

SUMMARY
Mutations in Kv1.1 (Kcna1) voltage-gated potassium channels in humans and mice generate network hyperexcitability, enhancing aberrant postnatal neurogenesis in the dentate subgranular zone, resulting in epilepsy and hippocampal hypertrophy. While Kcna1 loss stimulates proliferation of progenitor cell subpopulations, the identity of extrinsic molecular triggers linking network hyperexcitability to aberrant postnatal neurogenesis remains incomplete. System x-c (Sxc) is an inducible glutamate/cysteine antiporter that regulates extracellular glutamate. Here, we find that the functional unit of Sxc, xCT (Slc7a11), is upregulated in regions of Kcna1 knockout (KO) hippocampus, suggesting a contribution to both hyperplasia and epilepsy. However, Slc7a11 KO suppressed and rescued hippocampal enlargement without altering seizure severity in Kcna1-Sic7a11-KO mice. Microglial activation, but not astrocytosis, was also reduced. Our study identifies Sxc-mediated glutamate homeostasis as an essential non-synaptic trigger coupling aberrant postnatal neurogenesis and neuroimmune crosstalk, revealing that neurogenesis and epileptogenesis in the dentate gyrus are not mutually contingent events.
Graphical Abstract
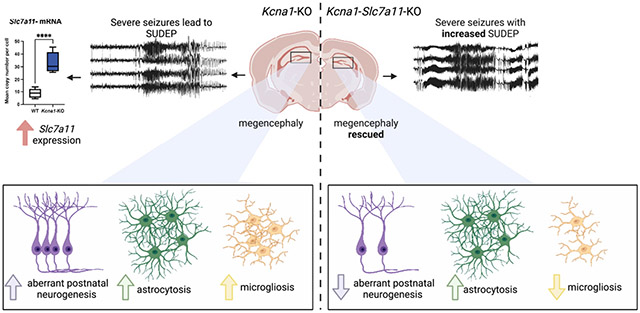
In brief
Aloi, et al. identified the glutamate antiporter System x-c (Sxc) as an essential, non-synaptic trigger that couples aberrant postnatal neurogenesis and neuroimmune crosstalk. Deletion of Sxc impairs neurogenesis, but not epileptogenesis, in the dentate gyrus of Knca1-KO mice, suggesting that they are not mutually contingent events.
INTRODUCTION
Mutations in the Kv1.1 (Kcna1) subunit of voltage-gated potassium channels result in episodic ataxia type I and refractory temporal lobe epilepsy in humans.1,2 Mouse models recapitulate this phenotype. The mceph spontaneous mutant mouse model presents with aberrant postnatal neurogenesis in the dentate that leads to a robust megencephalic phenotype,1,2 which may be partially rescued by antiseizure treatment.3 Recently, studies showed that defects in Kv1.1 directly contribute to the aberrant postnatal neurogenesis underlying hippocampal enlargement. Aberrant postnatal neurogenesis is undetectable at the conclusion of the formative phase of neurogenesis (embryonic day 16.5 [E16.5]) and as late as postnatal day 10 (P10) in the granule cell layer.4 However, non-spiking type 2b late-stage neural progenitor cells expressing doublecortin (DCX+) that lack Kcna1 are abnormally depolarized by P14,5 a time point overlapping with seizure onset.6 Extrinsic sources of cellular depolarization of the developing dentate progenitors by neurotransmitter release, e.g., NMDA receptor activation7 or synaptic8 and non-synaptic9 depolarizing GABA signaling, may also accelerate the cell cycle in both hippocampal neuroblasts and glia.10 Aberrant postnatal neurogenesis in the dentate gyrus subgranular zone (SGZ) occurs early in the aftermath of prolonged convulsant-induced seizures, a period of intense epileptogenesis leading to pharmacoresistant epilepsy in the temporal lobe.4 If inclusion of these newborn cells into dentate local circuits promotes aberrant synchronization, recurrent bursting could stimulate further neurogenesis and a self-regenerative trajectory leading to a persistent epileptic focus.11 Isolating the molecular triggers for aberrant postnatal neurogenesis amid the complex changes in gene expression, microcircuitry, and excitotoxic cell death incurred by convulsant models has been challenging since interventions that impede neurogenesis also reduce seizures, and vice versa. Further insight into the potential therapeutic implications of selectively targeting postnatal neurogenesis requires a clean dissociation of the two processes in a naturally occurring model of temporal lobe epilepsy.
Excitotoxicity and aberrant postnatal neurogenesis are inextricably linked in epileptogenesis. One molecule contributing to all these processes is System x-c (Sxc). Sxc is a sodium-independent antiporter of cysteine and glutamate. Sxc is expressed on glial and neuronal membranes, taking up cysteine in exchange for glutamate that is released to the extracellular space.12,13 Functional Sxc is highly inducible and important for the defense against oxidative stress and reactive oxygen species (ROS)14 that can arise from neuronal damage and inflammatory responses initiated by microglia. Microglia, the innate immune cells of the brain, engulf developmentally apoptotic, immature neurons and prune excessive synaptic connections at the neonatal and juvenile stages. Furthermore, microglia activation similarly influences the adult hippocampal circuitry. However, the mechanisms converging on Sxc function during epileptogenesis and reactive postnatal neurogenesis in pharmacoresistant models of developmental encephalopathy are unexplored. Recent work has begun to uncover the role of Sxc in epileptogenesis, where seizure thresholds were found to be decreased in Sxc knockout mice with acute chemoconvulsant administration.15 Previous work focused on understanding how increased glutamate in the extracellular space contributed to hyperexcitability in healthy brain. Unlike other glutamate transporters, Sxc is expressed at very low levels in healthy brain but is upregulated during epileptogenesis and neoplasia in a glioblastoma model.16 We recently showed that Sxc is upregulated in this pharmacoresistant epilepsy model and that prolonged pharmacological block of Sxc using sulfasalazine (SAS) exerted a protective antiepileptic effect.16 Due to the potential role of Sxc in linking hyperexcitability and aberrant postnatal neurogenesis, we set out to identify a role for Sxc in a genetic mouse model of developmental epileptic encephalopathy.
Here, we identify a critically permissive role of Sxc in the stimulation of progenitor cells in the Kcna1 knockout (KO) mouse megencephaly model. Sxc is upregulated in the Kcna1-KO hippocampus during epileptogenesis. However, genetic deletion of Sxc in Kcna1-KO mice results in decreased doublecortin-expressing progenitors in the dentate, preventing hippocampal enlargement, despite a markedly severe epilepsy phenotype. Thus, it is possible that the absence of extrinsic glutamate/cystine homeostasis overrides the cell-autonomous proliferative effects of Kv1.1 loss. Our finding presents evidence that aberrant postnatal neurogenesis in the dentate granule cell layer, long considered an important destabilizing network excitability mechanism, is not a prerequisite for epileptogenesis in this model.
