

Abstract
Synthetic biology approaches living systems with an engineering perspective and promises to deliver solutions to global challenges in healthcare and sustainability. A critical component is the design of biomolecular circuits with programmable input–output behaviors. Such circuits typically rely on a sensor module that recognizes molecular inputs, which is coupled to a functional output via protein-level circuits or regulating the expression of a target gene. While gene expression outputs can be customized relatively easily by exchanging the target genes, sensing new inputs is a major limitation. There is a limited repertoire of sensors found in nature, and there are often difficulties with interfacing them with engineered circuits. Computational protein design could be a key enabling technology to address these challenges, as it allows for the engineering of modular and tunable sensors that can be tailored to the circuit’s application. In this article, we review recent computational approaches to design protein-based sensors for small-molecule inputs with particular focus on those based on the widely used Rosetta software suite. Furthermore, we review mechanisms that have been harnessed to couple ligand inputs to functional outputs. Based on recent literature, we illustrate how the combination of protein design and synthetic biology enables new sensors for diverse applications ranging from biomedicine to metabolic engineering. We conclude with a perspective on how strategies to address frontiers in protein design and cellular circuit design may enable the next generation of sense-response networks, which may increasingly be assembled from de novo components to display diverse and engineerable input-output behaviors.
Keywords: Biological sensor-actuators, cellular therapies, computational protein design, de novo design, molecular sensors, protein engineering, protein–small-molecule interactions, Rosetta, synthetic biological signaling systems, synthetic biology
This article reviews the field of computational protein design, focusing on the advances in the engineering of synthetic small-molecule-binding protein sensors as well as sensor–actuator proteins.
I. INTRODUCTION
The approaches and foundational principles used in the engineering of electrical circuits, such as Boolean logic, modularity, standardization, and design-build-test cycles, have not only enabled some of the most important technological developments of the last century but also inspired life scientists and bioengineers to approach their aims in a similar way. With clear analogy to electrical engineering, early landmark studies in the field of “synthetic biology” have shown that it is possible to implement a toggle switch [1] or emergent behaviors, such as oscillations [2], using known biological components in new genetic circuits in simple bacterial model cells. Bottom-up approaches aim to engineer life-like systems and their critical subprocesses, such as cell division to reveal minimal requirements and principles through a “learning-by-creating” approach [3]. Conversely, top-down approaches have reengineered complex living cells for applications in sustainability and biomedicine. For example, the metabolism of microorganisms has been rewired to produce high-value chemicals, including drug precursors [4] or biomaterials [5]. In the biomedical context, a prominent approach that has been translated into the clinic used engineered immune cells equipped with a chimeric antigen receptor (CAR) to sense tumor markers and activate cell-killing responses [6].
Biomolecular circuits that produce a defined output in response to a customized input are central to many synthetic biology applications [7]. Typically, the output is the expression of a gene, which is regulated by an upstream binding event in a sensor molecule (see Fig. 1). While synthetic biology aims at engineering such circuits in a modular and predictable fashion, inspiration is provided by nature. A classic example of a small-molecule-controlled gene regulatory circuit is the lac operon [8]. Here, binding of inducers, such as the small-molecule metabolite allolactose or the chemical analog isopropyl β-d-1-thiogalactopyranoside (IPTG) to the regulator LacI, activates the expression of downstream metabolic genes [9]. Life scientists have long taken advantage of such systems to express genes and the encoded proteins in a controlled fashion. Moreover, many synthetic gene circuits in bacteria, including the pioneering “Repressilator,” have used well-understood transcriptional regulators, such as the lac repressor, or similarly well-characterized repressors [2].
Fig. 1.

Common biological engineering framework for modular input–output circuits. Diverse small-molecule inputs (red) can be sensed by dedicated protein-based binders (light blue). This signal is then interpreted by a cellular circuit, which often regulates the expression of an output-specific target gene.
While gene expression was the basis of the first generation of synthetic biological circuits, it is important to note that other modes of input–output circuits have been developed. In particular, cellular networks have recently been designed that act solely on the protein level [10], [11], which has been recently reviewed [12]. Here, protein–protein and protein–small-molecule interactions are used as triggers in protein-level switches or logic gates, which controls signaling networks mediating substantial changes in cell states. While networks with a gene-based output may be easier to engineer and diversify via the exchange of DNA parts, a major advantage of such protein-level circuits is that they can respond to inputs on substantially shorter timescales.
Moreover, input sensing can occur both inside the cell or on the cell surface [12]. This area has seen several exciting developments in the last decade, and different chimeric and modular protein architectures have been engineered. When extracellular inputs are recognized by engineered cell surface receptors, the signal has to be relayed to the intracellular environment. Different molecular mechanisms for signal transduction have been employed, including ligand-induced dimerization and/or posttranslational modifications, which are ultimately interfaced with intracellular pathways controlling gene expression [13]–[15].
In this review article, we focus on small-molecule-responsive systems, as they have been used as inputs in early synthetic biology milestones [1], [2], and continue to represent a critical modality to regulate synthetic biological circuits. As a “small molecule,” we define an organic, nonpolymeric compound with a molecular weight of typically below 1 kDa. Nevertheless, we note that a variety of other inputs have been used in synthetic sensor–actuator circuits, including proteins and nucleic acids, as well as physical triggers, such as light, mechanical force, or temperature [16], [17].
While outputs of biological circuits are often easy to engineer (most commonly, via expression of a user-defined gene), input sensing is much more difficult to engineer. Here, there are two main challenges: first, input sensing requires physical interaction with the sensor that is specific to each input small molecule; second, sensing needs to be coupled to a functional output that is ideally modular, such as gene expression. For many small molecule targets, there is no known binding protein that could be repurposed as a sensor. Moreover, even when a natural binding domain could be exploited for sensing, it may not be as modular as desired, i.e., proteins that evolved to function in specific natural environments may perform erratically or not at all when used in different contexts. In addition, few binders naturally couple interaction with the input to a modular output, such as gene regulation. Therefore, input modularity in synthetic biological circuits has been limited by the availability of binding modules and generalizable approaches to couple binding to functional outputs in a modular fashion. However, input modularity is critical for customizing synthetic circuits for a given application. For instance, bespoke sensor-actuators could be used to activate specific output responses based on the presence of diverse molecules, such as disease biomarkers or drugs approved by the U.S. Food and Drug Administration (FDA) for biomedical applications or pollutants or toxins for environmental applications (see Fig. 1).
Computational protein design represents an engineering approach to generate proteins with modified or completely new structural and functional properties. As such, it appears ideally positioned to address the limitation of insufficient input modularity in synthetic biological circuits. In the last decades, methods for computational protein design have seen dramatic improvements [18]. Starting with the first automated computational design of a new amino acid sequence for an existing protein [19], an early focus of the field was the design of proteins with folds that are not found in nature [20]. Structure-oriented approaches, with the Rosetta software suite representing a widely applied design framework [18], [21], have since advanced to the design of supramolecular protein architectures, such as defined 2-D lattices [22] and 3-D assemblies, such as icosahedra [23] from first principles. Moreover, computational protein design has substantially advanced toward building proteins with new functions, including small-molecule binding [24], protein binding [25], and fold switching [26] (see Fig. 2). Such functionalities have been harnessed for applications, including the design of potential therapeutic inhibitors [25], immune modulators [27], and vaccines [28] or the development of protein-based switches [29], sensors [30], and sensor-actuators [31] (see Fig. 2). With a particular focus on approaches using Rosetta, we review the major developments in engineering artificial small-molecule-binding sensors and sensor-actuators while highlighting their relevance for synthetic biology. We emphasize the increasing role of computational protein design in advancing biological engineering and end with a perspective on future avenues.
Fig. 2.

Schematic depicting key advances in computational protein design. These advances enable important applications in their own right and form the basis for engineering small-molecule-controlled protein circuits, as indicated on the bottom. While the design of protein–ligand interactions allows for small-molecule sensing, the design of protein–protein interactions and conformational changes enables coupling of sensing to a functional output. The schematic for conformational switching was inspired by latching orthogonal cage-key proteins (LOCKR) [29]. The protein structural representation corresponds to a caffeine-binding protein (PDB entry 3rfm).
II. COMPUTATIONAL DESIGN OF NOVEL LIGAND-BINDING PROTEINS
The main area where computational protein design opens new avenues is in de novo design, i.e., the design of a protein with a new structure or function from design principles without starting from a natural protein with a similar function. An alternative approach to computational design is experimental mutagenesis and selection—sometimes referred to as “directed evolution”—which is typically performed in multiple iterations and can be done in high throughput. Experimental techniques have been extremely successful in improving the performance of existing sensors or reengineering proteins to bind to similar molecules [32], [33]. However, this approach is typically more difficult or not feasible for ligands for which no binder for a closely related molecule is known. Here, computational design can be used to generate a de novo binder for arbitrary user-defined ligands. However, achieving high affinity and selectivity has remained challenging for computational design [34]. Thus, a common theme that we will illustrate below when describing specific studies is that computational design is often (although not always) followed by high-throughput experimental optimization to generate novel binders with high affinity.
Several methodologies exist to design proteins de novo,1 from the rational, parametric design of specific folds, such as helical bundles or coiled-coil proteins [35], [36], to the assembly of new structures from helical, beta-strand, and loop elements [37], [38]. Given a design target objective, computational protein design is typically formulated as an optimization problem. In an inverse approach to the structure prediction problem, which seeks to predict structures given a sequence, protein design in Rosetta searches for sequences that adopt a given structure and function. In this process, candidate sequence solutions are evaluated with a scoring function that estimates the energetics of atomic-level interactions within proteins and between proteins and their interaction partners [39]. To allow for rapid evaluation of large numbers of sequence-structure combinations (as the search space is typically enormous), the applied scoring or energy functions make considerable approximations. As a consequence, top-ranking solutions are often not functional. To alleviate this problem, design approaches often apply a variety of heuristic metrics and filters to final sequence outputs to identify the most promising variants for experimental testing [see Fig. 3(a)].
