

ABSTRACT
The genus Chlamydia consists of diverse, obligate intracellular bacteria that infect various animals, including humans. Although chlamydial species share many aspects of the typical intracellular lifestyle, such as the biphasic developmental cycle and the preference for invasion of epithelial cells, each chlamydial strain also employs sophisticated species-specific strategies that contribute to an extraordinary diversity in organ and/or tissue tropism and disease manifestation. In order to discover and understand the mechanisms underlying how these pathogens infect particular hosts and cause specific diseases, it is imperative to develop a mutagenesis approach that would be applicable to every chlamydial species. We present functional evidence that the region between Chlamydia trachomatis and Chlamydia muridarum pgp6 and pgp7, containing four 22-bp tandem repeats that are present in all chlamydial endogenous plasmids, represents the plasmid origin of replication. Furthermore, by introducing species-specific ori regions into an engineered 5.45-kb pUC19-based plasmid, we generated vectors that can be successfully transformed into and propagated under selective pressure by C. trachomatis serovars L2 and D, as well as C. muridarum. Conversely, these vectors were rapidly lost upon removal of the selective antibiotic. This conditionally replicating system was used to generate a tarP deletion mutant by fluorescence-reported allelic exchange mutagenesis in both C. trachomatis serovar D and C. muridarum. The strains were analyzed using in vitro invasion and fitness assays. The virulence of the C. muridarum strains was then assessed in a murine infection model. Our approach represents a novel and efficient strategy for targeted genetic manipulation in Chlamydia beyond C. trachomatis L2. This advance will support comparative studies of species-specific infection biology and enable studies in a well-established murine model of chlamydial pathogenesis.
KEYWORDS: Chlamydia, plasmid, mutagenesis, Tarp
INTRODUCTION
Members of the genus Chlamydia are diverse, Gram-negative, obligate intracellular bacteria that include 11 species infecting a wide range of hosts: Chlamydia muridarum (mice and hamsters), C. suis (pigs), C. pecorum (livestock), C. abortus (ruminants), C. felis (cats), C. caviae (guinea pigs), and C. avium, C. gallinacea, and C. psittaci (birds) (1–3). C. trachomatis and C. pneumoniae are prevalent human pathogens, causing a variety of acute as well as chronic diseases. C. trachomatis infects the mucosal surfaces of the human urogenital tract (UGT) or ocular tissue. Chlamydia-mediated genital infections represent the leading cause of bacterial sexually transmitted diseases (STDs) in the United States and worldwide. C. trachomatis isolates are currently divided into serovariants (4) that include serovars A to C and D to K, which infect the epithelia of the eye and the genital tract, respectively. In contrast to these noninvasive strains, the lymphogranuloma venereum (LGV) serovars C. trachomatis L1 to L3 transiently infect genital tract epithelial cells but then progress to the submucosae to infect macrophages, which facilitates dissemination of the bacteria to regional lymph nodes. Although infections with LGV serovars are more systemic and severe, the majority of chlamydial STDs worldwide are caused primarily by serovars D, E, and F (4). C. trachomatis infections with any UGT serovar can also lead to the development of chronic diseases, such as PID (pelvic inflammatory disease) in women, which can progress into ectopic pregnancy and infertility (5–7). Furthermore, C. trachomatis serovariants A, B, Ba, and C are causative agents of conjunctivitis, which if left untreated, can lead to severe scarring of the inside of the eyelid. Trachoma is the world’s leading cause of preventable blindness (8). Although not affecting humans, intravaginal infection of female mice with murine-specific C. muridarum has been used extensively as a small animal model to elucidate mechanisms of chlamydial disease (9). Critical pathogenesis parameters (10), such as chlamydial infectivity, ascension into the upper genital tract, and the subsequent pathology and immune response, can be readily studied.
Although it remains unclear which chlamydial determinants govern the host range, tissue tropism, and disease outcome, all chlamydiae undergo a typical developmental cycle, which is initiated by invasion of a host cell by the infectious elementary body (EB). Shortly after internalization, EBs begin to reorganize and differentiate into reticulate bodies (RBs), which are vegetative cell types specialized in bacterial multiplication. Late in the cycle, logarithmic growth of chlamydiae ceases, and RBs asynchronously reorganize back into EBs, which are then released upon lysis of the host cell (11) or via chlamydial extrusion (12). The chlamydial intracellular lifestyle posed a significant barrier to the achievement of specific inactivation of genes within the chlamydial genome. Successful and reproducible transformation of C. trachomatis L2 with the shuttle vector pGFP::SW2 was achieved almost a decade ago, leading to the development of a tractable genetic system in this chlamydial species (13). This progress has facilitated a more rapid appreciation of the molecular mechanisms governing the cellular infection biology of this model species (14).
All Chlamydia spp. employ a type III secretion system to remodel key biology of the infected host cells (15, 16). The application of genetics in C. trachomatis L2 has provided insight into how chlamydiae employ type III secreted effector proteins to create and maintain the parasitophorous vacuole. The translocated actin-recruiting phosphoprotein (Tarp) represents one well-characterized example (17). Tarp is a multifunctional effector contributing directly to chlamydial invasion by nucleating actin polymerization (18) and bundling F-actin filaments (19). Actin-binding domains are present in Tarp from all Chlamydia species (20). C. trachomatis Tarp contains a tyrosine-rich repeat domain phosphorylated by host kinases (21) and serving as a signaling platform leading to Arp2/3-mediated actin polymerization (22, 23). This region is absent from Tarp in other chlamydial species, such as C. muridarum and C. pneumoniae (20). A C. trachomatis L2 strain lacking tarP exhibits a decrease in invasion efficiency that is phenocopied by a strain expressing a Tarp allele lacking only the C-terminal actin-binding domains (24).
Although highly conserved, chlamydial plasmids exhibit a species-specific host range (25). For example, an engineered plasmid derived from C. pneumoniae can be used to transform C. pneumoniae but not C. trachomatis, C. muridarum, C. caviae, C. pecorum, or C. abortus (26). Pgp8 is at least one determinant implicated in this specificity, since replacement of pgp8 in a pL2-derived plasmid with the corresponding pgp8 from C. muridarum pNigg enabled the transformation of C. muridarum with an otherwise L2-derived plasmid (27). We investigated the potential role of iteron sequences located between the genes encoding CDS1/Pgp7 and CDS8/Pgp6. The engineered plasmid pKW-L2ori was constructed by mobilization of the entire region between these two genes, containing the four 22-bp tandem repeats, from pL2 into a pUC19-based plasmid. This otherwise Escherichia coli vector enabled the transformation of C. trachomatis L2 and was stably maintained when propagated under selective pressure. These data confirm that the functional ori in the chlamydial native plasmid is indeed located within that region. We also show that pKW-L2ori can be transformed into and maintained by C. trachomatis serovar D. However, pKW-L2ori could not be transformed into C. muridarum. Therefore, we modified this vector to include the species-specific iteron from C. muridarum, generating pKW-CMori, which was then successfully transformed and propagated by C. muridarum. Importantly, we demonstrate that pKW-CMori can be utilized as a shuttle vector in production of a tarP deletion mutant and a cis-tarp-complemented strain in C. muridarum by allelic exchange via homologous recombination. Finally, we show that in a murine infection model, Δtarp chlamydiae are eradicated from the mouse genital tract at a higher rate than wild-type (WT) or complemented C. muridarum strains, and animals infected with the tarP knockout chlamydiae exhibited significantly less severe pathologies of the upper genital tract.
