

Abstract
Pancreatitis is currently the leading cause of gastrointestinal hospitalizations in the US. This condition occurs in response to abdominal injury, gallstones, chronic alcohol consumption or, less frequently, the cause remains idiopathic. CD73 is a cell surface ecto‐5′‐nucleotidase that generates extracellular adenosine, which can contribute to resolution of inflammation by binding adenosine receptors on infiltrating immune cells. We hypothesized genetic deletion of CD73 would result in more severe pancreatitis due to decreased generation of extracellular adenosine. CD73 knockout (CD73 −/−) and C57BL/6 (wild type, WT) mice were used to evaluate the progression and response of caerulein‐induced acute and chronic pancreatitis. In response to caerulein‐mediated chronic or acute pancreatitis, WT mice display resolution of pancreatitis at earlier timepoints than CD73 −/− mice. Using immunohistochemistry and analysis of single‐cell RNA‐seq (scRNA‐seq) data, we determined CD73 localization in chronic pancreatitis is primarily observed in mucin/ductal cell populations and immune cells. In murine pancreata challenged with caerulein to induce acute pancreatitis, we compared CD73 −/− to WT mice and observed a significant infiltration of Ly6G+, MPO+, and Granzyme B+ cells in CD73 −/− compared to WT pancreata and we quantified a significant increase in acinar‐to‐ductal metaplasia demonstrating sustained metaplasia and inflammation in CD73 −/− mice. Using neutrophil depletion in CD73 −/− mice, we show neutrophil depletion significantly reduces metaplasia defined by CK19+ cells per field and significantly reduces acute pancreatitis. These data identify CD73 enhancers as a potential therapeutic strategy for patients with acute and chronic pancreatitis as adenosine generation and activation of adenosine receptors is critical to resolve persistent inflammation in the pancreas.
Keywords: acinar‐to‐ductal metaplasia, CD73, inflammation, purinergic signaling
Abbreviations
- ADM
acinar‐to‐ductal metaplasia
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- CCK
cholecystokinin
- CD39
ectonucleotidase triphosphate diphosphohydrolase‐1
- EEC
enteroendocrine
- HPLC
high‐performance liquid chromatography
- IHC
immunohistochemistry
- TNF
tumor necrosis factor α
- NECA
5′‐N‐Ethylcarboxamidoadenosine
1. INTRODUCTION
Pancreatitis is an inflammatory condition of the pancreas characterized by severe abdominal pain and the prevalence of acute and chronic pancreatitis is increasing worldwide. 1 The incidence and morbidity of pancreatitis are increased significantly over the past few decades and acute pancreatitis is now the leading cause of gastrointestinal diagnosis for inpatient hospitalizations in the United States. 1 , 2 This condition occurs in response to chronic alcohol consumption, gallstones, abdominal injury, or, less frequently, the cause remains idiopathic. 3 In alcohol‐associated pancreatitis, alcohol induces the aberrant intracellular activation of trypsin within the pancreatic acini resulting in autodigestion of the organ. 4 Additionally, alcohol releases the secretagogue cholecystokinin (CCK) from duodenal cells that stimulates secretion of zymogen granules in pancreatic acinar cells resulting in a profound systemic inflammatory response, acinar‐to‐ductal metaplasia (ADM), and fibrosis. 4 Animal models of acute and chronic pancreatitis have been developed to study mechanisms of acinar cell responses to injury by administering supraphysiological concentrations of caerulein, a CCK analog. 2
Adenosine (ADO) has been recognized for decades as an important physiologic and pharmacologic regulator that signals through cell surface receptors to regulate cellular functions. 5 , 6 More recently, adenosine has been described as paracrine regulator of the tumor microenvironment (TME) and immunosuppressive metabolite elevated in a number of solid tumors including pancreatic ductal adenocarcinoma (PDAC). 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 While inhibitors targeting CD73 or adenosine receptors are therapeutic targets for PDAC patients, 18 under acute or chronic inflammatory conditions, adenosine can promote fibrosis or reduce inflammation, both critical components of wound healing and repair necessary for tissue regeneration. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 Adenosine triphosphate (ATP) is the primary source of energy for cellular processes localized in the intracellular space; however, in response to inflammation or hypoxia, ATP is released into the extracellular space and has been shown to promote pancreatitis. 29 , 30 In the presence of ectonucleotidase triphosphate diphosphohydrolase‐1, NTPDase1, (CD39), extracellular ATP is rapidly converted to adenosine diphosphate (ADP) and monophosphate (AMP), which is subsequently converted to adenosine by a cell surface ecto‐5′‐nucleotidase, CD73. 31 , 32 , 33 Generation of extracellular adenosine by CD73 and stimulation of adenosine receptors are shown to promote fibrosis by signaling through A2A and A2B receptors in the lung, skin, and liver. In contrast, acute stimulation of the A2B receptor restrains fibrosis in the heart through reduced fibroblast proliferation and decreased collagen synthesis; however chronic A2B stimulation may promote cardiac fibrosis. 34 , 35 , 36 CD73‐generated adenosine can promote resolution of inflammation by inhibiting the inflammatory function of neutrophils by signaling at higher concentrations via A1 or A2 receptors to prevent tissue injury from prolonged inflammatory responses. 27 , 32 , 37 , 38 , 39 , 40 In models of sepsis, agents with nucleoside triphosphate hydrolase activity targeting CD39 reduced platelet–leukocyte–endothelium interactions, reduced pro‐inflammatory cytokines, and prolonged survival. 41 The adenosine ENT1 transporter facilitates the uptake of extracellular adenosine across the cell membrane and is one mechanism to downregulate extracellular concentrations of adenosine. 42 Repressing ENTs in the lung has been shown to reduce inflammatory lung injury. 43 Adenosine signals via G‐protein coupled receptors: A1, A2A, A2B, and A3. 44 , 45 , 46 , 47 The A1 Gi‐coupled receptor has a high affinity for adenosine, while the A2A and A2B Gs‐coupled receptors have a lower affinity. Additionally, A3 receptor affinity for adenosine was shown to rank higher than A2B; however, discrepancies in the role of A3 receptors have also been observed in the literature, presenting both anti‐inflammatory and pro‐inflammatory effects. 44 , 48 , 49 , 50 , 51 , 52 , 53 , 54 Therefore, at early stages of inflammation, low local concentrations of adenosine may promote immune recruitment via the A1 receptor while later high adenosine concentrations can suppress immune activity via the A2A, A2B, or A3 receptors. 44 , 52 , 55
Adenosine binding to adenosine receptors modulates both the innate and the adaptive immune response to hypoxia, inflammation, and tissue repair. 33 The A2A and A2B receptors exert anti‐inflammatory effects by inhibiting neutrophil chemotaxis, attachment to vascular endothelial cells, and phagocytosis. 14 , 44 , 56 , 57 In the pancreas, A2A has been shown to protect against pancreatic dysfunction, islet size, and insulin context in the context of high‐fat diet‐induced diabetes and obesity. 58 Inflammatory macrophages are inhibited through A2A and A2B activation resulting in decreased production of cytokines including IL‐1β, IL‐18, IL‐6, and TNF‐α. 14 , 32 Additionally, CD4+ T cell activation and proliferation and natural killer cell cytotoxic functions are inhibited by A2A receptor activation. 14 Additionally, A3 enhancement was reported to inhibit macrophage inflammatory protein (MIP)‐1α in a model of collagen‐induced arthritis, 53 and two other murine studies of colitis showed reduced inflammation and increased survival following A3 activation. 54
In contrast, decreased activity of CD73 and extracellular adenosine is associated with amplified activation and chemotactic functions of immune cell populations. 32 The purinergic P2X and P2Y family of receptors are expressed on neutrophils and promote neutrophil‐mediated oxidative burst‐induced tissue injury in the presence of ATP. 59
In response to injury, pancreatic acini can undergo acinar‐to‐ductal metaplasia (ADM), a metaplastic event that limits pancreatic tissue damage via a rapid decline in zymogen production. 60 , 61 , 62 Experimental studies have shown injured acinar cells activate a shift in gene expression regulated by Mist1 and Ptf1α to transdifferentiate away from their specified cell type and function, which consists of highly specialized cells involved in the production and secretion of digestive enzymes, toward a ductal phenotype. 57 , 63 ADM trans‐differentiation is also triggered by innate and pro‐inflammatory immune cells, including neutrophils and macrophages, that infiltrate the pancreas resulting in elevated secretion of inflammatory cytokines including RANTES and tumor necrosis factor α (TNF). 64 Differentiation into a cell type with ductal characteristics demonstrates a pancreatic repair process under strong positive selection in pancreatitis. 23 , 26 These mucinous populations can subsequently seed tuft cell and enteroendocrine lineages as further reparative mechanisms. 65
2. MATERIALS AND METHODS
2.1. Animal model
All mouse model procedures are in compliance with UTHealth's CLAMC Animal Welfare Committee Review and approved on Dr. Bailey‐Lundberg's AWC protocol. To evaluate the role of CD73 in pancreatitis, CD73 knockout (CD73 −/−) and C57BL/6 (wild type) mice were used. Full‐body CD73KO mice were purchased from The Jackson Laboratory strain 018986. Controls for each experiment were derived from wild‐type crosses. Caerulein injections were performed for each experiment during Spring months (March–June) 2022 and mixed genders were equally included for all groups. In the acute pancreatitis model, CD73 −/− and wild‐type mice were intraperitoneally injected on alternating flanks with 70 μg/kg of caerulein (Sigma Aldrich 17650‐98‐5) 8× a day for two consecutive days. Mice were euthanized at 1, 4, and 7 days after the last caerulein injection to evaluate ADM abundance, inflammatory progression, and organ repair. For the chronic model, CD73 −/− and wild‐type mice were injected intraperitoneally on alternating flanks with 250 μg/kg of caerulein (Sigma Aldrich 17650‐98‐5) twice a day, 5 days a week, for 2 weeks. The mice were allowed to recover for 2 days after the last injection before euthanasia by isoflurane overdose.