RESULTS
Cells in the Kcna1-KO hippocampus show increased expression of the functional unit of Sxc
We examined the expression levels of the functional unit of Sxc, xCT, in the hippocampus of P21 (not shown) and P30 Kcna1-wild-type (WT) and Kcna1-KO mice with RNAScope (Figure 1) and immunostaining (Figure S3). Although there is no significant difference between the levels of xCT-expressing neurons (Figure 1B; Neun+ Slc7a11+ cells, unpaired two-tailed t test, p = 0.2135) or astrocytes (Figure 1C; Gfap+ Slc7a11+ cells, unpaired two-tailed t test, p = 0.0687), we observed a significant increase in xCT mRNA copy number per cell in the Kcna1-KO hippocampus relative to WT littermates (Figure 1D; unpaired two-tailed t test, p < 0.0001). In addition, there was a significant increase in xCT staining throughout the neuropil of the dentate gyrus molecular layer and cell bodies in the subgranual zone (SGZ), hilus, and CA1 pyramidal cell layer in Kcna1-KO mice relative to WT (p = 0.0488; n = 3 mice, 4–6 sections per group; Figures S3A and S3B). At this age, seizure activity, reactive neurogenesis, and hippocampal enlargement are already well established in the Kcna1-KO brain,17 thus the trigger and causal contribution of Sxc induction to these phenotypes is unclear. To further determine its role, we crossed lines deficient for each gene and examined Kcna1-KO/Slc7a11-KO double-mutant mice (Kcna1-Slc7a11-KO) for selective effects upon seizure severity and hippocampal hyperplasia.
Figure 1. The functional unit of Sxc, xCT, is upregulated in the hippocampus, and seizure severity in Kcna1-KO mice is independent of Sxc expression.

(A) RNAScope images (40× stitched) of hippocampi from Kcna1-WT (n = 3; 2 males/1female) and Kcna1-KO mice (n = 3; 2 male/1 female; littermates). Sections were labeled with probes to detect mRNA levels of NeuN, Gfap, and Slc7a11.
(B and C) There is no difference in Slc7a11 expression in (B) NeuN-positive cells or (C) Gfap-positive cells between Kcna1-WT and Kcna1-KO mice.
(D) There is a significant increase in Slc7a11 mRNA copy numbers of Slc7a11-expressing cells in Kcna1-KO mice relative to Kcna1-WT littermates (3–4 sections per group).
(E–J) Representative EEG recordings from mice monitored at P21–P25. (E) Kcna1-WT (n = 5; 3 males/2 females), (F) Slc7a11-KO (n = 4; 2 males/2 females), (G) Kcna1-KO (n = 3; 2 males/1 female), and (H) Kcna1-Slc7a11-KO mice (n = 4; 2 males/2 females). Kcna1-WT and Slc7a11-KO mice (E and F) show absence of EEG hyperactivity. Seizures in Kcna1-KO, in Kcna1-KO-Slc7a11-Het (n = 5; 2 males/3 females), and in Kcna1-Slc7a11-KO mice were similar in (I) severity (seizure duration) and
(J) postictal depression duration and did not significantly differ between genotypes.
(K) Interictal power spectral densities did reveal the absence of an elevated 6–8 Hz power band in Kcna1-Slc7a11-KO mice. Scale bars: 250 μm. Data are represented as mean ± SEM.
Seizure severity in Kcna1-KO mice is independent of Sxc
Pharmacological Sxc blockade raises the evoked seizure threshold in both chemoconvulsant and low-frequency (6 Hz) stimulation paradigms.18,19 A similar effect is seen in mice with genetic deletion of functional Sxc.20 However, these models reflect convulsant thresholds of adult WT brain rather than the development of spontaneous seizures as seen in genetic epilepsy models. Therefore, we assessed Kcna1-Slc7a11-KO mice for an expected reduction in seizure burden. We monitored WT (n = 5), Slc7a11-KO (n = 4), Kcna1-KO (n = 3), Kcna1-KO-Slc7a11-heterozygous (Het) (n = 5), and Kcna1-Slc7a11-double KO mice (Kcna1-Slc7a11-KO, n = 4) (Figures 1E-1H) with video electroencephalography (EEG) from the age of P22–P25 for 3 weeks or until sudden death. We found no significant difference in total seizure duration between Kcna1-KO, Kcna1-KO-Slc7a11-Het, and Kcna1-Slc7a11-KO mice. No seizures were observed in control Slc7a11-KO or WT groups (Figure 1I; p = 0.4373). In addition, we found no significant differences in postictal depression of EEG amplitude (Figure 1J; p = 0.1178), a potential sudden unexpected mortality in epilepsy (SUDEP) biomarker.21 Comparison of power spectral densities during interictal periods revealed only a loss of power in the theta frequency band in Kcna1-Slc7a11-KO mice (Figure 1K; p = 0.4371, n = 3–5 animals per group). Therefore, at P30 when the Kcna1-KO line shows increased SUDEP events, seizure severity in Kcna1-KO mice is independent of Sxc.
Hippocampal enlargement in Kcna1-KO mice is prevented by developmental loss of Sxc
Complete lack of Kv1.1 significantly enlarges the ventral cortex and hippocampus, while loss of a single Kv1.1 allele does not alter brain size.1,21 We examined WT, Slc7a11-KO, Kcna1-KO, and Kcna1-Slc7a11-KO mice to determine whether loss of Sxc in homozygous Kcna1-KO mutants altered the megencephaly phenotype (Figures 2A-2D). We observed a significant increase in the total area of the granule cell layer (GCL) in Kcna1-KO relative to WT mice (Figure 2E; p = 0.0006; WT versus Kcna1-KO: p = 0.0236). Surprisingly, Kcna1-Slc7a11-KO mice showed a significant reduction in GCL area relative to Kcna1-KO mice (p = 0.0020). This reduction was similar to WT and Slc7a11-KO GCL (for WT versus Kcna1-Slc7a11-KO: p = 0.5094; for WT versus Slc7a11-KO: p = 0.2287; for Slc7a11-KO versus Kcna1-Slc7a11-KO: p = 0.9282). Importantly, WT and Slc7a11-KO GCL areas were not significantly different from each other (p = 0.3702), suggesting that loss of Sxc alone does not result in these changes. This pattern was also observed in the total area of the dentate gyrus (DG). While Kcna1-KO mice showed a significant increase in total DG area relative to WT mice (Figure 2F; p = 0.0064; WT versus Kcna1-KO: p = 0.0270), Kcna1-Slc7a11-KO mice showed a significant reduction in total DG area (Kcna1-KO versus Kcna1-Slc7a11-KO mice: p = 0.0069) to a value comparable to WT (WT versus Kcna1-Slc7a11-KO: p = 0.9084) and Slc7a11-KO (Slc7a11-KO versus Kcna1-Slc7a11-KO: p = 0.9497). Loss of Sxc did not affect total DG area (p = 0.9991). Overall, the percentage area of the GCL was significantly larger in Kcna1-KO mice compared with WT mice (Figure 2G; p = 0.0256; WT versus Kcna1-KO: p = 0.0344; Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.0499), while Kcna1-Slc7a11-KO mice showed a normalized GCL area comparable to WT (WT versus Kcna1-Slc7a11-KO mice: p > 0.999). While the loss of functional Sxc failed to reduce seizure severity, we observed a full rescue of the dorsal hippocampus enlargement seen in Kcna1-KO mice.
