

Abstract
Influenza infection induces lung epithelial cell injury via programmed cell death. Glutathione, a potent antioxidant, has been reported to be associated with influenza infection. We hypothesized that lung epithelial cell death during influenza infection is regulated by glutathione metabolism. Eight-week-old male and female BALB/c mice were infected with influenza (PR8: A/PR/8/34 [H1N1]) via intranasal instillation. Metabolomic analyses were performed on whole lung lysate after influenza infection. For in vitro analysis, Beas-2B cells were infected with influenza. RNA was extracted, and QuantiTect Primer Assay was used to assess gene expression. Glutathione concentrations were assessed by colorimetric assay. Influenza infection resulted in increased inflammation and epithelial cell injury in our murine model, leading to increased morbidity and mortality. In both our in vivo and in vitro models, influenza infection was found to induce apoptosis and necroptosis. Influenza infection led to decreased glutathione metabolism and reduced glutathione reductase activity in lung epithelial cells. Genetic inhibition of glutathione reductase suppressed apoptosis and necroptosis of lung epithelial cells. Pharmacologic inhibition of glutathione reductase reduced airway inflammation, lung injury, and cell death in our murine influenza model. Our results demonstrate that glutathione reductase activity is suppressed during influenza. Glutathione reductase inhibition prevents epithelial cell death and morbidity in our murine influenza model. Our results suggest that glutathione reductase-dependent glutathione metabolism may play an important role in the host response to viral infection by regulating lung epithelial cell death.
Keywords: influenza, pneumonia, glutathione metabolism, cell death
Influenza, a viral infection that attacks the respiratory system, is caused by influenza A and influenza B viruses in humans (1). It is estimated that influenza has resulted in between 9 million and 45 million illnesses, 140,000 and 810,000 hospitalizations, and 12,000 and 61,000 deaths in the United States annually since 2010 (2). Risk factors for influenza infection include metabolic derangements seen in conditions such as diabetes, obesity, and advanced age (3–5).
The replication of the influenza virus depends on host cellular metabolic pathways involving nucleic acids, lipids, and glycoproteins (6, 7). In mice, influenza infection affects more than 100 metabolites in serum, lung, and BAL fluid (8). On a mechanistic level, influenza virus infection interferes with the cellular metabolic pathways of glycolysis, glutaminolysis, pentose phosphate, and fatty acid synthesis to generate ATP (adenosine triphosphate) and structural materials of viral replication (9). Previous work has demonstrated that altering metabolic processes can improve the outcomes of influenza. Specifically, suppression of glycolysis can result in reduced viral replication (10). In another study, inhibition of the mTOR pathway, a key regulatory pathway involved in protein synthesis and viral replication, significantly reduced viral replication and mortality in murine models of influenza infection (11, 12).
This paper investigates the role of glutathione metabolism in influenza infection. Glutathione, a key antioxidant that neutralizes reactive oxygen species induced in host cells during RNA virus infection, regulates innate immunity at various degrees during influenza infection (13, 14). The cellular content of glutathione is inversely related to influenza virus replication in the cell (15). Glutathione-dependent antiviral pathways appear to be pivotal in the immune response against influenza. Amatore and colleagues showed that influenza infection induces increased glutathione synthesis in cells, which subsequently triggers the Th1 (T helper type 1) cellular response downstream (15). Previously, it has been shown that the administration of glutathione in drinking water can reduce viral titers in the murine lung during influenza (16).
Little is known about the pathogenetic effects of glutathione metabolism in the lung in response to influenza infection. In our study, we hypothesize that glutathione metabolism regulates influenza-induced epithelial cell death and that inhibition of glutathione reductase will ameliorate programmed cell death, airway inflammation, and lung injury during influenza infection in our murine model.
Methods
Detailed methods are provided in the data supplement.
Results
Glutathione Metabolism Is Altered in Murine Lung during Influenza Infection
To investigate metabolic changes in the lung during influenza infection, we performed metabolomic analyses (Figure 1A) on both influenza-infected and control mice over multiple time points. Young mice were infected intranasally with a sublethal dose of the H1N1 influenza A virus Puerto Rico/8/34 strain, and metabolic profiling was performed on lung lysates. Principal–component analysis revealed a markedly altered metabolic profile in the lung on influenza infection (Figure 1B). Of these metabolic alterations, we focused on changes in the glutathione metabolism pathway that were identified in mice during the course of influenza infection (Figure 1C). Specifically, we examined changes in the tissue concentrations of both the reduced (GSH) and oxidized (GSSG) forms of glutathione, in addition to the ratio of GSH to GSSG. When compared with control samples, a significant decrease in both GSH and GSSG concentrations was observed by Day 7 after influenza infection (Figure 1D). It is important to note that the GSH to GSSG ratio was also significantly decreased, indicating a comparatively greater reduction in GSH than GSSG.
Figure 1.