Fig. 3.

Computational (re)design of small-molecule binding proteins. (a) Design-build-test cycle for computational binder design. (b) Key steps in different approaches to design small-molecule binding proteins starting from either an existing binder or scaffold (left), a defined binding motif (middle), or the ligand (right). Structural representations correspond to the caffeine ligand and a caffeine-binding protein (PDB entry 3rfm). (c) Future directions to increase design success and efficiency. Colored circles in (a) and (b) highlight steps in the design process that would benefit from the respective direction.
In Sections II-A–II-D, we summarize recent developments in designing physical interactions between proteins and small molecules. Of central relevance to all approaches is the design of a binding “motif,” that is, a set of amino acid residues that make key interactions with the ligand. Moreover, a protein backbone that accommodates a motif can be regarded as a “scaffold” for the functional site. Starting from the redesign of proteins with preexisting binding motifs (A), we will discuss increasingly complex engineering approaches toward novel protein binders. Such approaches range from the incorporation of binding motifs into existing scaffolds with preexisting cavities (B) or creating new binding sites in existing scaffolds (C) to the de novo design of both binding motifs and scaffolds (D). A summary of the computational approaches to build binding motifs into scaffolds is given in Table 1.
Table 1.
Major Computational Approaches to Build Binding Motifs Into Protein Scaffolds
| Method | Purpose | Core functionality | References |
|---|---|---|---|
| Rosetta Match | Incorporation of defined motifs into suitable scaffolds | Checks suitability of scaffold backbones to satisfy geometric constraints of motif side chains relative to a ligand | [44] |
| Motif Graft | Incorporation of defined motifs into suitable scaffolds | Checks suitability of scaffold backbones to accommodate motif side chains or insertion of extended backbone segments | [86] |
| Phoenix Match | Favorable ligand placement within a scaffold | Explores a ligand’s orientations and conformations (simultaneously to protein sequence optimization) | [40, 58] |
| Rotamer Interaction Field (RIF) docking | Motif generation & incorporation of motifs into suitable scaffolds | Generates “inverse” rotamers for binding a ligand and identifies suitable scaffolds by docking them into the field of generated rotamers | [50] |
| Convergent Motifs for Binding Sites (COMBS) | Motif generation & incorporation of motifs into suitable scaffolds | Uses PDB-derived information to identify suitable backbone coordinates relative to ligand atoms (“van der Mers”) | [51] |
| Binding Sites From Fragments (BSFF) | Motif generation & assistance with context design | Uses PDB-derived information to identify contacts between proteins and ligand sub-structures to assemble new binding motifs and complement the subsequent design process | [43] |
A. Redesign of an Existing Binding Motif
If there is a known protein that binds a chemically similar compound to the target small molecule, its binding pocket can be redesigned computationally for altered specificity [see Fig. 3(b) (left)], provided that an atomic-level structure exists for the protein or a good model can be generated. This “redesign” approach has successfully been applied to transcription factors [40], [41], which will be discussed in more detail in Section III. Moreover, two different redesign approaches were applied to a lysine/arginine/ornithine binding protein (LAOBP), which enabled it to bind L-glutamine after identifying a single mutation that was shared in the predictions of both approaches [42]. While redesign can substantially simplify the design process overall, redesign relies on a protein already binding a ligand closely related to the target as a starting point.
B. Incorporation of Binding Motifs Into Scaffolds With Existing Binding Pockets
If there is no known protein binding a similar target ligand, a binding site for the target can be built computationally into a set of protein scaffolds [see Fig. 3(b) (middle)]. First, a binding motif is defined that consists of key amino acids for binding, and their orientation relative to the ligand is defined. If a high-resolution protein-target-ligand complex structure is available in the protein data bank (PDB), the binding motif may be (partially) extracted from this structure [31]. Alternatively, binding motifs can be defined manually by chemical intuition [24] and/or assembled de novo in an automated fashion that is described in Section II-D [43]. In a second step, the defined binding motif is built (“matched”) into a protein scaffold. An obvious choice for scaffolds is known proteins with preexisting binding pockets. To maximize the success rate of matching, one usually tests a library of scaffolds. While several techniques can be used for matching, an important computational tool is the Rosetta Match application [44] (see Table 1) that iterates through specified positions in protein scaffolds for satisfying distance and angular constraints of defined motif residue atoms with respect to defined ligand atoms.
When binding motifs are incorporated into scaffolds, they often show a suboptimal fit in the backbone. Moreover, the scaffold may be destabilized upon the incorporation of the motif residues. To address these issues and stabilize the protein-motif match, the sequence of the spatially surrounding (“second shell”) amino acids is redesigned. After diversifying sequences for the redesigned match, promising candidates are determined based on different metrics. While the precise determinants for experimental success have not been systematically discerned, various properties have been successfully employed in design selection. In addition to maintaining protein stability, design studies have used criteria, such as the estimated protein–ligand interaction energy and shape complementarity, preorganization of the binding site, and compatibility between the obtained sequence and local backbone structure [24], [45]. Designs filtered in this way are then characterized and often further optimized experimentally. Experimental insights, particularly from structural studies, can then be used to inform further rounds of design.
A landmark example for a binder designed by matching a binding motif to scaffolds, followed by sequence redesign and experimental optimization, focused on the steroidal compound digoxigenin (DIG) [24]. The binder featured an idealized binding site for DIG composed of hydrogen bonding interactions between amino acid side chains and the ligand’s polar groups in addition to atomic packing interactions. The computational designs initially bound DIG with only a modest affinity of around 12 μM,2 but the affinity could be experimentally optimized via mutagenesis and selection of beneficial mutants via yeast surface display3 [46] to below 6 nM [24]. The final optimized DIG-binding design was subsequently used as a starting point for engineering various functional responses via ligand-dependent protein stabilization [47], as will be discussed in Section III.
In a different study, a binder for the synthetic opioid fentanyl was designed [48]. As this small molecule is hydrophobic and overall more “featureless” than one defined by polar functional groups, the authors sought to design a binding site based on shape complementarity with the ligand [48]. For this, the ligand was docked to a set of scaffolds with predetermined cavities, followed by sequence design with Rosetta. Site-saturation mutagenesis and selection via yeast display were then applied to improve the affinity from 6.9 μM to 64 nM in the case of the tightest binder.
A similar approach was used to design binders for 17α-hydroxyprogesterone (17-OHP), which is a biomarker for congenital adrenal hyperplasia [49]. Here, protein–ligand shape complementarity was intended via docking the small molecule to a set of “nuclear transport factor 2” (NTF2)-like scaffolds. In a subsequent step, sequence design with Rosetta was applied to obtain specific binding interactions, including hydrogen bonds. Characterization of initial computationally designed binders showed micromolar affinities, which were subsequently improved via mutagenesis and selection to 5.1 nM for the best binder [49]. In contrast to the designed DIG [24] and fentanyl [19] binders, which showed close structural agreement between the design model and experimentally determined high-resolution structures, there were substantial disagreements for the 17-OHP design. Strikingly, the ligand showed a “flipped” orientation in the designed binding site [49]. This disagreement was attributed to subtle backbone changes and an underestimated desolvation penalty for the ligand’s polar groups. These findings highlight remaining challenges in small-molecule-binder design and potential room for improvement in both design energy functions and approaches to optimize (sample) conformations in designed binding sites [49].
C. Incorporation of Binding Motifs Into Scaffolds Without Preexisting Binding Pockets
While the availability of preexisting binding cavities within scaffolds can simplify the design process, pockets for ligand binding can also be introduced into proteins or protein complexes de novo, i.e., built into proteins not harboring cavities by default. This approach, which expands the functional design space, was demonstrated for a binding site that was built de novo into a protein–protein interface [31], allowing coupling to function via conditional assembly of fused actuator domains, as will be discussed in more detail in the following (see Section III-C and Fig. 4). In the design workflow, a small library of binding motifs was built into more than 1000 protein–protein heterodimer interfaces using the Rosetta Match application [44]. The modeled binding sites of initial approximate matches were then optimized using flexible backbone design approaches that generated ensembles of thousands of modeled binding site backbones for design.
Fig. 4.