Figure 2. Loss of functional Sxc rescues hippocampal enlargement in Kcna1-KO mice.

(A–D) Low power (10×) images of DAPI stained hippocampi from biological replicates (A) Kcna1-WT (n = 5; 2 males/3 females), (B) Slc7a11-KO (n = 3; 2 males/1 female), (C) Kcna1-KO (n = 3; 2 males/1 female), and (D) Kcna1-Slc7a11-KO mice (n = 3; 1 male/2 female) were used to quantify area of granule cell layer (GCL) and dentate gyrus (DG).
(E–G) GCL area (E), DG area (F), and normalized %GCL of DG (G) show rescue of enlargement in Kcna1-Slc7a11-KO mice.
Scale bars: 250 μm. Data are represented as mean ± SEM.
Sxc deletion depresses aberrant postnatal neurogenesis in Kcna1-KO mice
The reduction in hippocampal area is explained by a reduction of the pool of newborn DCX+ cells in the DG of Kcna1-Slc7a11-KO mice (Figures 3A-3I). As expected, we found an increase in the total number of DCX+ late-stage neural progenitor cells at the DG apex in Kcna1-KO relative to WT mice at P30 (Figure 3J; p = 0.0009; WT versus Kcna1-KO: p = 0.0180). There was a significant reduction in DCX+ late-stage progenitor cell numbers in the DG apex in Kcna1-Slc7a11-KO mice relative to Kcna1-KO mice (p = 0.0007). Kcna1-Slc7a11-KO mice showed similar levels of DCX+ late-stage progenitor cell numbers as WT mice (p = 0.4177). Previous studies of seizure-induced neurogenesis report the presence of DCX+ cells in abnormal or ectopic locations.4,22,23 We did not identify DCX+ staining in ectopic locations like the hilus or molecular layer. These findings support a role for Sxc in regulating the pool of DCX+ late-stage neural progenitor cells in the epileptic brain independently of seizure severity.
Figure 3. Aberrant postnatal neurogenesis is significantly reduced in the hippocampal SGZ of Kcna1-Slc7a11-KO mice.

(A–I) Low power (10×) images of doublecortin (DCX)-labeled hippocampi from biological replicates (A) Kcna1-WT (n = 5; 2 males/3 females), (B) Kcnal-KO (n = 3; 1 male/2 females), and (C) Kcnal-Slc7a11-KO mice (n = 3; 1 male/2 females). Representative higher power (40×) images of (D and E) Kcna1-WT, (F and G) Kcna1-KO, and (H and I) Kcna1-Slc7a11-KO mice. DCX staining is essentially absent in SGZ of Kcna1-Slc7a11-KO mice.
(J) Quantification of DCX+ cells at the DG apex per group (HALO, Indica Labs).
Scale bars: 250 μm. Data are represented as mean ± SEM.
Loss of Sxc in the Kcna1-KO brain decreases microglia density in the DG
Microglia are innate immune cells that play crucial roles during brain development and defense against injury or disease.24 Microglia density and morphology change rapidly upon inflammatory activation or tissue-reparative states.24 In chemoconvulsant models of epilepsy, microglia proliferate locally after seizures with enlarged somas and less ramified processes.25 Using microglia/macrophage-specific calcium-binding protein (IBA-1) as a marker, we compared the number and state of microglia in the DG and SGZ of WT (Figures 4A and 4B), Kcna1-KO (Figures 4C and 4D), and Kcna1-Slc7a11-KO (Figures 4E and 4F) mice. At P30, microglia numbers in the DG of the Kcna1-KO brain trended higher but did not differ significantly from WT DG (Figure 4G; p = 0.0154; WT versus Kcna1-KO: p = 0.5929; see Figure S2 for quantification details). Similarly, loss of Sxc in Kcna1-Slc7a11-KO mice did not significantly alter microglia numbers in the DG relative to WT (WT versus Kcna1-Slc7a11-KO: p = 0.1733); however, a reduction in microglia numbers was found when compared with the Kcna1-KO brain (Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.0140). Microglia density in the SGZ of both WT and Kcna1-KO hippocampus did not differ (Figure 4H; p = 0.0039; for WT versus Kcna1-KO: p = 0.9991; for WT and Kcna1-Slc7a11-KO: p = 0.0178; for Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.0097). Furthermore, although we observed a reduction in IBA-1 cell size in Kcna1-Slc7a11-KO in the DG relative to Kcna1-KO (Figure S4A; p = 0.0218) and WT (p = 0.0184) in this annotation layer, we did not observe differences in IBA-1 labeling intensity. We did not observe differences in IBA-1 staining intensity in the DG between genotypes (Figure S4B; WT versus Kcna1-KO: p = 0.05043; Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.7442; WT versus Kcna1-Slc7a11-KO: p = 0.8739). In the SGZ, we observed that IBA-1 cells were significantly larger in average area (μm2) in the Kcna1-KO brain relative to WT (p = 0.0472) and Kcna1-Slc7a11-KO (p = 0.0318), suggesting that ramified-to-ameboid cell changes during epileptogenesis are blunted by lack of functional Sxc (Figure S4C). We observed that IBA-1 staining intensity was elevated in the WT SGZ relative to Kcna1-KO and Kcna1-Slc7a11-KO (Figure S4D; WT versus. Kcna1-KO: p = 0.0074; WT versus Kcna1-Slc7a11-KO: p = 0.0017). We did not observe differences in IBA-1 staining intensity in the SGZ between Kcna1-KO and Kcna1-Slc7a11-KO (p = 0.9934). Therefore, as seen in the DCX+ newborn population, loss of Sxc may limit the microglial response to oxidative stress and/or glutamate excess, reducing microglia activation phenotypes in the epileptic brain of Kcna1-KO mice.
Figure 4. Microglia numbers are elevated in SVZ of the Kcna1-KO DG and strikingly absent in this zone in Kcna1-Slc7a11-KO mice.