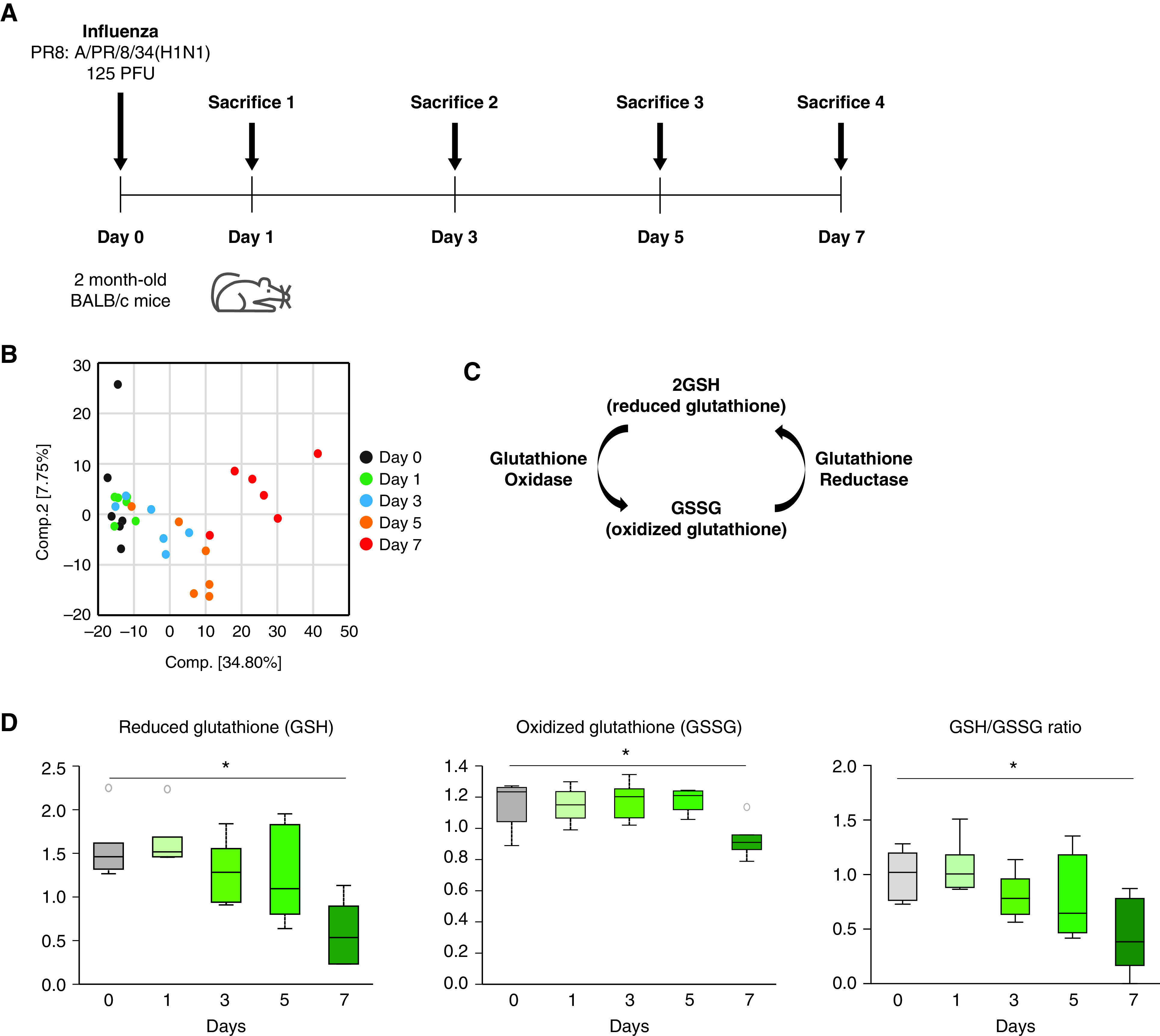
Glutathione metabolism is decreased in the murine lung during influenza infection. (A) Experimental layout of influenza-induced lung injury (Day 0 intranasal instillation of 125 PFU of influenza, PR8: A/Puerto Rico/8/1934 H1N1, 50 μl volume in PBS per mouse). (B) Two-dimensional PLS-DA score plot. (C) Glutathione metabolism pathway. (D) Relative concentrations for the GSH, GSSG, and GSH to GSSG ratio in mouse lung lysates after influenza infection. Data are median ± interquartile range. *P < 0.05 by ANOVA. GSH = reduced form of glutathione. GSSG = oxidized glutathione; PLS-DA = partial least squares-discriminant analysis.
With aging being a major risk factor for mortality and morbidity in influenza infection, we assessed if these changes in glutathione metabolism after infection are different in aged versus young mice. Interestingly, unlike in young mice, there were no observed significant changes in concentrations of GSH and GSSG during influenza infection in aged mice lungs (Figures E1A and E1B in the data supplement). In addition, both GSH and GSSG appear to be consistently lower in elderly mice. This demonstrates that storage of glutathione at baseline is lower in elderly mice compared with young control samples, which may contribute to impaired glutathione metabolism in older mice in response to influenza infection. This suggests that glutathione metabolism may play a protective role in pathogenesis during influenza infection. Taken together, there appears to be selective dysregulation of glutathione metabolism in response to influenza infection as a function of age.
Glutathione Reductase Activity Is Decreased during Influenza Infection
To expand on these results, we assessed for changes in enzymatic activity that control glutathione metabolism in our murine influenza model. We first used a conventional colorimetric assay to confirm a reduction of GSH and GSSG concentrations in influenza-infected lungs (Figure 2A). We then measured the activity of glutathione reductase, an enzyme that catalyzes the reduction of GSSG to GSH, in mouse lungs 7 days after infection. A significant decrease in glutathione reductase activity was observed in influenza-infected lungs compared with healthy control samples (Figure 2B). We further examined if these observations were reproducible in an in vitro model system of human lung epithelial cells. Similar to our in vivo murine studies, there was a significant decrease in GSH, GSH to GSSG ratio, and glutathione reductase activity in influenza-treated Beas-2B cells, a human lung epithelial cell line (Figures 2C and 2D). In addition, the gene expression of glutathione reductase was significantly reduced in influenza-infected epithelial cells compared with control samples (Figure 2E). We next examined enzymes that oxidize GSH to GSSG by performing PCR arrays on panels of glutathione peroxidase enzymes. The gene expression of glutathione peroxidases was found to be comparable between influenza-infected and control mice, in contradistinction to glutathione reductase (Figures E2A and E2B).
Figure 2.