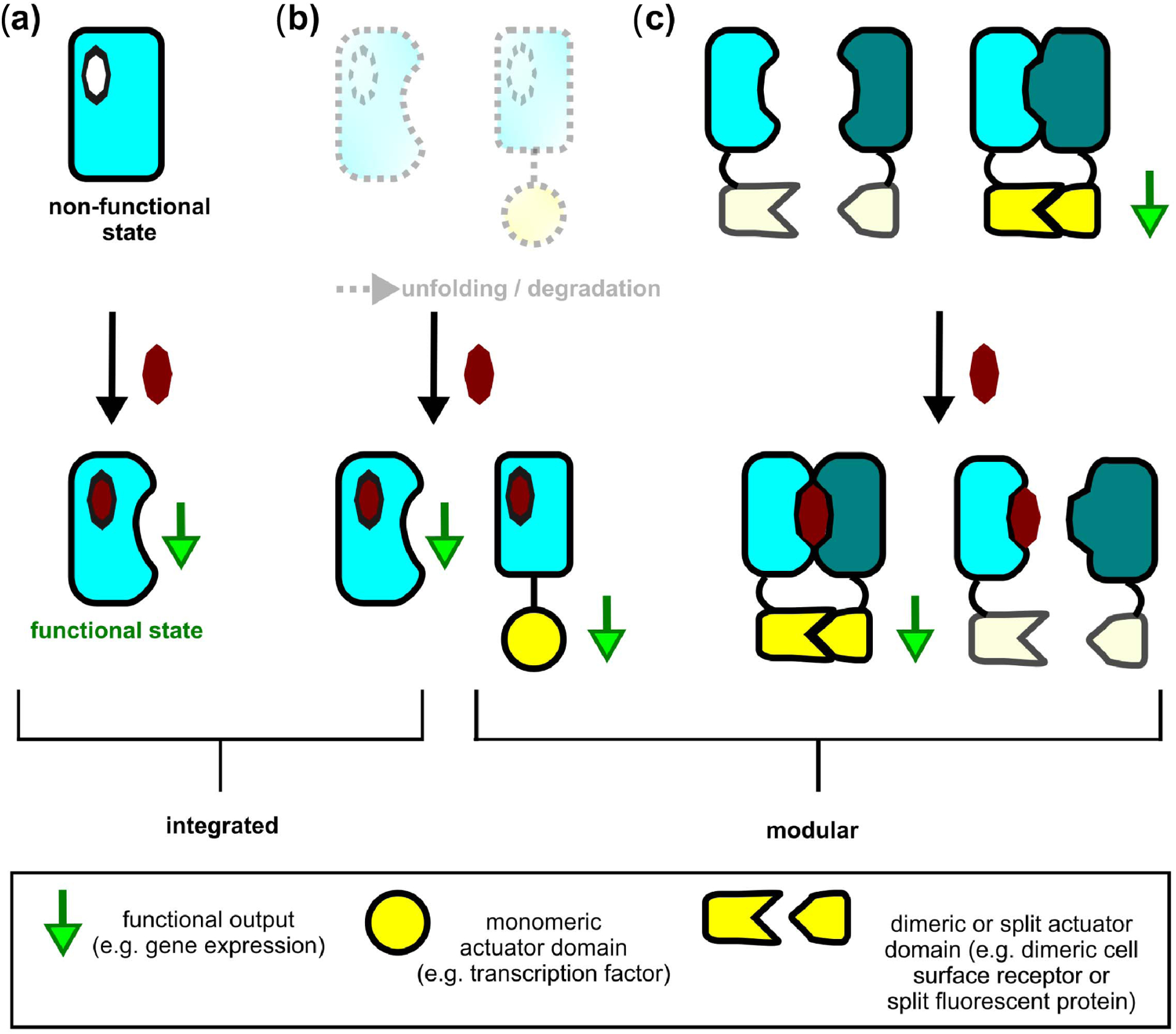
Schematic of mechanisms to couple small-molecule binding to protein function (specific examples for each mechanism are detailed in Table 2). (a) Allostery couples ligand binding to function through a conformational perturbation that changes a protein’s local or global structure. (b) Conditional stabilization via ligand binding rescues a protein’s structure from unfolding and/or degradation, which can be exploited in integrated (left) or modular (fusion-)proteins (right). (c) Conditional assembly controls function by reconstituting (CID; left) or disrupting (CDHs; right) split actuator domains that are fused to a protein pair, which associates (left) or dissociates (right) in response to ligand binding.
Nine computational designs based on three different scaffold types were selected for experimental characterization in a bacterial growth rescue assay. Here, the two parts of the heterodimer harboring the binding motif were fused to two complementary fragments of a split enzyme, whose functional dimerization in response to small-molecule binding increases cell growth under otherwise growth-inhibited conditions. Two variants of one of the scaffold types showed activity in this assay. Remarkably, experimentally screening libraries generated via computational design or mutagenesis did not lead to an increase in activity over the top-ranked individual computational designs, which highlights the power of the computational design method [31]. Subsequent single-site mutagenesis suggested two sites for beneficial mutations that stabilized the ternary complex, which was then crystallized for structure determination. The structure showed overall excellent agreement with the design model but suggested one additional mutation in the binding site, which resulted in an effective sensitivity to the target ligand of 180 nM without further optimization.
D. Approaches That Generate Binding Motifs De Novo and Place Them Into De Novo Designed Scaffolds
Besides the use of naturally occurring scaffolds, recent studies have successfully designed small-molecule-binders using not only de novo binding sites but also scaffolds designed entirely de novo [50]–[52]. One key advantage of de novo scaffolds is that they are often exceptionally stable [25] and, hence, tolerate the incorporation of functional sites that typically are destabilizing to proteins. Moreover, de novo proteins can have the advantage of increased scaffold diversity and improved room for customization compared to natural scaffolds. One potential concern may be the immunogenicity of de novo scaffolds, but this issue has not been found to be a problem at least in studies available to date [25], possibly due to the high stability of de novo designed proteins.
A remarkable demonstration of this de novo approach was the computational design of a β-barrel protein that binds and activates fluorescence of the small molecule DFHBI by restricting it to a planar conformation upon binding [50]. First, the authors designed an ensemble of β-barrel scaffolds, a protein topology that had not been successfully de novo designed before. β-barrel scaffolds feature an internal cavity as a potential binding site. Second, a binding site for the target ligand was defined with a computational method that builds a large set of amino acid side-chain conformations (“rotamers”) making polar and nonpolar interactions with the ligand. Third, the pregenerated β-barrel scaffolds were docked into the “rotamer interaction field” (RIF) of amino acid side chains coordinating the ligand [50] (see Table 1). Finally, Rosetta sequence design was performed on the docked scaffolds to stabilize the binding site in the protein [see Fig. 3(b) (right)]. The most promising of the initial computational designs bound DFHBI with an affinity of 12.8 μM and activated fluorescence 12-fold. Additional computational design and experimental optimization via mutagenesis and selection resulted in variants with 80-fold and 60-fold fluorescence activations and affinities of 0.56 and 0.18 μM, respectively. Fluorescence activation in bacteria, yeast, and mammalian cells validated their functionality for cell-based applications [50].
A common topology that has been used for de novo scaffold design is α-helical bundles, which can be easily parameterized mathematically to generate a variety of bundle backbone structures [53]. In one example, a hydrophobic, nonnatural porphyrin molecule was manually docked into a parameterized helical bundle, and Rosetta design was applied to design both the binding site and stabilize the distant protein core [52]. This process resulted in a highly stable protein, which bound its target ligand with an affinity of 45 nM and whose experimentally determined structure agreed well with the design model [52].
More recently, helical bundles were used as scaffolds to bind a polar molecule, the small-molecule anticoagulant drug apixaban [51]. Here, the ligand-binding site and protein backbone were designed in an intertwined manner using a “Convergent Motifs for Binding Sites” (COMBS) approach (see Table 1). COMBS uses a new structural unit, termed “van der Mer.” Van der Mers link ligand chemical groups to protein backbone coordinates that can position protein atoms in orientations optimal for interactions with the ligand. After searching the PDB for frequently occurring instances of van der Mers, binding sites are optimized to favor statistically preferred contacts between chemical groups on the ligand and the proteins. In the first step, chemical groups relevant for binding the ligand are defined. In the second step, a search algorithm is applied to identify helical bundle proteins from a precomputed ensemble, for which van der Mers display favorable protein–ligand interaction. In particular, hits are determined by exhibiting a threshold number of van der Mers that cooperatively interact with the ligand in an identical position and orientation relative to the backbone. Finally, the binding site and protein core are designed for stabilization. Without further experimental optimization, this approach resulted in apixaban binding with affinities in the high nanomolar and low micromolar range [51].
Another computational workflow that leverages contacts between a ligand’s chemical groups and protein residues was recently developed [43]. In this method termed “Binding Sites From Fragments” (BSFF) (see Table 1), chemical substructures of the ligand are manually selected and used to identify highly represented contacts between these chemical fragments and protein side chains from the PDB to assemble a “contact pool”. In a subsequent step, energetically favorable composite binding sites are assembled from the members of the contact pool using the Rosetta score function. In contrast to the classical approach of matching a handful of binding sites that were defined by chemical intuition or extracted from an existing protein–ligand complex, this method can generate hundreds of thousands of composite binding sites in an automated manner. This approach facilitates the design of binders for target ligands for which there is no known binding site. Moreover, the higher number of potential binding sites available for matching to scaffolds was shown to markedly increase the success rate of matching [43]. Finally, the contact pool of highly represented protein side chains for contacting ligand fragments can be harnessed for supplementing the repertoire of rotamers used in subsequent Rosetta design steps [43]. It will be interesting to experimentally test these potential improvements in the design of novel small-molecule-binding proteins.
III. APPROACHES TO COUPLE LIGAND BINDING TO FUNCTIONAL OUTPUTS
The second overarching problem in designing sensor–actuator systems, after designing input binding proteins, is linking sensing of small-molecule inputs to (synthetic) biological outputs. Strategies to do so can be classified into: 1) integrated and 2) modular approaches [54] (see Fig. 4). In integrated systems, the functional output is typically contained within the scaffold harboring the ligand-binding site. As we outline in the following, these systems are prevalent in nature but more difficult to engineer for two main reasons. First, the mechanisms for coupling inputs to outputs in integrated systems are generally incompletely understood. Second, integrated systems are intrinsically less modular. In contrast, in modular systems, the functional output is mediated by a fused output (actuator) domain that can be coupled to different input-sensing ligand-binding domains (see Fig. 4). In the following, we will discuss specific types of both integrated and modular sensor-actuators, which can be interfaced with synthetic biological circuits (see Fig. 1 and Table 2).
Table 2.
Recent Examples of Engineered Protein-Based Sensor-Actuators
| Input | Output | Engineering Starting Point | Engineering Method | Coupling Mechanism | References |
|---|---|---|---|---|---|
| fucose, lactitol, sucralose, gentiobiose | Gene regulation | LacI | Computational design, single-site saturation mutagenesis | Allostery | [41] |
| Vanillin | Gene regulation | QacR | Computational design | Allostery | [40] |
| Indole | Enzymatic activity, gene regulation, fluorescence | β-glycosidase, β-glucuronidase, cI, +5 GFP, | Rational / Computational design | Conditional stabilization | [64,65] |
| Digoxin, progesterone, fentanyl | Fluorescence, gene regulation, enzymatic activity | Computationally designed ligand-binding domains | Computational design / experimental optimization | Conditional stabilization | [24, 47, 48] |
| ABT-737 | Gene regulation, CAR regulation | Bcl-xL/ABT-737 complex | Experimental selection | Conditional assembly | [81] |
| A1120 | CAR regulation | hRBP4-Al120 complex | Experimental selection | Conditional assembly | [82] |
| Danoprevir, grazoprevir, asunaprevir | Gene regulation, cell signaling | NS3a-drug complexes | Computational design / experimental optimization | Conditional assembly | [84] |
| A1155463 | CAR regulation | Bcl-xL | Computational design | Conditional assembly | [85] |
| Farnesyl pyrophosphate | Enzymatic activity, fluorescence, luminescence | Famesyl pyrophosphate binding motif & library of >1000 protein-protein scaffolds | Computational design / structure-guided design | Conditional assembly | [31] |
A. Allostery
Allostery is commonly defined as the modulation of protein function via a conformational change that is triggered by interaction at a site distant from the functional region [55], [56] [see Fig. 4(a)]. An allosteric conformational change can also be accompanied by quaternary structural transitions, e.g., by controlling the association state of the allosteric protein with another macromolecule in a ligand-dependent manner. Allostery is a prevalent strategy in nature and allosteric coupling controls diverse processes, such as gene expression and metabolic flux. Although the concept of allostery was introduced more than 50 years ago [55], [56], the physicochemical principles and mechanisms of this phenomenon are incompletely understood and remain an object of active research [57].