(A–F) Representative images of higher power (40×) stitched images of whole hippocampi (A, C, and E) and single field of view (B, D, and F) in (A and B) Kcna1-WT (n = 3; 1 male/2 females, 12 stitched images), (C and D) Kcna1-KO (n = 3; 1 male/2 females, 12 stitched images), and (E and F) Kcna1-Slc7a11-KO (n = 3; 1 male/2 females, 12 stitched images) mice immunolabeled with anti-IBA-1 to mark microglia.
(G and H) Quantification of IBA-1+ microglia in the DG (G) and SGZ (H) of the GCL (HALO, Indica Labs).
Scale bars: 250 μm. Data are represented as mean ± SEM.
Astrogliosis in Kcna1-KO hippocampus is unaffected by developmental loss of functional Sxc
Astrocyte activation is a consistent response to neuronal hyperactivity during seizures, resulting in enlarged cell bodies, extension of processes, and induction of reactive genes such as glial fibrillary acidic protein (GFAP),12,26 and Kcna1-KO mice show a numerical increase of astrocytes.21 We compared reactive astrocytosis via immunostaining for GFAP in Kcna1-KO, Kcna1-Slc7a11-KO, and WT mice at P30 ([n = 3; Figures S5A and S5B] Kcna1-KO mice [n = 3; Figures S5C and S5D], and Kcna1-Slc7a11-KO mice [n = 3; Figures S5D and S5E]). We identified a significant increase in the number of activated astrocytes in the DG of Kcna1-KO mice relative to WT, as expected (Figure S5F; p < 0.0001; for Kcna1-KO versus WT: p < 0.0001). We observed a similar increase in GFAP+ astrocytes in the Kcna1-Slc7a11-KO relative to WT mice (Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.0035) but not relative to the Kcna1-KO mice (Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.0978). A similar pattern was observed in the SGZ layer, where DCX+ progenitors arise (Figure S5G; p = 0.0006; for Kcna1-KO versus WT: p = 0.0004; for Kcna1-KO versus Kcna1-Slc7a11-KO: p = 0.1761; for WT versus Kcna1-Slc7a11-KO: p = 0.0165). Interestingly, these findings indicate that unlike progenitor and microglial cell proliferation, developmental loss of Sxc does not significantly blunt astrogliosis in the epileptic brain.
Genetic loss of functional Sxc modifies sudden death risk in Kcna1-KO mice
Kcna1-KO mice are a well-studied model of SUDEP related to temporal lobe epilepsy.27 Homozygous Kcna1-KO mice exhibit a ~50% death rate in the first postnatal month, while the remainder show a normal lifespan,28,29 as do Slc7a11-KO mice.30 We examined the survival of Kcna1-Slc7a11-KO mice relative to heterozygous and homozygous Kcna1-KO littermate controls. Unexpectedly, we observed a significant negative impact on survival in Kcna1-Slc7a11-KO mice, despite equivalent severity of the seizure disorder. Double mutants with homozygous loss of both Kcna1 and Slc7a11 displayed the same early onset of SUDEP but rapidly exhibited fully penetrant mortality, with no mice surviving beyond 10 weeks of age (Figure S6). There was no difference in survival between Kcna1-KO-Slc7a11-Het or Kcna1-Slc7a11-KO mice, and no sex difference in survival of the Kcna1-KO, Kcna1-KO-Slc7a11-Het, and Kcna1-Slc7a11-KO mice (data not shown). Interestingly, in a previous study, we found that deletion of Mapt/tau from Kcna1-KO mice fully suppressed seizures, rescued megencephaly, and prolonged survival nearly 4-fold, indicating that the sudden death phenotype is seizure dependent and not linked to hippocampal hyperplasia.31 Taken together, we conclude that Sxc in the Kcna1-KO mouse plays an important epistatic protective role in SUDEP.
DISCUSSION
Ion channelopathy, both with and without epilepsy, has emerged as a significant category of early developmental encephalopathy, focusing attention on the role of membrane excitability during cellular division, migration, and laminar positioning in developing brain.32,33,34 Even in the absence of action potentials, there is evidence for the control of embryonic cell number and fate by glutamate- and GABA-mediated membrane depolarization.35-37 However, less is known about voltage-dependent firing properties and postnatal mitosis in progenitor cell regions, as first illustrated by Kv1.1 mutations.1 Despite its seniority among ion channels leading to both aberrant network synchronization and cellular proliferation phenotypes, the exact signals that stimulate excess neurogenesis in Kcna1-KO mice have been obscured by their hyperexcitable network environment. Chou et al.5 found that loss of Kv1.1 channels can be considered an intrinsic, cell-autonomous mechanism underlying progenitor cell proliferation, since type 2b (Sox2+, DCX+) progenitor cells were depolarized and showed elevated TrKb signaling in the hyperproliferative Kcna1-KO model, while TrKb blockade reduced postnatal neurogenesis. We did not observe a surplus of DCX+ cells at P10 (not shown) in the SGZ, an age believed to predate the first detectable seizures.6 However, since the major neurogenic wave overlaps with the emergence of seizure activity, the effects of extrinsic network activation were still present. A serial study of BrdU labeling in the Kcna1-Slc7a11-KO mice will be important to distinguish between proliferation versus accelerated death of the DCX+ pool. Here, we identify Sxc, a key regulator of glutamate and cystine homeostasis, as a regulator of an activity-induced pathway that may play a role in aberrant postnatal neurogenesis. Hyperexcitability due to deletion of Kcna1 stimulates hippocampal expression of Sxc and is accompanied by an increase in DCX+ cells. Yet, co-deletion of Kv1.1 and Sxc led to a reduction of DCX+ cells, suggesting a suppression of aberrant postnatal neurogenesis, and rescue of hippocampal enlargement even in the continuing presence of seizures. Sxc may be the key molecule that could exclude aberrant postnatal neurogenesis as critical to the underlying epileptogenic process of temporal lobe epilepsy in this model, where network hyperexcitability is enhanced by widespread loss of Kv1.1. In addition, it demonstrates that seizure activity alone is insufficient to promote postnatal neurogenesis, which requires Sxc.