2.2. Neutrophil depletion in vivo
CD73 −/− mice were intraperitoneally administered 300 μg of anti‐Ly6G antibody (clone1A8, BioXCell, West Lebanon, NH) at Day ‐2, 0, and 1 day after the last caerulein injection. IgG2a isotype (clone 2A3, BioXCell, West Lebanon, NH) was used for control. Mice were euthanized at Day 4.
2.3. NECA in vivo
CD73 −/− and wild‐type mice under acute pancreatitis protocol of eight injections per day during two consecutive days (Days ‐1 and 0) were administered a single injection of 80 μg/kg of 5′‐N‐Ethylcarboxamidoadenosine (NECA) (MedChemExpress HY‐103173) after the sixth injection of caerulein on day zero. Mice were euthanized at Day 1. NECA injection was performed intraperitoneally.
2.4. Immunohistochemistry and ImageJ analysis
Tissues were fixed in zinc‐buffered formalin, processed according to standard protocols, and embedded in paraffin. The unstained sections were baked at 60°C for 45 min. The sections were deparaffinized with Histoclear and rehydrated stepwise. Heat‐mediated antigen retrieval was followed with a pH 6 unmasking solution (Vector Laboratories, H‐3300) and a pH 9 unmasking solution (Abcam 100X Tris‐EDTA). All sections were blocked for 1 h in 10% FBS in PBST. Primary antibodies were used at a 1:200 dilution and incubated overnight at 4°C. Secondary antibodies were used at a 1:500 dilution and incubated at room temperature for 30 min. The Vectastain ABC kit Peroxidase Standard (Vector Laboratories, PK4000) and DAB Peroxidase (HRP) Substrate kit (Vector Laboratories, SK‐4100) were used. Human pancreatitis tissues were obtained from a TMA array (US Biomax, Inc. BIC14011b), where all tissues were fixed in 10% neutral formalin for 24–48 h, dehydrated with gradient ethanol, cleared with xylene, and embedded in paraffin. All human tissues from US Biomax TMA BIC14011b were collected under HIPPA‐approved protocols and approved for commercial product development. Six independent chronic pancreatitis cores were analyzed for CD73 expression (Abcam, cat. Ab133582) following the IHC protocol mentioned above.
Image analysis was performed using ImageJ (http://imagej.nih.gov/ij/) software and three to five representative fields per tissue were used depending on the size of the tissue. The color threshold tool was used to determine positive staining and ensure normalization of all samples. Freehold selection tool was used to isolate mild and severe pancreatitis from normal tissue. Pancreatitis areas were defined by the presence of swelling between pancreatic lobes, significant infiltrating immune cells, ADM cells, and loss of normal cellular histology.
2.5. Nucleoside purification
Pancreas tissue was collected at the end of the experiment and flash frozen. At time of experiment, tissue was homogenized in perchloric acid using a Beadbug microtube homogenizer (BeadBug™, cat. SKUD1036). Pancreas homogenates were then centrifuged at 14 000 rpm at 4°C for 10 min and the supernatant was collected. A Pierce™ BCA Protein Assay Kit (Thermo Scientific, cat. 23225) was performed to determine protein concentration following manufacturer instructions. Samples were then neutralized with phenol red and KHCO3/KOH, vortexed, acidified with (NH4)3PO4 and H3PO4, then vortexed. Samples were centrifuged at 14 000 rpm for 5 min and 1 ml of supernatant was collected and filtered for further analysis.
2.6. High‐performance liquid chromatography (HPLC)
Filtered supernatants were analyzed by high‐performance liquid chromatography using the Waters Breeze 2 HPLC System (Waters 2489 UV/Visible Detector and Waters 1525 Binary HPLC Pump). Flow rate was 1 ml/min and 100 μl per sample was injected in the column (XSElect HSS C18 SB 5 μm 4.6 × 250 mm) with a mobile phase 100% A (0.02M NH4H2PO4) for 0–4 min, which then was switched to 100% B (0.02M NH4H2PO4 containing 20% methanol) from 4 to 8 min, then stayed in 100% B from 8 to 18 min, and finally switched back to 100% A from 18 to 20 min. Absorbance was measured at a wavelength of 260 and 280 nm, and adenosine and AMP peaks were determined using a standard HPLC curve. Pancreas tissue adenosine and AMP levels were normalized to lysate protein levels.
2.7. Analysis of published single‐cell RNA sequencing datasets
Processed count matrices for scRNA‐seq datasets from Ma et al. were downloaded from the Gene Expression Omnibus (GEO) database (accession number GSE172380). The processed human pancreas sNuc‐seq dataset from Tosti et al. was obtained from http://singlecell.charite.de/pancreas/. 65 , 66 Low‐quality cells were filtered on read counts, the number of genes expressed, and the ratio of mitochondrial reads following the thresholds described in the respective publications. Filtered gene count matrices were log‐normalized, and the top 2000 variable features were further scaled prior to dimension reduction by PCA and being embedded in UMAP using the R package Seurat. 67 Seurat cell clusters were labeled with major cell types using marker genes provided by the authors.
3. RESULTS
3.1. CD73 is expressed on ductal cells in human chronic pancreatitis
Recent literature has described that CD73 is elevated in PDAC epithelial cells resulting in adenosine generation and an immune‐suppressive tumor microenvironment. 9 , 10 , 11 In the normal pancreas, CD73 is expressed in vascular cells, but limited to no expression is observed in acinar or ductal cells. In contrast, NTPdase1 (CD39) is expressed in acinar cells and blood vessels, which have ATPase activity; yet CD39 is expressed at low levels in normal pancreatic ducts. 68 , 69 As chronic pancreatitis is a risk factor for development of PDAC, 70 and divergent roles for adenosine have been described in inflammatory diseases, we wanted to investigate the cell‐type‐specific localization of CD73 in human and murine chronic pancreatitis and determine if CD73 is an important determinant of pancreatitis severity. To evaluate cellularity of CD73 expression, we analyzed previously published single‐nucleus RNA‐sequencing data (sNuc‐seq) generated from two patients with chronic pancreatitis (totaling 2726 nuclei). 65 These data revealed NT5E, the gene encoding for CD73, is highly expressed in a MUC5B+ ductal cell population (Figure 1A). To confirm ductal cells expressed CD73, we used a human tissue microarray to evaluate cellularity of CD73 immunolabeling in chronic pancreatitis. An immunohistochemistry (IHC) stain for CD73 in 6 patient samples from human chronic pancreatitis (Figure 1B) revealed a strong positive staining in ADM cells (green arrows), ductal cells (red arrows), and infiltrating immune cells (yellow arrows).
FIGURE 1.
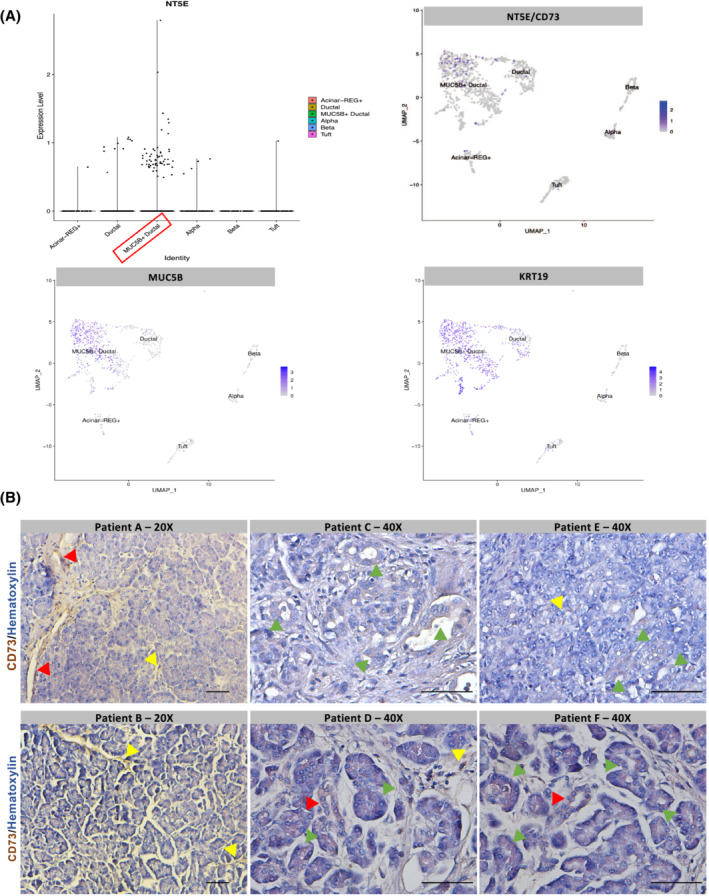
CD73 is expressed on ductal cells and infiltrating immune cells in human chronic pancreatitis. Single‐nuclear RNA sequencing of human chronic pancreatitis was analyzed. IHC for CD73 was performed in a TMA with six human cases of chronic pancreatitis. (A) Single‐nuclear RNA sequencing 65 and associated UMAP from human chronic pancreatitis. NT5E (CD73) is expressed in a MUC5B+ ductal cell population (MUC5B+ and KRT19+ cells). (B) Human chronic pancreatitis tissue (n = 6) demonstrating positive CD73 staining in ADM cells (green arrows), ductal cells (red arrows), and infiltrating immune cells (yellow arrows). Bars represent 50 μM.
