

Richard O'Connell and colleagues report the genomes and transcriptomes of two Colletotrichum plant fungal pathogens. C. higginsianum infects Arabidopsis thaliana, and C. graminicola infects maize (Zea mays); comparative genomics in both species lead to molecular insights into the transition from biotrophic to necrotrophic life stages.
Supplementary information
The online version of this article (doi:10.1038/ng.2372) contains supplementary material, which is available to authorized users.
Subject terms: Gene expression profiling, Transcriptomics, Plant genetics
Abstract
Colletotrichum species are fungal pathogens that devastate crop plants worldwide. Host infection involves the differentiation of specialized cell types that are associated with penetration, growth inside living host cells (biotrophy) and tissue destruction (necrotrophy). We report here genome and transcriptome analyses of Colletotrichum higginsianum infecting Arabidopsis thaliana and Colletotrichum graminicola infecting maize. Comparative genomics showed that both fungi have large sets of pathogenicity-related genes, but families of genes encoding secreted effectors, pectin-degrading enzymes, secondary metabolism enzymes, transporters and peptidases are expanded in C. higginsianum. Genome-wide expression profiling revealed that these genes are transcribed in successive waves that are linked to pathogenic transitions: effectors and secondary metabolism enzymes are induced before penetration and during biotrophy, whereas most hydrolases and transporters are upregulated later, at the switch to necrotrophy. Our findings show that preinvasion perception of plant-derived signals substantially reprograms fungal gene expression and indicate previously unknown functions for particular fungal cell types.
Supplementary information
The online version of this article (doi:10.1038/ng.2372) contains supplementary material, which is available to authorized users.
Main
The genus Colletotrichum (Sordariomycetes, Ascomycota; Fig. 1a) comprises ∼600 species1 attacking over 3,200 species of monocot and dicot plants (ARS Fungal Databases, see URLs). These pathogens use a multistage hemibiotrophic infection strategy2: dome-shaped appressoria first puncture host surfaces using a combination of mechanical force and enzymatic degradation, bulbous biotrophic hyphae enveloped by an intact host plasma membrane then develop inside living epidermal cells, and finally, the fungus switches to necrotrophy and differentiates thin, fast-growing hyphae that kill and destroy host tissues (Fig. 1b,c). We sequenced two Colletotrichum species with different host specificities and infection strategies: C. higginsianum attacks several members of Brassicaceae, including Arabidopsis, and has emerged as a tractable model for studying fungal pathogenicity and plant immune responses3,4,5. Biotrophy in this fungus is confined to the first invaded host cell and is followed by a complete switch to necrotrophy5 (Supplementary Fig. 1). In contrast, C. graminicola primarily infects maize (Zea mays), causing annual losses of approximately 1 billion dollars in the United States alone6. In this species, biotrophy extends into many host cells and persists at the advancing colony margin while the center of the colony becomes necrotrophic7 (Supplementary Fig. 2).
Figure 1. Phylogeny and infection of the two Colletotrichum species analyzed in this study.
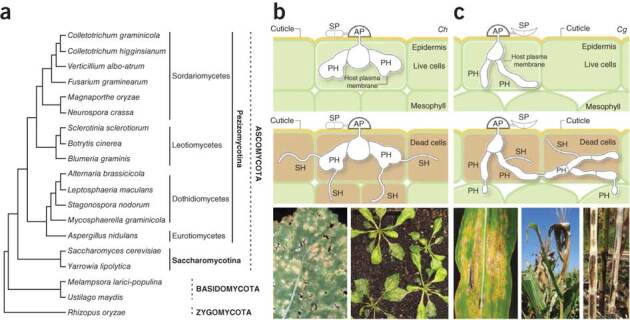
(a) Cladogram showing the phylogenetic relationship of Colletotrichum to other sequenced fungi, including 13 species used for comparative analyses (see Fig. 3). The unscaled tree was constructed using CVTree34 with Rhizopus oryzae as the outgroup. (b) Infection process of C. higginsianum (Ch) and leaf anthracnose symptoms on Brassica and Arabidopsis. The Brassica image is reproduced with permission of University of Georgia Plant Pathology Archive (bugwood.org/). (c) Infection process of C. graminicola (Cg), and leaf-blight, top die-back and stalk-rot symptoms on maize. SP, spore; AP, appressorium; PH, biotrophic primary hyphae; SH, necrotrophic secondary hyphae.