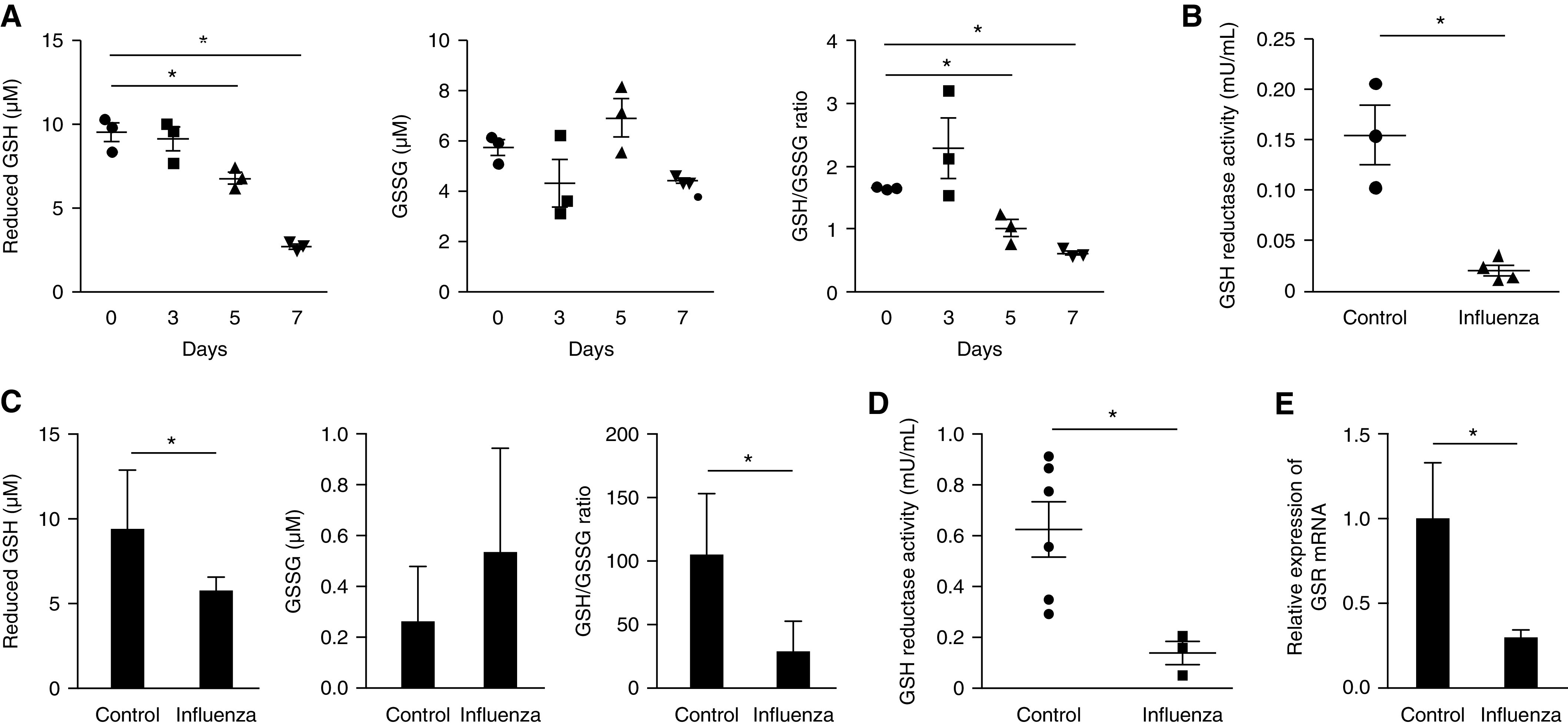
Influenza infection induces alteration of glutathione metabolism in vivo and in vitro. (A) Total, reduced, and oxidized forms of glutathione and the ratio of reduced and oxidized glutathione measured in lung tissue lysate of influenza-infected mice (125 PFU, n = 3 mice per group). (B) Glutathione reductase activity in murine lung 7 days after influenza infection. (C) Total, reduced, and oxidized forms of glutathione and the ratio of reduced and oxidized glutathione measured in Beas2B cells after treatment with influenza (1.25 × 104 PFU) for 48 hours. (D) Glutathione reductase activity and (E) gene expression of glutathione reductase in Beas2B cells 48 hours after influenza infection. Data are mean ± SEM. *P < 0.05 by ANOVA. Results are representative of three or more independent experiments.
Inflammation, Lung Injury, and Cell Death Are Increased in Influenza Infection
In prior work, we demonstrated that influenza infection increases morbidity in a mouse model (Stout-Delgado, JI, 2012). Here we furthered our investigation and showed that markers of inflammation (cell count) and lung injury (lactate dehydrogenase [LDH], and total protein) in BAL fluid are significantly increased in mice as the course of influenza infection progresses (Figures 3A–3C and E3). We then assessed cell death during influenza infection using TUNEL staining. A TUNEL+ cell death response was increased after influenza infection (Figure 4A). We also evaluated representative markers of apoptosis and necroptosis by Western blot (Figure 4B). During influenza infection, there was a significant increase in the expression of the programmed cell death markers, such as cleaved caspase 3, cleaved PARP, RIPK3 (receptor-interacting serine/threonine-protein kinase 3), phospho RIPK3, and MLKL (mixed lineage kinase domain-like protein).
Figure 3.