Taken together, we present here an adaptable and efficient approach for deletion mutagenesis in non-LGV chlamydiae, utilizing an engineered shuttle vector containing both E. coli and Chlamydia plasmid origins of replication. This strategy can potentially be utilized with other plasmids of nonchlamydial origin and could be applied to any chlamydial species. Importantly, the work opens the door to work with chlamydial species, where applicable small-animal infection models allow direct investigation of pathogenic mechanisms.
RESULTS
pKW-L2ori and pKW-CMori are propagated by C. trachomatis and C. muridarum, respectively.
Native plasmids of C. trachomatis and C. muridarum contain four 22-bp tandem repeats in the intergenic region between coding sequences for Pgp6 and Pgp7 (28), yet the precise sequences (29) vary between the two species (Fig. 1A). It has been proposed that in C. trachomatis pL2, the plasmid origin of replication (ori) is located close to the four 22-bp tandem repeats (30). We utilized a pUC19 plasmid, which is routinely employed in various E. coli cloning procedures, as a backbone for generation of a recombinant vector for chlamydial transformation, as well as plasmid maintenance by the bacteria (Fig. 1B). This modified pUC19 vector contains aadA to confer spectinomycin (Spec) resistance. The genes gfp and mCherry, both expressed by the constitutive Neisseria meningitidis promoter (NmPro) from pGFP::SW2 (13), provide fluorescence reporting. In addition, loxP sites, flanking the aadA-gfp cassette, were introduced to enable the downstream floxed cassette allelic exchange mutagenesis approach (31). Although not required for the current studies, incorporation of loxP sites in the vector backbone enables excision of the marker cassette with Cre recombinase. This capability is useful when working with operon genes to relieve potential polar effects (31). Unique SalI and SbfI restriction sites were also added for efficient Gibson assembly-based cloning of the chlamydial DNA necessary for homologous recombination into the plasmid (32). Finally, the entire intergenic DNA sequence (231 bp) between pgp6 and pgp7 of pL2 (L2 ori) was introduced, generating the 5.45-kb vector pKW-L2ori. C. trachomatis serovar L2 was readily transformed with pKW-L2ori, using CaCl2 treatment (13), as determined by the presence of green and red fluorescent chlamydial inclusions (Fig. 2A). Moreover, chlamydiae were able to maintain and replicate pKW-L2ori throughout numerous passages in the presence of Spec. These data demonstrate that the putative ori in the chlamydial native plasmid is indeed located between pgp6 and pgp7. Upon removal of the selective antibiotic, loss of pKW-L2ori from C. trachomatis quickly followed. Almost 90% of serovar L2 lost the vector after just 2 passages in the absence of Spec, as determined by quantitative PCR (qPCR) (Fig. 2B).
FIG 1.

Generation of pKW-L2ori. (A) Species-specific iterons from C. trachomatis pL2 and C. muridarum pNigg showing 4 (I to IV) 22-bp tandem repeats (yellow) and immediate flanking DNA (blue). (B) Schematic of the pKW-L2ori shuttle vector, constructed as a backbone for downstream gene deletion applications. An aadA-gfp cassette is flanked by restriction sites (SbfI/SalI) for the subsequent introduction of ~3 kb of genomic sequences flanking the target gene. The constitutively expressed mCherry gene is included as a fluorescence marker of vector maintenance. The 265-bp region containing the pL2 iteron represents the putative chlamydial plasmid ori, and pBR322 is present to enable cloning manipulations in E. coli. LoxP sites were included to enable the subsequent CRE-mediated excision of the integrated aadA-gfp cassette from the chlamydial genome.
FIG 2.

Maintenance of pKW-L2ori and pKW-CMori by chlamydiae. (A) Fluorescent images of live C. trachomatis serovar L2 inclusions transformed with pKW-L2ori in McCoy cells at 30 h postinfection. (B) qPCR analysis of pKW-L2ori vector maintenance and elimination in four consecutive passages of L2 in the presence or absence of spectinomycin. (C) Fluorescent images of live C. trachomatis serovar D inclusions transformed with pKW-L2ori in McCoy cells at 40 h postinfection. (D) qPCR analysis of the pKW-L2ori vector in four consecutive passages of serovar D, incubated with or without spectinomycin. (E) Fluorescent images of live C. muridarum inclusions transformed with pKW-CMori in McCoy cells at 24 h postinfection. (F) qPCR analysis of pKW-CMori vector maintenance and elimination in four consecutive passages of C. muridarum in the presence or absence of penicillin G. The gfp gene copy number was relative to the 16S rRNA region. Scale bar = 10 μM.
The four 22-bp tandem repeats are identical among sequenced Chlamydia trachomatis serovars (28). Therefore, we tested the possibility that pKW-L2ori can be transformed into and replicated by C. trachomatis serovar D as well. Similar to L2, serovar D chlamydiae were successfully transformed with pKW-L2ori, and the vector was propagated in the presence of Spec. Likewise, removal of the selective antibiotic led to a rapid loss of the vector (Fig. 2C and D). We also tested whether just the tandem repeats (88 bp) were sufficient for plasmid replication in C. trachomatis. Unfortunately, pKW-L2repeats did not result in successful transformation of C. trachomatis L2, under the same conditions as described above, implying that the tandem repeats are necessary but not sufficient to function as the pL2 origin of replication. Transformation and propagation of pKW-L2ori in a plasmid-free C. trachomatis L2 strain (25667R) could not be achieved either, indicating that trans production of plasmid-encoded proteins from the native chlamydial plasmid is essential for maintenance of pKW-L2ori by chlamydiae. Interestingly, transformation of C. muridarum with pKW-L2ori was also unsuccessful. Thus, the pL2 ori region in pKW-L2ori was replaced with CMori, amplified from the C. muridarum native plasmid pNigg, generating pKW-CMori. The aadA gene was replaced with bla, and C. muridarum was successfully transformed with pKW-CMori. This rodent-specific chlamydial species maintained and replicated the vector in the presence of penicillin G (Fig. 2E). Removal of the selective antibiotic resulted in a rapid loss of pKW-CMori in a manner similar to that detected with C. trachomatis (Fig. 2F). Furthermore, unlike with previously published transformation and propagation of C. trachomatis L2 with the pGFP::SW2, pSU6, or pSUmC vectors (27, 32), the fluorescence signal was heterogeneous within inclusions harboring pKW plasmids. This uneven abundance of the vector by the bacteria likely resulted in a varying gfp copy number, relative to 16S rRNA, in consecutive passages of chlamydiae with selective antibiotics (Fig. 2B, D, and F). Taken together, we have shown that different serovars and species of Chlamydia are transformable with a plasmid containing only the species-specific iteron-containing region and that the vector propagates in chlamydiae only under selective pressure. Rapid curing of the vector upon removal of the selective antibiotic makes this an ideal platform for allelic exchange mutagenesis.
Generation of Δtarp in C. trachomatis serovar D and C. muridarum employing the pKW shuttle vector.