Optical mapping showed that the genomes of the two species are similar in size and structure. C. graminicola has a 57.4-Mb genome that is distributed among 13 chromosomes, including three minichromosomes less than 1 Mb in size, whereas C. higginsianum has a 53.4-Mb genome comprising 12 chromosomes, including two minichromosomes (Supplementary Table 1). We sequenced the C. graminicola genome using Sanger and 454 platforms, which provided a high-quality reference assembly of 50.9 Mb. We sequenced the C. higginsianum genome using 454 and Illumina platforms, yielding an assembly of 49.3 Mb (Supplementary Table 2). Repetitive DNA comprises 12.2% of the C. graminicola genome assembly and 1.2% of the C. higginsianum assembly (Supplementary Tables 2 and 3). The repeats clustered in genomic regions with low GC content in C. graminicola (Supplementary Fig. 3), similar to the AT-rich isochores found in Leptosphaeria maculans8. Including unassembled genomic regions (mostly repeats, such as ribosomal DNA, telomeres, centromeres and transposons), repetitive DNA was estimated to total 22.3% of the C. graminicola genome and 9.1% of the C. higginsianum genome. The two Colletotrichum species diverged relatively recently (∼47 million years ago), after the separation of monocots and dicots 140–150 million years ago9 (Supplementary Fig. 4). Although C. graminicola and C. higginsianum belong to sister clades within the genus (Supplementary Fig. 5), only 35% of the two genomes are syntenic (Supplementary Table 4), which is less than the synteny between Botrytis cinerea and Sclerotinia sclerotiorum10. Nevertheless, an analysis of synteny between the two Colletotrichum genomes identified homologous chromosomes and revealed that major intrachromosomal rearrangements have occurred in one or both species (Fig. 2a and Supplementary Table 5). The minichromosomes do not contain homologous sequences (Fig. 2b and Supplementary Table 4), suggesting that they are lineage-specific innovations, and in C. graminicola, the minichromosomes are enriched with repetitive DNA (averaging 23%) compared to the core genome (averaging 5.5%).
Figure 2. Conservation of synteny between the genomes of C. graminicola and C. higginsianum.
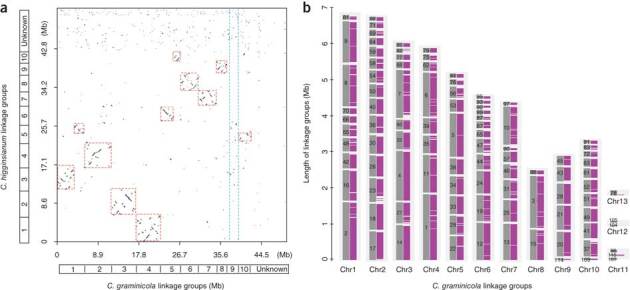
(a) Dot plot showing the syntenic blocks between the 13 chromosomes (optical linkage groups) of C. graminicola (horizontal axis) and the 12 chromosomes of C. higginsianum (vertical axis). Homologies between chromosomes of each species are highlighted in red dashed boxes. Homologous sequences of C. graminicola chromosome 9, indicated between the blue dashed lines, are dispersed among many C. higginsianum chromosomes. (b) Global view of syntenic alignments between the genomes of C. graminicola and C. higginsianum. Linkage groups of C. graminicola are shown as the reference, with linkage group lengths defined by the C. graminicola optical map. For each chromosome, numbered genomic scaffolds (dark gray) positioned on the optical linkage groups are separated by scaffold breaks. The magenta blocks show syntenic mapping of the C. higginsianum sequences; notably, there is a near absence of homologous sequences among the minichromosomes.
We predicted the existence of 12,006 protein-coding genes in C. graminicola compared to 16,172 in C. higginsianum (Supplementary Table 2). Having been compiled from short-read data only, the C. higginsianum assembly is more fragmented than that of C. graminicola, resulting in some genes (5.2%) being split into two or more gene models, whereas others (4%) are truncated versions of the complete gene (Supplementary Note). After correcting for this fragmentation, the estimated gene content of C. higginsianum (15,331) is still markedly larger than that of C. graminicola. The two species share 9,795 orthologous genes. Using Markov clustering (MCL)11 to analyze the proteomes, we found that 10,077 C. higginsianum genes belong to multicopy gene clusters, compared to 5,342 genes in C. graminicola, suggesting that the greater gene content of C. higginsianum results partly from gene duplication (Supplementary Table 6). The MCL analysis also revealed that gene clusters encoding serine proteases, methyl transferases, polyketide synthases, cytochrome P450 enzymes and small-molecule efflux pumps are expanded in C. higginsianum compared to C. graminicola (Supplementary Fig. 6), which we verified by manual inspection (Supplementary Tables 7 and 8 and Supplementary Fig. 7). Clusters that are expanded in C. graminicola relative to C. higginsianum include a family of genes encoding atypical cellulases (glycoside hydrolase GH61, described below) and another encoding secreted histidine acid phosphatases, which probably mobilize phytic acid, the main form of stored phosphorus in plants12.
