

Abstract
Polymeric nanoscale materials able to target and accumulate in the tumor microenvironment (TME) offer promising routes for a safer delivery of anticancer drugs. By reaching their targets before significant amounts of drug are released, such materials can reduce off-target side effects and maximize drug concentration in the TME. However, poor drug loading capacity and inefficient nanomaterial penetration into the tumor can limit their therapeutic efficacy. Herein, we provide a novel approach to achieve high loading profiles while ensuring fast and efficient drug penetration in the tumor. This is achieved by co-polymerizing light-sensitive paclitaxel with monomers responsive to tumor-associated enzymes, and assembling the resulting di-block copolymers into spherical micelles. While light exposure enables paclitaxel to decouple from the polymeric backbone into light-activated micelles, enzymatic digestion in the TME initiates its burst release. Through a series of in vitro cytotoxicity assays, we demonstrate that these light-switch micelles hold greater potency than covalently linked, non-triggered micelles, and enable therapeutic profiles comparable to that of the free drug.
Graphical Abstract
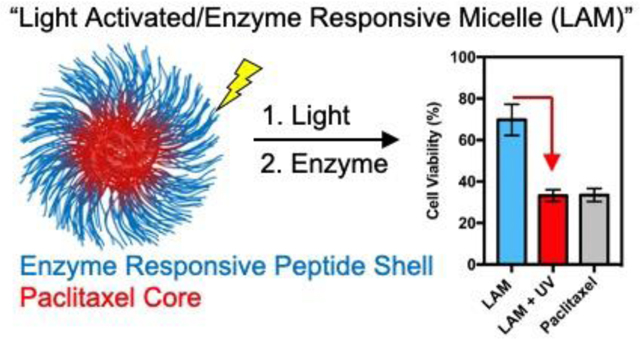
“High efficiency encapsulation is coupled with drug release via a light switch followed by particle disassembly via enzymatic action.”
Introduction
Polymeric nanocarriers have the potential to enhance drug solubility and improve selectivity thereby reducing off-target side effects.1–4 In some applications, formulations of this kind are proposed for the storage of otherwise toxic small molecule therapeutics in the nanocarrier’s core. By reaching their targets before significant amounts of drug are released, such nanomaterials have the potential to reduce off-target side effects and maximize drug concentration in the tumor microenvironment (TME). Therefore, targeting and retention of nanoparticles in the TME has recently emerged as a potential route toward safer delivery of anticancer drugs, immunotherapeutics or combinations.5–9 However, although TME targeting can enhance selectivity, nanoparticle accumulation mainly remains confined at the edge of the tumor mass. Simply, nanomaterials typically suffer from low penetration into the tumor, resulting in low uptake by cancer cells and poor treatment outcomes.10–14 Although smaller nanoparticles tend to diffuse better, they are also more susceptible to fast clearance from the TME,13 and end up being less active than their readily-internalized small-molecule counterparts.
Our laboratory has pioneered a polymeric nanoparticle delivery system that responds to matrix metalloproteinases (MMPs),15–21 which are overexpressed in the TME of many cancer types and other inflammatory diseases including myocardial infarction.22 20, 23 The response involves a phase transition that localizes and retains such drug depots within diseased tissues.15–20 These materials have been prepared through the assembly of amphiphilic di-block copolymers to generate spherical micelles. While the therapeutic of interest can be covalently conjugated to the hydrophobic block, the hydrophilic portion of the polymer, forming the shell of the micelle, is comprised of MMP-responsive peptides. Enzymatic cleavage of the outer shell of the nanoparticles in the TME results in a transition from small (20–40 nm) spherical nanoparticles to micrometer-scale scaffolds that can be retained for several weeks in the TME, where they can slowly release the loaded therapeutics.18 While such approach can selectively localize therapeutics in the TME, a faster drug release is desirable to enable efficient chemotherapy penetration within the tumor.