Next, we investigated whether pKW can be utilized as a conditionally replicating plasmid sufficient for the generation of deletion mutants via allelic exchange. As a proof of principle, we chose to delete the tarP gene, since it has been previously demonstrated that the Tarp effector is a monocistronic, nonessential gene in C. trachomatis L2 (24) but varies in domain structure across chlamydial serovars and species (20). We first tested whether pKW-L2ori could support mutagenesis in C. trachomatis serovar D. Gibson assembly was employed to insert ~3 kb of upstream and downstream tarP-flanking genomic DNA into pKW-L2ori at the SalI and SbfI sites, respectively. pKW-L2ori-Δtarp (see Fig. S1A in the supplemental material) was mobilized into C. trachomatis serovar D, and Spec was used to select for transformants. The transformants were passaged in the presence of the selective antibiotic until ~60% of inclusions displayed dim red and green fluorescence, indicating the presence of merodiploids. Spec was then removed for two consecutive passages, resulting in a rapid loss of the pKW-L2ori plasmid, as indicated by the loss of red fluorescence in chlamydial inclusions. Reintroduction of Spec in the following passages selected for chlamydiae where allelic exchange had occurred to yield a GFP+ deletion strain. Several isolates were then purified using the sequential limiting dilution method as previously described (32) to generate clonal strains. The absence of tarP in C. trachomatis serovar D was confirmed by qPCR (Fig. S2A) and in a Tarp-specific immunoblot of material derived from HeLa cells equally infected with WT or Δtarp chlamydiae (Fig. 3A). As expected, the manipulations did not result in curing of the endogenous plasmid from the mutant strain, since plasmid was detectible by qPCR (Fig. 3B). Whole-genome sequencing indicated several nonsynonymous point mutations in the chromosome (Table S2) but no alterations within the endogenous plasmid. Loss of Tarp in C. trachomatis L2 manifests as a modest decrease in invasion efficiency (24). We therefore tested whether Tarp-deficient C. trachomatis D would phenocopy this effect. Differentially stained external and internal EBs were enumerated in HeLa cells, fixed after allowing 30 min for invasion of the attached chlamydiae. Internalization of the Δtarp strain decreased significantly compared to the WT (Fig. 3C). In aggregate, these data confirm the efficacy of our approach for the mutagenesis of non-LGV C. trachomatis and confirm the conservation of a Tarp requirement for efficient C. trachomatis invasion.
FIG 3.
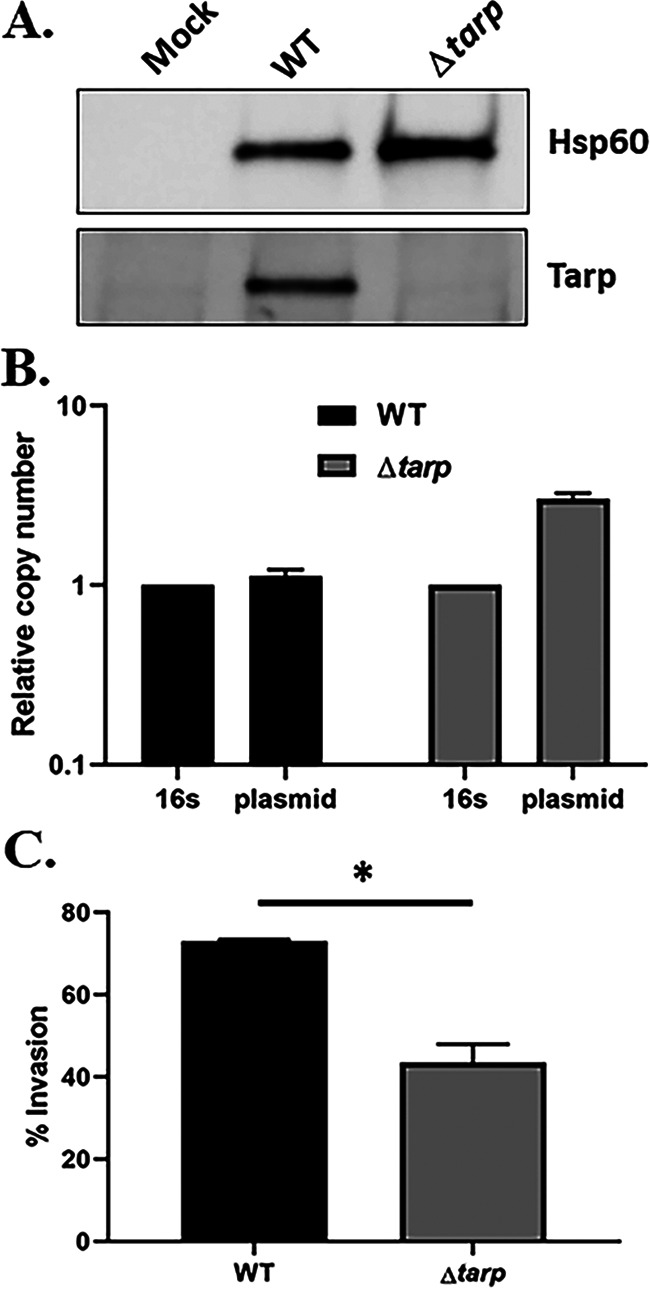
C. trachomatis serovar D Δtarp. (A) Immunoblot of whole-cell culture lysates from mock-infected HeLa cells or cells infected with WT or Δtarp C. trachomatis D for 24 h. Tarp was detected using Tarp-specific antibodies, and Hsp60-specific antibodies were used as a loading control. (B) qPCR analysis of Δtarp- and WT C. trachomatis D-infected cells showing the presence of the native chlamydial plasmid in both chlamydial strains. The copy number of genes pgp7-pgp8 was normalized to the 16S rRNA-specific signal. (C) HeLa monolayers were infected for 1 h at 4°C with WT or mutant strains at a multiplicity of infection (MOI) of 10. The cultures were shifted to 37°C for 30 min and then paraformaldehyde fixed and processed for inside-out staining to assess the invasion efficiency. Data are represented as the mean values for the percentage of internalized chlamydiae and are shown with standard deviations. Statistical significance was computed using a Student’s t test with Welch’s correction (*, P < 0.01).
We next shifted our strategy to focus on the model organism C. muridarum, with the goal of leveraging the murine infection model to test the in vivo role of Tarp. As done previously for C. trachomatis L2 (24), our approach was to delete the entire tarP gene to create a null mutant and subsequently use that strain as a parent to restore tarP for a cis-complemented strain using allelic replacement (Fig. 4A). To construct the shuttle vector, pKW-CMori was digested with SalI restriction enzyme, and ~3 kb of sequence upstream of the tarP gene was cloned into the vector by Gibson assembly (33). After that, pKW-CMori was linearized at the SbfI site prior to the insertion of ~3 kb downstream DNA to produce pKW-CMori-Δtarp (Fig. S1B). C. muridarum was transformed and a mutant strain isolated by sequential cultivation in the presence or absence of antibiotic selection, as described above. A similar approach was employed to generate the cis-tarp-complemented C. muridarum strain using pKW-CMori-cis-tarp (Fig. S1C). In both cases, clonal isolates were obtained by limiting dilution. Whole-genome sequencing indicated no changes in either the chromosome or endogenous plasmid compared to the parent. The absence or presence of Tarp in the respective strains was confirmed via tarP-specific qPCR (Fig. S2B). The abundance of tarP in the C. muridarum mutant and complemented chlamydiae, compared to the WT, was confirmed by immunoblot analysis (Fig. 4B). The presence of the native plasmid pNigg in Δtarp C. muridarum was confirmed via qPCR (Fig. 4C), demonstrating that the endogenous plasmid was not lost during manipulations in this species either. Enumeration of Δtarp and cis-tarp-complemented C. muridarum progeny EBs in 24-h McCoy cultures demonstrated that neither strain was impaired in chlamydial development compared to the WT chlamydiae (Fig. 5A). Differences in infectious particle abundance were also not detected in higher resolution studies, when cultures were sampled at multiple time points, ranging from 12 to 48 h (Fig. S3A). Unlike with C. trachomatis L2 (24), or C. trachomatis D, deletion of tarP from the C. muridarum genome did not significantly decrease the invasion of murine cells (Fig. 5B). Both the mutant and cis-complemented strains exhibited WT levels of entry at 60 min postinfection. Invasion was also not impacted during the similar infection of HeLa cells (Fig. S3B). In aggregate, we have demonstrated here that the pKW-based plasmid can be successfully utilized as a shuttle vector to support allelic replacement for production of specific deletion mutants. Unlike with previously developed approaches that are primarily applicable for C. trachomatis serovar L2, pKW has a great potential in the mutagenesis of other chlamydial strains and species, such as C. trachomatis serovar D and C. muridarum. These data also suggest that TarP-mediated actin polymerization may be dispensable for invasion of cells by C. muridarum.