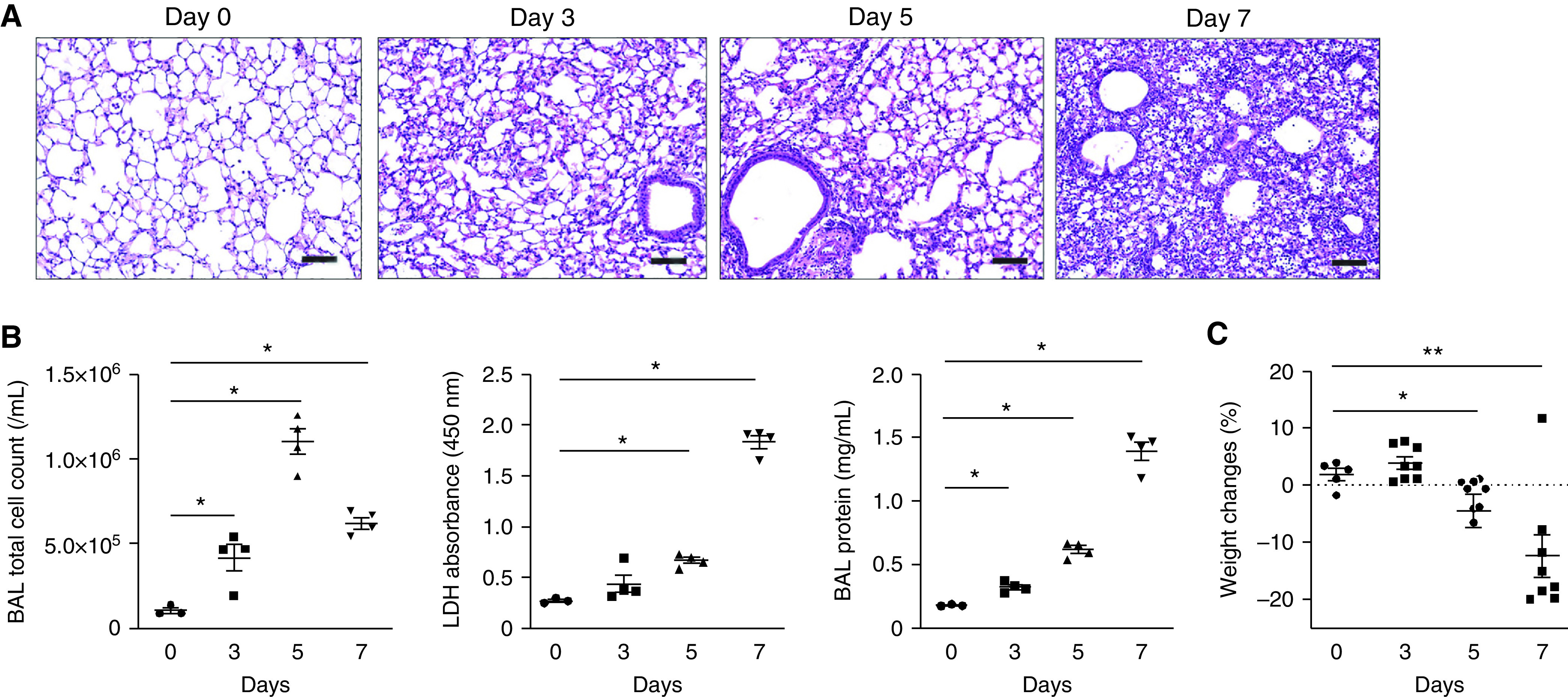
Inflammation, lung injury, and morbidity are increased in influenza-infected mice. (A) Lung sections of influenza-infected mice on Days 0, 3, 5, and 7. Hematoxylin and eosin stain, 40× magnification. Scale bar, 200 μm. (B) Total BAL cell count LDH cytotoxicity and BAL protein. (C) Weight changes in mice during influenza infection (influenza Day 0, n = 3; Day 3, n = 8; Day 5, n = 8; Day 7, n = 8). Data are mean ± SEM. *P < 0.05 by ANOVA. Results are representative of three or more independent experiments. LDH = lactate dehydrogenase.
Figure 4.

Cell death is increased during influenza infection. (A) TUNEL staining of mouse lung section 7 days after influenza infection (1.25 × 104 PFU). Scale bar, 100μm. (B) Immunoblot analysis for markers of PARP, cleaved PARP, caspase 3, cleaved caspase 3, RIPK3 (receptor-interacting serine/threonine-protein kinase 3), phospho RIPK3, and MLKL (mixed lineage kinase domain-like protein) in whole lung tissue lysate 7 days after influenza infection. Results are representative of three or more independent experiments.
Influenza Induces Epithelial Cell Death In Vitro
We investigated if influenza activates cell death pathways in cultured human epithelial cells (Beas-2B). Beas-2B cells treated with influenza were less confluent and exhibited morphological features of cell death, including cell shrinkage and fragmentation, compared with control cells (Figure 5A). Cell cytotoxicity after incremental doses of influenza infection was then analyzed. Cytotoxicity was found to be increased in a dose-dependent manner in Beas-2B cells as measured by LDH concentrations and viral toxglo assay (Figures 5B and 5C). We then assessed for apoptosis and necroptosis in our in vitro influenza model. Protein concentrations of the programmed cell death markers PARP, cleaved PARP, caspase 3, cleaved caspase 3, RIPK3, phospho-RIPK3, and MLKL were found to be increased in Beas-2B cells after influenza treatment. These results show apoptosis and necroptosis and possible mechanisms for cell death in our model.
Figure 5.
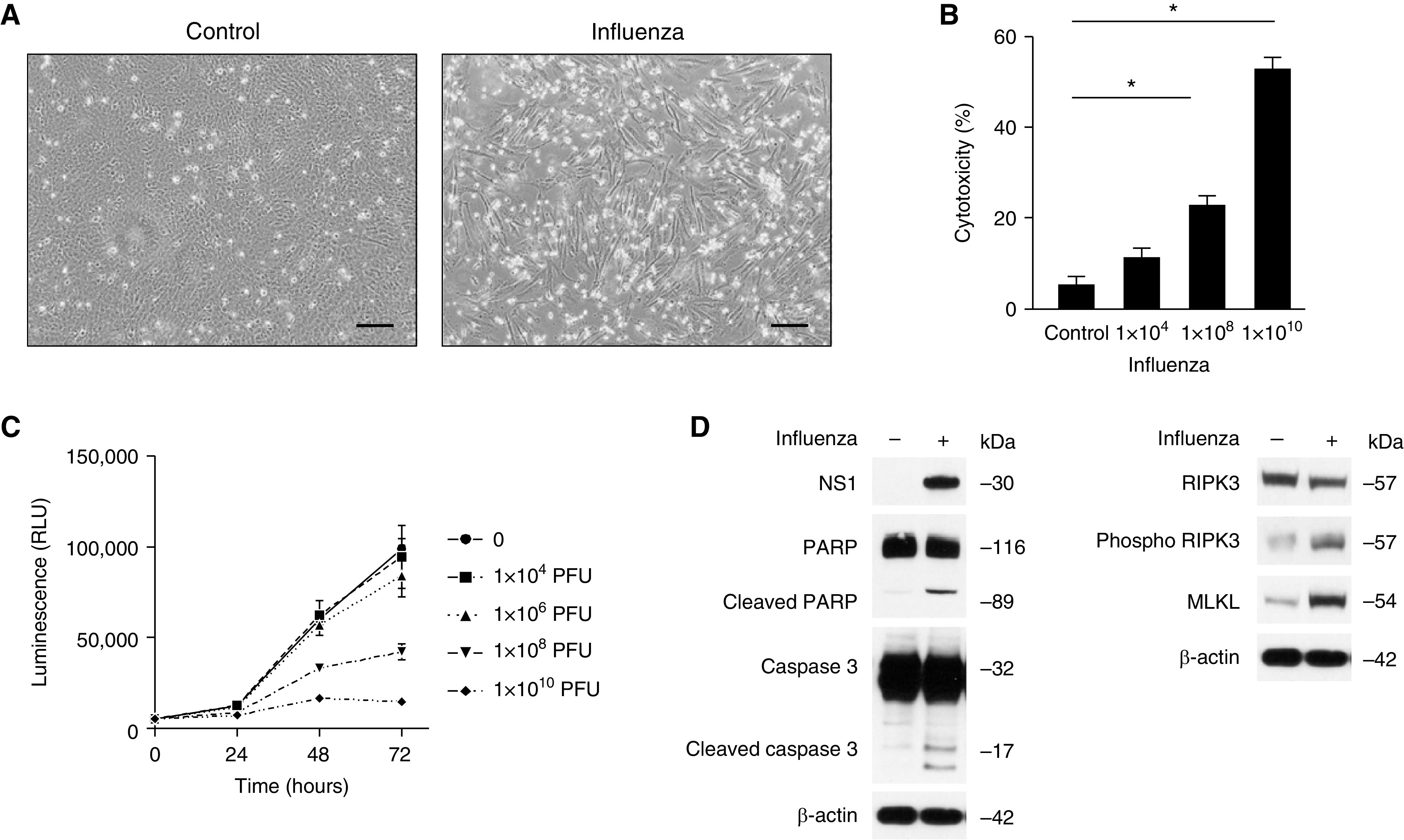
Influenza induces epithelial cell death in vitro. (A) Bright field microscopy of Beas2B cells treated with influenza (1.25 × 104 PFU, 72 h), 20× magnification. Scale bar, 100 μm. (B) Cytotoxicity assay of cell culture (Beas2B) media after influenza infection. Data are mean ± SEM. *P < 0.05 by ANOVA. (C) Increased viral-induced cytopathic effect on Beas2B cells in a dose-dependent manner after influenza infection. Data are mean ± SEM. (D) Immunoblot analysis for PARP, cleaved PARP, caspase 3, cleaved caspase 3, RIPK3, phospho RIPK3, and MLKL in Beas2B cells 48 hours after treatment with influenza. Results are representative of three or more independent experiments.
Genetic Inhibition of Glutathione Reductase Decreases Apoptotic and Necroptotic Cell Death In Vitro