Herein, we developed an MMP-responsive delivery system in which the model drug paclitaxel (PTX) is conjugated to the polymer backbone via a light-sensitive linker and incorporated by direct polymerization of a paclitaxel-modified monomer (Figure 1). Such polymers assemble into inactive micelles (IMs) with high drug loading (52 % by weight per polymer) when in aqueous solution. As a result, micelles activation prior to administration can be triggered using UV irradiation,24–26 allowing PTX release from the backbone of the polymer and generating a non-covalently encapsulated drug within the core of the micelle. Proteolytic cleavage of the micelle shell (at peptide substrates) in the TME subsequently drives micelle disruption, transition into microscale scaffolds and burst PTX release.
Figure 1. Light-activable micelles (LAMs) for the delivery of paclitaxel (PTX) to the tumor microenvironment (TME).

Schematic representation of the enzyme-responsive LAMs. UV irradiation of inactive micelles (IMs) causes PTX cleavage and release into the hydrophobic pocket switching IMs to active micelles (AMs). Cleavage of the outer peptide shell by proteases results in a morphological switch and burst PTX release.
Overall, the decoupling-disassembling two-stage approach presented in this work provides a novel generalized route benefiting from the upsides of direct polymerization and characterized by high efficiency drug release in response to tumor associated enzymes. In particular, we investigate this novel class of responsive micelles using multiple imaging techniques and determine their activity upon exposure to both endogenous and exogenous triggers in a model cancer cell line.
Result and discussion
Light-activatable block co-polymers were designed such that the hydrophobic drug PTX is conjugated to the norbornene monomer via the UV-cleavable, 4-(hydroxymethyl)-3-nitrobenzoic acid group (Figure 2 in red, Scheme S1, Figure S1–S3).24, 27, 28 The peptide sequence GPLGLAGGERDG was employed as the hydrophilic moiety and as a MMP recognition sequence (Figure 2 in blue and Figure S4–S5).19 Ring opening metathesis polymerization (ROMP) allowed precise control over the polymerization in particular owing to the functional group tolerance, which allow peptide incorporation in a precise and controlled manner and subsequent enzymatic response (Figure 2, Figure S6 and Table S1).29–32 Lengths of the di-blocks were optimized to obtain stable nanoparticle suspensions that could be stored for several months. An additional monomer containing a NIR Cy5.5 fluorophore (Nor-Cy5.5) was also incorporated at the end of the polymerization allowing imaging of the conformational switch by fluorescence microscopy (Figure 2 and Figure S7–S8).19
Figure 2. Synthesis of light-activable block copolymers (LAPs).

Ring-opening metathesis polymerization (ROMP) of functionalized monomers lead to the incorporation of (i) (PTX) containing the UV-cleavable linker (red), (ii) MMP-responsive peptide (blue) and (iii) NIR Cy5.5 fluorescent dye (green). Polymerizations were terminated via the addition of (iv) ethyl vinyl ether
Upon polymerization, the resulting block co-polymers were dissolved in DMF and DPBS was added slowly under ultrasonication over a period of a minute. The resulting solutions were dialyzed against PBS over two days to form micellar nanoparticles (Figure 3). Both dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Figure 3A and Figure 3D) confirmed the formation of nano-sized micelles (inactive micelles, IMs) by self-assembly of the block copolymers. Upon 30 min irradiation (365 nm) to cleave PTX from the polymer backbone and release it within the hydrophobic core of the micelles, the generated activated micelles (AMs) maintained their spherical shape and comparable size as determined by both DLS and TEM (Figure 3B and 3E). While AMs maintained their structure and morphology after UV irradiation (365 nm), a change in morphology occurred upon AM treatment with proteolytic enzyme (Figure 3C, Figure 3F and Figure S9). For these studies, thermolysin was used as a model enzyme as it cleaves in the same position as MMP-9, but it is a more robust enzyme for in vitro studies.19
Figure 3. Enzyme-triggered size and morphology switch of LAMs.