FIG 4.

Generation of the tarP-null mutant and cis-complemented strain in C. muridarum. (A) Schematic representation of the strategy employed in deletion of the entire tarP gene from the chlamydial genome and generation of a cis-complemented strain by allelic replacement. (B) Western blot analysis with anti-Tarp polyclonal antiserum, demonstrating the absence of the tarP gene in C. muridarum-infected cell cultures compared the WT or cis-tarp-complemented strains. Probing with genus-specific anti-Hsp60 monoclonal antibody was included as a loading control. (C) qPCR analysis of Δtarp- and WT C. muridarum-infected cells showing the presence of the native plasmid in both chlamydial strains. The copy number of genes pgp7-pgp8 is presented relative to a 16S rRNA region.
FIG 5.
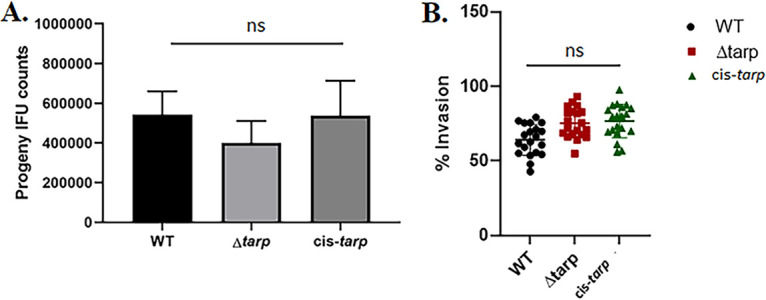
Analysis of tarP deletion in C. muridarum host cell invasion. (A) Murine McCoy cells or (B) mouse L929 cells were infected with either the tarP deletion mutant, cis-tarp-complemented strain, or WT C. muridarum. Progeny EB production was quantitated at 24 h postinfection (A) and chlamydial invasion was analyzed at 1 h postinfection (B). Data are represented as the mean values for progeny EBs or percentage of internalized chlamydiae and are shown with standard deviations. No significant differences (ns) in chlamydial replication or entry were detected among the tested strains.
Attenuation of C. muridarum Δtarp in a murine infection model.
Loss of C. trachomatis L2 tarP exhibits a modest negative impact on invasion in a tissue culture model but manifests as a pronounced attenuation of fitness in the murine infection model (24). We therefore extended our analysis of C. muridarum Δtarp to the murine infection model, given the utility of directly assessing genital tract pathogenesis with a host-appropriate pathogen. Groups of 6 female C57BL/6 mice were challenged cervicovaginally with 105 inclusion-forming units (IFUs) of WT, Δtarp, or cis-tarp C. muridarum, and the shed infectious particles were enumerated from vaginal swabs taken every 4 days. The Δtarp exhibited significant attenuation compared to the WT and the cis-complemented control (Fig. 6A and B). Shed Δtarp IFUs were ca. 100-fold less abundant as early as day 3, and overall shedding was significantly less for Δtarp by two-way ANOVA (P = 0.0004). Resolution of infection was also diminished significantly (P = 0.0007), with a mean time to clearance of day 16 (±3.26) for Δtarp compared to day 24 (±2.58) for WT and 23 (±2.85) for cis-tarp. These observations are reproducible, since we observed similar attenuation and kinetics in a preliminary study comparing WT and Δtarp that was conducted prior to construction of the cis-tarp strain (Fig. S4). The use of C. muridarum in the murine model enables assessment of upper genital tract pathology (34). The mice were therefore euthanized on day 60, and the gross pathology of the upper genital tracts was evaluated for the presence and severity of tubal dilation and hydrosalpinx (Fig. 6C). The incidence of tubal dilation and hydrosalpinx did not differ significantly among strains. However, pathologies were less severe in mice infected with Δtarp. Quantitation of hydrosalpinx formation (Fig. 6D) revealed a significant decrease in severity induced by Δtarp. Restoration of Tarp in the cis-tarp strain restored hydrosalpinx severity. In aggregate, these data clearly indicate the important nature of Tarp during chlamydial infection and the ensuing disease.
FIG 6.

Attenuation of Δtarp C. muridarum in a mouse infection model. Groups of 6 mice were challenged intravaginally with equal infectious units of WT, Δtarp, or cis-tarp C. muridarum, and the shed infectious particles were enumerated from days 3 to 28. Average progeny numbers shed (A) and percentage of mice actively shedding chlamydiae (B) at different days postinfection. Significance was determined by two-way analysis of variance (ANOVA) (***, P < 0.0004). Reproductive tracts were excised on day 60 postinfection and assessed for gross pathology. (C) Representative images are shown from each group, and areas of hydrosalpinx and tubal dilation are highlighted in insets (black or white arrows, respectively). (D) Plot of hydrosalpinx severity score data for each mouse. A Wilcoxon rank-sum test was performed to address significance (*, P < 0.01; **, P < 0.002).
DISCUSSION
Members of the genus Chlamydia are prevalent bacterial parasites of a diverse range of eukaryotic hosts. Different chlamydial species share many aspects of the typical intracellular lifestyle, such as the biphasic developmental cycle and the preference to primarily invade epithelial cells. However, each chlamydial strain also employs sophisticated species-specific strategies that significantly contribute to an extraordinary diversity in organ and/or tissue tropism and disease manifestation. For example, variation in gene content within the chlamydial plasticity zone represents an obvious source of species-specific biology (35). Despite highly conserved gene synteny outside the plasticity zone, significant alterations in the primary sequence of conserved proteins (36) likely also contributes. In order to discover and understand the mechanisms underlying how these pathogens infect particular hosts, utilize nutrients, respond to the eukaryotic intracellular environment, communicate with the host cell, and most importantly, cause specific diseases, it is imperative to utilize a genetic manipulation approach applicable to multiple chlamydial species.
Although human infections with the LGV serovars L1, L2, L2a, and L3 are rare in the developed world, C. trachomatis L2 is the least fastidious and most tractable to work with in laboratory conditions. Thus, this chlamydial serovariant is commonly utilized as a model organism in chlamydial research. Successful transformation and maintenance of the shuttle vector pGFP::SW2, performed with C. trachomatis L2, was the first, essential step leading to the development of tractable genetic systems in this chlamydial species (13). Random mutagenesis approaches using ethyl methanesulfonate (EMS) treatment or transposon mutagenesis of C. trachomatis L2 and C. muridarum were successfully achieved (37–41); however, generation of mutants in specific, target genes, utilizing TargeTron-based insertion mutagenesis (reviewed in reference 42) or FRAEM systems (24, 32), is limited primarily to C. trachomatis L2. Since this obligate intracellular pathogen is unable to stably maintain vectors that are typically propagated by E. coli and vice versa, numerous studies have employed various constructs for knockout or knock-in of a particulate gene(s), using the entire pL2 as a backbone. There are often obstacles when working with the entire native chlamydial plasmid. Routine cloning with pL2 can be challenging, since it appears that at least one of the eight open reading frame (ORF) products might be toxic to some E. coli strains, particularly in conjunction with other chlamydial and/or nonchlamydial sequences that are introduced into the construct. In addition, a particular sequence composition and/or the final size of the construct, which can reach ≥18 kb in the pSUmC-based FRAEM system (32), may contribute to instability of the shuttle vector and low transformation efficiency for C. trachomatis (unpublished observations).