Interestingly, the sparing of epileptogenesis in this model contrasts with the acute antiseizure effects of pharmacologic Sxc blockade in adult-onset convulsant drug models.15,16,38 Genetic Sxc deletion alone,39 which decreases glutamate levels without inducing oxidative stress,15 elevates limbic seizure thresholds tested in adult Slc7a11-KO mice with acute chemoconvulsant administration (pilocarpine, kainic acid, N-acetylcysteine,38 or kindling18), consistent with a protective effect against abnormal network synchronization. Our findings support the idea that aberrant postnatal neurogenesis is not required for epileptogenesis in this model, as Kcna1-Slc7a11-KO mice without postictal neurogenesis display spontaneous seizures with similar severity of Kcna1-KO mice. Deletion of Sxc uncouples aberrant postnatal neurogenesis from ambient seizure activity, thereby excluding neurogenesis as a critical element of the epileptogenic process. Previous attempts to dissect neurogenesis from epileptogenesis after the onset of seizures have been explored in chemoconvulsant seizure models. Conditional ablation of newborn cells generated in the adult after pilocarpine treatment using diphtheria toxin did not prevent seizures but greatly slowed their progression.40 Conditional knockout of NeuroD1 in granule cell progenitors following pilocarpine treatment diminished reactive neurogenesis but not spontaneous seizure frequency.41 These models show that aberrant postnatal neurogenesis contributes to the plasticity of network instability, even after the onset of established seizure activity. Our findings are further highlighted by Lin King et al.,42 where conditional, tamoxifen-induced selective depletion of Kv1.1 in adult neural stem cells causes their over-proliferation and the depletion of radial glia-like neural stem cells, preventing proper adult-born granule cell maturation and integration into the DG.42 In our study, we went further and identified that Sxc deletion resulted in a similar depletion of DCX+ cells and examined the effect on epileptogenesis and megencephaly in the Kcna1-KO model. Conversely, a reduction of DCX+ cell numbers could reflect a change in survival of immature neurons and not a direct result of a change in proliferation. There could also be DCX+ cells that do not respond to seizure activity.41,43 Cells that can still arise in the postnatal Kcna1-Slc7a11-KO brain may lend some weight to the concept that their survival may be shortened rather than not newly born. Future studies should focus on further understanding the role of glutamate and Sxc in aberrant postanal neurogenesis and include mechanisms downstream of Sxc loss.
We additionally identify a positive feedback loop involving Sxc crosstalk with microglia. Excitotoxic glutamate release stemming from Sxc activity was first observed in stimulated microglia cultures.44 Microglia activation by pro-inflammatory stimuli results in a substantial release of inflammatory mediators, glutamate, and other potentially neuroprotective and neurotrophic signals.9,45,46 Since microglia also express glutamate receptors,47 activation of microglia may be sustained by repetitive seizure activity. These stimuli can also rapidly induce high levels of Sxc expression in microglia. However, we found that microglia area, but not density, in the DG of Kcna1-KO mice is not significantly higher than in WT at P30. In Kcna1-Slc7a11-KO mice, microglia area and number in the DG and SGZ were significantly decreased, indicating that absence of Sxc can blunt microglial proliferation along with neurogenesis in the epileptic brain. Since inflammatory activation via interleukin-1β (IL-1β) specifically induces Sxc expression in astrocytes,48 we also sought to elucidate the impact of Sxc loss on astrocyte activation in the Kcna1-KO brain. Although GFAP-expressing astrocytes were increased in the DG of both Kcna1-KO and Kcna1-Slc7a11-KO mice relative to WT, we found no significant differences, suggesting that seizures alone are sufficient to activate astrocytosis and that lack of functional Sxc did not impair inflammation of this glial subpopulation. Our findings provide evidence that Sxc up-regulation is a critical link coupling reactive neurogenesis, hippocampal hypertrophy, and seizures in the Kcna1-KO model of developmental epileptic encephalopathy.
Limitations of the study
A principal limitation of our study includes the absence of conditional targeting of Sxc in the Kv1.1 null mouse model. Future studies should contrast and compare inducible loss of Sxc in a cell-type-specific approach to pharmacological blockade with SAS and assess the therapeutic potential of targeting Sxc in this model. Although we observed a reduction in DCX+ cells upon Sxc loss, future work should query the cellular dynamics that occur in the DCX+ progenitor population (e.g., BrdU labeling). In addition, since Sxc is expressed in both neurons and glia, further investigations into the molecular signals underlying network-activity-dependent Sxc induction are needed to identify additional modulatory targets.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Jeffrey Noebels (jnoebels@bcm.edu).
Materials availability
Slc7a11-KO mice were generated on C57Bl/6 background as in Sato, et al.39 Mice were crossed to and maintained on a C57BL/6 background. The mice were a gift from Dr. Sato and were shared with our group by Dr. B. Gan at MD Anderson, with permission from Dr. Sato. Please contact Dr. Jeffrey Noebels for further details. There may be limitations to the availability of Slc7a11-KO mice due to lulls in breeding.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Kcna1-heterozygous mice (C57Bl6 background; Strain #:003,532, The Jackson Laboratory) were crossed with xCT-homozygous knock-out (Slc7a11-KO/C57BL6 39; see Materials availability) mice to generate doubly heterozygous Kcna1/Slc7a11 mice. These were then used to generate experimental animals. Litters were genotyped by postnatal day P14 and weaned at P21. All experiments were performed in accordance with a Baylor College of Medicine IACUC approved Protocol (AN-602). Both male and female mice were utilized for this study between the ages of P21 and P30 (see figure legends for further details). Mice were maintained in a specific pathogen-free facility and group housed with ad libitum access to food and water on a 12/12 light dark cycle.
METHOD DETAILS
Continuous in vivo video-EEG monitoring
In vivo video-EEG recordings were performed as reported Hatcher, et. al.16. At P21, mice were anesthetized by isoflurane vaporization pump during surgical implantation with bilateral silver wire electrodes (0.005-inch diameter, Omnetics) attached to a microminiature connector. Electrodes were implanted subdurally over the temporal and parietal cortices, with frontal reference and ground electrodes. Mice were allowed to recover 24-48 h before further study. LabChart (v8.1.13, AD Instruments) was used for video-EEG recordings. EEG signals were acquired at 200 Hz and filtered using a 0.3-Hz high-pass filter, 70-Hz low-pass filter, and a 60-Hz notch filter. Mice were housed singly for continuous recordings in a satellite facility, with weekly water and cage changes, for up to 4 weeks or until sudden death. Seizure activity defined by EEG waveform and corresponding video-recorded behavior were quantified by visual inspection and spectral frequency analysis (EEG-Lab49; MatLab). Male and female mice were utilized for this study.