A) Size of inactive micelles (IMs), B) size of active micelles (AMs) before and C) after 18 h incubation with the model enzyme thermolysin as determined by dynamic light scattering (DLS) measurements. Changes in micelles morphology was investigated via both (D-F) dry state transmission electron microscopy (TEM) imaging (scale bars 200 nm), as well as confocal fluorescence microscopy of Cy5.5 labeled micelles in PBS G) before and H) after enzymatic cleavage. All settings were kept constant between images. Scale bars 50 μm and 10 μm
RP-HPLC analysis (Figure 4A and Figure S15) confirmed that UV irradiation is required to cleave PTX from the polymer backbone and release it into the AM core. RP-HPLC traces of irradiated AM show a very distinct PTX peak (Figure 4B), also confirmed via ESI-MS spectrometry (Figure 4D and Figure S16). In addition, analysis of the filtrate solution upon centrifugal filtration of AMs suggests that UV irradiation did not result in PTX leakage from the AM (Figure 4C).
Figure 4. UV irradiation triggers PTX cleavage from the polymer backbone and results in micelles activation.

(A) RP-HPLC traces of IMs before and (B) after 30 min UV irradiation at 365 nm (AMs). (C) RP-HPLC traces of the filtrate deriving from AM centrifugal filtration. RP-HPLC gradient: 10 to 80 % acetonitrile in 45 min. (D) ESI-MS spectra of the peak eluted at Rt 24.5 min in the chromatogram reported in (C): PTX [M+H]+ = 854.35.
Efficient micelle disruption and concomitant polymer reorganization upon enzymatic cleavage of the peptide hydrophilic shell was further characterized by fluorescence microscopy of Cy5.5 labeled micelles. Small spherical micelles initially appeared as tiny punctate dots (Figure 3G) and transitioned to microscale fluorescent assemblies upon enzymatic digestion (Figure 3H). This result was further confirmed by the detection of the cleaved peptide fragment in the solution as demonstrated by HPLC and ESI-MS studies (Figure S9). All together these results validate the successful light-induced cleavage of PTX from the polymer backbone and its release and accumulation into the AM core. This represents the first step of a drug delivery strategy where the efficient PTX trapping within hydrophobic pockets is followed by enzyme-directed morphological transition and PTX release.
Next, in vitro activity experiments were performed to evaluate AM cytotoxicity in the presence of the enzymatic trigger. Efficacy was determined in vitro via cell viability assays performed using HT1080 fibrosarcoma cells, which overexpress MMPs. This cell line has been widely used to evaluate the performance of MMP-responsive drug delivery systems both in vitro as well as in vivo.10, 16, 33, 34 Ester-linked micelles (EMs), in which PTX is covalently conjugated to the hydrophobic portion of the polymer backbone via an ester bond were prepared as a control (Figure 5A, Schemes S4–S5, Figures S10–S12 and Table S2). Ester bonds have been previously employed to release drugs in a slow and sustained manner.18, 19, 21, 35 Both control EM and LAM were administered to HT1080 cells either before (inactive micelles, IMs) or after light irradiation (active micelles, AMs). MMPs released by HT1080 cells into the media were expected to trigger a morphological change of such formulations with consequent PTX release. Given that MMP expression within in vitro experiments is generally lower than the in vivo microenvironments, a positive control in which AMs were enzymatically pre-treated was included as a comparison (Figure 5B and Figure 5F). The PTX concentration range was selected based on previous studies and polymer dose was adjusted based on PTX functionalization.33 Fluorescence microscopy of HT-1080 cells incubated for 72 h with either Cy5.5-labeled LAMs or EMs, revealed the presence of microscale fluorescent aggregates within the cell media, appearing both intra- and extracellular (Figure 5C and Figure 5G and Figure S17). While both treatments had comparable appearance, cell viability, determined via Cell Titer Blue (CTB) assay, revealed a remarkable difference. While free PTX treatment resulted in a dramatic reduction of cell proliferation at low nanomolar concentrations, the control EM showed no effect on cell viability for all tested conditions and concentrations. This was observed even upon enzymatic pre-treatment. This result indicates that, independently of the formulation (nanoparticle vs microscale assembly), the slow PTX release from polymer backbones via ester bond hydrolysis limits efficacy. By comparison, UV irradiated AMs resulted in lower cell viability, which is comparable to that obtained for cells incubated with free PTX, especially at higher tested concentrations. This effect was further confirmed by analysis of cells incubated with AMs that underwent enzymatic pre-treatment.