We have developed an efficient and adaptable strategy utilizing a pUC19-based vector that can be employed in generation of targeted full gene deletion mutants in the chromosome of any member of the genus Chlamydia. Instead of placing an E. coli origin of replication into pL2-based constructs, we introduced a putative ori of the chlamydial native plasmid into the pUC19 vector and achieved successful transformation and propagation of the construct by the bacteria. Most importantly, we successfully utilized the resulting pKW shuttle vector for generation of tarP deletion mutants in C. trachomatis serovar D and C. muridarum. Selection with either spectinomycin or penicillin worked equally well during the mutagenesis procedures. In plasmids from other bacterial species, tandem repeats (or iterons) are typically involved in plasmid replication, control of copy numbers, and incompatibility (29, 43). We show that the pgp6/pgp7 region in pL2 and pNigg, containing four 22-bp tandem repeats, represents the functional ori of the chlamydial native plasmid. Pgp7 is encoded directly after the origin of replication (29). However, the repeats by themselves were insufficient for the transformation and/or propagation of pKW by chlamydiae. The precise sequences representing the pL2 and/or pNigg ori remains to be determined but likely corresponds to the footprint of replication machinery.
It has been shown that some of the eight ORFs within the native chlamydial plasmid in both C. trachomatis and C. muridarum regulate multiple chromosomal genes (44, 45). One of the plasmid-regulated loci encodes the enzyme glycogen synthase (GlgA) (44). Although ORF1 to ORF8 are highly conserved in C. trachomatis and C. muridarum, it has been demonstrated that the transformation of plasmid-free strains of C. trachomatis serovar A or C. muridarum was only possible when the chlamydial plasmid-based shuttle vector contained the backbone of the plasmid naturally found in those strains. This phenomenon was termed plasmid tropism (46). Our results show that the pKW-L2ori vector, containing the four 22-bp tandem repeats from pL2, could be transformed and propagated by C. trachomatis serovars L2 and D but not by C. muridarum. These data suggest that the previously described plasmid tropism might also involve the species-specific chlamydial plasmid origin of replication. Furthermore, the plasmid-free L2 strain (25667R) could not be transformed with pKW-L2ori, indicating that the presence of the native plasmid is essential for propagation of the vector by chlamydiae. Indeed, the 7.5-kb native plasmid was maintained during generation of both the C. trachomatis D and C. muridarum tarP strains. The ability to produce specific, targeted gene deletion mutants and cis-complemented strains in C. muridarum is imperative for studying the pathogenesis of chlamydial disease in vivo. There is no relevant animal model mimicking human disease for LGV serovars. Although a transcervical inoculation route has been developed for C. trachomatis D (47), intravaginal infection with murine-specific C. muridarum has historically proven to be the most appropriate small animal model for studying chlamydial disease (34). As a proof of principle, we therefore chose to utilize the pKW-CMori shuttle vector to construct a tarP mutant and complemented strains for animal model studies.
Tarp nucleates actin polymerization associated with chlamydial invasion (18). The C. trachomatis L2 deletion strain manifests a defect in invasion efficiency (24), and this phenotype was replicated by the C. trachomatis D Δtarp strain. We were therefore intrigued that the loss of Tarp in C. muridarum did not decrease the invasion efficiency. Numerous possibilities could explain this apparent divergence. First, we cannot exclude the possibility that technical differences in infectivity could influence the outcome. For example, highly efficient attachment and invasion of tissue culture cells by C. muridarum requires centrifugation or pretreatment with DEAE-dextran, whereas C. trachomatis L2 does not (48). This explanation seems unlikely, since our infections were carried out on untreated cells, and centrifugation did not alter the outcome. Likewise, a similar protocol was followed for C. trachomatis D, where an invasion defect was detected. Alternatively, it is possible that C. trachomatis and C. muridarum Tarp function differently, despite ca. 59% overall primary sequence identity (21, 49). Tarp orthologs contain multiple functional domains, including tyrosine-rich repeats that are targeted by host kinases, an oligomerization-mediating proline-rich domain, domains that interact with actin to promote polymerization, and domains interacting with structural elements of the host cytoskeleton (reviewed in reference 17). C. muridarum Tarp does lack the tyrosine-containing repeat domains and is not phosphorylated by host kinases (49). However, the phosphorylation state of Tarp does not affect C. trachomatis entry (49, 50). Instead, the C-terminal Tarp domains mediating actin bundling appear to be most important for chlamydial invasion (24). Importantly, C. muridarum Tarp retains its actin polymerization function (20) and contains domains necessary for actin bundling. Chlamydia spp. clearly exhibit unique requirements for host cell entry, and this could also explain differences in the Tarp requirements. For example, C. caviae, but not C. trachomatis, requires Cdc42 activity to achieve actin polymerization associated with entry (51). Finally, C. muridarum shares many common invasion requirements with C. trachomatis (16), yet it is possible that redundant mechanisms manifested by C. muridarum, but not C. trachomatis, might mask the impact of tarP deficiency in a tissue culture infection model. These interesting possibilities are under investigation.
Murine infection experiments clearly indicate a role for C. muridarum Tarp during in vivo infection. We have previously demonstrated that C. trachomatis L2 Δtarp sheds fewer EBs from the murine lower genital tract. Although these studies allude to a Tarp-specific fitness requirement, they do not directly address more complex and important questions regarding contributions to Chlamydia pathogenesis. Intravaginal C. trachomatis infections of mice are confined to the lower genital tract, do not yield overt upper genital tract pathology, and can be resolved by innate immunity (52). Our present study emphasizes the utility of extending mutagenesis capability beyond non-LGV Chlamydia. The use of C. muridarum leverages a well-established animal model (34) to investigate pathogenesis parameters such as chlamydial infectivity, ascension into the UGT, and the subsequent ability to induce pathology (10). Mice infected with tarP-null C. muridarum shed ca. 100-fold fewer chlamydiae than those infected with the WT or cis-tarp strains at day 3. Interestingly, shedding did not immediately continue to drop. Instead, Δtarp-infected mice continued to shed similar levels of bacteria at days 7 and 11, followed by a gradual decrease until the IFUs were below detection. These data suggest that Tarp may be important for establishing initial colonization but is less critical for maintaining ongoing infection. We did observe less severe hydrosalpinx in Δtarp-infected mice. Hydrosalpinx requires the ascension of chlamydiae into the upper genital tract (53). We suspect that less pathology correlates with an overall lower infectious burden of Δtarp. Additional possibilities include a requirement of Tarp to efficiently ascend beyond the cervical mucosae or a proinflammatory response linked to Tarp activity. Additional work will be required to discriminate among these possibilities.
In summary, we determined the functional origin of replication within chlamydial native plasmids. By mobilizing this ori region, in a Chlamydia species-specific manner, into the pUC19 plasmid, we achieved successful maintenance of this E. coli vector by the pathogen. The possibility of routinely utilizing a nonchlamydial vector(s) for generation of deletion mutants and complemented strains in any chlamydial species, by introducing the ori of the native chlamydial plasmid into that vector, represents a great leap forward in genetic manipulation of these obligate intracellular bacteria. This strategy would allow for a direct comparison of phenotype(s), in particular, chromosomal deletions, among various chlamydial species and/or serovariants and would thus reveal the sophisticated strategies employed by each chlamydial strain, leading to a specific pathogenesis outcome(s). Importantly, the ability to generate specific gene knockouts in C. muridarum is essential for discovery of disease mechanisms associated with genital chlamydial infections in vivo.
