Once this macromolecule can no longer react with another 5-HMF molecule, it can be acid attacked to generate a nucleophilic carbon again (as initially occurred with a 5-HMF molecule) and further react with another enol available in the reaction medium. It can also subsequently undergo acid-catalyzed keto–enol tautomerization.
Scheme 8. Extended Reaction Mechanism for Oligomeric Humin Formation from Dimer Derived from 5,6-Enol and 5-HMF.

This suggested mechanism supports aldol-addition and condensation as the main reaction for humin formation. The molecular structure corresponding to the oligomer shown in Scheme 8 reveals that as more 5-HMF molecules react, their carbonyl functional groups are lost, forming repeated double bonds with carbons initially belonging to the enol molecule. Experimentally, it has been observed through IR spectra of humins derived from 5-HMF that the peak corresponding to the carbonyl group of 5-HMF is absent in humins. On the other hand, the IR of humins derived from 5-HMF reveals that the structure of humins is rich in furan rings, which aligns with the structure of the oligomer shown in Scheme 8.
5-HMF-derived macromolecules, as shown in Scheme 8, would constitute humins observed in biomass reactions. However, it is important to highlight that humins derived from other biomass moieties, could be present too. Experimentally it has been shown that the aromatic ring of other organic molecules (i.e., benzaldehyde) added to the reaction system gets incorporated into the structure of humins, while the aldehyde group is lost. Additionally, benzaldehyde alone did not generate humins. These experimental observations suggest that regardless of the initial species, the aldol-addition mechanism would be the main pathway leading to humin formation.30,31
4. Conclusions
Pathways for acid-catalyzed reactions resulting in the formation of humins, from 5-HMF, are simulated using density functional theory (DFT) and the reaction free energies and the activation free energy barriers are computed. Elementary reaction steps involve the hydration of 5-HMF to 2,5-dioxo-6-hydroxy-hexanal (DHH), followed by the keto–enol tautomerization of DHH and the aldol-addition condensation reaction of the enols with 5-HMF. Depending on the location of the carbonyl group, four different enols can be formed from DHH. The rate-determining step in this mechanism is the aldol-addition reaction between the DHH-derived enols and 5-HMF, except for the pathway involving 1,2-enol, where the activation barrier is the highest for the keto–enol tautomerization step. The rehydration of 5-HMF to DHH (key promoter of humin formation) and the overall reaction from the keto–enol tautomerization of DHH to the formation of the dimers are found to be endergonic. The initiation reaction pathway computed in this work explains the subsequent chemistry of the further polymerization of the dimer by reacting with 5-HMF to form humins. The computed mechanism and the suggested polymerization scheme are in excellent agreement with the experimental spectroscopic evidence that suggests aldol-addition mechanism as the main pathway for humin formation. An implicit solvation model in DFT computations did not reveal a significant difference in reaction energetics for water and DMSO. Thus, to evaluate the effect of solvent on kinetics and thermodynamics of humin formation, the key reaction steps identified in this work need to be simulated with explicit solvent molecules, in the condensed phase, and possibly with finite temperature dynamics.
Acknowledgments
This research was supported by funding from the Canada First Research Excellence Fund as part of the University of Alberta’s Future Energy Systems Research Initiative, the Natural Sciences and Engineering Research Council of Canada NSERC, University of Alberta and the MITACS Graduate Fellowship Award. The Digital Research Alliance of Canada provided computational resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04783.