This limited understanding makes the predictive reverse and forward engineering of allosteric sensor-actuators one of the most challenging endeavors in protein design. Nevertheless, in some cases, allosteric ligand-controlled switches have been engineered successfully by the extensive experimental screening of domain-insertion libraries [32] or computational repurposing of existing allosteric proteins [40], [41]. One advantage of the allosteric coupling strategy is that the function is contained within the ligand-binding protein [see Fig. 4(a)], which alleviates the need for identifying and optimizing the type, orientation, or detailed linkage of fused actuator domains. On the other hand, mutating natural proteins can easily break the allosteric mechanism.
Allosteric transcription factor proteins represent a suitable starting point for engineering, as they couple ligand binding to gene expression. Typically, ligand binding allosterically controls the association state of the transcription factor protein with a DNA operator sequence upstream of the target gene, which, in turn, regulates the initiation of transcription. For example, the inducer specificity of the lac repressor was reengineered to respond to four different saccharide molecules [41]. For three of the four new inducers, Rosetta design was applied, and the best variants showed responses with a similar fold induction as for the routinely used inducer IPTG. In contrast, sequence variation introduced randomly (using error-prone PCR) was found to be less effective, producing only responses for two of the three molecules and with lower maximum induction. For a fourth inducer, single amino-acid-saturation mutagenesis was successfully applied, which revealed that mutations distant to the binding site were as beneficial as those closer to the ligand. As most engineered variants showed promiscuous induction, rather than ligand-specific responses, further experimental optimization based on shuffling and combinations of mutations was performed, which increased both specificity and induction [41].
In another example, the TetR-family repressor QacR was computationally designed to bind the unrelated molecule vanillin, which is a growth inhibitor that can negatively impact bioindustrial processes [40]. Here, design was guided by the structures of QacR in its ligand-bound conformation and its DNA-bound (and ligand-free) conformation. In particular, targeted ligand placement using Phoenix Match [58] (see Table 1) was performed to identify locations of the new ligand within the binding site that would sterically clash with the protein in its DNA-bound conformation while optimizing favorable interactions for ligand binding at nearby positions for the non-DNA-bound state. After rational mutagenesis to address an initial failure of repression, two promising designs were identified experimentally. However, these variants still responded to the original inducer of the wild-type repressor, dequalinium, and exhibited a higher background signal and lower fold induction than the wild type [40]. Improved specificity and induction may be achieved with single-site saturation mutagenesis starting from these computationally designed variants, as performed for the lac repressor [41]. Considering eventual experimental optimization, the authors suggest that the sensors provide a starting point for interesting applications in synthetic circuits (see Fig. 1), such as coupling of vanillin sensing to the expression of an efflux pump or an enzyme to export or degrade vanillin.
While the examples discussed here are focused on computationally redesigning the ligand specificity of existing allosteric proteins, a complementary approach to designed allostery is based on the insertion of ligand-sensitive domains into proteins of interest. For this strategy, it is important to first identify appropriate insertion sites in the target protein, which allows coupling from the ligand-binding domain to the protein’s functional site. Toward this end, different computational methods have been developed, which have recently been reviewed in more detail [59]. Notable approaches in this regard include, but are not limited to, the analysis of statistical interactions between residues based on evolutionary data [60] or a recent network-based method relying only on the target protein’s structure [61]. Several examples of the design of insertion-based allosteric proteins will be discussed in Section III-B because they utilize the principle of ligand-dependent stabilization.
B. Conditional Stabilization
A set of approaches to regulate protein activity is based on the ligand-dependent modulation of protein global stability or local structure [62]–[64].
An integrated approach that is closely related to the allosteric coupling of binding to function was developed by Deckert et al. [64] [see Fig. 4(b)]. In this rational strategy, termed “chemical rescue of structure,” a buried, hydrophobic residue (tryptophan) close to, but distinct from, the active site is mutated to a glycine residue. This mutation creates a cavity and locally perturbs the protein’s active site geometry, as illustrated by the structure of an accordingly mutated β-glycosidase enzyme [64]. Remarkably, the binding of the small-molecule indole to the cavity resulted in reconstitution of the original active site geometry and the rescue of enzymatic activity [64]. While this study heavily relied on detailed structural knowledge and success was attributed to a fairly distinct ligand-dependent conformational change, a follow-up study [65] provided important insights on the general applicability and various mechanisms underlying the “chemical rescue of structure” strategy. The authors first developed a cell-based assay, in which dimeric proteins are fused to DNA-binding domains, such that a reporter gene is repressed by stable dimers [65]. Various tryptophan-to-glycine mutations were introduced into three different homodimers. For a subset of mutants, the authors observed gene repression in the absence and (partial) rescue in the presence of indole. Moreover, computed differences in stability between wild-type and mutants were predictive of the mutants’ experimentally determined capability of repression. In contrast to the previous, single case of a well-defined alteration of an active site’s geometry [64], the tryptophan-to-glycine mutations led to different outcomes including global or local unfolding or increased fluctuations between different functional states, all of which could be (partially) rescued by filling the generated cavity with indole [65].
These findings have important implications from an engineering perspective because the approach of impairment and chemical rescue is likely more generalizable than the design of a defined conformational change affecting active site geometry. However, while detailed structural knowledge may not be strictly required, it would certainly be beneficial to use it to reliably predict the effects of mutations. One potential limitation of the chemical rescue strategy in its current form arises from the use of indole for rescuing protein structure and function. While this compound is ideally suited to replace a buried tryptophan residue, it is bioactive in different cellular contexts, and its administration in a biomedical, or even biomanufacturing application, may, thus, be problematic [64]. Therefore, it would be intriguing to apply protein design methodologies and develop strategies that can achieve similar impairment and rescue of protein structure for inputs that are customized for a given application.
Conditionally stabilized ligand-binding domains have also been used to regulate fused proteins in modular systems [62], [63] [see Fig. 4(b)]. An early experimental study of note was based on the F36V mutant of the human protein “FK506 binding protein 12” (FKBP12), which served as a starting point for random mutagenesis to select further destabilized variants that are degraded in mammalian cells [62]. The binding of the customized ligand Shld-1 protects the destabilized FKPB12 variants from degradation. This strategy allows reversible modulation of the levels of proteins fused to FKPB12, which are decreased in the absence and increased in the presence of Shld-1.
A related strategy, which bridges aspects of conditional stabilization and allostery, entails the insertion of domains that undergo ligand-dependent disorder-order transitions into proteins of interest such that these proteins now become ligand-responsive. This domain insertion approach was successfully applied to generate a version of Src kinase regulated by the small molecule rapamycin upon insertion of an engineered rapamycin-stabilized domain termed uniRapR into a site in the kinase that was found to be coupled allosterically to the active site [66]. Similarly, mammalian cell morphology and motility could be controlled chemically or optically upon insertion of the uni-RapR domain or a photosensitive domain, respectively, into cell signaling proteins after computationally identifying appropriate insertion sites [67]. Moreover, the insertion of circularly permuted fluorescent proteins into ligand-binding proteins, such as periplasmic binding proteins, has resulted in sensors for several small molecules, from the sugar maltose [68] to neurotransmitters [69], [70].
Other rationally developed, experimental strategies for the conditional (de)stabilization of proteins used small molecules to regulate the accessibility of “degrons,” short peptide motifs that target fused proteins for proteasomal degradation [71], [72]. One notable application of this approach fused an FKBP12-based “ligand-induced degradation domain” to a CAR, whose surface expression in T cells could be downregulated in a dose-dependent manner with the ligands Shld-1 and AS-1 [73]. Such biomedical applications of conditional protein destabilization highlight a promising role of computational protein design. While current strategies repurpose known protein–ligand complexes, computational design could be used to generate synthetic systems responding to small-molecule drugs that can be chosen at will for a given application.
Indeed, chemical rescue approaches have been extended successfully to computationally designed small-molecule-binding proteins such that they are destabilized in the absence of ligands and conditionally stabilized in their presence [47]. Computationally designed ligand-binding domains for digoxin and progesterone [24] initially did not confer ligand-dependent stability changes detectable by a fused fluorescent reporter in yeast [47]. Random mutagenesis via error-prone PCR, followed by selections for high signal in the presence of ligand, and low signal in the absence of ligand, resulted in destabilization and ligand-dependent rescue of stability with around fivefold activation. To further amplify the signal, the ligand-binding domains were fused with an N-terminal DNA-binding domain and a C-terminal transcriptional activation domain. Performing another round of random mutagenesis in this reporter architecture identified mutations in the DNA-binding domain, which increased the dynamic range for both ligand-binding domains and enabled around 60-fold activation of gene expression in yeast and up to 100-fold activation in mammalian cells [47]. This coupling of ligand binding to transcriptional responses was then applied to monitor and optimize the bioproduction of progesterone in yeast. Furthermore, performing a gene repair assay with a fluorescent readout, the authors showed that the fusion of an engineered ligand-binding domain enables regulation of Cas9-based gene editing by the digoxin sensor with an 18-fold increase in the population of GFP-positive cells [47]. These applications of computationally designed and experimentally optimized proteins suggest that the forward engineering of conditional stabilization in response to new ligands may be a promising strategy to couple customized ligand inputs to diverse outputs in synthetic biological circuits.