Transfection of Beas-2B with siRNA targeting glutathione reductase (GSR) resulted in suppression of GSR gene expression and reduction in glutathione reductase activity (Figures E4A and E4B) and was protective against programmed cell death during influenza infection (Figure 6A). It was also protective against apoptotic cell death in the presence of the apoptosis inducer staurosporine (Figure 6B). In addition, the expression of the apoptotic markers PARP, cleaved PARP, caspase 3, and cleaved caspase 3 were decreased in Beas-2B cells transfected with GSR siRNA compared with control siRNA after treatment with staurosporine (Figure 6C). We then tested the effect of glutathione reductase on necroptotic cell death by analyzing cell cytotoxicity after treatment with the necroptosis inducers TNF, smac mimetic, and Z-VAD (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone) treatment. As in apoptosis, there was reduced necroptotic cell death in Beas-2B cells transfected with GSR siRNA compared with control siRNA (Figure 6D). In addition, expression of the necroptosis markers phospho RIPK3 and phospho MLKL were decreased in Beas-2B cells transfected with GSR siRNA compared with control siRNA after treatment with the necroptosis inducers TNF, smac mimetic, and Z-VAD (Figure 6E). Taken together, these results demonstrate the critical role of glutathione reductase in apoptotic and necroptotic cell death.
Figure 6.
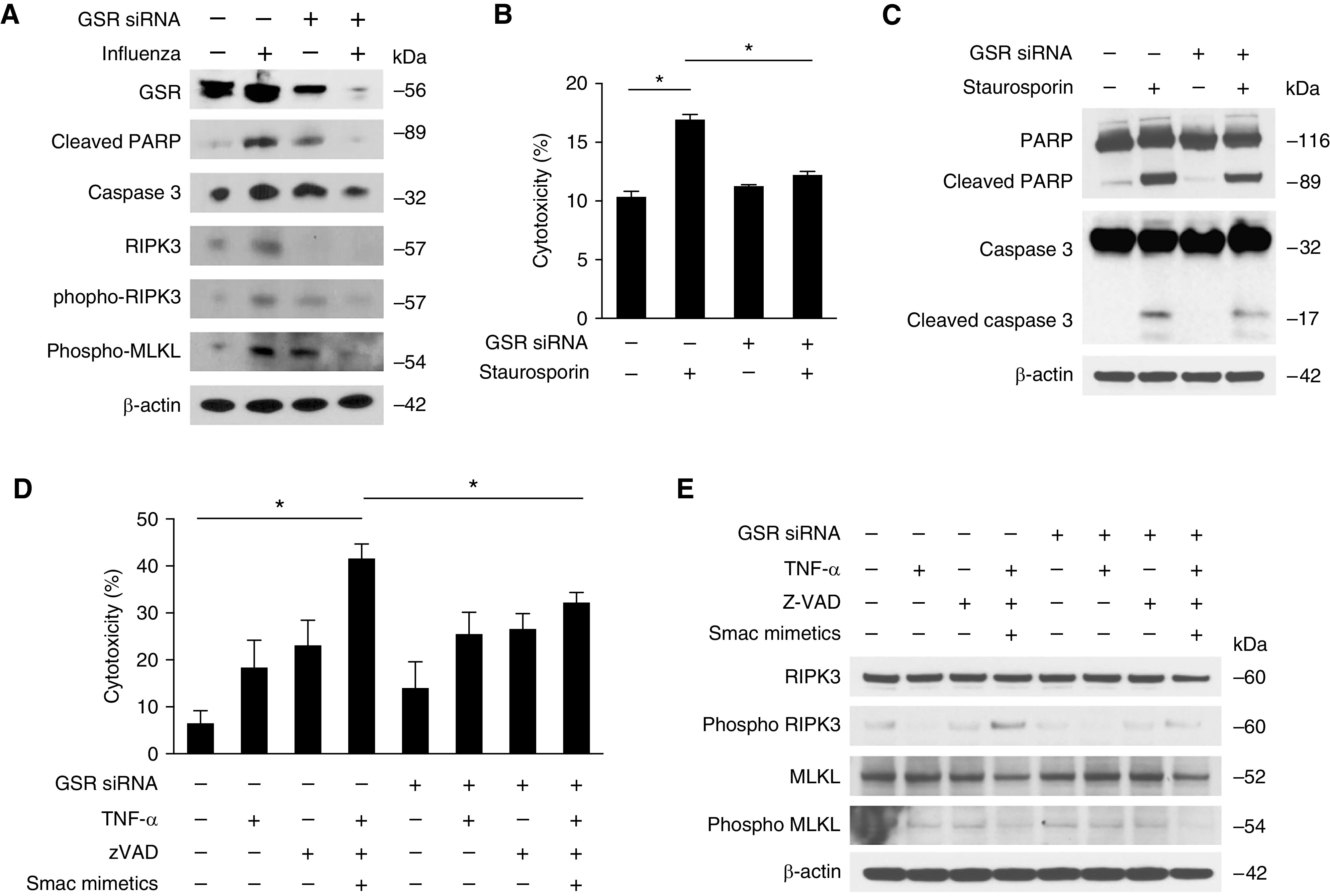
Inhibition of glutathione reductase decreases epithelial cell death. (A) Immunoblot analysis for cleaved PARP, caspase 3, RIPK3, phospho RIPK3, and MLKL in Beas2B cells transfected with GSR siRNA or control siRNA after treatment with influenza. (B) Cytotoxic assay and (C) immunoblot analysis for PARP, cleaved PARP, caspase 3, and cleaved caspase 3 in Beas2B cells transfected with GSR siRNA or control siRNA after treatment with staurosporine (500 nM) for 4 hours. (D) Cytotoxicity and (E) immunoblot analysis for RIPK3, phospho RIPK3, MLKL, and phospho MLKL in Beas2B cells transfected with GSR siRNA or control siRNA after treatment with TNFα (10 ng/ml), Smac mimetic (10 mM), and zVAD (10 mM), or vehicle (DMSO) for 4 hours. Data are mean ± SEM. *P < 0.05 by ANOVA. Results are representative of two or more independent experiments. DMSO = dimethyl sulfoxide; zVAD = (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone).
Pharmacologic Inhibition of Glutathione Reductase Reduces Airway Inflammation, Lung Injury, and Cell Death
To investigate the role of glutathione reductase in influenza-induced lung injury in vivo, we examined the impact of lomustine, a potent glutathione reductase inhibitor, on mice after influenza infection (Figure 7A). To confirm the effect of lomustine on glutathione metabolism, we measured GSH, GSSG, and GSH to GSSG ratio in influenza-infected lungs after lomustine treatment (Figure E5A). We noted a significant decrease in GSH and GSH to GSSG ratio, whereas GSSG was comparable between lomustine-treated lungs and control samples. Lomustine acts as a DNA-alkylating agent, and it also inhibits various key enzymatic reactions by carbamylating proteins. Alkylation could inhibit genome