Localization of Slc7a11 in the Kcna1-KO hippocampus
RNAScope was performed according to Massey, et al50. Tissue sections containing hippocampus were cryo-sectioned using a CM1950 cryostat (Leica Biosystems, Buffalo Gove, IL) at 15 μm thick sections and mounted onto Thermofisher Superfrost Plus slides, then stored at −80C until further use. RNAscope Multiplex Fluorescent V2 kit was used to perform RNAscope hybridization (Advanced Cell Diagnostics, Newark, CA). We used probes targeting xCT (Mm-Slc7a11-C2; Cat No. 422511-C2), Neun (Mm-Rbfox3; Cat. No. 313311), and Gfap (Mm-Gfap-C3; Cat. No. 313211-C3) mRNA species. Channel two and three probes were diluted into channel one probes at a 1:1:50 ratio. Probes were labeled using Opal fluorescent dyes (Akoya Biosciences, Marlborough, MA) at a 1:1500 dilution. We used Opal dye 570 for channel one probes (Mm-Rbfox3 Cat. No. 313311, Cat. No. FP1488001KT), Opal dye 690 for channel two probes (Mm-Slc7a11-C2; Cat No. 422511-C2, Cat. No. FP1497001KT) and Opal dye 520 for channel three probes (Mm-Gfap-C3; Cat. No. 313211-C3), Cat. No. FP1487001KT). 40,6-Diamidino-2-phenylindole (DAPI; RNAscope Multiplex Fluorescent V2 kit) was used to label nuclei, and slides were mounted with coverslips using Prolong Gold Antifade Mountant (Cat. No. P36930, Thermo Fisher Scientific). Fluorescent images were taken using a Keyence Fluorescence Microscope BZ-X800 with 4 channels: DAPI (nuclei), green fluorescent protein (Gfap), Cyanine 5 (Slc7a11), TRITC (Neun). Full scan images were taken at 40X magnification with uniform exposure times, light transmission, and other filter settings and stitched at 10% overlap for all samples. We used ImageJ51 and HALO Image Analysis software (Indica Labs, Albuquerque, NM) to assess hippocampal Regions for positive probe signal.
Histology and Immunofluorescence
At P30, mice were anesthetized and perfused intracardially with cold 1x PBS followed by cold 4% paraformaldehyde/1x PBS solution. Brains were extracted and post-fixed in 4% PFA overnight, then immersed in 30% sucrose solution until sunk. Brains were embedded in Tissue-Tek O.C.T. compound (Sakura) and frozen at −80°C. Cryopreserved brains were cut into 20-μm coronal free-floating sections using a Leica CM1950 cryostat, washed twice with 0.3% Triton X-100 in 1x PBS, and then blocked with 1% BSA, 0.3% Triton X-100/PBS for 2 h at room temperature. Sections were incubated for 18 h at 4°C with primary antibodies (mouse-anti-Doublecortin (DCX; 1:250, Santa Cruz, sc-271390), rabbit-anti-xCT (xCT-P3; 1:500, Zogenix, Inc.), mouse-anti-GFAP (1:500; Millipore, IF03L), or rabbit-anti-IBA-1 (1:500, Wako Chemicals, 011-27991). Sections were washed with 0.01% Tween 20 in 1x PBS, incubated with Alexa Fluor–labeled secondary antibodies for 2 h at room temperature (Thermo Fisher Scientific, 1:1,1000), washed twice with 0.01% Tween 20 in 1x PBS and once with 1x PBS, then mounted onto slides with media containing DAPI (Prolong Gold, Cell Signaling) and cover slipped.
Quantification of dentate gyrus area and Automated cell detection
Low magnification images (10X) of DAPI stained sections were captured using a Nikon Eclipse TE200-S inverted microscope, and TIFs (16-bit) imported into ImageJ. The dentate gyrus (DG) and granule cell layer (GCL) were manually delineated using the wand tool and their areas calculated as a percent of the area encompassing the dorsal hippocampus. 4-6 sections were quantified from each animal. Group data were compiled and analyzed using Prism v 9.2.0. Representative images and traces were created in Adobe Illustrator. Higher (40X) magnification images of hippocampus were collected using a Nikon Eclipse TE200-S inverted microscope or Keyence Fluorescence Microscope BZ-X800. Images were separated by channel then transformed to 16-bit single channel images with Image J. Images were imported to Halo Software (v 3.1.1076.264, Indica Labs). High magnification images were used to quantify Doublecortin (DCX)-DAPI + cells at the DG apex (mouse anti-DCX, Santa Cruz, (E–6): sc-271390). Background signal values were determined manually and subtracted uniformly per filter across images analyzed to maximize nuclei detection and minimize background artifacts. The SGZ was manually selected to exclude hilar and GCL cells for each image analyzed. The number of DCX-positive and DAPI-positive cells per layer were detected in each image from 4-6 sections per animal of each group (Figures S1A and S1B). For GFAP (Millipore, IF03L. 1:500 dilution) and IBA-1 (Wako Chemicals, 011-27991, 1:500 dilution) quantification, images were processed as before, and cells were quantified per biological replicate (Figures S2A and S2B). To quantify GFAP and IBA-1 expressing cells, higher magnification images (40x) were stitched to reveal the entirety of the dorsal hippocampus, and cells were quantified as described above. Number of DAPI+ and Cy5+ cells were compiled and graphed (Prism v.9.2.0).
QUANTIFICATION AND STATISTICAL ANALYSIS
Seizure duration was quantified manually using notations in AD Instruments LabChart 8.1.13. We used a One-way ANOVA with Tukey’s post hoc correction for comparisons of seizure duration and post-ictal depression duration. Power spectral densities were compared with a 2-way ANOVA with Tukey’s post hoc correction. RNAScope quantifications were tested with an unpaired two-tailed t test while histological quantifications were tested with a One-way ANOVA with Tukey’s post hoc correction. Survival data were analyzed by Log Rank test with Pairwise comparisons with Bonferroni-Holm post hoc correction with RStudio52 using the survfit function for survival curve analysis, then plotted in Prism v 9.0. All results are displayed using boxplots, mean, and error bars represent the standard error.