C. Chemically Induced and Chemically Disrupted Heterodimers
While allostery and conditional stabilization modulate protein function primarily via intra-chain effects, binding of a small molecule can also control: 1) assembly or 2) disassembly of interacting proteins in a protein (hetero)dimer [see Fig. 4(c)]. When both protein partners of the dimer are fused to complementary split actuator domains, ligand binding can: 1) increase or 2) decrease actuator function. The more traditional of these two approaches is chemically induced dimerization (CID).
Several CID systems have been rationally repurposed using naturally occurring proteins. Two such systems, which respond to the plant hormones abscisic acid [74] and gibberellin [75], respectively, are based on a ligand-dependent conformational change in one protein partner, which allows binding to the other protein partner [74], [75]. In contrast, in the most commonly used CID system, the two partner proteins FKBP and FRB [the “FKBP Rapamycin binding” domain of the mammalian target of rapamycin (mTOR)] dimerize upon binding of the small molecule rapamycin directly in the interface [76], [77]. Rapamycin can also be replaced by a range of chemical analogs (rapalogs) [78]. Rapamycin binding to FKBP increases the affinity of FRB for the ligand by over three orders of magnitude [77]. Remarkably, this interaction of FRB with the FKBP-rapamycin complex was estimated to occur at subpicomolar affinity, while there was no detectable interaction of the two proteins in the absence of the ligand [77]. These findings highlight the potential sensitivity that can be obtained with CID systems.
Because CID systems allow the control of diverse functional responses via fusion to modular split reporters, CID systems have been widely used for both basic science and engineering applications [79], [80]. For example, reconstitution of a split CAR can be controlled “remotely” via ligand-dependent dimerization of fused rapalog- or gibberellic-acid-controlled CID modules [80]. While such applications of CID systems represent an important step toward avoiding harmful toxicities of engineered cell therapies via small-molecule-based control, they also highlight limitations of classical CID systems. In particular, the handful of ligands (and proteins) available for natural CID systems often show suboptimal properties for biotechnology or medicine. For example, biomedical applications often necessitate specific demands on favorable pharmacodynamics and pharmacokinetics of small molecules and minimal immunogenicity of proteins. Therefore, methods to design and engineer CID systems with tailored properties for a given application are highly sought after, as discussed in more detail in the following.
To engineer new CID systems for therapeutic applications, two studies [81], [82] based their strategies on human proteins and FDA-approved small-molecule drugs, whose well-documented physiological properties could facilitate translation into the clinic. Both studies used an existing drug-protein complex as a starting point. One approach experimentally engineered an antibody binding to the complex of the antiapoptotic protein “B-cell lymphoma–extra large” (Bcl-xL) with the drug ABT-737, which together provides a large composite solvent-accessible surface for recognition [81]. The second approach used randomized versions of two different scaffolds to select experimentally for binders of the complex of a lipocalin protein, which undergoes a conformational change when bound to the orally available drug A1120 [82]. A third experimental method to generate CID pairs termed “combinational binders-enabled selection of CID” (COMBINES-CID) does not require an existing protein–ligand complex as the starting point [83]. Here, an “anchor binder” is first selected experimentally to bind the target ligand. In a subsequent step, a “dimerization binder” protein is experimentally selected to bind the anchor-ligand complex. The authors validated this method by generating a nanobody-based CID system that responds to cannabidiol [83].
The examples above all relied on either rational mutagenesis or extensive experimental selection of binders from large libraries. Recent strategies increasingly apply computational design methods to engineer sensor–actuator systems using ligand-responsive assembly. Two of these studies set out to specifically develop ligand-controlled switches for cell-based therapies [84], [85]. In the first example, an existing protein served as a starting point, as it was a drug target for which several drugs binding to it were already known [84]. To avoid side effects in mammalian cells, a viral protease (NS3a) and drugs without known human targets were chosen. A major conceptual difference to the previous approaches was that different “reader” proteins were designed to bind the NS3a “receiver” protein in various states, i.e., in complex with different known drugs [84]. In this way, the presence of multiple inputs at the same time could, in principle, enable pleiotropic responses, such as graded and proportional outputs, in contrast to the simpler output behavior of a single-input–single-output system. Potential binder scaffolds were computationally docked to the structure of the drug-receiver-protein complex structure, and the predicted binding interfaces were designed with Rosetta to stabilize the interaction. Successful binders, which were based on designed helical repeat scaffolds, were then experimentally optimized for affinity and specificity by mutagenesis and selection via yeast surface display. The authors then validated their multi-input approach by controlling cellular responses. In one example, the receiver protein was fused to a nuclease-deficient Cas9 variant (dCas9) for guide RNA–regulated DNA targeting, while a drug-specific reader protein was fused to a transcriptional activation domain. When the reader-targeted drug competed with a different drug for binding the receiver, a more graded or “analog,” response was obtained compared to a more “digital” response in the presence of the reader-specific drug alone. This level of control makes intermediate levels of gene expression more accessible and was suggested to facilitate the fine-tuning of pathway activity in synthetic biology applications [84]. Furthermore, to demonstrate that proportional responses can be enabled by multi-input sensing, the receiver was fused to dCas9, and two different readers were fused to orthogonal RNA-hairpin-binding proteins, which allowed for the control of expression levels for two different genes simultaneously, dependent on the concentrations of two drugs that were recognized by the different readers. These examples illustrate the emergent outputs that can be obtained by sensing multiple inputs, in particular, when interfaced with cellular processes, such as gene expression, which forms the basis of most synthetic biology applications.
Another study that aimed at developing a conditional-assembly based switch for cell therapy based their approach on the ligand-controlled disruption of a CAR to reversibly downregulate its activity [85]. Such transient pausing of receptor activity is desirable for dynamically managing adverse effects in cell therapies. Moreover, in immunotherapy, continuous antigen exposure can cause “exhaustion” of engineered immune cells. In this case, it would be advantageous to temporally control immune receptor activity in engineered cells by transiently downregulating receptor activity in response to a drug but reactivating function upon drug withdrawal. To realize this goal, a “STOP-CAR” was designed, which harbors its extracellular antigen-binding functionality on one protein chain and the intracellular signaling functionality on the other chain [85]. To enable pausing of CAR activity with small molecules, the authors designed a “chemically disruptable heterodimer” (CDH) [85] [see Fig. 4(c)]. As a starting point, they chose the antiapoptotic protein Bcl-xL, which serves as the target for various small-molecule drugs, and which forms a complex with the protein BIM (“Bcl-2-like protein 11”). As BIM does not fold into a stable structure in the absence of its binding partner, as would be desired for fusion within the context of a STOP-CAR application, a computational design approach was applied to generate a protein binder to a Bcl-xL-drug complex based on a human protein scaffold. In particular, the authors applied a method termed “MotifGraft” [86] (see Table 1) to search for scaffold backbones that, on their surface, can present BIM’s binding motif for Bcl-xL. The approach then proceeds by transplanting the binding motif into suitable scaffold variants, redesigning their binding interface, and selecting promising candidates. Two out of three leads generated in this way tightly bound Bcl-xL when measured in vitro with purified components with an affinity of up to 3.9 pM. Importantly, this interaction could be disrupted with two different drugs with an apparent half-maximal inhibitory concentration of 101 nM for the more potent drug. Next, the authors incorporated the designed CDH into a STOP-CAR, whose efficacy in immune cells was comparable to a nonsplit version of the receptor. In line with the design goal, the addition of one of the two drugs reversibly impaired receptor function in vitro and in vivo.
In contrast to the two approaches above, a strategy to computationally design CID systems without the need for an existing protein–ligand complex as the starting point was recently developed in our lab [31], as described on a methodological level in Section II-C. The key advantage of this strategy is that all components of the CID system can, in principle, be customized. Most importantly, the strategy can be applied to input signals for which no known sensor exists. Moreover, in contrast to the COMBINES-CID method for experimental selection of nanobody-based binders for a small molecule [83], computational design, in principle, allows for the engineering of components whose properties can be tuned predictively in terms of their binding parameters. In addition, the computationally designed CID system was linked to three different modular outputs using split reporters, demonstrating user-defined control over both inputs and outputs of the sensor–actuator system. The approach was demonstrated by developing a CID system for FPP [31]. This small molecule is a toxic intermediate, which occurs during the production of high-value chemicals in metabolically engineered microorganisms [87]. Therefore, a sensor may enable the monitoring of pathway activity and/or interface with regulatory networks to actuate feedback on pathway flux.
The examples above highlight that conditional-assembly-based switches can function as powerful, modular sensor–actuator systems, which can be applied in engineered cells in the framework of synthetic biology (see Fig.1). Nevertheless, important challenges remain. For example, the dynamic range of the sensors can be limited by both the intrinsic (ligand-independent) affinity of the CID components and the split actuators for each other. Improving this property is challenging because the dimer-affinity would need to be reduced without sacrificing the stability of the small-molecule-bound ternary complex. While the CID pair’s background affinity may need to be reduced via negative design on a case-by-case basis depending on the scaffold and binding motif, a recent method promises to streamline the generation of split actuator systems with reduced propensity of background reconstitution while maintaining stability and function [88]. Likewise, a computational method for identifying suitable split sites within proteins promises to facilitate the generation of novel split proteins [89]. This strategy has successfully been applied to generate a “chemically induced trimerization” system through the computationally informed splitting of one of the two protein components of the rapamycin-sensitive FKBP–FRB pair [90]. Due to the modular nature of CID systems, optimized split actuator systems may broadly benefit the performance of computationally and/or experimentally engineered CID systems.