expression and replication and might cause base-mispairing to occur, resulting in an increased mutation rate which can affect pro- or antiproliferation ability. We thus examined if lomustine has any cytotoxic effects apart from that because of influenza infection. We found that the cytotoxic effect was comparable between lomustine-treated and untreated Beas-2B cells after influenza infection (Figure E5B). Although the serum half-life of lomustine ranges from 16 hours to 2 days, on the basis of our measurements of lung tissue GSR activity, its effect on GSR appears to last at least 72 hours after the first injection (Figure E5C).
Figure 7.
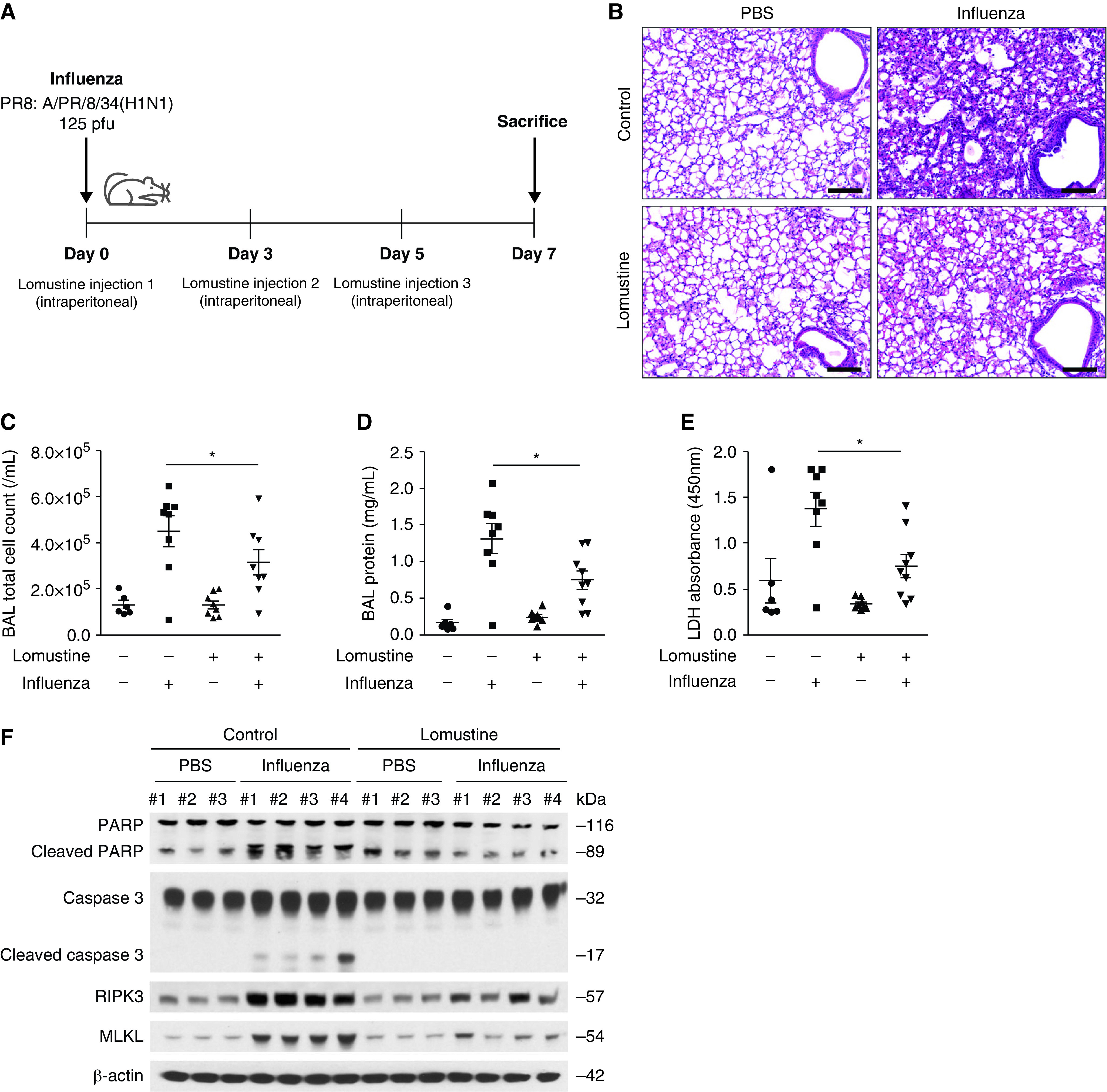
Pharmacologic inhibition of glutathione reductase reduced airway inflammation, lung injury, and cell death. (A) Experimental layout of influenza-induced lung injury in lomustine-treated mice (20 mg/kg, intraperitoneal injection on Days 0, 3, and 5). Lung tissue and BAL fluids were obtained on Day 7. (B) Representative lung sections of influenza-infected mice after lomustine treatment. Stained with hematoxylin and eosin staining. Scale bars, 200 μm. (C) BAL cell count, (D) the protein concentration in BAL fluid, and (E) lactate dehydrogenase cytotoxicity in influenza-infected mice after lomustine treatment (n = 69 mice/group). Data are mean ± SEM. *P < 0.05 by ANOVA. (F) Immunoblot analysis for PARP, cleaved PARP, caspase 3, cleaved caspase 3, RIPK3, and MLKL in influenza-infected lung after lomustine treatment. Results are representative of two independent experiments.
When compared with nontreated control samples, lomustine treatment reduced inflammation in the airways, subepithelial area, and alveolar septum (Figure 7B). Inflammatory cell count and total protein concentration in BAL fluid were significantly suppressed by lomustine treatment when compared with vehicle control samples (Figures 7C and 7D). LDH concentrations in BAL fluid were also decreased after lomustine treatment (Figure 7E). Lomustine further decreased protein expression of the programmed cell death markers cleaved PARP, cleaved caspase 3, phospho-RIPK3, and MLKL (Figure 7F). These results point to a protective effect of glutathione reductase inhibition in influenza-infected lungs by regulating apoptosis and necroptosis.
Discussion
Metabolic derangement accompanying influenza infection has been increasingly recognized as having an important role in the pathogenesis of virus-induced lung injury. In this study, we demonstrate that metabolites of glutathione metabolism undergo significant alterations during the course of influenza infection and that these alterations arise from reduced glutathione reductase activity in influenza-infected lungs. Moreover, we showed that inhibition of glutathione reductase protects epithelial cells from apoptosis and necroptosis, pointing to the potential ameliorative effects of glutathione reductase inhibition during influenza-induced lung injury.