C. higginsianum and C. graminicola are particularly well equipped with genes encoding carbohydrate-active enzymes (CAZymes)13 that potentially degrade the plant cell wall14 (Fig. 3a and Supplementary Table 9) and modify the fungal cell wall (Supplementary Tables 10 and 11). Both species encode more CAZymes than 13 other fungal genomes we examined. These expanded CAZyme arsenals are more similar to those of other hemibiotrophic and necrotrophic pathogens than to the highly reduced set found in biotrophs such as Melampsora and Blumeria (Fig. 3a). The exceptionally large and diverse inventory of CAZymes encoded by both Colletotrichum genomes provides a rich source of enzymes for potential commercial exploitation15. C. higginsianum encodes over twice as many pectin-degrading enzymes as does C. graminicola (Fig. 3b), the majority (62%) of which are activated during necrotrophy (Supplementary Fig. 8 and Supplementary Note). Conversely, although both species encode similar numbers of cellulases and hemicellulases, C. graminicola activates many more of these genes during necrotrophy (48%) than does C. higginsianum (26%), including 22 GH61 copper-dependent oxygenases, which act in concert with classical cellulases to enhance lignocellulose hydrolysis16,17. Thus, C. graminicola and C. higginsianum use very different strategies to deconstruct plant cell walls, reflecting their host preferences: dicot cell walls are enriched with pectin (35% in dicots compared to 10% in maize), whereas the cell walls of grasses contain more hemicellulose (60% in grasses compared to 30% in dicots) and phenolics (up to 5%)18.
Figure 3. Comparison of fungal carbohydrate-active enzyme (CAZyme) repertoires.
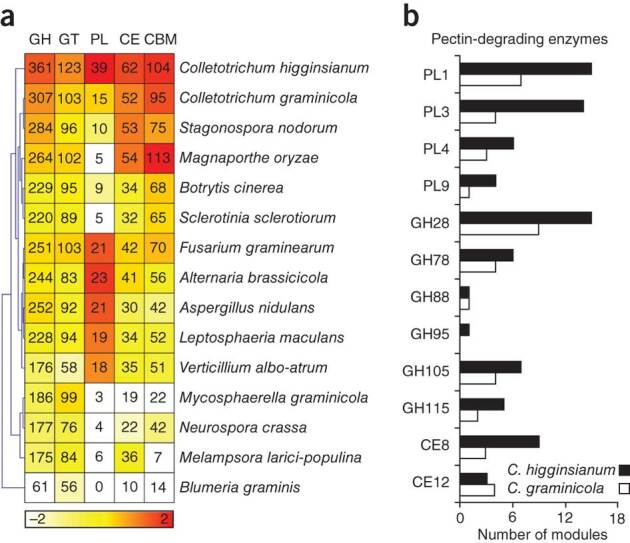
(a) Hierarchical clustering of CAZyme classes from Colletotrichum and 13 other fungal genomes. GH, glycoside hydrolase; GT, glycosyltransferase; PL, polysaccharide lyase; CE, carbohydrate esterase; CBM, carbohydrate-binding module. The numbers of enzyme modules in each genome are shown. Overrepresented (orange to red) and underrepresented modules (pale yellow to white) are depicted as fold changes relative to the class mean. (b) Comparison of the pectin-degrading enzyme repertoires of C. higginsianum and C. graminicola shown as the number of modules in each CAZyme family11. In total, C. higginsianum encodes 86 such modules, whereas C. graminicola encodes only 42.
Many phytopathogens secrete proteins known as effectors that facilitate infection by reprogramming host cells and modulating plant immunity19. By defining candidate secreted effectors (CSEPs) as predicted extracellular proteins without any homology to proteins outside the genus Colletotrichum, we found 177 CSEP-encoding genes in C. graminicola, 85 (48%) of which were species specific. In contrast, C. higginsianum encodes twice as many CSEPs (365), including more species-specific proteins (264, or 72%) (Supplementary Fig. 9a). The CSEPs are mostly small proteins (averaging 110 residues and 175 residues in C. higginsianum and C. graminicola, respectively) and are more cysteine rich than the total proteome (Supplementary Fig. 9b). CSEP-encoding genes are randomly distributed across the chromosomes of C. graminicola, with no evidence for clustering, enrichment on particular chromosomes or localization near transposable elements or telomeres, as has been reported for some other plant pathogens8,20,21,22 (Supplementary Note). An MCL analysis revealed that relatively few Colletotrichum CSEPs (14% in both species) belong to small multigenic families with two to five members (Supplementary Fig. 9c). The larger, more diversified CSEP repertoire of C. higginsianum might be an adaptation to invade a broader range of host plants than C. graminicola, which is restricted to infection of Zea under field conditions23.