IV. CONCLUSION, CHALLENGES, AND FRONTIERS
To build synthetic sense-and-response systems for arbitrary small-molecule ligand inputs, both ligand recognition and its coupling to a functional output need to be engineered. While a variety of new ligand-binding proteins have successfully been designed computationally, several challenges remain. Design is not as predictive as desired. Typically, several design candidates have to be screened to identify functional variants. Moreover, the affinity of these initial designs is often relatively weak (in the micromolar range) and, in the majority of cases, requires experimental optimization.
Obvious areas for improvement in computational protein design methods are the accuracy of scoring, efficiency of sampling, and better knowledge of quality determinants during computational filtering and selection of design candidates [see Fig. 3(c)]. While limitations in these areas practically affect all applications of protein design, addressing them is particularly critical for designing ligand-binding sites, which depends on the precise orientation of side chains in the binding pocket. For example, reported disagreements between design models and experimentally solved structures [49] imply that better sampling of backbone flexibility and more accurate capturing of factors, such as desolvation penalties during scoring, could decrease the gap between computed and observed structures. Advances in score functions [91] show improved performance in contexts such as ligand binding, and it will be interesting to test potential design improvements by such score functions in practice.
While properties such as ligand-protein shape complementarity, preorganized binding sites, and local sequence/structure compatibility have repeatedly been featured in successful designs [24], [31], [45], [51], a systematic understanding of the quality determinants of designed ligand-binding proteins is still missing. As experimental characterization poses a larger burden on resources than generating a large number of designs during upstream computational design, more efficient filtering could have a highly beneficial impact on the design process overall. One route toward a more comprehensive understanding would be the high-throughput experimental characterization of designed proteins featuring different values in the filters’ parameter space. An analogous approach has been taken to characterize the stability of designed miniproteins via yeast surface display and limited proteolysis [92]. Extending such approaches to ligand-binding proteins could produce data-derived insights that could, in turn, be used to improve the design selection process.
It would also be interesting to systematically test if the computational design of small-molecule-binding proteins would benefit from favoring designed binding motifs with high similarity to recurring motifs found in existing protein structures. Moreover, diversifying both the binding motifs and protein scaffolds [93] would increase the success rate during incorporation of binding sites into protein backbones [see Fig. 3(b) and (c)], which would ultimately increase the number of unique variants for sequence design and, therefore, the likelihood of success overall. With regards to motif diversification, a promising direction is the application of a recent method describing the automated design of large numbers of potential binding sites [43] upstream in the design process.
Design success can also be limited by the number of available scaffolds. For example, from a library of more than 1000 scaffolds, binding motifs for FPP could only be computationally accommodated in three scaffolds, of which only one showed a robust sensor signal [31]. The availability of a higher number and diversity of scaffolds would be expected to increase design success through an increase in the sampling of backbones that could accommodate a given binding motif. In addition, the incorporation of binding motif residues into scaffolds can be particularly detrimental if the starting scaffolds are only marginally stable (as common for many naturally occurring proteins) and, thus, easily unfolded by additional mutations. De novo scaffolds can be designed for high stability (although some flexibility may be necessary for shape complementarity when binding the ligand) and can additionally be subjected to methods to tune structural features for increased scaffold diversity. Toward this end, two methods have recently been developed that generate variants of a given protein fold that has tunable variations in backbone conformation [94], [95]. Going forward, new methods could be used to design de novo scaffolds around a predefined motif (instead of starting with a predefined library of scaffolds). Similar approaches have recently been developed for protein–protein interactions [28], [96]. Finally, machine learning methodologies could be applied to inform designs based on data from already characterized natural proteins or newly designed proteins [97], [98].
The application of computational methodologies for the binding of arbitrary small molecules is also limited by the molecular properties of the ligand. Binder design is expected to be easier for more rigid small molecules with a low number of rotatable bonds and, consequently, a lower number of possible conformations to minimize the decrease in entropy upon binding. The achievable affinity of protein–ligand interactions should generally increase with the ligand’s molecular weight and, in particular, a larger hydrophobic interaction surface. However, the size of the ligand is practically limited by algorithms such as Rosetta Match [44] that can typically only place three to four motif residues in the desired geometry given the existing scaffold backbones and the shape of designable cavities in the scaffold. Here, the ability to finely tune the shapes of de novo designed scaffolds [94], [95] could help with both problems [99]. In addition, highly polar ligands pose considerable challenges as hydrogen bond donor and acceptor capabilities of the ligand should ideally be satisfied upon binding. These issues place additional requirements on the precision with which the shapes of the scaffolds can be engineered and may expose additional difficulties with the accuracy of modeling the energetics of polar interactions. While these considerations of ligand properties are intuitive from a physicochemical perspective, no study has yet systematically varied ligand properties and evaluated the success of computational binder design.
Further challenges lie in addressing the problem of coupling ligand binding by computationally designed proteins to a functional output. Allostery, for example, is notoriously difficult to engineer. A more systemic understanding of the underlying principles would help in the design of new allosteric proteins and could be obtained by reverse engineering existing allosteric proteins. In contrast to allostery, systems based on conditional protein assembly are more modular and allow the regulation of diverse outputs. However, the dynamic range of these systems can be limited by the intrinsic affinities of the dimerizing domains in the absence of the small molecule input. One way to address the resulting background activity without compromising the stability of the functional ligand-bound signaling complex would be to add additional layers of regulation, such as conditional stabilization or allosteric control.
In the broader context of synthetic biology, computationally designed sensor–actuator systems have already shown promise for regulating outputs on the transcriptional [47], [84] and protein level [85]. An exciting prospect for the future is the computational design of interlinked protein–small-molecule and protein–protein interactions that together constitute protein-level circuits to control biological processes. Protein circuits could give rise to emergent behaviors, controlled by custom inputs, which are more complex than the control of a single protein pair and would act on faster timescales than transcriptional regulation. Recent studies have developed protein-level circuits using existing proteins [10], [11] that can exhibit bistability [11] or interface with gene expression to define cell states [100]. The de novo design of all circuit components could provide unique advantages for engineerability. Recent studies have already described the design of logic gates [101], solely on the protein level, and feedback mechanisms [102]. These systems could be endowed with the ability to sense arbitrary user-defined inputs created by computational design. Such efforts could ultimately give rise to signaling networks that are based entirely on de novo designed components with tunable properties that can be recombined in modular and predictable ways to create diverse signaling behaviors. De novo systems have the potential to operate in engineered cells with maximal orthogonality. Moreover, such systems could help test our understanding of the design principles of natural protein networks in a bottom-up fashion. Taken together, we believe that computational protein design will be an enabling technology to realize the next generation of highly customized signaling circuits in engineered biological systems.
Acknowledgment
The authors would like to thank Amy Guo [University of California at San Francisco (UCSF)] for comments on the manuscript.
This work was supported by the U.S. National Institutes of Health (NIH) under Grant GM110089 and additionally by a Postdoctoral Independent Research Grant of the UCSF “Program for Breakthrough Biomedical Research” (PBBR), which is partially funded by the Sandler foundation. Tanja Kortemme is a Chan Zuckerberg Biohub Senior Investigator.
Biographies

Simon Kretschmer received the B.Sc. degree in chemistry and biochemistry and the M.Sc. degree in biochemistry from the Ludwig Maximilian University of Munich, Munich, Germany, in 2009 and 2012, respectively, and the Ph.D. degree in biochemistry from the Graduate School of Quantitative Biosciences, Ludwig Maximilian University of Munich, in 2018, for a thesis on protein self-organization in the Laboratory of Prof. Petra Schwille at the Max Planck Institute of Biochemistry, Martinsried, Germany.
His current research interests in Prof. Kortemme’s Laboratory at the University of California at San Francisco, San Francisco, CA, USA, lie at the intersection of protein design and synthetic biology. He is particularly interested in designing novel small-molecule-controlled protein switches for applications in cell therapy.

Tanja Kortemme received the Ph.D. degree from the University of Hannover, Hannover, Germany, for graduate work in protein biophysics at the European Molecular Biology Laboratory (EMBL) in Heidelberg, Germany. As an EMBO and Human Frontiers Science Program Postdoctoral Fellow, she pioneered computational methods for the design of proteins and their interactions.
She is currently a Professor of Bioengineering and Therapeutic Sciences at the University of California at San Francisco, San Francisco, CA, USA, where her group advances and invents new approaches to engineer biological functions at multiple scales.
Footnotes
As “de novo design,” we define the generation of proteins or their structural or functional elements from physical principles without relying on a closely related natural protein.
Biochemical affinities are typically expressed in terms of the dissociation constant (KD), which is defined as the concentration ratio of a complex’s dissociated components to the intact complex at equilibrium. Thus, a lower KD implies tighter complex formation.
Yeast surface display is a high-throughput technology for testing proteins, in particular, their binding to target molecules. Typically, yeast cells are transformed with a library of genes such that each cell displays multiple copies of a distinct protein variant on the surface. Adding a fluorescently labeled target molecule to the cells and sorting cells based on fluorescence then allow for characterization of binding for all displayed protein variants simultaneously.