Free energy values for all of the intermediate species and transition states for the dehydration of 5-HMF to DHH, keto-tautomerization of DHH, aldol-addition, and condensation reactions are reported in kJ/mol; the free energy values are reported using both water and DMSO as implicit solvents; additionally, the Cartesian coordinates of all of the reactants, transition state intermediates, and the products optimized at M06-2X/6-311++G (2d, p) in the presence of an implicit environment of water and DMSO are shown (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- McKendry P. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. 10.1016/S0960-8524(01)00118-3. [DOI] [PubMed] [Google Scholar]
- McKendry P. Energy production from biomass (part 2): conversion technologies. Bioresour. Technol. 2002, 83, 47–54. 10.1016/S0960-8524(01)00119-5. [DOI] [PubMed] [Google Scholar]
- Huber G. W.; Iborra S.; Corma A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. 10.1021/cr068360d. [DOI] [PubMed] [Google Scholar]
- Christian D. G. Biomass for Renewable Energy, Fuels, and Chemicals. J. Environ. Qual. 2000, 29, 662–663. 10.2134/jeq.2000.00472425002900020040x. [DOI] [Google Scholar]
- Serrano-Ruiz J. C.; West R. M.; Dumesic J. A. Catalytic conversion of renewable biomass resources to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 79–100. 10.1146/annurev-chembioeng-073009-100935. [DOI] [PubMed] [Google Scholar]
- Huber G. W.; Dumesic J. A. An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery. Catal. Today 2006, 111, 119–132. 10.1016/j.cattod.2005.10.010. [DOI] [Google Scholar]
- Chheda J. N.; Huber G. W.; Dumesic J. A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem., Int. Ed. 2007, 46, 7164–7183. 10.1002/anie.200604274. [DOI] [PubMed] [Google Scholar]
- Chheda J. N.; Dumesic J. A. An overview of dehydration, aldol-condensation and hydrogenation processes for production of liquid alkanes from biomass-derived carbohydrates. Catal. Today 2007, 123, 59–70. 10.1016/j.cattod.2006.12.006. [DOI] [Google Scholar]
- Rosatella A. A.; Simeonov S. P.; Frade R. F. M.; Afonso C. A. M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. 10.1039/c0gc00401d. [DOI] [Google Scholar]
- Werpy T.; Petersen G.. Top Value Added Chemicals from Biomass Volume I; U.S. NREL, 2004; p 76.
- Román-Leshkov Y.; Dumesic J. A. Solvent effects on fructose dehydration to 5-hydroxymethylfurfural in biphasic systems saturated with inorganic salts. Top. Catal. 2009, 52, 297–303. 10.1007/s11244-008-9166-0. [DOI] [Google Scholar]
- Chheda J. N.; Román-Leshkov Y.; Dumesic J. A. Production of 5-hydroxymethylfurfural and furfural by dehydration of biomass-derived mono- and poly-saccharides. Green Chem. 2007, 9, 342–350. 10.1039/b611568c. [DOI] [Google Scholar]
- Tucker M. H.; Alamillo R.; Crisci A. J.; Gonzalez G. M.; Scott S. L.; Dumesic J. A. Sustainable solvent systems for use in tandem carbohydrate dehydration hydrogenation. ACS Sustainable Chem. Eng. 2013, 1, 554–560. 10.1021/sc400044d. [DOI] [Google Scholar]
- Weingarten R.; Rodriguez-Beuerman A.; Cao F.; Luterbacher J. S.; Alonso D. M.; Dumesic J. A.; Huber G. W. Selective Conversion of Cellulose to Hydroxymethylfurfural in Polar Aprotic Solvents. ChemCatChem 2014, 6, 2229–2234. 10.1002/cctc.201402299. [DOI] [Google Scholar]
- van Zandvoort I.; Koers E. J.; Weingarth M.; Bruijnincx P. C. A.; Baldus M.; Weckhuysen B. M. Structural characterization of 13C-enriched humins and alkali-treated 13C humins by 2D solid-state NMR. Green Chem. 2015, 17, 4383–4392. 10.1039/c5gc00327j. [DOI] [Google Scholar]