3.2. Genetic loss CD73 increases severity of chronic pancreatitis
To determine the role of CD73 in pancreatitis, we utilized a murine 2‐week chronic pancreatitis model (Figure 2A). Wild type and CD73 −/− mice were used for the study, and both genotypes were subjected to a caerulein‐induced pancreatitis protocol consisting of two 250 μg/kg injections per day, 5 consecutive days a week, for 2 weeks. The mice were then euthanized after a 2‐day recovery period to determine expression of CD73 in murine pancreatitis and to evaluate if loss of CD73 resulted in any histopathologic changes in the pancreas compared to wild‐type pancreata. We determined the localization of CD73 in vivo by CD73 IHC staining and observed a strong positive expression of CD73 on infiltrating immune cells as well as ductal cells only in wild‐type mice under chronic pancreatitis, whereas the absence of CD73 expression was confirmed in pancreata from the in CD73 −/− mice (Figure 2B, Top panels, black arrows).
FIGURE 2.
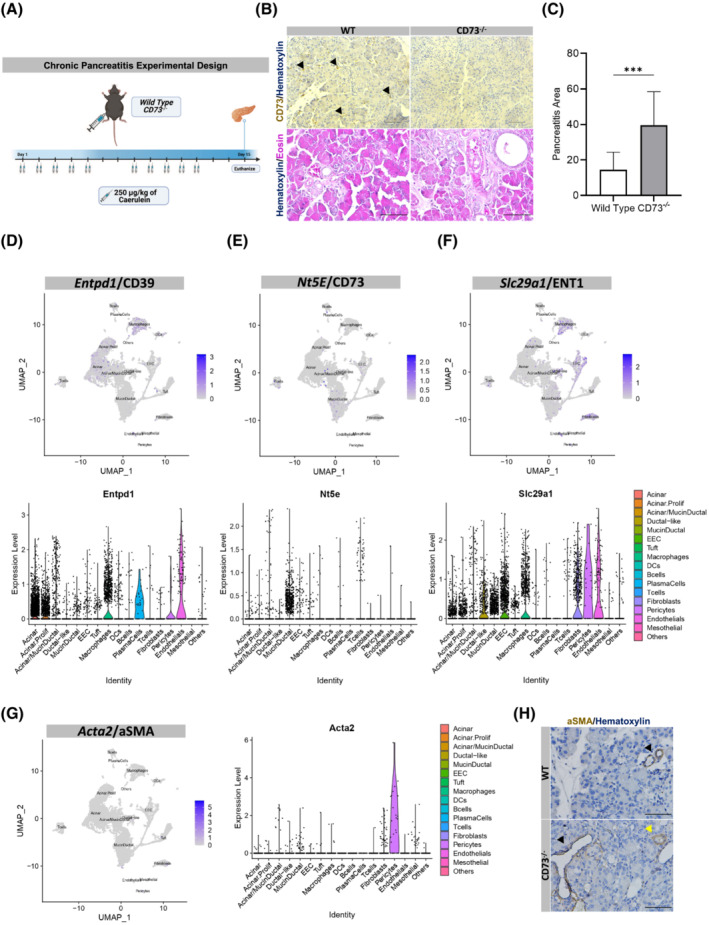
Genetic loss of CD73 increases severity of chronic pancreatitis. An in vivo model of chronic pancreatitis was performed in WT and CD73 −/− mice. Histopathology and purinergic enzymes and stellate cell marker expression were analyzed. (A) Experimental design for the chronic pancreatitis model in vivo. (B) IHC stain for CD73 in WT and CD73 −/− mice demonstrating positive staining on infiltrating immune cells as well as ductal cells only in WT mice (Top panel, black arrows). H&E stain of chronic pancreatitis in WT and CD73 −/− mice (Bottom panel). (C) ImageJ quantification of severe chronic pancreatitis revealed significantly increased severe pancreatitis area CD73 −/− mice compared to WT. Data were analyzed by Student's t test. (D–F) Single‐cell RNA sequencing and associated UMAP from a chronic pancreatitis mouse model at 2 and 4 weeks 65 showed Entpd1/CD39 is highly expressed in acinar cells and macrophages (D), Nt5e/CD73 is highly expressed in mucin/ductal cell populations (E), Slc29a1/ENT1 adenosine transporter is highly expressed in ductal‐like, endothelial, EEC cell populations and macrophages (F) and (G) Acta2/aSMA is expressed mainly by pericytes, mucin‐ductal, and fibroblast cells. (H) IHC showing detectable aSMA expression in small vessels (black arrows) and duct‐like structures (yellow arrows) but undetected in fibroblast cells in both WT and CD73 −/− mice. Scale bars 500 μm. Error bars, SEM. ***p ≤ .001.
To evaluate tissue injury and pathology in the context of chronic caerulein treatment, we used Hematoxylin & Eosin (H&E) to stain wild‐type and CD73 −/− mice (Figure 2B, Bottom panels). For comparison, severe pancreatitis was quantified and defined as the presence of significant infiltrating immune cells, ADM cells, and loss of normal cellular histology. Under caerulein‐mediated chronic pancreatitis conditions, CD73 −/− mice displayed significantly increased severe pancreatitis area per field (p < .001) (Figure 2C) suggesting the loss of extracellular adenosine generation exacerbates and sustains tissue injury as well as inhibits tissue regeneration.
As we observed such a prominent difference in pancreatic injury in wild type compared to CD73 −/− mice after chronic injection of caerulein, we wanted to evaluate the cellular expression of CD73, CD39, and ENT1 in caerulein‐mediated murine chronic pancreatitis. We analyzed single‐cell RNA sequencing data from a chronic caerulein‐mediated mouse model recently published by Ma et al. encompassing ~21 140 cells from four mice. 65 The results demonstrated the enzyme CD39, responsible for catalyzing the conversion of ATP to ADP and AMP, is highly expressed on macrophages, pericytes, endothelial cells, and acinar cells (Figure 2D); whereas, similar to what we identified in human chronic pancreatitis, CD73 is highly expressed in a mucin/ductal cell population as well as in T cells, macrophages, and B cells (Figure 2E). Additionally, we observed the ENT1 adenosine transporter that facilitates the movement of extracellular adenosine across the cell membrane is highly expressed in macrophages, fibroblasts, pericytes, enteroendocrine cells, endothelial cells, and ductal‐like cells (Figure 2F). Lastly, given its role in chronic pancreatitis and fibrotic development, 71 the expression of aSMA, a common marker of activated stellate cells, was studied. Single‐cell RNA sequencing analysis showed Acta2, the gene encoding for aSMA, expression mainly in pericytes, mucin‐ductal, and fibroblast cells during chronic pancreatitis (Figure 2G); however, when analyzed by IHC (Figure 2H) in both WT and CD73 −/− mice, aSMA protein expression was not detected in fibroblasts; however, aSMA expression was clearly observed in small vessels (black arrows) and mucin‐ductal structures (yellow arrow), suggesting little or undetected activation of stellate cells in this experimental mouse model of chronic pancreatitis by the above‐mentioned technique.
3.3. Purinergic signaling modulates response to acute pancreatitis
As we observed such a significant difference in pancreatitis area in the chronic pancreatitis model, we wanted to evaluate the role of CD73 in a caerulein‐mediated acute pancreatitis model, which allows for histologic visualization of pancreatic repair over a time frame of 7 days after acute injury. 72 Wild type and CD73 −/− mice were used for the study and underwent a caerulein‐induced acute pancreatitis protocol which consisted of eight injections of 70 μg/kg caerulein per day for two consecutive days (Figure 3A). Mice were then euthanized at 1, 4, and 7 days after the last caerulein injection to evaluate the timing of AMP and adenosine generation, which were also compared to chronic exposure to caerulein levels. Under acute pancreatitis, high‐performance liquid chromatography (HPLC) revealed AMP levels acutely decrease from Day 1 to Day 4, and increase from Day 4 to Day 7 in wild‐type mice; in contrast, in CD73 −/− mice these levels increase from Day 1 to Day 4 and decrease from Day 4 to Day 7, suggesting a transient accumulation of AMP in the context of no CD73 activity (Figure 3B). In wild‐type mice, acute pancreatitis showed AMP variations that were accompanied by a significant increase in ADO levels between Day 1 and Day 4, followed by a decrease between Day 4 and Day 7; however, no significant variations were observed in CD73 −/− mice (Figure 3C). Chronic exposure to caerulein showed increased AMP levels in CD73 −/− mice but decreased ADO levels when compared with wild‐type pancreata.
FIGURE 3.