MATERIALS AND METHODS
Cell culture and organisms.
C. trachomatis serovars L2 (LGV 434) and D (D/UW-3/CX) and C. muridarum (Nigg) were purchased from the American Type Culture Collection (Manassas, VA). Chlamydiae were propagated in either HeLa 229 cells (CCL-2.1; ATCC) or McCoy cells (CRL-1696; ATCC) and purified using MD-76R (diatrizoate meglumine and diatrizoate sodium injection USP; Mallinckrodt Pharmaceuticals) as previously described (48). All tissue culture cells were grown in RPMI 1640 containing 2 mM l-glutamine supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Thermo Fisher) and incubated at 37°C in an atmosphere of 5% CO2 and 95% humidified air.
Cloning.
pUC19-aadA-gfp was previously generated by cloning the aadA-gfp cassette into pUC19 (New England Biolabs; NEB) by iPCR (insertion/deletion PCR) (32, 54). The mCherry gene and SalI-loxP and SbfI-loxP sites were amplified from the pSUmC vector (32) and introduced into pUC19-aadA-gfp by insertion/deletion PCR. Finally, sequences between pgp6 and pgp7 (total, 265 bp) containing four 22-bp tandem repeats (L2 ori) were amplified from C. trachomatis L2 and cloned into the vector by iPCR, generating pKW-L2ori (see Table S1 in the supplemental materials). For generation of pKW-CMori, the region between the genes pgp6 and pgp7 of pNigg was amplified from C. muridarum. The vector was amplified by divergent PCR, and the CMori sequences were introduced into pUC19 by Gibson assembly (HiFi DNA assembly cloning kit; NEB). A similar approach was used for replacing aadA with the bla gene in pKW-CMori. For generation of the shuttle vector pKW-L2ori-Δtarp, pKW-L2ori was first linearized with SalI restriction enzyme, and ~3-kb 5′ arm sequences upstream of the tarP gene, amplified from C. trachomatis D, were introduced into the vector using the HiFi DNA assembly cloning kit (NEB). Then, ~3-kb 3′ arm sequences, downstream of the tarP gene, were cloned into the plasmid at the SbfI site as described above. pKW-CMori-Δtarp was similarly constructed using DNA PCR amplified from C. muridarum template DNA and inserted into pKW-L2ori. The pKW-CMori-cis-tarp construct was generated by replacing the bla-gfp cassette with aadA and full-length tarP in pKW-CMori-Δtarp, as described above. E. coli strain NEB-10β (NEB) was utilized in all cloning procedures, and the dam− dcm− strain (NEB) was used as a host to generate unmethylated plasmid DNA prior to chlamydial transformations. E. coli growth was carried out at 30°C. All primers employed in the cloning procedures were custom DNA oligonucleotides from Integrated DNA Technologies (Table S1). Q5 high-fidelity polymerase, SalI-HF and SbfI-HF restriction enzymes, and the HiFi DNA assembly cloning kit were purchased from New England Biolabs. KOD hot-start DNA polymerase, used in divergent PCRs, was obtained from Millipore.
Chlamydial transformation.
C. trachomatis serovars L2 and D and C. muridarum were transformed with pKW-L2ori and pKW-CMori, respectively, as previously described (32). Chlamydial transformants were selected with either 500 μg/mL for L2 or 400 μg/mL for D of spectinomycin or 0.6 µg/mL of penicillin G sodium salt for C. muridarum. DNA from McCoy-infected cell cultures with chlamydial transformants, with or without the selective antibiotic, was extracted from four consecutive passages, as previously published (37). qPCR analyses were performed with primer sets targeting chlamydial 16S rRNA and plasmid gfp genes (32) using iTaq universal SYBR green supermix and the CFX96 real-time thermal cycler (Bio-Rad). tarP mutants were obtained by continuous passaging of chlamydiae, transformed with the pKW-L2ori-Δtarp (C. trachomatis D) or pKW-CMori-Δtarp (C. muridarum) shuttle vectors, in the presence of antibiotic until approximately 60% of inclusions became dim green, with no detectable red fluorescence. The mutant tarP populations were then enriched by performing two consecutive passages of chlamydiae in the absence of the selective antibiotic, followed by adding the respective antibiotic back into the culture. Isolation of clonal populations was achieved by the limiting dilution method, as previously described (32). A similar approach was employed using pKW-CMori-cis-tarp to generate a cis-tarp-complemented strain of C. muridarum. The loss of tarP and retention of the endogenous plasmid were confirmed via qPCR of strain-specific DNA, as previously described (28). Amplification of 16S DNA from both C. trachomatis serovar D and C. muridarum was accomplished using the primers 16S-s (5′-CCTGGTAGTCCTTGCCGTAAAC-3′) and 16S-as (5′-TACTCCTCAGGCGGCATACTTA-3′). The primers Dtarp-s (5′-ACACTACTGCCTTCACCACCTC-3′) and Dtarp-as (5′-TCCTCCCATCATCAAGGATGTGG-3′) or CMtarp-s (5′-CCGGAGGAATCATAGCTCATAC-3′) and CMtarp-as (5′-CGCTGCATCATAGAGAGAGAC-3′) were used to probe for tarP from C. trachomatis serovar D or C. muridarum, respectively. The serovar D endogenous plasmid was detected using the primers pgp7 s (5′-GACAACGCTTCAAAGAAGATGGCTCTAATATAG-3′) and pgp8 a-s (5′-CCTCTAGTACAAACACCCCCAATATTGTG-3′), spanning pgp7-pgp8. The primers CMpgp7 s (5′-CTGTCGCAAATATACCAACTATATTGTG-3′) and CMpgp8 (5′-GAC AAT GCC TCA AAG AAG ATG GCC C-3′) were also used. The signals were normalized to those from the respective 16S amplifications. Whole-genome DNA was extracted from the parent and engineered strains, followed by sequencing and variant analyses (SeqCenter).
Δtarp C. muridarum growth and immunoblotting analysis.
McCoy or HeLa 229 cells were seeded into 6-well plates (2 × 105 cells/well) 24 h prior to infection. Individual replicate wells were infected either with WT, Δtarp, or cis-tarp-complemented C. muridarum. To facilitate the infection, cultures were centrifuged at 1,000 × g for 1 h, followed by incubation at 37°C. Progeny EBs were enumerated at 24 h by passage of lysates onto fresh McCoy cells. For one-step growth curves, cultures were harvested 0, 12, 24, 36, and 48 h postinfection. Cycloheximide was supplemented to the culture medium at a concentration of 1 μg/mL. The cell lysates were frozen at −80°C until samples from all time points had been collected. The cell lysates were then thawed on ice and diluted, and fresh HeLa cell monolayers were infected as described above. All progeny cell cultures were fixed and immune-stained with an anti-Chlamydia antibody and Alexa 488 conjugated secondary antibody, and the inclusions were counted using the BioTek Cytation 5 cell imaging multimode reader. The averages were plotted using GraphPad Prism software.
Protein samples were obtained from lysates of cells infected with WT, Δtarp (C. trachomatis D or C. muridarum), or cis-tarp-complemented strains and concentrated by adding trichloroacetic acid (Millipore) to 10% (vol/vol). Collected protein pellets were analyzed by SDS-PAGE electrophoresis, followed by immunoblot analysis using anti-Tarp polyclonal rabbit antiserum and peroxidase-conjugated secondary antibody (Millipore). Blotting with genus-specific anti-Hsp60 MAb (Santa Cruz) was included as a loading control. Proteins were visualized by probing with horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescence detection using Amersham ECL Plus reagent (GE Healthcare UK Limited).