Both Colletotrichum species encode markedly more secondary metabolism enzymes (103 in C. higginsianum and 74 in C. graminicola) than other sequenced fungi (2–58 in ascomycetes24,25) (Supplementary Fig. 10a and Supplementary Table 8). In fungi, secondary metabolism genes are typically located in clusters26; we found 42 of these clusters in C. graminicola and 39 in C. higginsianum, surpassing the numbers found in most other sequenced ascomycetes (Supplementary Fig. 10b). Only 11 secondary metabolism gene clusters are shared between the two Colletotrichum species (Supplementary Table 12), and only 6 of these clusters show limited synteny (Fig. 4). This cluster diversity seems to result from gene duplication or loss and chromosomal rearrangements and may be related to the association of secondary metabolism gene clusters (71% in C. graminicola) with repetitive DNA (Supplementary Note). Because each secondary metabolism gene cluster is probably involved in the biosynthesis of a specific metabolite24, each Colletotrichum species can be expected to produce unusually large and divergent spectra of secondary metabolites, some of which may be previously unknown bioactive molecules.
Figure 4. Structure and transcription of a secondary metabolism gene cluster.
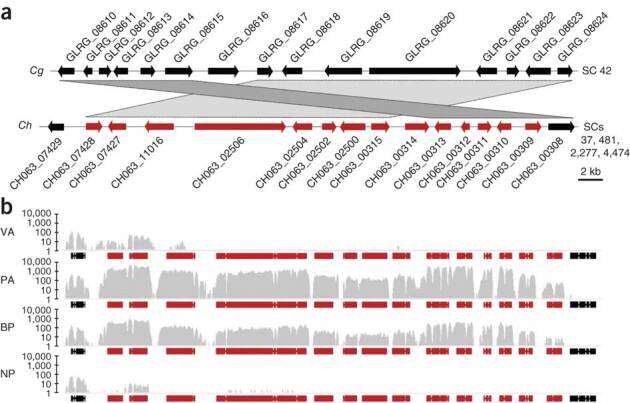
(a) Gene cluster 18 from C. graminicola (Cg) is orthologous to cluster 10 from C. higginsianum (Ch). The latter is split between four small scaffolds in the Broad Institute genome annotation (supercontigs (SCs) 37, 481, 2,277 and 4,474) and was reconstructed based on an improved genome assembly (Supplementary Note). Microsynteny is indicated by gray bars. The 14 genes highlighted in red in the C. higginsianum cluster are co-regulated. Functional annotation for the cluster genes is provided in Supplementary Table 12. (b) Visualization of RNA-Seq coverage across the C. higginsianum polyketide biosynthesis cluster. The gray curves indicate read coverage (log scale) for the four samples. Co-regulated gene models are highlighted in red. VA, in vitro appressoria; PA, in planta appressoria; BP, biotrophic phase; NP, necrotrophic phase.
To investigate how the fungal genetic program is deployed during host infection, we applied Illumina RNA sequencing to both pathosystems (Supplementary Tables 13 and 14). We collected samples from infected Arabidopsis or maize leaves at intervals corresponding to pre-penetration appressoria, the early biotrophic phase and the transition to necrotrophy and from C. higginsianum appressoria formed in vitro (Fig. 5a). Almost all the gene models were transcribed in planta (14,972 C. higginsianum genes, or 92%, and 10,812 C. graminicola genes, or 90%). However, this transcription was highly dynamic, particularly in C. higginsianum, where 7,162 genes (44%) were differentially regulated (log2 fold change >2, P < 0.05) between one or more of the infection stages (Supplementary Tables 15a and 16). Fewer genes (2,619, or 22%) were differentially regulated in C. graminicola, which may reflect the contrasting biology of this species, where biotrophic and necrotrophic growth occur simultaneously (Supplementary Tables 15b and 17). The more clearly defined infection stages of C. higginsianum provided better temporal and spatial resolution of expression changes, and we therefore highlight our results for this species.
Figure 5. Expression profiling of pathogenicity-related genes in C. higginsianum.