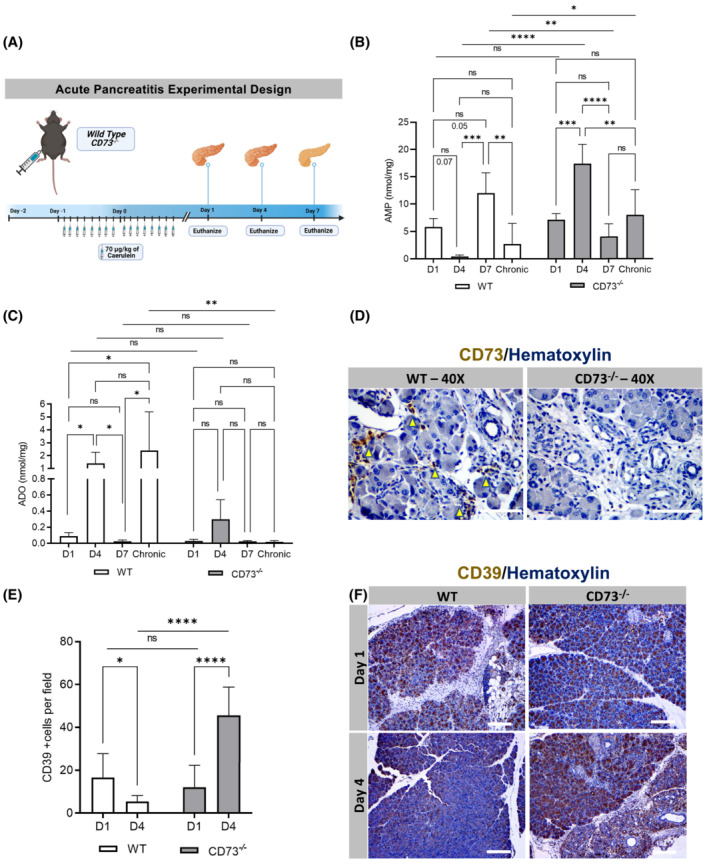
Purinergic signaling modulates response to acute pancreatitis. AMP and ADO levels were analyzed by HPLC from pancreatic tissues for AMP in WT mice at Days 1 (n = 4), 4 (n = 4), 7 (n = 3) and chronic model (n = 5) and CD73 −/− mice at Days 1 (n = 5), 4 (n = 4), 7 (n = 4), and chronic model (n = 5). CD39 and CD73 expressions were analyzed by IHC. (A) Experimental design for the acute pancreatitis model in vivo. (B) AMP levels acutely decrease from Day 1 to Day 4 and increase from Day 4 to Day 7 in WT mice; in contrast, in CD73 −/− mice these levels increase from Day 1 to Day 4 and decrease from Day 4 to Day 7. Chronic exposure to caerulein showed increased AMP levels in CD73 −/− mice. (C) ADO levels increased in WT mice between Day 1 and Day 4, followed by a decrease between Day 4 and Day 7; however, no significant variations were observed in CD73 −/− mice. Chronic exposure to caerulein showed decreased ADO levels when compared with WT group. D1 and D4 were compared with Student's t test. (D) CD73 at Day 4 revealed positive staining on infiltrating immune cells (yellow arrows) in the WT only. (E) ImageJ quantification of CD39 showed decreased expression between Day 1 and Day 4 in WT mice and increased in CD73 −/− murine pancreata. Additionally, at Day 4 a significant increase in CD39 expression was observed in CD73 −/− compared to the WT pancreata. (F) IHC stain for CD39 in WT and CD73 −/− mice at Day 1 and 4. Data were analyzed by two‐way ANOVA. Error bars, SEM. *p ≤ .05; **p ≤ .01; ***p ≤ .001; ****p ≤ .0001; n.s., not significant. Scale bars 50 μm.
ADO levels were significantly elevated at Day 4 during acute pancreatitis so we wanted to determine the localization of CD73 at Day 4. IHC showed CD73 expression in infiltrating immune cells as well as ductal cells (Figure 3D, yellow arrows) in wild‐type mice. CD73 staining was also performed on CD73 −/− mice to confirm the genotype, which correctly demonstrated negative staining of the tissue.
To compare the initial modulation in nucleotide generation under acute conditions in wild‐type and CD73 −/− mice, an IHC antibody stain for CD39 was performed at Day 1 and Day 4. CD39 expression decreased between Day 1 and Day 4 in wild‐type mice and increased in CD73 −/− murine pancreata (Figure 3E,F). Additionally, at Day 4 a significant increase in CD39 expression was observed in CD73 −/− compared to the wild‐type pancreata. These data support the HPLC data that CD73 −/− mice are experiencing enhanced nucleotide or purinergic signaling at Day 4, which may be a major determinant of sustained tissue injury in CD73 −/− mice, while the wild‐type mice utilize CD73 to convert AMP to adenosine.
3.4. Genetic loss of CD73 promotes metaplasia in acute pancreatitis
In order to better comprehend the initial tissue injury response and resolution in the acute pancreatitis model, we evaluated immune infiltration and metaplasia. To correlate tissue injury to the amount of ADM cells present, an IHC antibody stain for Cytokeratin‐19, a marker for ductal cells, was performed in wild‐type and CD73 −/− mice at Day 1 and Day 4 (Figure 4A). At Day 1, the number of ductal cells per field between experimental groups was similar, indicating comparable initial tissue injury and metaplasia (p = n.s.) (Figure 4B). However, at Day 4, CD73 −/− mice displayed a significant increase in the amount of Cytokeratin‐19+ areas per field, indicating a significant increase in metaplastic ducts and ADM, compared to wild‐type pancreata (p < .001). Interestingly, from Day 1 to Day 4 there was a significant increase in Cytokeratin‐19+ cells in both wild‐type mice (p < .01) and CD73 −/− mice (p < .0001) (Figure 4B). These findings indicate the ADM process is a reparative mechanism concurrent with peak pancreatic adenosine generation.
FIGURE 4.
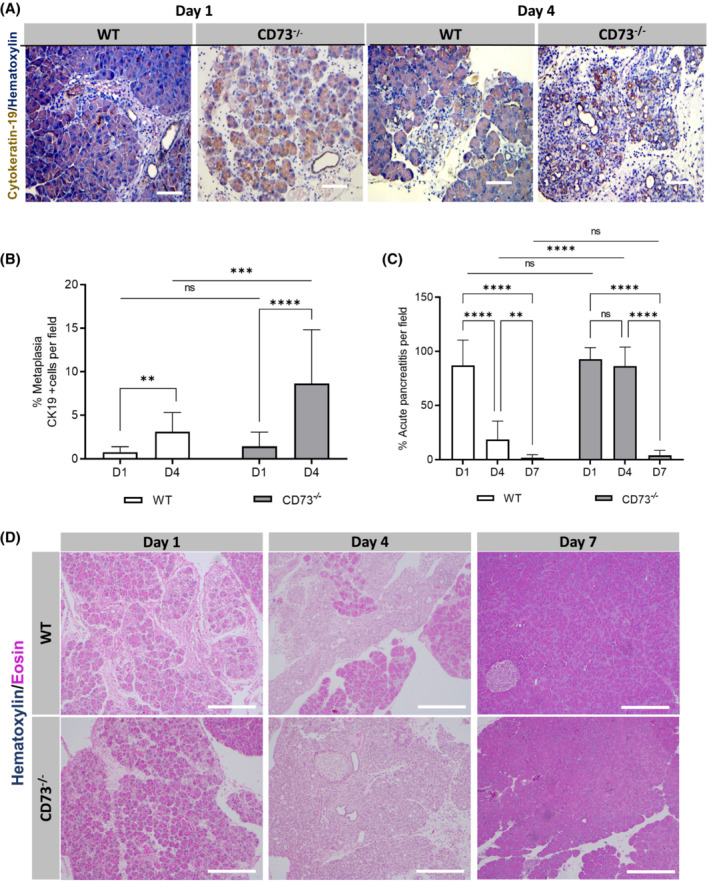
Genetic loss of CD73 promotes metaplasia in acute pancreatitis. Metaplasia was studied by Cytokeratin 19 IHC staining in WT and CD73 −/− mice at Days 1 and 4. Pancreatitis area was evaluated in WT mice at Days 1 (n = 12) and 4 (n = 14) and in CD73 −/− mice at Days 1 (n = 12) and 4 (n = 21). (A) IHC stain for Cytokeratin‐19 in WT and CD73 −/− mice at Days 1 and 4. (B) ImageJ quantification of Cytokeratin‐19‐positive areas per field a significant increase from Day 1 to Day 4 in both genotypes. Additionally, at Day 4, the presence of Cytokeratin‐19+ cells was significantly increased in CD73 −/− mice compared to WT. Student's t test was used to compare WT timepoints. (C) ImageJ quantification of acute pancreatitis revealed increased persistent pancreatitis in CD73 −/− mice compared to WT. By Day 7, both genotypes demonstrated similar histology with no significant difference in pancreatitis. (D) H&E stain of acute pancreatitis in wild type and CD73 −/− mice at Days 1, 4, and 7. Data were analyzed by two‐way ANOVA. Error bars, SEM. **p ≤ .01; ***p ≤ .001; ****p ≤ .0001; n.s., not significant. Scale bars 50 μm.
To evaluate severity of pancreatitis at Day 4 and Day 7, an H&E stain was performed in wild type and CD73 −/− mice at Day 1, 4, and 7. At Day 1, both experimental groups showed increased fluid between the pancreatic lobes and acinar cells, a similar presence of ADM cells, and infiltrating immune cells (Figure 4C,D). At Day 4, wild‐type mice demonstrated a return to normal histology characterized by the disappearance of ADM cells and infiltrating immune cells and reduction in excess fluid, while CD73 −/− mice showed significantly increased residual pancreatitis areas (p < .0001). Lastly, at Day 7 both experimental groups demonstrated near‐complete return to normal histology with no significant difference between them (p = n.s.). These data suggest CD73 −/− mice exhibit sustained tissue injury and require more time for tissue regeneration compared to the wild‐type mice.
3.5. Loss of adenosine increases immune infiltration in acute pancreatitis
To evaluate mediators of sustained inflammation in the CD73 −/− mice, we used IHC to stain for Granzyme B, Myeloperoxidase (MPO) and NIMPR14, a neutrophil marker. IHC experiments were performed in wild‐type and CD73 −/− mice at Day 1 and Day 4. At Day 1, the number of Granzyme B+ cells was similar between experimental groups (p = n.s.) (Figure 5A,D). However, at Day 4 CD73 −/− mice demonstrated significantly increased Granzyme B+ cells compared to the wild‐type mice (p < .0001). Additionally, from Day 1 to Day 4 there was a significant increase in Granzyme B+ cells in CD73 −/− mice (p < .05) and a significant decrease in the wild type (p < .05). These data suggest Granzyme B expressing cells are a major determinant of pancreatitis severity in the absence of extracellular adenosine.
FIGURE 5.