REFERENCES
- [1].Gardner TS, Cantor CR, and Collins JJ, “construction of a genetic toggle switch in Escherichia coli,” Nature, vol. 403, no. 6767, pp. 339–342, Jan 2000 [DOI] [PubMed] [Google Scholar]
- [2].Elowitz MB and Leibler S, “A synthetic oscillatory network of transcriptional regulators,” Nature, vol. 403, no. 6767, pp. 335–338, Jan. 2000 [DOI] [PubMed] [Google Scholar]
- [3].Kretschmer S, Ganzinger KA, Franquelim HG, and Schwille P, “Synthetic cell division via membrane-transforming molecular assemblies,” BMC Biol., vol. 17, no. 1, p. 43, Dec. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ro D-K et al. , “Production of the antimalarial drug precursor artemisinic acid in engineered yeast,” Nature, vol. 440, no. 7086, pp. 940–943, Apr. 2006. [DOI] [PubMed] [Google Scholar]
- [5].Gilbert C and Ellis T, “Biological engineered living materials: Growing functional materials with genetically programmable properties,” ACS Synth. Biol, vol. 8, no. 1, pp. 1–15, Jan. 2019. [DOI] [PubMed] [Google Scholar]
- [6].Lim WA and June CH, “The principles of engineering immune cells to treat cancer,” Cell, vol. 168, no. 4, pp. 724–740, Feb. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kitada T, DiAndreth B, Teague B, and Weiss R, “Programming gene and engineered-cell therapies with synthetic biology,” Science, vol. 359, no. 6376, Feb. 2018, Art. no. eaad1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jacob F and Monod J, “Genetic regulatory mechanisms in the synthesis of proteins,” J. Mol. Biol, vol. 3, no. 3, pp. 318–356, Jun. 1961. [DOI] [PubMed] [Google Scholar]
- [9].Lewis M, “Allostery and the lac operon,” J. Mol. Biol, vol. 425, no. 13, pp. 2309–2316, Jul. 2013. [DOI] [PubMed] [Google Scholar]
- [10].Gao XJ, Chong LS, Kim MS, and Elowitz MB, “Programmable protein circuits in living cells,” Science, vol. 361, no. 6408, pp. 1252–1258, Sep. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mishra D, Bepler T, Teague B, Berger B, Broach J, and Weiss R, “An engineered protein-phosphorylation toggle network with implications for endogenous network discovery,” Science, vol. 373, no. 6550, Jul. 2021, Art. no. eaav0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen Z and Elowitz MB, “Programmable protein circuit design,” Cell, vol. 184, no. 9, pp. 2284–2301, Apr. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Morsut L et al. , “Engineering customized cell sensing and response behaviors using synthetic notch receptors,” Cell, vol. 164, no. 4, pp. 780–791, Feb. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Scheller L, Strittmatter T, Fuchs D, Bojar D, and Fussenegger M, “Generalized extracellular molecule sensor platform for programming cellular behavior,” Nature Chem. Biol, vol. 14, no. 7, pp. 723–729, Jul. 2018. [DOI] [PubMed] [Google Scholar]
- [15].Schwarz KA, Daringer NM, Dolberg TB, and Leonard JN, “Rewiring human cellular input–output using modular extracellular sensors,” Nature Chem. Biol, vol. 13, no. 2, pp. 202–209, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].English MA, Gayet RV, and Collins JJ, “Designing biological circuits: Synthetic biology within the operon model and beyond,” Annu. Rev. Biochem, vol. 90, no. 1, pp. 221–244, Jun. 2021. [DOI] [PubMed] [Google Scholar]
- [17].Mansouri M and Fussenegger M, “Therapeutic cell engineering: Designing programmable synthetic genetic circuits in mammalian cells,” Protein Cell, Sep. 2021, doi: 10.1007/s13238-021-00876-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Leman JK et al. , “Macromolecular modeling and design in Rosetta: Recent methods and frameworks,” Nature Methods, vol. 17, no. 7, pp. 665–680, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dahiyat BI and Mayo SL, “De novo protein design: Fully automated sequence selection,” Science, vol. 278, no. 5335, pp. 82–87, Oct. 1997. [DOI] [PubMed] [Google Scholar]
- [20].Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, and Baker D, “Design of a novel globular protein fold with atomic-level accuracy,” Science, vol. 302, no. 5649, pp. 1364–1368, Nov. 2003. [DOI] [PubMed] [Google Scholar]
- [21].Koehler Leman J et al. , “Better together: Elements of successful scientific software development in a distributed collaborative community,” PLOS Comput. Biol, vol. 16, no. 5, May 2020, Art. no. e1007507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gonen S, DiMaio F, Gonen T, and Baker D, “Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces,” Science, vol. 348, no. 6241, pp. 1365–1368, Jun. 2015. [DOI] [PubMed] [Google Scholar]
- [23].Hsia Y et al. , “Design of a hyperstable 60-subunit protein dodecahedron. [corrected],” Nature, vol. 535, no. 7610, pp. 136–139, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tinberg CE et al. , “Computational design of ligand-binding proteins with high affinity and selectivity,” Nature, vol. 501, no. 7466, pp. 212–216, Sep. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chevalier A et al. , “Massively parallel de novo protein design for targeted therapeutics,” Nature, vol. 550, no. 7674, pp. 74–79, Oct. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ambroggio XI and Kuhlman B, “Computational design of a single amino acid sequence that can switch between two distinct protein folds,” J. Amer. Chem. Soc, vol. 128, no. 4, pp. 1154–1161, Feb. 2006. [DOI] [PubMed] [Google Scholar]
- [27].Silva D-A et al. , “De novo design of potent and selective mimics of IL-2 and IL-15,” Nature, vol. 565, no. 7738, pp. 186–191, Jan. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sesterhenn F et al. , “De novo protein design enables the precise induction of RSV-neutralizing antibodies,” Science, vol. 368, no. 6492, May 2020, Art. no. eaay5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Langan RA et al. , “De novo design of bioactive protein switches,” Nature, vol. 572, no. 7768, pp. 205–210, Aug. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quijano-Rubio A et al. , “De novo design of modular and tunable protein biosensors,” Nature, vol. 591, no. 7850, pp. 482–487, Mar. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Glasgow AA et al. , “Computational design of a modular protein sense-response system,” Science, vol. 366, no. 6468, pp. 1024–1028, Nov. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guntas G, Mansell TJ, Kim JR, and Ostermeier M, “Directed evolution of protein switches and their application to the creation of ligand-binding proteins,” Proc. Nat. Acad. Sci. USA, vol. 102, no. 32, pp. 11224–11229, Aug. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Herud-Sikimić O et al. , “A biosensor for the direct visualization of auxin,” Nature, vol. 592, no. 7856, pp. 768–772, Apr. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Day AL et al. , “Unintended specificity of an engineered ligand-binding protein facilitated by unpredicted plasticity of the protein fold,” Protein Eng., Des. Selection, vol. 31, no. 10, pp. 375–387, Oct. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dawson WM, Rhys GG, and Woolfson DN, “Towards functional de novo designed proteins,” Current Opinion Chem. Biol, vol. 52, pp. 102–111, Oct. 2019. [DOI] [PubMed] [Google Scholar]
- [36].Korendovych IV and DeGrado WF, “De novo protein design, a retrospective,” Quart. Rev. Biophys, vol. 53, p. e3, Feb. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Höcker B, “Design of proteins from smaller fragments—Learning from evolution,” Current Opinion Struct. Biol, vol. 27, pp. 56–62, Aug. 2014. [DOI] [PubMed] [Google Scholar]
- [38].Pan X and Kortemme T, “Recent advances in de novo protein design: Principles, methods, and applications,” J. Biol. Chem, vol. 296, Jan. 2021, Art. no. 100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alford RF et al. , “The Rosetta all-atom energy function for macromolecular modeling and design,” J. Chem. Theory Comput, vol. 13, no. 6, pp. 3031–3048, Jun. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].de los Santos ELC, Meyerowitz JT, Mayo SL, and Murray RM, “Engineering transcriptional regulator effector specificity using computational design and in vitro rapid prototyping: Developing a vanillin sensor,” ACS Synth. Biol, vol. 5, no. 4, pp. 287–295, Apr. 2016. [DOI] [PubMed] [Google Scholar]
- [41].Taylor ND et al. , “Engineering an allosteric transcription factor to respond to new ligands,” Nature Methods, vol. 13, no. 2, pp. 177–183, Feb. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Banda-Vazquez J, Shanmugaratnam S, Rodriguez-Sotres R, Torres-Larios A, Hocker B, and Sosa-Peinado A, “Redesign of LAOBP to bind novel L-amino acid ligands,” Protein Sci, vol. 27, no. 5, pp. 957–968, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lucas JE and Kortemme T, “New computational protein design methods for de novo small molecule binding sites,” PLOS Comput. Biol, vol. 16, no. 10, Oct. 2020, Art. no. e1008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zanghellini A et al. , “New algorithms and an in silico benchmark for computational enzyme design,” Protein Sci, vol. 15, no. 12, pp. 2785–2794, Dec. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tinberg CE and Khare SD, “Computational design of ligand binding proteins,” Methods Mol. Biol, vol. 1529, pp. 363–373, Sep. 2017. [DOI] [PubMed] [Google Scholar]
- [46].Boder ET and Wittrup KD, “Yeast surface display for screening combinatorial polypeptide libraries,” Nature Biotechnol, vol. 15, no. 6, pp. 553–557, Jun. 1997. [DOI] [PubMed] [Google Scholar]
- [47].Feng J et al. , “A general strategy to construct small molecule biosensors in eukaryotes,” eLife, vol. 4, Dec. 2015, Art. no. e10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bick MJ et al. , “Computational design of environmental sensors for the potent opioid fentanyl,” eLife, vol. 6, Sep. 2017, Art. no. e28909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dou J et al. , “Sampling and energy evaluation challenges in ligand binding protein design,” Protein Sci, vol. 26, no. 12, pp. 2426–2437, Dec. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dou J et al. , “De novo design of a fluorescence-activating β-barrel,” Nature, vol. 561, no. 7724, pp. 485–491, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Polizzi NF and DeGrado WF, “A defined structural unit enables de novo design of small-molecule–binding proteins,” Science, vol. 369, no. 6508, pp. 1227–1233, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Polizzi NF et al. , “De novo design of a hyperstable non-natural protein–ligand complex with sub-Å accuracy,” Nature Chem., vol. 9, no. 12, pp. 1157–1164, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Grigoryan G and DeGrado WF, “Probing designability via a generalized model of helical bundle geometry,” J. Mol. Biol, vol. 405, no. 4, pp. 1079–1100, Jan. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stein V and Alexandrov K, “Synthetic protein switches: Design principles and applications,” Trends Biotechnol, vol. 33, no. 2, pp. 101–110, Feb. 2015. [DOI] [PubMed] [Google Scholar]