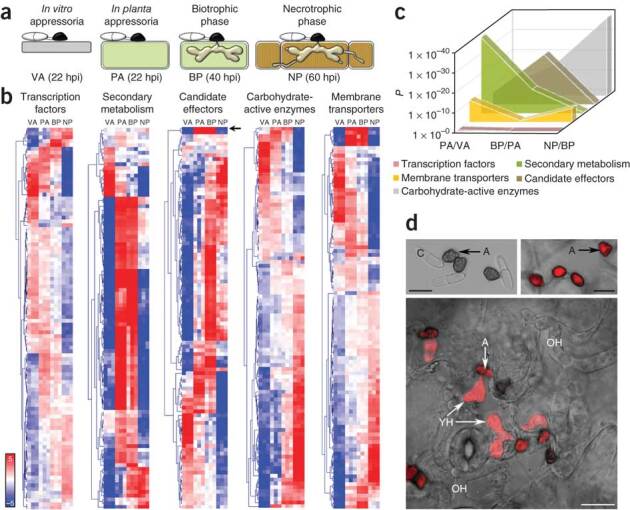
(a) Schematic representation of the four C. higginsianum developmental stages selected for RNA sequencing. Gray indicates polystyrene, green indicates living plant cell, and brown indicates dead plant cell. Hpi, hours post-inoculation. (b) Heatmaps of gene expression showing the 100 most highly expressed and significantly regulated genes (log2 fold change >2, P < 0.05) in five functional categories. Overrepresented (pale red to dark red) and underrepresented transcripts (pale blue to dark blue) are shown as log2 fold changes relative to the mean expression measured across all four stages. The arrow indicates the CSEP-encoding gene ChEC6 (CH063_01084). (c) The statistical significance of gene induction (y axis) in five functional categories during fungal developmental transitions (x axis). The P values were calculated using a one-sided Fisher's exact test and represent the probability of observing the number of significantly induced genes for a specific category during a transition given the total number of significantly induced genes during that transition (log2 fold change >2, P < 0.05) and the total number of genes in the category. (d) Transcriptional regulation of the effector gene ChEC6 by plant-derived signals. Confocal micrographs showing C. higginsianum expressing the mCherry reporter gene under the native ChEC6 promoter (overlays of bright-field and fluorescence channels). Appressoria (A) formed on polystyrene are unlabeled (top left), whereas those on the leaf surface (top right) have fluorescent cytoplasm. After host penetration, labeling is visible in young biotrophic hyphae (YH) but not older biotrophic hyphae (OH) (bottom). Scale bars, 10 μm. C, conidium.
Five gene categories relevant to pathogenicity (encoding transcription factors, secondary metabolism enzymes, CSEPs, CAZymes and transporters) had markedly different expression patterns during infection (Fig. 5b and Supplementary Fig. 11). We distinguished three waves of gene activation corresponding to pathogenic transitions (Fig. 5c). Among the genes upregulated at the appressorial phase were those encoding CAZymes that are predicted to degrade cutin, cellulose, hemicellulose and pectin, which may contribute to initial host penetration, together with a larger set of enzymes that potentially remodel the fungal cell wall (Fig. 5b and Supplementary Fig. 8a). However, early during infection, the transcriptome of C. higginsianum was dominated by secondary metabolism genes, with 12 different secondary metabolism gene clusters being induced before penetration and during biotrophy (Fig. 5b,c and Supplementary Table 13). This indicates previously unsuspected roles for appressoria and biotrophic hyphae in synthesizing an array of small molecules for delivery to the first infected plant cells. Because these cells initially remain alive, such molecules are probably not toxins and may instead function in host manipulation, similar to protein effectors27. Remarkably, the C. higginsianum secondary metabolism gene cluster with the strongest activation at this stage was silent in C. graminicola at all the infection stages we examined (Fig. 4 and Supplementary Fig. 12), suggesting that additional metabolite diversity is generated through transcriptional regulation.
Different sets of CSEP-encoding genes were expressed at each infection stage, but the majority of these genes were strongly induced during biotrophy (Fig. 5b,c and Supplementary Table 16). This suggests Colletotrichum requires a maximum capacity for host manipulation during intracellular colonization and that biotrophic hyphae provide a major interface for effector delivery to host cells. These specialized hyphae morphologically resemble the haustoria of obligate biotrophs, which function both as platforms for effector secretion and feeding structures for the uptake of sugars and amino acids28,29. However, we found no evidence for specific transcriptional reprogramming of nutrient transporters in C. higginsianum during biotrophy (Fig. 5b,c and Supplementary Table 16), suggesting that the biotrophic hyphae of this pathogen function primarily to deliver protein effectors and secondary metabolites to the plant cell.
Transcripts encoding a vast array of lytic enzymes are induced at the transition to necrotrophy, when the pathogen uses dead and dying host cells as a nutrient source to support rapid colonization and sporulation (Fig. 5b,c and Supplementary Fig. 8). These enzymes include 44 putative secreted proteases and 146 CAZymes that potentially cleave all major polysaccharides in the host wall (Supplementary Figs. 8 and 11). Concomitantly, numerous genes encoding plasma membrane transporters that may be required for assimilating the products of this degradative activity, for example, oligopeptides, amino acids and sugars, are also induced (Fig. 5b,c). In fungi, genes encoding secreted proteases, CAZymes and permeases are often subject to pH regulation30. Consistent with this, we found evidence that necrotrophy in C. higginsianum is associated with local alkalinization of Arabidopsis tissue, probably resulting from fungal ammonia secretion31, but tissue alkalinization was less pronounced in maize colonized by C. graminicola at this stage (Supplementary Fig. 13).