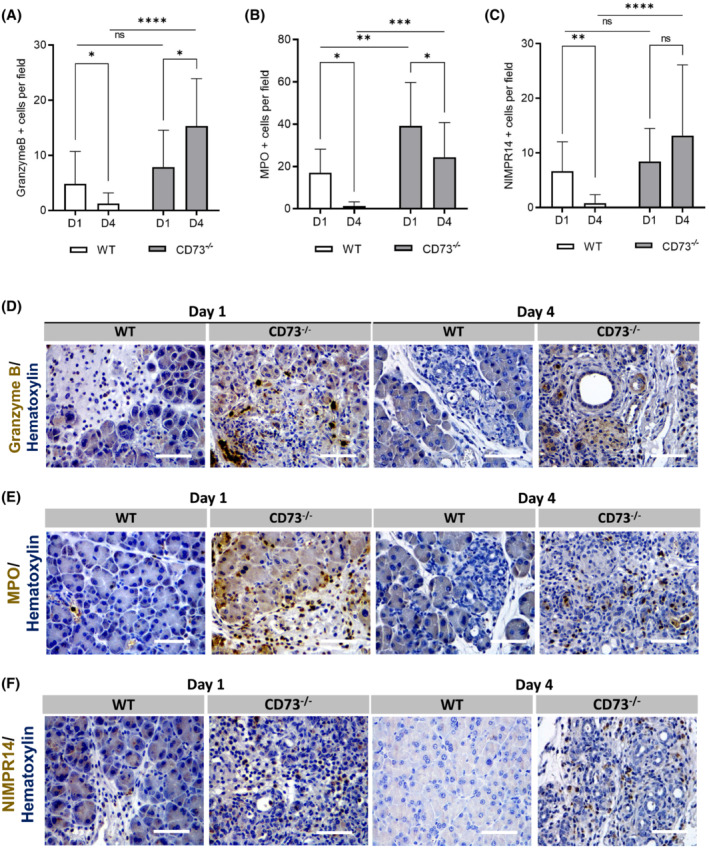
Loss of adenosine increases immune infiltration in acute pancreatitis. Immune cell infiltration was analyzed by IHC in WT and CD73 −/− mice at days 1 and 4. (A) ImageJ quantification of Granzyme B+ cells per field, a marker for T‐cells and Natural Killer cells, was increased only in CD73 −/− mice at D4 when compared with D1 and WT D4 measurements. WT mice presented decreased Granzyme B+ cells at D4 compared to D1. Student's t test was used to analyze WT timepoints. (B) ImageJ quantification of MPO+ cells per field showed a decrease from D1 to D4 in both genotypes. Additionally, CD73 −/− mice presented increased MPO‐positive cells at both timepoints when compared with WT mice. (C) ImageJ quantification of NIMPR‐14+ cells showed significantly increased staining in CD73−/− mice when compared with WT. In WT mice, levels decreased from D1 to D4. Student's t test was used to analyze WT timepoints. Representative images for Granzyme B (D), MPO (E), and NIMPR14 (F) IHC staining. Data were analyzed by two‐way ANOVA. Error bars, SEM. *p ≤ .05; **p ≤ .01; ***p ≤ .001; ****p ≤ .0001; n.s., not significant. Scale bars 50 μm.
To determine the innate immune system's role in initial tissue injury and resolution, an IHC stain for MPO, a marker for inflammatory neutrophils, was performed in wild‐type and CD73 −/− mice at Day 1 and Day 4 (Figure 5B,E). From Day 1 to Day 4 there was a significant decrease in MPO+ cells in both wild‐type and CD73 −/− mice (p < .05). Interestingly, CD73 −/− mice demonstrated a significantly increased amount of MPO+ cells per field compared to the wild‐type mice at both Day 1 and Day 4 measurements (p < .01; p < .001, respectively), suggesting the absence of extracellular adenosine in CD73 −/− mice promotes early tissue injury via innate immune cell infiltration.
Due to the potent induction of the innate immune system at Day 1 as shown in the MPO IHC, we decided to specifically evaluate neutrophil activity. An IHC stain for NIMPR‐14, a marker for Ly6G+ and Ly6C+ neutrophils, was performed in wild‐type and CD73 −/− mice at Day 1 and Day 4 (Figure 5C,F). At Day 1, the number of neutrophils per field was similar between experimental groups (p = n.s.); however, at Day 4 CD73 −/− mice demonstrated significantly increased neutrophils per field compared to the wild‐type (p < .0001). From Day 1 to Day 4 there was a significant decrease in neutrophils in wild‐type mice (p < .01); while no difference was observed in neutrophil abundance in the CD73 −/− (p = n.s.). These results suggest that the sustained tissue injury seen in CD73 −/− mice at Day 4 is primarily due to the continued induction and activation of neutrophils in the absence of extracellular adenosine and possibly due to heightened ATP or AMP‐dependent purinergic signaling.
3.6. Adenosine restrains neutrophil‐mediated tissue injury
To confirm that adenosine generation promoted neutrophil‐mediated tissue injury, we modified the caerulein‐induced acute pancreatitis model by treating CD73 −/− mice with a Ly6G neutrophil depletion antibody or vehicle (Figure 6A) and euthanized the mice at Day 4 post last caerulein injection. To confirm neutrophil depletion, an IHC stain for NIMPR‐14 was performed which demonstrated the presence of neutrophils in the vehicle‐treated and the absence of neutrophils in pancreata from the neutrophil‐depleted mice (Figure 6B). We performed an H&E stain to assess tissue injury, and found that at Day 4, neutrophil‐depleted CD73 −/− mice demonstrated significantly less pancreatitis per field compared to vehicle‐treated (p < .05) (Figure 6C,D), indicating enhanced neutrophil activity from loss of CD73 dependent adenosine generation promotes sustained inflammation.
FIGURE 6.
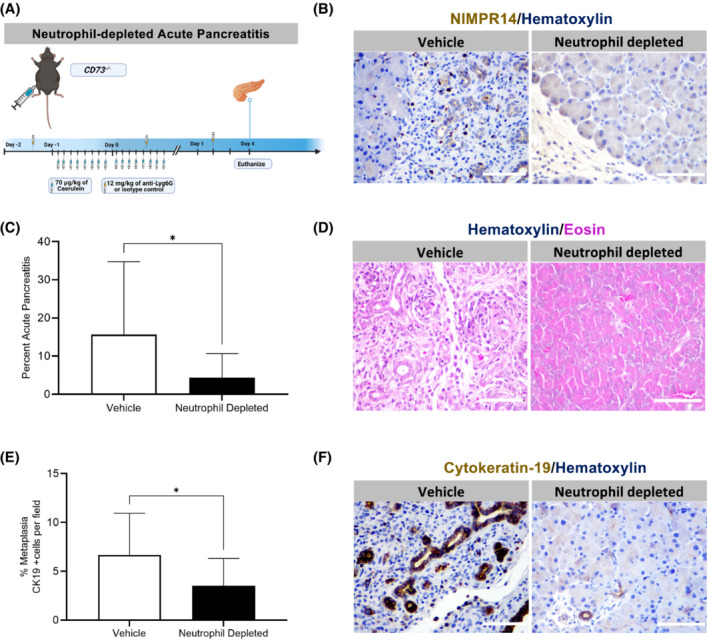
Genetic loss of adenosine generation by CD73 increases neutrophil‐mediated oxidative stress‐induced tissue injury. A neutrophil depletion in vivo experiment was performed in WT and CD73 −/− mice subjected to acute pancreatitis. Mice were euthanized at Day 4. Pancreatitis and cytokeratin 19 areas were assessed by histopathology and IHC. (A) Experimental design for the neutrophil‐depleted acute pancreatitis model in vivo. (B) IHC stain for NIMPR‐14 was performed in vehicle‐treated and neutrophil‐depleted CD73 −/− mice at Day 4 to confirm depletion. (C) ImageJ quantification at day 4 revealed decreased pancreatitis area per field in neutrophil‐depleted mice compared to vehicle‐treated. (D) H&E stain in neutrophil‐depleted acute pancreatitis mice model at Day 4. (E) ImageJ quantification of Cytokeratin‐19‐positive areas per showed decreased levels in neutrophil‐depleted mice compared to vehicle‐treated. (F) IHC stain for Cytokeratin‐19 in neutrophil‐depleted acute pancreatitis mice model at Day 4. Data were analyzed by Student's t test. Error bars, SEM. *p ≤ .05. Scale bars 50 μm.
To determine if neutrophil‐depletion could prevent metaplasia in acute pancreatitis, an IHC stain for Cytokeratin‐19 was performed in vehicle‐treated and neutrophil‐depleted CD73 −/− mice at Day 4, which showed vehicle‐treated mice, compared to neutrophil‐depleted mice, present significantly increased amounts of metaplasia per field, suggesting the absence of neutrophils restrains metaplasia in acute pancreatitis (p < .05) (Figure 6E,F).
3.7. Enhanced adenosine receptor activation reduces caerulein‐induced acute metaplasia
To determine if enhanced adenosine receptor activation would reduce tissue injury and metaplasia, wild‐type and CD73 −/− mice were administered NECA, a high‐affinity adenosine receptor enhancer, in an acute pancreatitis model (Figure 7A). We assessed tissue injury in H&E staining and found CD73 −/− mice demonstrate significantly less pancreatitis area per field compared to their caerulein‐only‐treated genotype comparison (p < .01) (Figure 7B,C). To evaluate the effect of enhanced adenosine signaling on metaplasia pancreatitis area, an IHC stain for Cytokeratin‐19 was conducted to investigate the amount of metaplastic ductal cells present at Day 1 in NECA‐treated wild‐type and CD73 −/− mice. Our analysis revealed NECA‐treated CD73 −/− mice present significantly decreased amount of metaplasia compared to the caerulein‐only‐treated CD73 −/− mice (p < .05) (Figure 7D,E). In contrast, there were similar levels of Cytokeratin‐19‐positive cells in NECA‐treated and caerulein‐only‐treated wild‐type mice at Day 1 (p = n.s.), suggesting that the most significant therapeutic effect was seen in animals that were previously adenosine depleted. Similarly, when MPO protein expression was evaluated by IHC (Figure 7F,G), we observed increased levels in CD73 −/− pancreata compared to WT mice after caerulein‐only treatment; however, NECA administration promoted a markedly decrease in MPO levels in CD73 −/− pancreata when compared with caerulein‐only CD73 −/− treated mice only, suggesting both neutrophils and MPO secretion may participate in the metaplastic process during an acute pancreatitis setting.