- Hoang T. M. C.; Lefferts L.; Seshan K. Valorization of humin-based byproducts from biomass processing - A route to sustainable hydrogen. ChemSusChem 2013, 6, 1651–1658. 10.1002/cssc.201300446. [DOI] [PubMed] [Google Scholar]
- Weingarten R.; Tompsett G. A.; Conner W. C.; Huber G. W. Design of solid acid catalysts for aqueous-phase dehydration of carbohydrates: The role of Lewis and Brønsted acid sites. J. Catal. 2011, 279, 174–182. 10.1016/j.jcat.2011.01.013. [DOI] [Google Scholar]
- Carlini C.; Giuttari M.; Galletti A. M. R.; Sbrana G.; Armaroli T.; Busca G. Selective saccharides dehydration to 5-hydroxymethyl-2-furaldehyde by heterogeneous niobium catalysts. Appl. Catal., A 1999, 183, 295–302. 10.1016/S0926-860X(99)00064-2. [DOI] [Google Scholar]
- Armaroli T.; Busca G.; Carlini C.; Giuttari M.; Galletti A. M. R.; Sbrana G. Acid sites characterization of niobium phosphate catalysts and their activity in fructose dehydration to 5-hydroxymethyl-2-furaldehyde. J. Mol. Catal. A: Chem. 2000, 151, 233–243. 10.1016/S1381-1169(99)00248-4. [DOI] [Google Scholar]
- Benvenuti F.; Carlini C.; Patrono P.; Galletti A. M. R.; Sbrana G.; Massucci M. A.; Galli P. Heterogeneous zirconium and titanium catalysts for the selective synthesis of 5-hydroxymethyl-2-furaldehyde from carbohydrates. Appl. Catal., A 2000, 193, 147–153. 10.1016/S0926-860X(99)00424-X. [DOI] [Google Scholar]
- Li N.; Tompsett G. A.; Huber G. W. Renewable high-octane gasoline by aqueous-phase hydrodeoxygenation of C5 and C6 carbohydrates over Pt/zirconium phosphate catalysts. ChemSusChem 2010, 3, 1154–1157. 10.1002/cssc.201000140. [DOI] [PubMed] [Google Scholar]
- Román-Leshkov Y.; Chheda J. N.; Dumesic J. A. Phase modifiers promote efficient production of hydroxymethylfurfural from fructose. Science 2006, 312, 1933–1937. 10.1126/science.1126337. [DOI] [PubMed] [Google Scholar]
- Weingarten R.; Rodriguez-Beuerman A.; Cao F.; Luterbacher J. S.; Alonso D. M.; Dumesic J. A.; Huber G. W. Selective conversion of cellulose to hydroxymethylfurfural in polar aprotic solvents. ChemCatChem 2014, 6, 2229–2234. 10.1002/cctc.201402299. [DOI] [Google Scholar]
- He J.; Liu M.; Huang K.; Walker T. W.; Maravelias C. T.; Dumesic J. A.; Huber G. W. Production of levoglucosenone and 5-hydroxymethylfurfural from cellulose in polar aprotic solvent-water mixtures. Green Chem. 2017, 19, 3642–3653. 10.1039/c7gc01688c. [DOI] [Google Scholar]
- Motagamwala A. H.; Huang K.; Maravelias C. T.; Dumesic J. A. Solvent system for effective near-term production of hydroxymethylfurfural (HMF) with potential for long-term process improvement. Energy Environ. Sci. 2019, 12, 2212–2222. 10.1039/c9ee00447e. [DOI] [Google Scholar]
- van Zandvoort I.; Wang Y.; Rasrendra C. B.; van Eck E. R. H.; Bruijnincx P. C. A.; Heeres H. J.; Weckhuysen B. M. Formation, molecular structure, and morphology of humins in biomass conversion: Influence of feedstock and processing conditions. ChemSusChem 2013, 6, 1745–1758. 10.1002/cssc.201300332. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Everhart J. L.; Tsilomelekis G.; Nikolakis V.; Saha B.; Vlachos D. G. Structural analysis of humins formed in the Brønsted acid catalyzed dehydration of fructose. Green Chem. 2018, 20, 997–1006. 10.1039/c7gc03054a. [DOI] [Google Scholar]
- Sumerskii I. V.; Krutov S. M.; Zarubin M. Y. Humin-Like substances formed under the conditions of industrial hydrolysis of wood. Russ. J. Appl. Chem. 2010, 83, 320–327. 10.1134/S1070427210020266. [DOI] [Google Scholar]