Notably, although appressoria in vitro are morphologically indistinguishable from those in planta, their transcriptomes are substantially different, with 1,515 genes significantly induced by host contact (Supplementary Table 15). One of these, the CSEP-encoding gene called ChEC6 (CH063_01084)32, is the most highly and significantly induced of all C. higginsianum genes (>50,000-fold) compared to appressoria in vitro. To experimentally verify this expression pattern at the cellular level, we generated transgenic C. higginsianum strains expressing a reporter gene under the control of the ChEC6 promoter (Fig. 5d). Using this method, we confirmed that the transcription of ChEC6 was plant specific, starting in the appressorium before penetration and continuing in young biotrophic hyphae, but it was switched off before the hyphae were fully expanded, indicating that its expression is transient and tightly regulated. The large-scale reprogramming of appressorial gene expression in planta shows that these specialized cells are highly responsive to host-derived cues that are perceived before penetration. Long regarded as organs of attachment and penetration33, our findings assign a previously unsuspected sensory function to fungal appressoria, enabling the pathogen to prepare for the subsequent invasion of living host cells.
Major hemibiotrophic plant pathogens such as Colletotrichum and the rice blast fungus Magnaporthe oryzae undergo major transformations in cell morphology and infection mode when switching from growth on the plant surface to intracellular biotrophy and from biotrophy to necrotrophy. Genome sequencing combined with high-throughput transcriptome sequencing revealed the transcriptional dynamics underlying these transitions and led us to redefine the functions of appressoria and intracellular hyphae. Despite their similar morphologies, a genomic comparison of C. higginsianum and C. graminicola uncovered major differences in their gene content. We propose that the diversification of functions required for host interaction, notably, the secretion of small-molecule and protein effectors and the degradation of plant polymers, allows C. higginsianum to colonize a wider range of plant species. In contrast, C. graminicola, a pathogen that is adapted to a narrow range of hosts, has maintained a more targeted arsenal of virulence factors.
URLs.
Broad Institute Colletotrichum Genome Database, http://www.broadinstitute.org/annotation/genome/colletotrichum_group; Max Planck Institute for Plant Breeding Research Fungal Genomes Database, http://www.mpipz.mpg.de/14157/fungal_genomes; Fungal Transcription Factor Database, http://ftfd.snu.ac.kr/; Fungal Cytochrome P450 Database, http://p450.riceblast.snu.ac.kr/species.php; Transporter Classification Database, http://www.tcdb.org/; Saccharomyces Genome Database, http://www.yeastgenome.org/; NCBI Conserved Domains Database, http://www.ncbi.nlm.nih.gov/cdd; SAMtools, http://samtools.sourceforge.net/; Broad Institute Integrative Genomics Viewer (IGV) browser, http://www.broadinstitute.org/igv/; Geneious v5.5., http://www.geneious.com/; RepeatMasker Open-3.0, http://www.repeatmasker.org/; InterProScan, http://www.ebi.ac.uk/Tools/pfa/iprscan/; ARS Fungal Databases, http://nt.ars-grin.gov/fungaldatabases/.
Methods
Sequencing and assembly.
C. graminicola strain M1.001 (M2) was collected in Missouri from infected maize (Fungal Genetics Stock Center culture 10212). C. higginsianum strain IMI349063 was isolated from Brassica campestris in Trinidad and Tobago (CABI culture collection, Wallingford, UK). The genome assemblies of C. graminicola were generated at the Broad Institute by combining data from Sanger and 454 pyrosequencing using a Newbler hybrid approach. Paired-end reads from 468,734 plasmids and 67,151 fosmids improved the continuity of the assembly (Supplementary Table 2). In the assembled genome, more than 98.5% of the sequence bases had quality scores >40. The C. higginsianum genome assembly was generated by GATC Biotech AG (Konstanz, Germany) by combining 454 GS-FLX shotgun reads and Illumina GAII mate-pair reads. Additionally, 864 fosmids were end-sequenced with Sanger technology. After removing dinucleotide repeats, the 454 reads and the fosmid end sequences were coassembled using the SeqMan NGen assembler (DNAStar Inc., USA). Contigs were then sorted into scaffolds using the paired-end information derived from an Illumina 3-kb–insert mate-pair library (2 × 36 bp reads). Scaffolds were manually edited to correct falsely joined contigs and falsely arranged scaffolds. To correct homopolymer sequencing errors in the 454 data, the Illumina GA data (76-fold coverage) were mapped to the scaffolded contigs, and the depth of coverage was used to create a final corrected consensus sequence (Supplementary Table 2).
Gene annotation.