FIGURE 7.
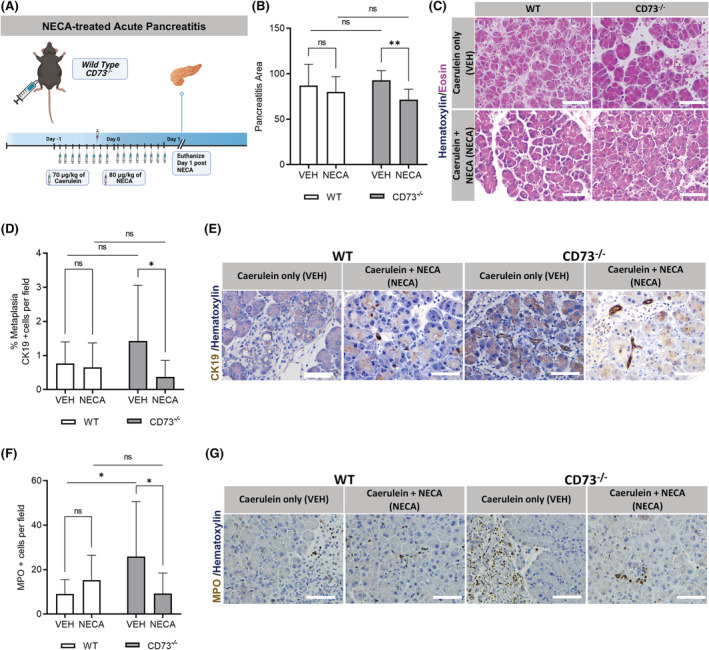
Enhanced adenosine receptor activation reduces inflammation after acute caerulein treatment. An acute pancreatitis in vivo experiment was performed in CD73 −/− mice treated with NECA or vehicle. Pancreatitis and Cytokeratin 19+ areas were assessed by histopathology and IHC. (A) Experimental design for the NECA‐treated acute pancreatitis model in vivo. (B) ImageJ quantification of pancreatitis per field revealed NECA‐treated CD73 −/− presented less pancreatitis area than vehicle‐treated mice after acute pancreatitis induction. (C) H&E stain of VEH and NECA‐treated WT and CD73 −/− mice at Day 1 in the acute pancreatitis model. (D) ImageJ quantification of Cytokeratin‐19‐positive areas per field showed increased metaplastic ductal cells per field in caerulein‐only‐treated CD73 −/− mice compared to NECA‐treated. (E) IHC stain for Cytokeratin‐19 in VEH‐ and NECA‐treated WT and CD73 −/− mice at Day 1. (F) ImageJ quantification of MPO‐positive cells per field showed increased levels in cerulean‐only treated CD73 −/− mice compared to WT mice. Additionally, NECA treatment significantly reduced MPO‐positive cells only in CD73 −/− mice. (G) IHC stain for MPO in VEH and NECA‐treated WT and CD73 −/− mice at Day 1. Data were analyzed by two‐way ANOVA. Error bars, SEM. *p ≤ .05; **p ≤ .01; n.s., not significant. Scale bars 50 μm.
4. DISCUSSION
Acinar and ductal cells comprise the exocrine component of pancreatic parenchymal function. Acinar cells are highly specialized cells characterized by zymogen granules and abundant rough endoplasmic reticulum. Acinar cells are responsible for synthesizing, storing, and secreting digestive enzymes including amylase, lipase, peptidase, and nucleases. In addition, acinar cells are the major source of trypsinogen, a component of pancreatic juice that is cleaved to trypsin by enteropeptidases in the intestinal mucosa. 73 , 74 The main function of the pancreatic ducts is to carry fluid containing digestive enzymes secreted from acinar cells. Ductal cells are a major source of sodium bicarbonate (NaHCO3) rich fluid which flushes out and neutralizes pH from digestive enzymes. 75 In pancreatitis, the ductal epithelium does not appropriately regulate pH and more neutral pH or acidic pH is generated, causing obstruction and dilation of the duct lumen. 76 The ductal system is intricate, with many small peripheral ducts all channeling to the main pancreatic duct. When gallstones, calcification or intraductal lesions block the exocrine component of the pancreas, patients develop pain and inflammation as there is an abnormal release of digestive enzymes, which can contribute to pancreatic fibrosis, calcification, and downstream pathophysiology. Neutrophil infiltration is one of the first pathogenic responses in early phases of pancreatitis 77 and neutrophil accumulation is thought to prematurely activate trypsinogen release and aid in progression from acute to severe acute and chronic pancreatitis through production of reactive oxygen species (ROS) and hydrolases (reviewed in Ref. [77]). Neutrophil depletion significantly reduces serum amylase and reduces pancreatic injury in models of severe acute pancreatitis. 78 , 79 , 80 The coordinated role of innate immune cells, including macrophages, and intrapancreatic cytokines and chemokines in pancreatic injury is important for resolution of injury and restoration of organ function.
In this manuscript, we describe that ductal cells and possibly subsets of ADM can express CD73 in the context of caerulein‐mediated acute or chronic pancreatitis. Through analysis of published single nuclear or single‐cell RNA‐seq datasets and immunohistochemistry, we show ductal cells and immune cells within interstitial spaces that express CD73 and we demonstrate acinar‐to‐ductal metaplasia may be a CD73‐mediated reparative process in the pancreas. We show CD73 promotes adenosine generation, a critical nucleoside to resolve tissue injury in response to pancreatitis, and suggest the increased acinar‐to‐ductal metaplastic cells seen in CD73 −/− mice during acute pancreatitis may arise as an attempted tissue repair process. Under chronic conditions, we observed the expression of CD39 in vivo on macrophages which may indicate that the innate immune system is contributing to sustained injury, while the relatively high expression of CD73 in vivo on immune cells including T cells may indicate a divergent immune‐mediated mechanism is contributing to CD73‐mediated tissue injury resolution. Future studies delineating the role of specific adenosine receptors in stromal and immune cells in pancreatitis models will establish the mechanistic consequences of elevated intrapancreatic adenosine on development and resolution of pancreatitis.
Sustained enzymatic activity of CD39 in CD73 −/− mice implicates enhanced AMP and loss of extracellular adenosine which contribute to enhanced disease severity in pancreatitis. Extracellular AMP is necessary for the initial response to acute injury in the pancreas by promoting inflammation, as seen by similar levels of CD39 in both wild‐type and CD73 −/− mice at Day 1. However, sustained AMP levels promote a more severe phenotype. Thus, after initial insult, in wild‐type mice at Day 4, CD73 enzymatic activity is increased to promote adenosine generation and signaling through adenosine receptors, which promotes inflammation resolution. However, in CD73 −/− mice, sustained CD39 activity at Day 4 demonstrates reduced capacity to generate extracellular adenosine in the absence of CD73. This switch in modulation is also seen by HPLC in wild‐type mice by the upregulation of purinergic signaling at initial tissue injury and then significant downregulation of AMP levels at Day 4 while simultaneously promoting adenosine generation after sustained tissue injury to promote resolution.
While both genotypes showed similar tissue injury and histologic change at Day 1 in the acute pancreatitis model, by Day 4 the wild‐type mice showed a near complete resolution of inflammation, while the CD73 −/− mice demonstrated significant residual pancreatitis injury. The increased presence of acinar‐to‐ductal metaplastic cells at Day 4 in CD73 −/− mice compared to the wild type suggests that without adenosine generation, reparative processes are still necessary to mitigate pancreatic injury. Additionally, increased staining of Cytokeratin‐19 in pancreatitis areas compared to normal histologic areas in both wild type and CD73 −/− mice, but even greater still in CD73 −/− mice, demonstrates the response to injury is regulated by adenosine receptor activation. This suggests the enhanced CD73 activity partially by ductal cells or ADM and most prominently by extra‐epithelial cells in wild‐type mice is allowing for an immune‐mediated rapid resolution of pancreatitis. However, at Day 7 in CD73 −/− mice there is also a near complete resolution of tissue injury. In addition, we observe by HPLC adenosine in CD73 −/− mice at Day 4 indicating intracellular conversion of AMP to adenosine and subsequent transport of adenosine into the microenvironment by ENT1 transporters may have been mechanistically why CD73 −/− mice were able to resolve caerulein‐induced pancreatitis by Day 7. Further studies will be required to investigate cellularity of adenosine receptor expression to determine how adenosine specifically reduces pancreatic inflammation.
In these experiments, in addition to histological changes and differences in ADM abundance, we quantified a significant increase in MPO+ Ly6G+ and Granzyme B+ cells in CD73 −/− mice at Day 4, which demonstrates a potent pro‐inflammatory response in the absence of adenosine in response to caerulein‐induced injury. The binding of ATP to the P2X and P2Y families of receptors, expressed on neutrophils in humans and in vivo, 59 enhances neutrophil phagocytosis, chemotaxis, and oxidative burst. Under conditions of enhanced purinergic signaling, the resulting sustained activity and chemotactic ability of neutrophils promote sustained tissue injury and slower inflammation resolution. We experimentally show the significant impact of neutrophils in our genetic model using neutrophil deletion experiments which significantly reduced inflammation in CD73 −/− mice. In addition, in NECA‐treated mice, we predict the high adenosine concentration at Day 1 potentially promotes adenosine‐dependent activation of A2A, A2B, and A3 adenosine receptors over the high‐affinity A1 receptor to promote tissue regeneration and restrain MPO accumulation and metaplasia in acute pancreatitis. 44 , 55 This identifies adenosine receptor enhancers as potential therapeutic targets for patients with acute pancreatitis as a mechanism to rapidly eliminate persistent inflammation via activation of A2 and A3 receptors.