It is well established that advanced age is a significant risk factor for morbidity in influenza infection in humans as well as in mouse models (17, 18). A strength of our study was that we used both young and aged mice influenza models to identify glutathione metabolism as an important metabolic pathway in influenza infection. In young mice, progressive reduction in both GSH and GSSH concentrations, as well as GSH to GSSH ratio, were observed during the course of influenza infection, whereas GSH and GSSH remained unchanged in aged mice. We believe the relatively dynamic changes in glutathione metabolism seen in young mice may underlie their superior clinical outcomes compared with aged mice during influenza infection. In one of our prior studies, young mice began to recover clinically on Day 7 after influenza inoculation, whereas aged mice began to further clinically deteriorate at this time point. This divergence in clinical course parallels has been observed in human influenza, in which young persons without significant comorbidities tend to recover by Day 7, whereas older persons with comorbidities often clinically deteriorate.
The mechanism by which changes in glutathione metabolism alter the course of influenza infection appears to be multifactorial. It is known that glutathione detoxifies cells by restoring enzymatic and nonenzymatic antioxidants and serves as a nonenzymatic antioxidant itself. Indeed, higher concentrations of the reduced form of glutathione have been considered protective against influenza infection. In our study, however, we found that reduction in absolute concentrations of GSH that accompany glutathione reductase inhibition correlates paradoxically with superior clinical outcomes. On the basis of our results, it is our belief that dynamic changes in GSH/GSSH balance result from glutathione reductase activity and not necessarily the absolute concentrations of these metabolites that are important in influenza infection.
Glutathione reductase inhibition ameliorating clinical outcomes in influenza infection is a central finding in our study. Mechanistically, we demonstrated a reduction in apoptosis and necroptosis in lung epithelial cells on inhibition of glutathione reductase. Whether mitochondria, which are classically implicated in programmed cell death pathways (19), are affected by glutathione reductase inhibition is unknown. Interestingly, a study has reported that cell death is promoted by the extent of depletion of mitochondrial glutathione rather than the changes in the cytoplasmic pool (20). These findings are consistent with the potential role of glutathione reductase in regulating apoptotic cell death by altering concentrations of glutathione in mitochondria.
The direct effect of glutathione reductase inhibition on necroptosis is also not well established. It is possible that dysregulated glutathione metabolism in mitochondria may promote necroptosis by altering the function of PGAM5 (phosphoglycerate mutase family member 5) and DRP1 (dynamin-related protein 1), mitochondrial proteins that are known to be associated with the necrosome (21, 22).