ADDITIONAL RESOURCES
See Key resources table for further details.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse-anti-Doublecortin (1:500 dilution) | Santa Cruz | sc-271390 (E-6); RRID:AB_10610966 |
| Rabbit-anti-IBA-1 (1:250 dilution) | Wako Chemicals | 011-27991; RRID:AB_839504 |
| Rabbit-anti-GFAP (1:500 dilution) | Millipore | AB5804; RRID:AB_2109645 |
| Anti-astroglial xCT (1:500 dilution) | Zogenix, Inc. Emeryville, California, USA | gift of Thadd Reeder |
| Prolong Gold Antifade Mountant | Thermo Fisher Scientific | Cat. No. P36930 |
| Critical commercial assays | ||
| For Slc7a11 (Mm-Slc7a11-C2) | Advanced Cell Diagnostics, Newark, CA | Cat No. 422511-C2 |
| For Neun (Mm-Rbfox3) | Advanced Cell Diagnostics, Newark, CA | Cat. No. 313311 |
| For Gfap (Mm-Gfap-C3) | Advanced Cell Diagnostics, Newark, CA | Cat. No. 313211-C3 |
| Opal dye 570 for channel one probes (for Mm-Slc7a11-C2; Cat No. 422511-C2) | Akoya Biosciences, Marlborough, MA | Cat. No. FP1487001KT |
| Opal dye 520 for channel three probes (for Mm-Gfap-C3; Cat. No. 313211-C3) | Akoya Biosciences, Marlborough, MA | Cat. No. FP1497001KT |
| Opal dye 570 for channel one probes (for Mm-Rbfox3 Cat. No. 313311) | Akoya Biosciences, Marlborough, MA | Cat. No. FP1487001KT |
| 40,6-Diamidino-2-phenylindole (DAPI; RNAscope Multiplex Fluorescent V2 kit) | Advanced Cell Diagnostics, Newark, CA | Cat. No. 323100 |
| Experimental models: Organisms/strains | ||
| Kcna1-KO (maintained in C57BL/6J background) | The Jackson Laboratory | Strain #:003532 |
| Slc7a11-homozygous knock-out (maintained in C57BL/6J background) | Sato, et al. (39) | The mice were a gift from Dr. Sato and were shared with our group by Dr. B. Gan at MD Anderson, with permission from Dr. Sato. |
| C57BL/6J mice | The Jackson Laboratory | Strain #:000664 |
| Software and algorithms | ||
| Image J | Schneider et al. (50) | https://imagej.nih.gov/ij/ |
| HALO Software | Indica Labs, Albuquerque, NM | v 3.1.1076.264 |
| R studio | R Studio Team (51) | http://www.rstudio.com/ |
| Prism | Graphpad by Dotmatics | v.9.2.0 |
| EEG-Lab | Delorme and Makeig (49) | https://eeglab.org/ |
| LabChart | AD Instruments | v8.1.13 |
| Other | ||
| Female Silver Wire electrodes .025"/.64mm | Omnetics | NPS-09-WD-18.0-C-GS |
| Male Silver Wire electrodes .025"/.64mm | Omnetics | NSS-09-WD-18.0-C-GS |
| Keyence Fluorescence Microscope | Keyance | Model: BZ-X800 |
| Nikon Eclipse | Nikon | Model: TE200-S |
| CM1950 cryostat | Leica | Model: CM1950 |
Supplementary Material
Highlights.
Kcna1-KO mice exhibit seizures, megencephaly, and death in epilepsy (SUDEP)
System x-c (Slc7a11) is upregulated in the hippocampus of Kcna1-KO mice
Kcna1-Slc7a11-KO are normocephalic despite seizures and fully penetrant SUDEP
Slc7a11 deletion prevents aberrant postnatal neurogenesis and rescues megencephaly
ACKNOWLEDGMENTS
The authors thank Dr. Cory Massey, Dr. Qing-Long Miao, Dr. Isamu Aiba, Yao Ning, Timothy Abreo, and Luis E. Salazar León for their technical support and insightful discussions. This work was funded by Pediatric Brain Disorders and Development Fellowship T32NS043124-149 (M.S.A.), NINDS F31 NS124345-01 (S.J.T.), NINDS NS29709 and NCI CA223388 (J.L.N.), and the Blue Bird Circle Foundation.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111696.
REFERENCES
- 1.Persson A-S, Westman E, Wang F-H, Khan FH, Spenger C, and Lavebratt C (2007). Kv1.1 null mice have enlarged hippocampus and ventral cortex. BMC Neurosci. 8, 10. 10.1186/1471-2202-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersson S, Persson A-S, Johansen JE, Ingvar M, Nilsson J, Klement G, Århem P, Schalling M, and Lavebratt C (2003).Truncation of the Shaker-like voltage-gated potassium channel, Kv1.1, causes megencephaly. Eur. J. Neurosci 18, 3231–3240. 10.1111/j.1460-9568.2003.03044.x. [DOI] [PubMed] [Google Scholar]
- 3.Lavebratt C, Trifunovski A, Persson A-S, Wang F-H, Klason T, Öhman I, Josephsson A, Olson L, Spenger C, and Schalling M (2006). Carbamazepine protects against megencephaly and abnormal expression of BDNF and Nogo signaling components in the mceph/mceph mouse. Neurobiol. Dis 24, 374–383. 10.1016/j.nbd.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 4.Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, and Lowenstein DH (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat Hippocampus. J. Neurosci 17, 3727–3738. 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou S-M, Li K-X, Huang M-Y, Chen C, Lin King Y-H, Li GG, Zhou W, Teo CF, Jan YN, Jan LY, and Yang S-B (2021). Kv1.1 channels regulate early postnatal neurogenesis in mouse hippocampus via the TrkB signaling pathway. Elife 10, e58779. 10.7554/eLife.58779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rho JM, Szot P, Tempel BL, and Schwartzkroin PA (1999). Developmental seizure susceptibility of Kv1.1 potassium channel knockout mice. Dev. Neurosci 21, 320–327. 10.1159/000017381. [DOI] [PubMed] [Google Scholar]
- 7.Åmellem I, Yovianto G, Chong HT, Nair RR, Cnops V, Thanawalla A, and Tashiro A (2021). Role of NMDA receptors in adult neurogenesis and normal development of the dentate gyrus. eNeuro 8, ENEURO.0566-20.2021. 10.1523/ENEURO.0566-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tozuka Y, Fukuda S, Namba T, Seki T, and Hisatsune T (2005). GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47, 803–815. 10.1016/j.neuron.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Wang Q, Haydar TF, and Bordey A (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci 8, 1179–1187. 10.1038/nn1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J, Christian KM, Ming G, and Song H (2012). Modification of hippocampal circuitry by adult neurogenesis. Dev. Neurobiol 72, 1032–1043. 10.1002/dneu.22014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danzer SC (2018). Contributions of adult-generated granule cells to hippocampal pathology in temporal lobe epilepsy: a neuronal bestiary. Brain Plast. 3, 169–181. 10.3233/BPL-170056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel SA, Warren BA, Rhoderick JF, and Bridges RJ (2004). Differentiation of substrate and non-substrate inhibitors of transport system xc−: an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology 46, 273–284. 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, et al. (2013). The cystine/glutamate antiporter system x c − in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants Redox Signal. 18, 522–555. 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sagara J, Miura K, and Bannai S (1993). Maintenance of neuronal glutathione by glial cells. J. Neurochem 61, 1672–1676. 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- 15.De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, Vermoesen K, Bannai S, Sato H, Michotte Y, et al. (2011). Loss of system xFormula does not induce oxidative stress but decreases extracellular glutamate in Hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci 31, 5792–5803. 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatcher A, Yu K, Meyer J, Aiba I, Deneen B, and Noebels JL (2020). Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Invest 130, 2286–2300. 10.1172/JCI133316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wenzel HJ, Vacher H, Clark E, Trimmer JS, Lee AL, Sapolsky RM, Tempel BL, and Schwartzkroin PA (2007). Structural consequences of Kcna1 gene deletion and transfer in the mouse Hippocampus. Epilepsia 48, 2023–2046. 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leclercq K, Liefferinge JV, Albertini G, Neveux M, Dardenne S, Mairet-Coello G, Vandenplas C, Deprez T, Chong S, Foerch P, et al. (2019). Anticonvulsant and antiepileptogenic effects of system xc−inactivation in chronic epilepsy models. Epilepsia 60, 1412–1423. 10.1111/epi.16055. [DOI] [PubMed] [Google Scholar]