AUTHOR CONTRIBUTIONS
Conceptualization: Jennifer M. Bailey‐Lundberg, Baylee J. O'Brien, Erika Y. Faraoni, Kathleen E. DelGiorno; Data Curation: Baylee J. O'Brien, Lincoln N. Strickland, Erika Y. Faraoni, Tingting Mills, Samantha Mota, Victoria Mota, Xuebo Chen; Formal analysis: Baylee J. O'Brien, Erika Y. Faraoni, Kathleen E. DelGiorno, Jennifer M. Bailey‐Lundberg; Writing—Original Draft: Baylee J. O'Brien; Writing—Review and Editing: Erika Y. Faraoni, Jennifer M. Bailey‐Lundberg, Kathleen E. DelGiorno; Resources and Funding acquisition: Jennifer M. Bailey‐Lundberg, Holger K. Eltzschig, Kathleen E. DelGiorno.
FUNDING INFORMATION
Texas Medical Center Digestive Disease Center Pilot and Feasibility Award NIH‐NIDDK‐2P30 056338‐16 and R21CA249924‐01 (J.M.B‐L). National Institute of Health Grants R01HL154720, R01DK122796, R01HL133900 and Department of Defense Grant W81XWH2110032 to H.K.E. Vanderbilt Digestive Disease Research Center Pilot and Feasibility Grant (NIH‐NIDDK P30 058404), American Gastroenterological Association Research Scholar Award (AGA2021‐13‐02), and NIH‐NIGMS R35 GM142709 (K.E.D.).
DISCLOSURES
The authors have no conflicts of interest to disclose.
O’Brien BJ, Faraoni EY, Strickland LN, et al. CD73‐generated extracellular adenosine promotes resolution of neutrophil‐mediated tissue injury and restrains metaplasia in pancreatitis. The FASEB Journal. 2022;37:e22684. doi: 10.1096/fj.202201537R
Baylee J. O'Brien and Erika Y. Faraoni contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data, analytic methods, and study materials are stored on a University Lab Archives account and are available upon request.
REFERENCES
- 1. Yadav D, Lowenfels AB. Trends in the epidemiology of the first attack of acute pancreatitis: a systematic review. Pancreas. 2006;33(4):323‐330. doi: 10.1097/01.mpa.0000236733.31617.52 [DOI] [PubMed] [Google Scholar]
- 2. Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology. 2013;144(6):1180‐1193. doi: 10.1053/j.gastro.2012.12.043 [DOI] [PubMed] [Google Scholar]
- 3. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195‐203. doi: 10.1097/01.mcg.0000436438.60145.5a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sonnenday CJ. Disorders of the exocrine pancreas. In: Hammer GD, McPhee SJ, eds. Pathophysiology of Disease: An Introduction to Clinical Medicine. McGraw‐Hill Education; 2013:784. [Google Scholar]
- 5. Sattin A, Rall TW. The effect of adenosine and adenine nucleotides on the cyclic adenosine 3′, 5′‐phosphate content of guinea pig cerebral cortex slices. Mol Pharmacol. 1970;6(1):13‐23. [PubMed] [Google Scholar]
- 6. Drury AN, Szent‐Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68(3):213‐237. doi: 10.1113/jphysiol.1929.sp002608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6(1):57. doi: 10.1186/s40425-018-0360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King RJ, Shukla SK, He C, et al. CD73 induces GM‐CSF/MDSC‐mediated suppression of T cells to accelerate pancreatic cancer pathogenesis. Oncogene. 2022;41(7):971‐982. doi: 10.1038/s41388-021-02132-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao J, Soto LMS, Wang H, et al. Overexpression of CD73 in pancreatic ductal adenocarcinoma is associated with immunosuppressive tumor microenvironment and poor survival. Pancreatology. 2021;21:942‐949. doi: 10.1016/j.pan.2021.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh K, Faraoni E, Dai Y, et al. Pancreatic Cancer Ductal Cell of Origin Drives CD73‐Dependent Generation of Immunosuppressive Adenosine. CellPress Sneak Peek (SSRN); 2021. [Google Scholar]
- 11. Chen Q, Pu N, Yin H, et al. CD73 acts as a prognostic biomarker and promotes progression and immune escape in pancreatic cancer. J Cell Mol Med. 2020;24(15):8674‐8686. doi: 10.1111/jcmm.15500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shevchenko I, Mathes A, Groth C, et al. Enhanced expression of CD39 and CD73 on T cells in the regulation of anti‐tumor immune responses. Onco Targets Ther. 2020;9(1):1744946. doi: 10.1080/2162402X.2020.1744946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma XL, Shen MN, Hu B, et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1‐mediated membrane localization of P110β and predicts poor prognosis. J Hematol Oncol. 2019;12(1):37. doi: 10.1186/s13045-019-0724-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arab S, Hadjati J. Adenosine blockage in tumor microenvironment and improvement of cancer immunotherapy. Immune Netw. 2019;19(4):e23. doi: 10.4110/in.2019.19.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33(5):231‐237. doi: 10.1016/j.it.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 16. Antonioli L, Fornai M, Pellegrini C, et al. Adenosine signaling in the tumor microenvironment. Adv Exp Med Biol. 2021;1270:145‐167. doi: 10.1007/978-3-030-47189-7_9 [DOI] [PubMed] [Google Scholar]
- 17. Faraoni EY, Strickland LN, O'Brien BJ, et al. Radiofrequency ablation in combination with CD73 inhibitor AB680 reduces tumor growth and enhances anti‐tumor immunity in a syngeneic model of pancreatic ductal adenocarcinoma. Front Oncol. 2022;12:995027. doi: 10.3389/fonc.2022.995027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harvey JB, Phan LH, Villarreal OE, Bowser JL. CD73's potential as an immunotherapy target in gastrointestinal cancers. Front Immunol. 2020;11:508. doi: 10.3389/fimmu.2020.00508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo F, Le NB, Mills T, et al. Extracellular adenosine levels are associated with the progression and exacerbation of pulmonary fibrosis. FASEB J. 2016;30(2):874‐883. doi: 10.1096/fj.15-274845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karmouty‐Quintana H, Philip K, Acero LF, et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J. 2015;29(1):50‐60. doi: 10.1096/fj.14-260182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burnstock G, Vaughn B, Robson SC. Purinergic signalling in the liver in health and disease. Purinergic Signal. 2014;10(1):51‐70. doi: 10.1007/s11302-013-9398-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burnstock G, Novak I. Purinergic signalling in the pancreas in health and disease. J Endocrinol. 2012;213(2):123‐141. doi: 10.1530/JOE-11-0434 [DOI] [PubMed] [Google Scholar]
- 23. Imarisio C, Alchera E, Sutti S, et al. Adenosine a(2a) receptor stimulation prevents hepatocyte lipotoxicity and non‐alcoholic steatohepatitis (NASH) in rats. Clin Sci (Lond). 2012;123(5):323‐332. doi: 10.1042/CS20110504 [DOI] [PubMed] [Google Scholar]
- 24. Karmouty‐Quintana H, Zhong H, Acero L, et al. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 2012;26(6):2546‐2557. doi: 10.1096/fj.11-200907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou Y, Schneider DJ, Morschl E, et al. Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin‐induced lung injury. J Immunol. 2011;186(2):1097‐1106. doi: 10.4049/jimmunol.1002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng Z, Fernandez P, Wilder T, et al. Ecto‐5′‐nucleotidase (CD73) ‐mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 2008;22(7):2263‐2272. doi: 10.1096/fj.07-100685 [DOI] [PubMed] [Google Scholar]
- 27. Montesinos MC, Gadangi P, Longaker M, et al. Wound healing is accelerated by agonists of adenosine A2 (G alpha s‐linked) receptors. J Exp Med. 1997;186(9):1615‐1620. doi: 10.1084/jem.186.9.1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eckle T, Kewley EM, Brodsky KS, et al. Identification of hypoxia‐inducible factor HIF‐1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J Immunol. 2014;192(3):1249‐1256. doi: 10.4049/jimmunol.1100593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367(24):2322‐2333. doi: 10.1056/NEJMra1205750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dixit A, Cheema H, George J, et al. Extracellular release of ATP promotes systemic inflammation during acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2019;317(4):G463‐G475. doi: 10.1152/ajpgi.00395.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Leve S, Wirsdörfer F, Jendrossek V. Targeting the immunomodulatory CD73/adenosine system to improve the therapeutic gain of radiotherapy. Front Immunol. 2019;10:698. doi: 10.3389/fimmu.2019.00698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19(6):355‐367. doi: 10.1016/j.molmed.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno‐oncology. Nat Rev Clin Oncol. 2020;17(10):611‐629. doi: 10.1038/s41571-020-0382-2 [DOI] [PubMed] [Google Scholar]
- 34. Toldo S, Zhong H, Mezzaroma E, et al. GS‐6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. J Pharmacol Exp Ther. 2012;343(3):587‐595. doi: 10.1124/jpet.111.191288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan ES, Cronstein BN. Adenosine in fibrosis. Mod Rheumatol. 2010;20(2):114‐122. doi: 10.1007/s10165-009-0251-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bekisz JM, Lopez CD, Corciulo C, et al. The role of adenosine receptor activation in attenuating cartilaginous inflammation. Inflammation. 2018;41(4):1135‐1141. doi: 10.1007/s10753-018-0781-z [DOI] [PubMed] [Google Scholar]
- 37. Bono MR, Fernández D, Flores‐Santibáñez F, Rosemblatt M, Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. 2015;589(22):3454‐3460. doi: 10.1016/j.febslet.2015.07.027 [DOI] [PubMed] [Google Scholar]