- Dee S. J.; Bell A. T. A study of the acid-catalyzed hydrolysis of cellulose dissolved in ionic liquids and the factors influencing the dehydration of glucose and the formation of humins. ChemSusChem 2011, 4, 1166–1173. 10.1002/cssc.201000426. [DOI] [PubMed] [Google Scholar]
- Patil S. K. R.; Lund C. R. F. Formation and growth of humins via aldol addition and condensation during acid-catalyzed conversion of 5-hydroxymethylfurfural. Energy Fuels 2011, 25, 4745–4755. 10.1021/ef2010157. [DOI] [Google Scholar]
- Patil S. K. R.; Heltzel J.; Lund C. R. F. Comparison of structural features of humins formed catalytically from glucose, fructose, and 5-hydroxymethylfurfuraldehyde. Energy Fuels 2012, 26, 5281–5293. 10.1021/ef3007454. [DOI] [Google Scholar]
- Horvat J.; Klaić B.; Metelko B.; Šunjić V. Mechanism of levulinic acid formation in acid catalyzed hydrolysis of 2-(hydroxymethyl)furan and 5-(hydroxymethyl)-2-furancarboxaldehyde. Croat. Chem. Acta 2015, 59, 429–438. [Google Scholar]
- Frisch Æ.; Plata R. E.; Singleton D. A. Gaussian 09W Reference. J. Am. Chem. Soc. 2009, 137, 3811–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora J. S.; Chew J. W.; Mushrif S. H. Influence of Alkali and Alkaline-Earth Metals on the Cleavage of Glycosidic Bond in Biomass Pyrolysis: A DFT Study Using Cellobiose as a Model Compound. J. Phys. Chem. A 2018, 122, 7646–7658. 10.1021/acs.jpca.8b06083. [DOI] [PubMed] [Google Scholar]
- Mayes H. B.; Broadbelt L. J. Unraveling the reactions that unravel cellulose. J. Phys. Chem. A 2012, 116, 7098–7106. 10.1021/jp300405x. [DOI] [PubMed] [Google Scholar]
- Park M.; Shin I.; Singh N. J.; Kim K. S. Eigen and Zundel forms of small protonated water clusters: Structures and infrared spectra. J. Phys. Chem. A 2007, 111, 10692–10702. 10.1021/jp073912x. [DOI] [PubMed] [Google Scholar]
- Decka D.; Schwaab G.; Havenith M. A THz/FTIR fingerprint of the solvated proton: Evidence for Eigen structure and Zundel dynamics. Phys. Chem. Chem. Phys. 2015, 17, 11898–11907. 10.1039/c5cp01035g. [DOI] [PubMed] [Google Scholar]
- Yang Z. Z.; Ding Y. L.; Zhao D. X. Insight into Markovnikov reactions of alkenes in terms of ab initio and molecular face theory. ChemPhysChem 2008, 9, 2379–2389. 10.1002/cphc.200800364. [DOI] [PubMed] [Google Scholar]
- Hinchliffe A. Ab initio SCF MO study of the hydrogen-bonded dimers of cyanogen with HF and HCl. Chem. Phys. Lett. 1982, 87, 417–419. 10.1016/0009-2614(82)83002-9. [DOI] [Google Scholar]
- Kalyanashis J.; Bishwajit G. DFT Study To Explore the Importance of Ring Size and Effect of Solvents on the Keto–Enol Tautomerization Process of α- and β-Cyclodiones. ACS Omega 2018, 3, 8429–8439. 10.1021/acsomega.8b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacchioni G. Ketonization of Carboxylic Acids in Biomass Conversion over TiO2 and ZrO2 Surfaces: A DFT Perspective. ACS Catal. 2014, 4, 2874–2888. 10.1021/cs500791w. [DOI] [Google Scholar]
- Yamaguchi S.; Deguchi H.; Kawauchi S.; Motokura K.; Miyaji A.; Baba T. Mechanistic Insight into Biomass Conversion to Five–membered Lactone Based on Computational and Experimental Analysis. ChemistrySelect 2017, 2, 591–597. 10.1002/slct.201601888. [DOI] [Google Scholar]
- Arora J. S.; Ansari K. B.; Chew J. W.; Dauenhauer P. J.; Mushrif S. H. Unravelling the catalytic influence of naturally occurring salts on biomass pyrolysis chemistry using glucose as a model compound: a combined experimental and DFT study. Catal. Sci. Technol. 2019, 9, 3504–3524. 10.1039/C9CY00005D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.