- 19.Massie A, Boillée S, Hewett S, Knackstedt L, and Lewerenz J (2015). Main path and byways: non-vesicularglutamate release by system x c − as an important modifier of glutamatergic neurotransmission. J. Neurochem 135, 1062–1079. 10.1111/jnc.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilella L, Lacuey N, Hampson JP, Rani MRS, Sainju RK, Friedman D, Nei M, Strohl K, Scott C, Gehlbach BK, et al. (2019). Post-convulsive central apnea as a biomarker for sudden unexpected death in epilepsy (SUDEP). Neurology 92, e171–e182. 10.1212/WNL.0000000000006785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S-B, Mclemore KD, Tasic B, Luo L, Jan YN, and Jan LY (2012). Kv1.1-dependent control of hippocampal neuron number as revealed by mosaic analysis with double markers: Kv1.1-dependent control of hippocampal neuron number. J. Physiol 590, 2645–2658. 10.1113/jphysiol.2012.228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kron MM, Zhang H, and Parent JM (2010). The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J. Neurosci 30, 2051–2059. 10.1523/JNEUROSCI.5655-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessberger S, Nakashima K, Clemenson GD, Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, and Hsieh J (2007). Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci 27, 5967–5975. 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prinz M, Masuda T, Wheeler MA, and Quintana FJ (2021). Microglia and central nervous system–associated macrophages—from origin to disease modulation. Annu. Rev. Immunol 39, 251–277. 10.1146/annurev-immunol-093019-110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng L, Murugan M, Bosco DB, Liu Y, Peng J, Worrell GA, Wang H, Ta LE, Richardson JR, Shen Y, and Wu L (2019). Microglial proliferation and monocyte infiltration contributeto microgliosis following status epilepticus. Glia, glia, 23616. 10.1002/glia.23616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sofroniew MV (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glasscock E, Yoo JW, Chen TT, Klassen TL, and Noebels JL (2010). Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J. Neurosci 30, 5167–5175. 10.1523/JNEURO-SCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A, and Tempel BL (2007). Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J. Neurophysiol 98, 1501–1525. 10.1152/jn.00640.2006. [DOI] [PubMed] [Google Scholar]
- 29.Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, and Tempel BL (1998). Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. 10.1016/S0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 30.Conrad M, and Sato H (2012). The oxidative stress-inducible cystine/glutamate antiporter, system x c −: cystine supplier and beyond. Amino Acids 42, 231–246. 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 31.Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, and Noebels JL (2013).Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci 33, 1651–1659. 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, and Malenka RC (2004). Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42, 535–552. 10.1016/S0896-6273(04)00266-1. [DOI] [PubMed] [Google Scholar]
- 33.Smith RS, and Walsh CA (2020). ion channel functions in early brain development. Trends Neurosci. 43, 103–114. 10.1016/j.tins.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitali I, Fièvre S, Telley L, Oberst P, Bariselli S, Frangeul L, Baumann N, McMahon JJ, Klingler E, Bocchi R, et al. (2018). Progenitor hyperpolarization regulates the sequential generation of neuronal subtypes in the developing neocortex. Cell 174, 1264–1276, e15. 10.1016/j.cell.2018.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LoTurco JJ, Owens DF, Heath MJS, Davis MBE, and Kriegstein AR (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. 10.1016/0896-6273(95)90008-X. [DOI] [PubMed] [Google Scholar]
- 36.Platel J-C, Dave KA, and Bordey A (2008). Control of neuroblast production and migration by converging GABA and glutamate signals in the postnatal forebrain: GABA and glutamate control of postnatal neurogenesis. J. Physiol 586, 3739–3743. 10.1113/jphysiol.2008.155325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lancaster MA (2019). An electric take on neural fate and cortical development. Dev. Cell 48, 1–2. 10.1016/j.devcel.2018.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Alcoreza O, Tewari BP, Bouslog A, Savoia A, Sontheimer H, and Campbell SL (2019). Sulfasalazine decreases mouse cortical hyperexcitability. Epilepsia 60, 1365–1377. 10.1111/epi.16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T, et al. (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem 280, 37423–37429. 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 40.Hosford BE, Rowley S, Liska JP, and Danzer SC (2017). Ablation of peri-insult generated granule cells after epilepsy onset halts disease progression. Sci. Rep 7, 18015. 10.1038/s41598-017-18237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brulet R, Zhu J, Aktar M, Hsieh J, and Cho K-O (2017). Mice with conditional NeuroD1 knockout display reduced aberrant hippocampal neurogenesis but no change in epileptic seizures. Exp. Neurol 293, 190–198. 10.1016/j.expneurol.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin King Y-H, Chen C, Lin King JV, Simms J, Glasscock E, Yang S-B, Jan Y-N, and Jan LY (2022). K v 1.1 preserves the neural stem cell pool and facilitates neuron maturation during adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 119, e2118240119. 10.1073/pnas.2118240119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pun RYK, Rolle IJ, LaSarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piani D, and Fontana A (1994). Involvement of the cystine transport system x, in the macrophage-induced glutamate-dependent cytotoxicity to neurons’ 152, 3578. [PubMed] [Google Scholar]
- 45.Espey MG, Kustova Y, Sei Y, and Basile AS (2002). Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J. Neurochem 71, 2079–2087. 10.1046/j.1471-4159.1998.71052079.x. [DOI] [PubMed] [Google Scholar]
- 46.Shih J, Tatum, and Rudzinski L (2013). New drug classes for the treatment of partial onset epilepsy: focus on perampanel. Therapeut. Clin. Risk Manag 285. 10.2147/TCRM.S37317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noda M, Nakanishi H, Nabekura J, and Akaike N (2000). AMPA–Kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci 20, 251–258. 10.1523/JNEUR0SCI.20-01-00251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gajtkó A, Bakk E, Hegedűs K, Ducza L, and Holló K (2020). IL-1β induced cytokine expression by spinal astrocytes can play a role in the maintenance of chronic inflammatory pain. Front. Physiol 11, 543331. 10.3389/fphys.2020.543331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delorme A, and Makeig S (2004). EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J. Neurosci. Methods 134, 9–21. 10.1016/j.jneu-meth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Massey CA, Thompson SJ, Ostrom RW, Drabek J, Sveinsson OA, Tomson T, Haas EA, Mena OJ, Goldman AM, and Noebels JL (2021). X-linked serotonin 2C receptor is associated with a non-canonical pathway for sudden unexpected death in epilepsy. Brain Communications 3, fcab149. 10.1093/braincomms/fcab149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.RStudio T(n.d.). RStudio Team (2015). RStudio: Integrated Development Environment for R [Internet]. Available from: Http://www.rstudio.com/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.