In our study, glutathione reductase activity was suppressed with lomustine. We chose this pharmacologic inhibition as lomustine is already currently used to treat patients with oncologic conditions, including brain tumors and hematologic malignancies (23, 24), and thus this medication would be readily available for any future potential clinical trials on glutathione reductase inhibition in human patients. Another advantage of lomustine is that it can be administered orally, although we used intraperitoneal injection in our study to deliver an exact dose of lomustine to mice. It is important to mention that in our study, lomustine was administered before inoculation with influenza. Additional studies will be required to evaluate its therapeutic effects after influenza onset.
This is the first study we know to demonstrate that glutathione reductase activity directly affects clinical outcomes during influenza infection. The importance of glutathione metabolism and enzymes such as glutathione reductase that affect its delicate balance has been gaining traction in the scientific community. Although glutathione reductase has been an attractive target for drug development, no successful glutathione reductase-related therapeutics have been implemented in pulmonary disease to date. Our data gives support for the selective inhibition of glutathione reductase as a promising means of treating severe influenza infection.
Footnotes
Supported by grants R01 AG052530 (H.S.-D.), R01 AG056699 and K08 HL138285 (S.J.C.).
Author Contributions: Conception and design: K.S.H., K.P., R.H., J.Y., H.S.-D., and S.J.C. Analysis and interpretation: K.S.H., W.W., R.H., J.Y., H.S.-D., and S.J.C. Drafting the manuscript for important intellectual content: K.S.H., K.P., W.W., H.S.-D., and S.J.C.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0372OC on June 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Influenza. Nat Rev Dis Primers . 2018;4:3. doi: 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, et al. Annual estimates of the burden of seasonal influenza in the United States: a tool for strengthening influenza surveillance and preparedness. Influenza Other Respir Viruses . 2018;12:132–137. doi: 10.1111/irv.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mertz D, Kim TH, Johnstone J, Lam PP, Science M, Kuster SP, et al. Populations at risk for severe or complicated influenza illness: systematic review and meta-analysis. BMJ . 2013;347:f5061. doi: 10.1136/bmj.f5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allard R, Leclerc P, Tremblay C, Tannenbaum TN. Diabetes and the severity of pandemic influenza A (H1N1) infection. Diabetes Care . 2010;33:1491–1493. doi: 10.2337/dc09-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morgan OW, Bramley A, Fowlkes A, Freedman DS, Taylor TH, Gargiullo P, et al. Morbid obesity as a risk factor for hospitalization and death due to 2009 pandemic influenza A(H1N1) disease. PLoS One . 2010;5:e9694. doi: 10.1371/journal.pone.0009694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kido H, Indalao IL, Kim H, Kimoto T, Sakai S, Takahashi E. Energy metabolic disorder is a major risk factor in severe influenza virus infection: proposals for new therapeutic options based on animal model experiments. Respir Investig . 2016;54:312–319. doi: 10.1016/j.resinv.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 7. Tisoncik-Go J, Gasper DJ, Kyle JE, Eisfeld AJ, Selinger C, Hatta M, et al. Integrated omics analysis of pathogenic host responses during pandemic H1N1 influenza virus infection: the crucial role of lipid metabolism. Cell Host Microbe . 2016;19:254–266. doi: 10.1016/j.chom.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chandler JD, Hu X, Ko EJ, Park S, Lee YT, Orr M, et al. Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am J Physiol Regul Integr Comp Physiol . 2016;311:R906–R916. doi: 10.1152/ajpregu.00298.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keshavarz M, Solaymani-Mohammadi F, Namdari H, Arjeini Y, Mousavi MJ, Rezaei F. Metabolic host response and therapeutic approaches to influenza infection. Cell Mol Biol Lett . 2020;25:15. doi: 10.1186/s11658-020-00211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kohio HP, Adamson AL. Glycolytic control of vacuolar-type ATPase activity: a mechanism to regulate influenza viral infection. Virology . 2013;444:301–309. doi: 10.1016/j.virol.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 11. Kuss-Duerkop SK, Wang J, Mena I, White K, Metreveli G, Sakthivel R, et al. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late-stage replication. PLoS Pathog . 2017;13:e1006635. doi: 10.1371/journal.ppat.1006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smallwood HS, Duan S, Morfouace M, Rezinciuc S, Shulkin BL, Shelat A, et al. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep . 2017;19:1640–1653. doi: 10.1016/j.celrep.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox biology of respiratory viral infections. Viruses . 2018;10:392. doi: 10.3390/v10080392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghezzi P. Role of glutathione in immunity and inflammation in the lung. Int J Gen Med . 2011;4:105–113. doi: 10.2147/IJGM.S15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suliman HBRL, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol . 2001;280:L69–L78. doi: 10.1152/ajplung.2001.280.1.L69. [DOI] [PubMed] [Google Scholar]
- 16. Cai J, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP. Inhibition of influenza infection by glutathione. Free Radic Biol Med . 2003;34:928–936. doi: 10.1016/s0891-5849(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 17. Hernandez-Vargas EA, Wilk E, Canini L, Toapanta FR, Binder SC, Uvarovskii A, et al. Effects of aging on influenza virus infection dynamics. J Virol . 2014;88:4123–4131. doi: 10.1128/JVI.03644-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stout-Delgado HW, Vaughan SE, Shirali AC, Jaramillo RJ, Harrod KS. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol . 2012;188:2815–2824. doi: 10.4049/jimmunol.1103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol . 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 20. Lash LH. Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact . 2006;163:54–67. doi: 10.1016/j.cbi.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell . 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 22. Yu B, Ma J, Li J, Wang D, Wang Z, Wang S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat Commun . 2020;11:2549. doi: 10.1038/s41467-020-16312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weller M, Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev . 2020;87:102029. doi: 10.1016/j.ctrv.2020.102029. [DOI] [PubMed] [Google Scholar]
- 24. Musolino A, Perrone MA, Michiara M, Delnevo D, Franciosi V, Di Blasio B, et al. Lomustine (chloroethylnitrosourea [CCNU]), ifosfamide, bleomycin, vincristine, and cisplatin (CIBO-P) is an effective regimen for patients with poor prognostic refractory or multiple disease recurrent aggressive non-Hodgkin lymphoma. Cancer . 2005;103:2109–2117. doi: 10.1002/cncr.21024. [DOI] [PubMed] [Google Scholar]