Figure 5. LAMs cytotoxicity in HT1080 cell line is comparable to that of free PTX.

(A) Structure of the control ester di-block copolymer (EP) and B) DLS before (blue) and after (red) enzymatic cleavage. (C) Live fluorescence imaging of the resulting Cy 5.5-labeled EMs in cell medium after 24 h incubation with HT-1080 cells. Micelle aggregates are indicated in red (Cy5.5) and the cell nuclei are stained in blue (Hoechst). Scale bars 10 μm. Transmitted light images as well as more examples are reported in the Supporting Information. (D) HT1080 cell viability upon treatment with either EMs or free PTX. Cell viability was determined after 72 h via Cell Titer Blue (CTB) assay and the reported curves are the result of independent triplicate experiments with each condition performed in triplicate. The same results for LAMs before and after UV or both UV and enzymatic degradation are reported in the bottom panel (E-H).
Conclusion
Assemblies in which the drug is covalently bound to the polymer backbone in an inactive form and that can be readily activated and disassembled are highly desirable to obtain excellent safety profiles and therapeutic efficacy. In this work, such a delivery system was developed by combining direct polymerization of drug monomers, light-induced release of the cargo in the micelle core, and enzyme-driven micelle disruption. In vitro analysis of the light-activated micelles (AMs) in MMP-expressing HT-1080 fibrosarcoma model cell line led to toxicity comparable to that of free PTX. Such an effect could not be achieved without light switched unloading of the covalently bound, core linked drug. This work provides a proof-of-concept towards high loading capacity nanocarriers characterized by greater potency than covalently linked, non-triggered micelles, and enabling therapeutic profiles comparable to that of the free drug.
Supplementary Material
Acknowledgements
We acknowledge support of this research from the National Science Foundation (NSF, DMR-1710105) and the National Institutes of Health (NIH, R01HL139001) and a MURI from the Army Research Office (ARO, W911NF15–1-0568). C.B. thanks the Swiss National Science Foundation for the postdoctoral fellowship. This work made use of the Biological Imaging Facility, the BioCryo facility of Northwestern University’s NUANCE Center and the IMSERC at Northwestern University.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R. and Langer R, Nat Nanotechnol, 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- 2.Hare JI, Lammers T, Ashford MB, Puri S, Storm G. and Barry ST, Adv Drug Deliv Rev, 2017, 108, 25–38. [DOI] [PubMed] [Google Scholar]
- 3.Battistella C. and Klok HA, Macromol Biosci, 2017, 17. [DOI] [PubMed] [Google Scholar]
- 4.Ekladious I, Colson YL and Grinstaff MW, Nat Rev Drug Discov, 2019, 18, 273–294. [DOI] [PubMed] [Google Scholar]
- 5.Roma-Rodrigues C, Mendes R, Baptista PV and Fernandes AR, Int J Mol Sci, 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muntimadugu E, Kommineni N. and Khan W, Pharmacol Res, 2017, 126, 109–122. [DOI] [PubMed] [Google Scholar]
- 7.Dougan M. and Dougan SK, J Cell Biochem, 2017, 118, 3049–3054. [DOI] [PubMed] [Google Scholar]
- 8.Kzhyshkowska J, Bizzarri M, Apte R. and Cherdyntseva N, Curr Pharm Des, 2017, 23, 4703–4704. [DOI] [PubMed] [Google Scholar]
- 9.Leibovici J, Itzhaki O, Huszar M. and Sinai J, Immunotherapy, 2011, 3, 1385–1408. [DOI] [PubMed] [Google Scholar]