- [55].Monod J, Changeux J-P, and Jacob F, “Allosteric proteins and cellular control systems,” J. Mol. Biol, vol. 6, no. 4, pp. 306–329, Apr. 1963. [DOI] [PubMed] [Google Scholar]
- [56].Changeux JP, “The feedback control mechanism of biosynthetic L-threonine deaminase by L-isoleucine,” in Proc. Cold Spring Harbor Symposia Quant. Biol, vol. 26, 1961, pp. 313–318. [DOI] [PubMed] [Google Scholar]
- [57].Motlagh HN, Wrabl JO, Li J, and Hilser VJ, “The ensemble nature of allostery,” Nature, vol. 508, no. 7496, pp. 331–339, Apr. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lassila JK, Privett HK, Allen BD, and Mayo SL, “Combinatorial methods for small-molecule placement in computational enzyme design,” Proc. Nat. Acad. Sci. USA, vol. 103, no. 45, pp. 16710–16715, Nov. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Vishweshwaraiah YL, Chen J, and Dokholyan NV, “Engineering an allosteric control of protein function,” J. Phys. Chem. B, vol. 125, no. 7, pp. 1806–1814, Feb. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lockless SW and Ranganathan R, “Evolutionarily conserved pathways of energetic connectivity in protein families,” Science, vol. 286, no. 5438, pp. 295–299, Oct. 1999. [DOI] [PubMed] [Google Scholar]
- [61].Wang J, Jain A, McDonald LR, Gambogi C, Lee AL, and Dokholyan NV, “Mapping allosteric communications within individual proteins,” Nature Commun, vol. 11, no. 1, p. 3862, Dec. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL, and Wandless TJ, “A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules,” Cell, vol. 126, no. 5, pp. 995–1004, Sep. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tucker CL and Fields S, “A yeast sensor of ligand binding,” Nature Biotechnol, vol. 19, no. 11, pp. 1042–1046, Nov. 2001. [DOI] [PubMed] [Google Scholar]
- [64].Deckert K, Budiardjo SJ, Brunner LC, Lovell S, and Karanicolas J, “Designing allosteric control into enzymes by chemical rescue of structure,” J. Amer. Chem. Soc, vol. 134, no. 24, pp. 10055–10060, Jun. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xia Y et al. , “The designability of protein switches by chemical rescue of structure: Mechanisms of inactivation and reactivation,” J. Amer. Chem. Soc, vol. 135, no. 50, pp. 18840–18849, Dec. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dagliyan O et al. , “Rational design of a ligand-controlled protein conformational switch,” Proc. Nat. Acad. Sci. USA, vol. 110, no. 17, pp. 6800–6804, Apr. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dagliyan O et al. , “Engineering extrinsic disorder to control protein activity in living cells,” Science, vol. 354, no. 6318, pp. 1441–1444, Dec. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Marvin JS, Schreiter ER, Echevarria IM, and Looger LL, “A genetically encoded, high-signal-to-noise maltose sensor,” Proteins: Struct., Function, Bioinf, vol. 79, no. 11, pp. 3025–3036, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Marvin JS et al. , “An optimized fluorescent probe for visualizing glutamate neurotransmission,” Nature Methods, vol. 10, no. 2, pp. 162–170, Feb. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Marvin JS et al. , “A genetically encoded fluorescent sensor for in vivo imaging of GABA,” Nature Methods, vol. 16, no. 8, pp. 763–770, Aug. 2019. [DOI] [PubMed] [Google Scholar]
- [71].Bonger KM, Chen L-C, Liu CW, and Wandless TJ, “Small-molecule displacement of a cryptic degron causes conditional protein degradation,” Nature Chem. Biol, vol. 7, no. 8, pp. 531–537, Aug. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chung HK et al. , “Tunable and reversible drug control of protein production via a self-excising degron,” Nature Chem. Biol, vol. 11, no. 9, pp. 713–720, Sep. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Richman SA, Wang L-C, Moon EK, Khire UR, Albelda SM, and Milone MC, “Ligand-induced degradation of a CAR permits reversible remote control of CAR t cell activity in vitro and in vivo,” Mol. Therapy, vol. 28, no. 7, pp. 1600–1613, Jul. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Liang F-S, Ho WQ, and Crabtree GR, “Engineering the ABA plant stress pathway for regulation of induced proximity,” Sci. Signaling, vol. 4, no. 164, p. rs2, Mar. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Miyamoto T et al. , “Rapid and orthogonal logic gating with a gibberellin-induced dimerization system,” Nature Chem. Biol, vol. 8, no. 5, pp. 465–470, May 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Spencer DM, Wandless TJ, Schreiber SL, and Crabtree GR, “Controlling signal transduction with synthetic ligands,” Science, vol. 262, no. 5136, pp. 1019–1024, Nov. 1993. [DOI] [PubMed] [Google Scholar]
- [77].Banaszynski LA, Liu CW, and Wandless TJ, “Characterization of the FKBP·Rapamycin·FRB ternary complex,” J. Amer. Chem. Soc, vol. 127, no. 13, pp. 4715–4721, 2005. [DOI] [PubMed] [Google Scholar]
- [78].Kolos JM, Voll AM, Bauder M, and Hausch F, “FKBP ligands—Where we are and where to go?” Frontiers Pharmacol., vol. 9, p. 1425, Mar. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Straathof KC et al. , “An inducible caspase 9 safety switch for T-cell therapy,” Blood, vol. 105, no. 11, pp. 4247–4254, Jun. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wu CY, Roybal KT, Puchner EM, Onuffer J, and Lim WA, “Remote control of therapeutic T cells through a small molecule–gated chimeric receptor,” Science, vol. 350, no. 6258, 2015, Art. no. aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hill ZB, Martinko AJ, Nguyen DP, and Wells JA, “Human antibody-based chemically induced dimerizers for cell therapeutic applications,” Nature Chem. Biol, vol. 14, no. 2, pp. 112–117, Feb. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zajc CU et al. , “A conformation-specific ON-switch for controlling CAR t cells with an orally available drug,” Proc. Nat. Acad. Sci. USA, vol. 117, no. 26, pp. 14926–14935, Jun. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kang S et al. , “COMBINES-CID: An efficient method for de novo engineering of highly specific chemically induced protein dimerization systems,” J. Amer. Chem. Soc, vol. 141, no. 28, pp. 10948–10952, Jul. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Foight GW et al. , “Multi-input chemical control of protein dimerization for programming graded cellular responses,” Nature Biotechnol, vol. 37, no. 10, pp. 1209–1216, Oct. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Giordano-Attianese G et al. , “A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy,” Nature Biotechnol, vol. 38, no. 4, pp. 426–432, Apr. 2020. [DOI] [PubMed] [Google Scholar]
- [86].Silva DA, Correia BE, and Procko E, “Motif-driven design of protein–protein interfaces,” in Computational Design of Ligand Binding Proteins (Methods in Molecular Biology), vol. 1414. New York, NY, USA: Springer, 2016, pp. 285–304. [DOI] [PubMed] [Google Scholar]
- [87].Martin VJJ, Pitera DJ, Withers ST, Newman JD, and Keasling JD, “Engineering a mevalonate pathway in Escherichia coli for production of terpenoids,” Nature Biotechnol, vol. 21, no. 7, pp. 796–802, Jul. 2003. [DOI] [PubMed] [Google Scholar]
- [88].Dolberg TB et al. , “Computation-guided optimization of split protein systems,” Nature Chem. Biol, vol. 17, no. 5, pp. 531–539, May 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Dagliyan O et al. , “Computational design of chemogenetic and optogenetic split proteins,” Nature Commun., vol. 9, no. 1, p. 4042, Dec. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wu HD et al. , “Rational design and implementation of a chemically inducible heterotrimerization system,” Nature Methods, vol. 17, no. 9, pp. 928–936, Sep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Park H, Zhou G, Baek M, Baker D, and DiMaio F, “Force field optimization guided by small molecule crystal lattice data enables consistent sub-angstrom protein–ligand docking,” J. Chem. Theory Comput, vol. 17, no. 3, pp. 2000–2010, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rocklin GJ et al. , “Global analysis of protein folding using massively parallel design, synthesis, and testing,” Science, vol. 357, no. 6347, pp. 168–175, Jul. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pan X and Kortemme T, “De novo protein fold families expand the designable ligand binding site space,” PLoS Comput. Biol, vol. 17, no. 11, Nov. 2021, Art. no. e1009620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Basanta B et al. , “An enumerative algorithm for de novo design of proteins with diverse pocket structures,” Proc. Nat. Acad. Sci. USA, vol. 117, no. 36, pp. 22135–22145, Sep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pan X et al. , “Expanding the space of protein geometries by computational design of de novo fold families,” Science, vol. 369, no. 6507, pp. 1132–1136, Aug. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tischer D et al. , “Design of proteins presenting discontinuous functional sites using deep learning,” bioRxiv, Nov. 2020, doi: 10.1101/2020.11.29.402743. [DOI] [Google Scholar]
- [97].Anishchenko I et al. , “De novo protein design by deep network hallucination,” Nature, vol. 600, no. 7889, pp. 547–552, Dec. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gainza P et al. , “Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning,” Nature Methods, vol. 17, no. 2, pp. 184–192, Feb. 2020. [DOI] [PubMed] [Google Scholar]
- [99].Pan X and Kortemme T, “De novo protein fold families expand the designable ligand binding site space,” PLOS Comput. Biol, vol. 17, no. 11, Nov. 2021, Art. no. e1009620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhu R, del Rio-Salgado JM, Garcia-Ojalvo J, and Elowitz MB, “Synthetic multistability in mammalian cells,” Science, vol. 375, no. 6578, 2021, Art. no. eabg9765. [DOI] [PubMed] [Google Scholar]
- [101].Chen Z et al. , “De novo design of protein logic gates,” Science, vol. 368, no. 6486, pp. 78–84, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ng AH et al. , “Modular and tunable biological feedback control using a de novo protein switch,” Nature, vol. 572, no. 7768, pp. 265–269,Aug. 2019. [DOI] [PubMed] [Google Scholar]