A total of 28,424 expressed sequence tags (ESTs) from two C. graminicola complementary DNA libraries and 828,592 ESTs from six C. higginsianum libraries were used to provide a training set for the gene-calling pipeline and for validating the gene models (Supplementary Note). Protein-coding genes were annotated in C. graminicola using multiple lines of evidence from BLAST, PFAM searches and EST alignments, as described previously20. Gene structures were predicted using the Broad Institute automated gene-calling pipeline35 based on a combination of gene models predicted by the programs FGENESH (Softberry Inc., USA), GENEID36, GeneMark37, SNAP38 and Augustus39 together with EST-based and manually curated gene models. GENEID, FGENESH, SNAP and Augustus were trained using a set of high-confidence EST-based gene models generated by clustering Blat-aligned species-specific ESTs. By combining BLAST, EST and ab initio predictions, annotators manually built additional gene models that were otherwise missed by the automated annotation. C. graminicola was predicted to have 12,006 gene models, 39% of which were verified by the alignment of 13,600 Sanger EST reads. The C. higginsianum gene set was created similarly and was filtered using TBLASTN alignments from 10,661 of the C. graminicola gene models (<1 × 10−10). Another 1,564 gene models were based on evidence from C. higginsianum ESTs, and 600 were based on EVidenceModeler (EVM) models having BLAST hits to proteins in the UniRef90 database. C. higginsianum was predicted to have 16,172 protein-coding genes, 89% of which were validated by the alignment of 135,923 ESTs from 454 sequencing.
Optical mapping.
C. graminicola and C. higginsianum protoplasts40 were lysed and prepared for optical mapping41 using MluI (with an average fragment size for both genomes of 9.2 kb). Raw datasets comprising single DNA molecule maps (Rmaps; 300× coverage per genome) were assembled into genome-wide contig maps spanning each chromosome using divide-and-conquer41 and iterative assembly strategies42. PROmer from the MUMmer package43 was used to conduct pairwise comparisons between the C. graminicola and C. higginsianum genomes (Fig. 2a and Supplementary Table 4). The synteny map (Fig. 2b) was generated using the Argo browser44.
Transposable element analysis.
Repetitive DNA elements were identified by performing a self BLASTN of each genome and processing the output with a custom Perl script (available on request), which identified multicopy sequences and organized them into nonredundant families. Consensus sequences of these families were then used to generate a custom library for RepeatMasker (see URLs) to scan both genome assemblies. The distributions of the genes, the transposable elements and the GC content were examined within a 100-kb window, sliding 10 kb across each chromosome.
Orthology and multigene families.
To identify differences in gene family size between C. graminicola and C. higginsianum, we clustered their proteomes using the Markov clustering program MCL11. An all-versus-all BLASTP search was performed using default parameters, followed by clustering with MCL using an inflation value of 2.0. We also included the proteomes of 13 additional fungal species (Fig. 3). Sequences were aligned using MAFFT45, and phylogenetic trees were constructed using the neighbor-joining method, followed by a bootstrap test with 100 replications. Sequence editing and alignment and phylogenetic analyses were performed using Geneious Pro (version 5.5; see URLs).
Annotation of specific gene categories.
Secretomes of both Colletotrichum species were predicted using WoLF-PSORT46. CSEPs were defined as extracellular proteins with no significant BLAST homology (expect value <1 × 10−3) to sequences in the UniProt database (SwissProt and TrEMBL components). Homologs of proteins from outside the genus Colletotrichum were excluded. Genes encoding putative carbohydrate-active enzymes were identified using the CAZy annotation pipeline13. To identify secreted peptidase genes, sequences of predicted extracellular proteins were subjected to a MEROPS Batch BLAST analysis47. Membrane transporters were identified from BLAST searches against the Transporter Collection Database (see URLs) and Saccharomyces Genome Database (see URLs). Secondary metabolism genes were initially identified using MCL and gene family searches using the Broad Institute Colletotrichum database, BLAST searches against GenBank and InterproScan analysis (see URLs). The Secondary Metabolite Unknown Region Finder (SMURF)26 was used to predict secondary metabolism gene clusters. SMURF was applied to the Velvet assembly of C. higginsianum (Supplementary Note). Candidate genes identified using automated searches were inspected manually, including protein sequence alignments to known enzymes and searches against the NCBI Conserved Domain Database (see URLs). Further details of the secondary metabolism gene annotation are presented in the Supplementary Note. The Fungal Cytochrome P450 Database (see URLs) and Fungal Transcription Factor Database (see URLs) were used to annotate cytochrome P450 enzymes and transcription factors, respectively.
Whole-genome transcriptome profiling.
Arabidopsis leaves infected by C. higginsianum were obtained as described previously48. Sampling and RNA isolation of the pre-penetration stage (22 h hpi), the early biotrophic stage (40 hpi), the switch between biotrophy and necrotrophy (60 hpi) and in vitro appressoria (22 hpi) have been described previously32,49. Each experimental repetition of the in planta stages was based on RNA extracted from ∼300 leaves. Maize leaf sheaths infected by C. graminicola were obtained as described previously50. Sheaths from the maize inbred line Mo940 at the V3 stage were cut into 5-cm–long segments and inoculated with two 10-μl drops of spore suspension (5 × 105 spores per ml). Sheaths containing mature pre-penetration appressoria (24 hpi), intracellular biotrophic hyphae (36 hpi) and necrotrophic hyphae with water-soaked lesions (60 hpi) were sampled. Each leaf sheath was trimmed to include only the inoculated area, and total RNA was extracted as described previously51 (15 maize sheaths per experimental repetition). The RNA integrity of all samples was verified on an Agilent 2100 Bioanalyzer.