- 10.Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popovic Z, Jain RK, Bawendi MG and Fukumura D, Proc Natl Acad Sci U S A, 2011, 108, 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruan SB, Zhang L, Chen JT, Cao TW, Yang YT, Liu YY, He Q, Gao FB and Gao HL, Rsc Advances, 2015, 5, 64303–64317. [Google Scholar]
- 12.Cun XL, Chen JT, Ruan SB, Zhang L, Wan JY, He Q. and Gao HL, Acs Applied Materials & Interfaces, 2015, 7, 27458–27466. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZW, Wang H, Tan T, Li J, Wang ZW and Li YP, Advanced Functional Materials, 2018, 28. [Google Scholar]
- 14.Su YL and Hu SH, Pharmaceutics, 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ku TH, Chien MP, Thompson MP, Sinkovits RS, Olson NH, Baker TS and Gianneschi NC, J Am Chem Soc, 2011, 133, 8392–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chien MP, Thompson MP, Barback CV, Ku TH, Hall DJ and Gianneschi NC, Advanced Materials, 2013, 25, 3599–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlini A, Nguyen M, Chien MP, Sonnenberg S, Luo C, Braden R, Osborn K, Li YW, Christman K. and Gianneschi N, Abstracts of Papers of the American Chemical Society, 2016, 251. [Google Scholar]
- 18.Proetto MT, Callmann CE, Cliff J, Szymanski CJ, Hu DH, Howell SB, Evans JE, Orr G. and Gianneschi NC, Acs Central Science, 2018, 4, 1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battistella C, Callmann CE, Thompson MP, Yao S, Yeldandi AV, Hayashi T, Carson DA and Gianneschi NC, Adv Healthc Mater, 2019, 8, e1901105. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen MM, Carlini AS, Chien MP, Sonnenberg S, Luo CL, Braden RL, Osborn KG, Li YW, Gianneschi NC and Christman KL, Advanced Materials, 2015, 27, 5547–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callmann CE, Barback CV, Thompson MP, Hall DJ, Mattrey RF and Gianneschi NC, Advanced Materials, 2015, 27, 4611–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessenbrock K, Plaks V. and Werb Z, Cell, 2010, 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao Q, Kou L, Tu Y. and Zhu L, Trends Pharmacol Sci, 2018, 39, 766–781. [DOI] [PubMed] [Google Scholar]
- 24.Liu G, Liu W. and Dong CM, Polymer Chemistry, 2013, 4, 3431–3443. [Google Scholar]
- 25.Yu N, Huang L, Zhou Y, Xue T, Chen Z. and Han G, Adv Healthc Mater, 2019, 8, e1801132. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Ang CY, Li M, Tan SY, Qu Q, Luo Z. and Zhao Y, ACS Appl Mater Interfaces, 2015, 7, 18179–18187. [DOI] [PubMed] [Google Scholar]
- 27.Kim YK, Huang Y, Tsuei M, Wang X, Gianneschi NC and Abbott NL, Chemphyschem, 2018, 19, 2037–2045. [DOI] [PubMed] [Google Scholar]
- 28.Kammari L, Solomek T, Ngoy BP, Heger D. and Klan P, J Am Chem Soc, 2010, 132, 11431–11433. [DOI] [PubMed] [Google Scholar]
- 29.Kammeyer JK, Blum AP, Adamiak L, Hahn ME and Gianneschi NC, Polym Chem, 2013, 41, 3929–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gianneschi NC, Choi W, Sun H, Battistella C, Berger O, Vratsanos M. and Wang M, Angew Chem Int Ed Engl, 2020, DOI: 10.1002/anie.202005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu J, Sun H, Callmann CE, Thompson MP, Battistella C, Proetto MT, Carlini AS and Gianneschi NC, Chem Commun (Camb), 2020, 56, 6778–6781. [DOI] [PubMed] [Google Scholar]
- 32.Blum AP, Kammeyer JK and Gianneschi NC, Chem Sci, 2016, 7, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng J, Wan Y, Elhissi A, Zhang ZR and Sun X, Pharm Res-Dordr, 2014, 31, 2220–2233. [DOI] [PubMed] [Google Scholar]
- 34.Roomi MW, Kalinovsky T, Monterrey J, Rath M. and Niedzwiecki A, Int J Oncol, 2013, 43, 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavis LD, Acs Chem Biol, 2008, 3, 203–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.