Chlamydia invasion assay.
Fluorescent C. muridarum EBs were purified from cell cultures supplemented with CellTracker red CMTPX dye as previously described (54). Briefly, CMPTX-labeled C. muridarum EBs (multiplicity of infection, ~10) were added to McCoy or HeLa cells seeded into 24-well plates. To facilitate the infection, the cultures were centrifuged at 1,000 × g for 1 h, followed by incubation at 37°C for an additional hour. Cycloheximide was supplemented to the culture medium at a concentration of 1 μg/mL. For assays involving C. trachomatis D, unlabeled bacteria were used to infect HeLa monolayers at an MOI of 10. Infections were carried out by incubating cultures on ice for 1 h, followed by centrifugation for 5 min at 1,000 × g. The C. trachomatis D cultures were then shifted to 37°C for 30 min. All cultures were fixed with 4% paraformaldehyde at room temperature for 15 min and rinsed with phosphate-buffered saline (PBS). The cells were not permeabilized. Extracellular EBs were labeled for 1.5 h with a monoclonal antibody (MAb) specific for chlamydial lipopolysaccharide (LPS) clone F70G (Invitrogen). After three washes in PBS, secondary antibody conjugated to Alexa 488 was added for 1 h. The samples were washed three times in PBS. For C. trachomatis D, the samples were permeabilized with 0.01% Triton X-100 and stained with MOMP-specific antibodies. For each assay, the number of green (external) and red (total) EBs was determined over 10 fields of view. These data were then used to determine the percentage of internalized EBs. Data were collected from 3 individual replicate wells for each clone, and these percentages were then averaged together to give a final invasion rate.
Murine infection studies.
C. muridarum Nigg WT, Δtarp, and cis-tarp strains were used in murine infection studies. Groups of 6 female, 6- to 8-week-old C57BL/6 mice (Jackson Laboratory) were pretreated with 2.5 mg medroxyprogesterone delivered subcutaneously 5 days prior to infection. The mice were then intravaginally infected as described (55) with 105 IFUs of each chlamydial strain. Shed bacteria were recovered by vaginal swab beginning on day 3, then every 4 days until no chlamydiae were detected for all WT-infected mice. The recovered IFUs were enumerated on fresh HeLa cells as described above. The mice were sacrificed 60 days after intravaginal infection, and the gross pathology was assessed during necropsy. Complete genital tract tissues, representing the vagina to the ovaries, were isolated and photographed. Each uterine horn was examined individually for hydrosalpinx, as described (56). Individual horns were scored from 1 to 4, and cumulative scores were generated for each mouse, for a range of 0 to 8. Scores are defined as previously described (56), with 0 assigned for no visible hydrosalpinx. Severity scores were assigned based on whether the hydrosalpinx was visible only after amplification (score, 1), clearly visible to the naked eye but smaller than the ovary (score, 2), similar in size to the ovary (score, 3), or larger than the ovary (score, 4). The hydrosalpinx incidence and severity scores were analyzed for statistical significance of the differences between mice infected with different C. muridarum strains. Animal experiments were reviewed and approved by the University of Kentucky Institutional Animal Care and Use Committee.
ACKNOWLEDGMENTS
We thank Brian Stevenson for critical reading of the manuscript and R. W. Hayman for excellent technical assistance and critical reading of the manuscript. This work was supported by Public Health Service grants from the National Institutes of Health, NIAID, to K. Wolf (grant R21AI166271-01) and T. J. Jewett (R01AI139242-01).
Footnotes
This article is a direct contribution from Kenneth A. Fields, a member of the Infection and Immunity Editorial Board, who arranged for and secured reviews by Ian Clarke, University of Southhamptom, and Kyle Ramsey, CCOM-Midwestern University.
Supplemental material is available online only.
Contributor Information
Katerina Wolf, Email: kwo227@uky.edu.
Andreas J. Bäumler, University of California, Davis
REFERENCES
- 1.Everett KD. 2000. Chlamydia and Chlamydiales: more than meets the eye. Vet Microbiol 75:109–126. 10.1016/s0378-1135(00)00213-3. [DOI] [PubMed] [Google Scholar]
- 2.Sachse K, Laroucau K, Riege K, Wehner S, Dilcher M, Creasy HH, Weidmann M, Myers G, Vorimore F, Vicari N, Magnino S, Liebler-Tenorio E, Ruettger A, Bavoil PM, Hufert FT, Rossello-Mora R, Marz M. 2014. Evidence for the existence of two new members of the family Chlamydiaceae and proposal of Chlamydia avium sp. nov. and Chlamydia gallinacea sp. nov. Syst Appl Microbiol 37:79–88. 10.1016/j.syapm.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Wheelhouse N, Longbottom D. 2012. Endemic and emerging chlamydial infections of animals and their zoonotic implications. Transbound Emerg Dis 59:283–291. 10.1111/j.1865-1682.2011.01274.x. [DOI] [PubMed] [Google Scholar]
- 4.Lesiak-Markowicz I, Schotta AM, Stockinger H, Stanek G, Markowicz M. 2019. Chlamydia trachomatis serovars in urogenital and ocular samples collected 2014-2017 from Austrian patients. Sci Rep 9:18327. 10.1038/s41598-019-54886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schachter J. 1978. Chlamydial infections (first of three parts). N Engl J Med 298:428–435. 10.1056/NEJM197802232980805. [DOI] [PubMed] [Google Scholar]
- 6.Schachter J. 1978. Chlamydial infections (second of three parts). N Engl J Med 298:490–495. 10.1056/NEJM197803022980905. [DOI] [PubMed] [Google Scholar]
- 7.Schachter J. 1978. Chlamydial infections (third of three parts). N Engl J Med 298:540–549. 10.1056/NEJM197803092981005. [DOI] [PubMed] [Google Scholar]
- 8.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. 2004. Global data on visual impairment in the year 2002. Bull World Health Organ 82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 9.Dockterman J, Coers J. 2021. Immunopathogenesis of genital Chlamydia infection: insights from mouse models. Pathog Dis 79:ftab012. 10.1093/femspd/ftab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darville T, Hiltke TJ. 2010. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis 201 Suppl 2:S114–S125. 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moulder JW. 1966. The relation of the psittacosis group (Chlamydiae) to bacteria and viruses. Annu Rev Microbiol 20:107–130. 10.1146/annurev.mi.20.100166.000543. [DOI] [PubMed] [Google Scholar]
- 12.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci USA 104:11430–11435. 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolat L, Valdivia RH. 2019. A renewed tool kit to explore Chlamydia pathogenesis: from molecular genetics to new infection models. F1000Res 8:935. 10.12688/f1000research.18832.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller KE, Plano GV, Fields KA. 2014. New frontiers in type III secretion biology: the Chlamydia perspective. Infect Immun 82:2–9. 10.1128/IAI.00917-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400. 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caven L, Carabeo RA. 2019. Pathogenic puppetry: manipulation of the host actin cytoskeleton by Chlamydia trachomatis. Int J Mol Sci 21:90. 10.3390/ijms21010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. 2006. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci USA 103:15599–15604. 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiwani S, Alvarado S, Ohr RJ, Romero A, Nguyen B, Jewett TJ. 2013. Chlamydia trachomatis Tarp harbors distinct G and F actin binding domains that bundle actin filaments. J Bacteriol 195:708–716. 10.1128/JB.01768-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jewett TJ, Miller NJ, Dooley CA, Hackstadt T. 2010. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog 6:e1000997. 10.1371/journal.ppat.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutter EI, Bonner C, Holland MJ, Suchland RJ, Stamm WE, Jewett TJ, McClarty G, Hackstadt T. 2010. Phylogenetic analysis of Chlamydia trachomatis Tarp and correlation with clinical phenotype. Infect Immun 78:3678–3688. 10.1128/IAI.00515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. 2008. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4:e1000014. 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiwani S, Ohr RJ, Fischer ER, Hackstadt T, Alvarado S, Romero A, Jewett TJ. 2012. Chlamydia trachomatis Tarp cooperates with the Arp2/3 complex to increase the rate of actin polymerization. Biochem Biophys Res Commun 420:816–821. 10.1016/j.bbrc.2012.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh S, Ruelke EA, Ferrell JC, Bodero MD, Fields KA, Jewett TJ. 2020. Fluorescence-reported allelic exchange mutagenesis-mediated gene deletion indicates a requirement for Chlamydia trachomatis Tarp during in vivo infectivity and reveals a specific role for the C terminus during cellular invasion. Infect Immun 88:e00841-19. 10.1128/IAI.00841-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabo KV, O'Neill CE, Clarke IN. 2020. Diversity in chlamydial plasmids. PLoS One 15:e0233298. 10.1371/journal.pone.0233298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shima K, Wanker M, Skilton RJ, Cutcliffe LT, Schnee C, Kohl TA, Niemann S, Geijo J, Klinger M, Timms P, Rattei T, Sachse K, Clarke IN, Rupp J. 2018. The genetic transformation of Chlamydia pneumoniae. mSphere 3:e00412-18. 10.1128/mSphere.00412-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Cutcliffe LT, Skilton RJ, Ramsey KH, Thomson NR, Clarke IN. 2014. The genetic basis of plasmid tropism between Chlamydia trachomatis and Chlamydia muridarum. Pathog Dis 72:19–23. 10.1111/2049-632X.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comanducci M, Ricci S, Cevenini R, Ratti G. 1990. Diversity of the Chlamydia trachomatis common plasmid in biovars with different pathogenicity. Plasmid 23:149–154. 10.1016/0147-619x(90)90034-a. [DOI] [PubMed] [Google Scholar]
- 29.Thomas NS, Lusher M, Storey CC, Clarke IN. 1997. Plasmid diversity in Chlamydia. Microbiology (Reading) 143:1847–1854. 10.1099/00221287-143-6-1847. [DOI] [PubMed] [Google Scholar]
- 30.Tam JE, Davis CH, Thresher RJ, Wyrick PB. 1992. Location of the origin of replication for the 7.5-kb Chlamydia trachomatis plasmid. Plasmid 27:231–236. 10.1016/0147-619x(92)90025-6. [DOI] [PubMed] [Google Scholar]
- 31.Keb G, Fields KA. 2020. Markerless gene deletion by floxed cassette allelic exchange mutagenesis in Chlamydia trachomatis. J Vis Exp 30. 10.3791/60848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller KE, Wolf K, Fields KA. 2016. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. mBio 7:e01817-15. 10.1128/mBio.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 34.Murthy AK, Li W, Ramsey KH. 2018. Immunopathogenesis of chlamydial infections. Curr Top Microbiol Immunol 412:183–215. 10.1007/82_2016_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajaram K, Giebel AM, Toh E, Hu S, Newman JH, Morrison SG, Kari L, Morrison RP, Nelson DE. 2015. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun 83:2870–2881. 10.1128/IAI.00106-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nunes A, Gomes JP. 2014. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol 23:49–64. 10.1016/j.meegid.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 37.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci USA 108:7189–7193. 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, Bastidas RJ. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaBrie SD, Dimond ZE, Harrison KS, Baid S, Wickstrum J, Suchland RJ, Hefty PS. 2019. Transposon mutagenesis in Chlamydia trachomatis identifies CT339 as a ComEC homolog important for DNA uptake and lateral gene transfer. mBio 10:e01343-19. 10.1128/mBio.01343-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, LaBrie SD, Carrell SJ, Suchland RJ, Dimond ZE, Kwong F, Rockey DD, Hefty PS, Hybiske K. 2019. Development of transposon mutagenesis for Chlamydia muridarum. J Bacteriol 201:e00366-19. 10.1128/JB.00366-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Neill CE, Skilton RJ, Forster J, Cleary DW, Pearson SA, Lampe DJ, Thomson NR, Clarke IN. 2021. An inducible transposon mutagenesis approach for the intracellular human pathogen Chlamydia trachomatis. Wellcome Open Res 6:312. 10.12688/wellcomeopenres.16068.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber MM, Faris R. 2019. Mutagenesis of Chlamydia trachomatis using TargeTron. Methods Mol Biol 2042:165–184. 10.1007/978-1-4939-9694-0_12. [DOI] [PubMed] [Google Scholar]
- 43.Novick RP. 1987. Plasmid incompatibility. Microbiol Rev 51:381–395. 10.1128/mr.51.4.381-395.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, 3rd, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun 76:2273–2283. 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Connell CM, AbdelRahman YM, Green E, Darville HK, Saira K, Smith B, Darville T, Scurlock AM, Meyer CR, Belland RJ. 2011. Toll-like receptor 2 activation by Chlamydia trachomatis is plasmid dependent, and plasmid-responsive chromosomal loci are coordinately regulated in response to glucose limitation by C. trachomatis but not by C. muridarum. Infect Immun 79:1044–1056. 10.1128/IAI.01118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song L, Carlson JH, Zhou B, Virtaneva K, Whitmire WM, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2014. Plasmid-mediated transformation tropism of chlamydial biovars. Pathog Dis 70:189–193. 10.1111/2049-632X.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gondek DC, Olive AJ, Stary G, Starnbach MN. 2012. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol 189:2441–2449. 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scidmore MA. 2005. Cultivation and laboratory maintenance of Chlamydia trachomatis. Curr Protoc Microbiol Chapter 11:Unit 11A.1. 10.1002/9780471729259.mc11a01s00. [DOI] [PubMed] [Google Scholar]
- 49.Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T. 2005. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect Immun 73:3860–3868. 10.1128/IAI.73.7.3860-3868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jewett TJ, Dooley CA, Mead DJ, Hackstadt T. 2008. Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem Biophys Res Commun 371:339–344. 10.1016/j.bbrc.2008.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subtil A, Wyplosz B, Balana ME, Dautry-Varsat A. 2004. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J Cell Sci 117:3923–3933. 10.1242/jcs.01247. [DOI] [PubMed] [Google Scholar]
- 52.Sturdevant GL, Caldwell HD. 2014. Innate immunity is sufficient for the clearance of Chlamydia trachomatis from the female mouse genital tract. Pathog Dis 72:70–73. 10.1111/2049-632X.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhou Z, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geiser M, Cebe R, Drewello D, Schmitz R. 2001. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques 31:88–90, 92. 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- 55.Darville T, Andrews CW, Jr, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. 10.1128/iai.65.8.3065-3073.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Huang Y, Yang Z, Sun Y, Gong S, Hou S, Chen C, Li Z, Liu Q, Wu Y, Baseman J, Zhong G. 2014. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun 82:5327–5335. 10.1128/IAI.02576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4 and Tables S1 and S2. Download iai.00453-22-s0001.pdf, PDF file, 0.4 MB (414.8KB, pdf)