- 38. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol. 2013;171(1):1‐7. doi: 10.1111/j.1365-2249.2012.04623.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kubersky SM, Hirschhorn R, Broekman MJ, Cronstein BN. Occupancy of adenosine receptors on human neutrophils inhibits respiratory burst stimulated by ingestion of complement‐coated particles and occupancy of chemoattractant but not Fc receptors. Inflammation. 1989;13(5):591‐599. doi: 10.1007/BF00916765 [DOI] [PubMed] [Google Scholar]
- 40. Ehrentraut H, Clambey ET, McNamee EN, et al. CD73+ regulatory T cells contribute to adenosine‐mediated resolution of acute lung injury. FASEB J. 2013;27(6):2207‐2219. doi: 10.1096/fj.12-225201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Granja T, Körner A, Glück C, et al. Targeting CD39 toward activated platelets reduces systemic inflammation and improves survival in sepsis: a preclinical pilot study. Crit Care Med. 2019;47(5):e420‐e427. doi: 10.1097/CCM.0000000000003682 [DOI] [PubMed] [Google Scholar]
- 42. Ramadan A, Naydenova Z, Stevanovic K, Rose JB, Coe IR. The adenosine transporter, ENT1, in cardiomyocytes is sensitive to inhibition by ethanol in a kinase‐dependent manner: implications for ethanol‐dependent cardioprotection and nucleoside analog drug cytotoxicity. Purinergic Signal. 2014;10(2):305‐312. doi: 10.1007/s11302-013-9391-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morote‐Garcia JC, Köhler D, Roth JM, et al. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am J Respir Cell Mol Biol. 2013;49(2):296‐305. doi: 10.1165/rcmb.2012-0457OCx [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32(4):856‐864. doi: 10.1161/ATVBAHA.111.226845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manjunath S, Sakhare PM. Adenosine and adenosine receptors: newer therapeutic perspective. Indian J Pharmacol. 2009;41(3):97‐105. doi: 10.4103/0253-7613.55202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63(1):1‐34. doi: 10.1124/pr.110.003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olah ME, Stiles GL. Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol. 1995;35:581‐606. doi: 10.1146/annurev.pa.35.040195.003053 [DOI] [PubMed] [Google Scholar]
- 48. Wang J, Miao Y. Mechanistic insights into specific G protein interactions with adenosine receptors. J Phys Chem B. 2019;123(30):6462‐6473. doi: 10.1021/acs.jpcb.9b04867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen‐activated protein kinases. Cell Signal. 2003;15(9):813‐827. doi: 10.1016/s0898-6568(03)00058-5 [DOI] [PubMed] [Google Scholar]
- 50. Ohta A, Sitkovsky M. Role of G‐protein‐coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916‐920. doi: 10.1038/414916a [DOI] [PubMed] [Google Scholar]
- 51. Faraoni EY, Ju C, Robson SC, Eltzschig HK, Bailey‐Lundberg JM. Purinergic and adenosinergic signaling in pancreatobiliary diseases. Front Physiol. 2022;13:849258. doi: 10.3389/fphys.2022.849258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117(1):123‐140. doi: 10.1016/j.pharmthera.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 53. Szabó C, Scott GS, Virág L, et al. Suppression of macrophage inflammatory protein (MIP)‐1alpha production and collagen‐induced arthritis by adenosine receptor agonists. Br J Pharmacol. 1998;125(2):379‐387. doi: 10.1038/sj.bjp.0702040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mabley J, Soriano F, Pacher P, et al. The adenosine A3 receptor agonist, N6‐(3‐iodobenzyl)‐adenosine‐5′‐N‐methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466(3):323‐329. doi: 10.1016/s0014-2999(03)01570-x [DOI] [PubMed] [Google Scholar]
- 55. Truong LD, Trostel J, McMahan R, Chen JF, Garcia GE. Macrophage A2A adenosine receptors are essential to protect from progressive kidney injury. Am J Pathol. 2016;186(10):2601‐2613. doi: 10.1016/j.ajpath.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11(3):201‐212. doi: 10.1038/nri2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen NM, Singh G, Koenig A, et al. NFATc1 links EGFR signaling to induction of Sox9 transcription and acinar‐ductal transdifferentiation in the pancreas. Gastroenterology. 2015;148(5):1024‐1034.e9. doi: 10.1053/j.gastro.2015.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Csóka B, Törő G, Vindeirinho J, et al. A2A adenosine receptors control pancreatic dysfunction in high‐fat‐diet‐induced obesity. FASEB J. 2017;31(11):4985‐4997. doi: 10.1096/fj.201700398R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang X, Chen D. Purinergic regulation of neutrophil function. Front Immunol. 2018;9:399. doi: 10.3389/fimmu.2018.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang L, Xie D, Wei D. Pancreatic acinar‐to‐ductal metaplasia and pancreatic cancer. Methods Mol Biol. 2019;1882:299‐308. doi: 10.1007/978-1-4939-8879-2_26 [DOI] [PubMed] [Google Scholar]
- 61. Del Poggetto E, Ho IL, Balestrieri C, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science. 2021;373(6561):eabj0486. doi: 10.1126/science.abj0486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Strobel O, Dor Y, Alsina J, et al. In vivo lineage tracing defines the role of acinar‐to‐ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133(6):1999‐2009. doi: 10.1053/j.gastro.2007.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Means AL, Logsdon CD. Acinar ductal metaplasia: yap fills a gap. Gastroenterology. 2016;151(3):393‐395. doi: 10.1053/j.gastro.2016.07.022 [DOI] [PubMed] [Google Scholar]
- 64. Liou GY, Döppler H, Necela B, et al. Macrophage‐secreted cytokines drive pancreatic acinar‐to‐ductal metaplasia through NF‐κB and MMPs. J Cell Biol. 2013;202(3):563‐577. doi: 10.1083/jcb.201301001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma Z, Lytle NK, Chen B, et al. Single‐cell transcriptomics reveals a conserved metaplasia program in pancreatic injury. Gastroenterology. 2022;162(2):604‐620.e20. doi: 10.1053/j.gastro.2021.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tosti L, Hang Y, Debnath O, et al. Single nucleus and in situ RNA sequencing reveals cell topographies in the human pancreas. Gastroenterology. 2020;160:1330‐1344.e11. doi: 10.1053/j.gastro.2020.11.010 [DOI] [PubMed] [Google Scholar]
- 67. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single‐cell data. Cell. 2019;177(7):1888‐1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kittel A, Garrido M, Varga G. Localization of NTPDase1/CD39 in normal and transformed human pancreas. J Histochem Cytochem. 2002;50(4):549‐556. doi: 10.1177/002215540205000412 [DOI] [PubMed] [Google Scholar]
- 69. Kittel A, Pelletier J, Bigonnesse F, et al. Localization of nucleoside triphosphate diphosphohydrolase‐1 (NTPDase1) and NTPDase2 in pancreas and salivary gland. J Histochem Cytochem. 2004;52(7):861‐871. doi: 10.1369/jhc.3A6167.2004 [DOI] [PubMed] [Google Scholar]
- 70. Zhao Z, Liu W. Pancreatic cancer: a review of risk factors, diagnosis, and treatment. Technol Cancer Res Treat. 2020;19:1533033820962117. doi: 10.1177/1533033820962117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garcia PE, Scales MK, Allen BL, Pasca di Magliano M. Pancreatic fibroblast heterogeneity: from development to cancer. Cells. 2020;9(11):2464. doi: 10.3390/cells9112464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hendley AM, Wang YJ, Polireddy K, et al. p120 catenin suppresses basal epithelial cell extrusion in invasive pancreatic neoplasia. Cancer Res. 2016;76(11):3351‐3363. doi: 10.1158/0008-5472.CAN-15-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lugea A, Waldron RT, Mareninova OA, et al. Human pancreatic acinar cells: proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am J Pathol. 2017;187(12):2726‐2743. doi: 10.1016/j.ajpath.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Das SL, Kennedy JI, Murphy R, Phillips AR, Windsor JA, Petrov MS. Relationship between the exocrine and endocrine pancreas after acute pancreatitis. World J Gastroenterol. 2014;20(45):17196‐17205. doi: 10.3748/wjg.v20.i45.17196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grapin‐Botton A. Ductal cells of the pancreas. Int J Biochem Cell Biol. 2005;37(3):504‐510. doi: 10.1016/j.biocel.2004.07.010 [DOI] [PubMed] [Google Scholar]
- 76. Ishiguro H, Yamamoto A, Nakakuki M, et al. Physiology and pathophysiology of bicarbonate secretion by pancreatic duct epithelium. Nagoya J Med Sci. 2012;74(1–2):1‐18. [PMC free article] [PubMed] [Google Scholar]
- 77. Wan J, Ren Y, Yang X, Li X, Xia L, Lu N. The role of neutrophils and neutrophil extracellular traps in acute pancreatitis. Front Cell Dev Biol. 2020;8:565758. doi: 10.3389/fcell.2020.565758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Folias AE, Penaranda C, Su AL, Bluestone JA, Hebrok M. Aberrant innate immune activation following tissue injury impairs pancreatic regeneration. PLoS ONE. 2014;9(7):e102125. doi: 10.1371/journal.pone.0102125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Inoue S, Nakao A, Kishimoto W, et al. Anti‐neutrophil antibody attenuates the severity of acute lung injury in rats with experimental acute pancreatitis. Arch Surg. 1995;130(1):93‐98. doi: 10.1001/archsurg.1995.01430010095020 [DOI] [PubMed] [Google Scholar]
- 80. John DS, Aschenbach J, Krüger B, et al. Deficiency of cathepsin C ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of E‐cadherin. J Biol Chem. 2019;294(2):697‐707. doi: 10.1074/jbc.RA118.004376 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data, analytic methods, and study materials are stored on a University Lab Archives account and are available upon request.