Twelve C. higginsianum libraries (four developmental stages and three biological replicates) and nine C. graminicola libraries (three developmental stages and three biological replicates) were prepared with the Illumina TruSeq RNA Sample Preparation Kit and sequenced using the Illumina Genome Analyzer IIx (single reads, 100 bp for C. higginsianum and 76 bp for C. graminicola). Further details are provided in the Supplementary Note. The RNA-Seq reads were mapped to the annotated genomes with TopHat (a = 10, g = 5)52 and transformed into counts per annotated gene per sample with the 'coverageBed' function from the BEDtools suite53 and custom R scripts. Differentially expressed genes between two developmental stages were detected using the 'exactTest' function from the R package EdgeR54. To calculate fold changes, the number of reads for each gene in each library was normalized by the total number of mapped reads for the library, and direct ratios (log2) were calculated between the different developmental stages. Transcripts with a significant P value (<0.05) and more than a twofold change (log2) in transcript level were considered to be differentially expressed. All P values were corrected for false discoveries resulting from multiple hypothesis testing using the Benjamini-Hochberg procedure. Heatmaps of gene expression profiles were generated with the Genesis expression analysis package55. All codes for the RNA-Seq processing are available upon request. The C. higginsianum RNA-Seq data were also mapped onto the unannotated Velvet genome assembly (Supplementary Note) using bowtie56 and visualized with SAMtools (see URLs) and the IGV browser (see URLs). RNA-Seq expression profiles were validated by quantitative RT-PCR (Supplementary Note).
Molecular phylogeny and evolutionary divergence date estimation.
A whole-genome cladogram showing the phylogenetic relationships of C. graminicola and C. higginsianum to 17 other sequenced fungi was constructed with CVTree34 (Fig. 1). A phylogeny was generated for the genus Colletotrichum based on sequencing five genes in 28 selected isolates (Supplementary Fig. 5), as described in the Supplementary Note. To estimate the evolutionary divergence date for C. graminicola and C. higginsianum, a phylogenetic analysis was performed using the 13 species shown in Supplementary Figure 4. The proteomes were clustered using MCL, and proteins in each cluster were aligned using MUSCLE. Sixty-four clusters containing only one protein from each species and having at least 80% average pairwise nucleotide identity were used for further analyses. Sequence alignments were concatenated, and a phylogenetic tree was constructed with MrBayes57 using the WAG amino acid substitution model. Date estimates were computed using the program r8s58 with the nonparametric rate smoothing (NPRS) method using date estimates by Lücking et al.59.
Fluorescent reporter gene assay.
The promoter of the CSEP-encoding gene ChEC6 (CH063_01084) was fused to mCherry60 and a transcriptional terminator by overlap fusion PCR61 using the primer pairs shown in Supplementary Table 18. The genomic region between the ChEC6 start codon and the stop codon of its upstream gene (1,198 bp) was amplified with primer pair 1. The mCherry gene was amplified with primer pair 2. The transcriptional terminator of Aspergillus nidulans trpC was amplified from the plasmid pBin-GFP-hph5 with primer pair 3. After fusion, the insert was subcloned into the plasmid pENTR/D-TOPO (Invitrogen) and verified by sequencing. The insert was cut out with BamHI and EcoRI and ligated into the plasmid pBIGDR1, providing direct repeat recombination-mediated gene targeting62. A ku70 mutant of C. higginsianum strain IMI349063 (ref. 62) was used for Agrobacterium-mediated transformation3. Confocal images of transformants were obtained using a Leica TCS SP2 confocal laser scanning microscope. Excitation for imaging mCherry fluorescence was at 563 nm, and emission was detected at 566–620 nm.
Host tissue alkalinization.
The pH of the host cells during infection was measured using the cell-permeant pH-sensitive dye 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF) for analysis by epifluorescence microscopy31. Fluorescence intensity values were correlated with direct pH determinations obtained with a piercing-tip pH electrode (Eutech, Singapore). Ammonia concentrations in infected maize and Arabidopsis leaf tissues were measured using a photometric ammonium assay kit (Merck, Germany).
Accession codes.
The C. graminicola and C. higginsianum genome assemblies have been deposited in NCBI's Whole-Genome Shotgun Project with accession numbers ACOD0100000000 and CACQ0200000000, respectively. The RNA-Seq data for C. graminicola and C. higginsianum have been deposited in the NCBI Gene Expression Omnibus under GEO Series accession numbers GSE34632 and GSE33683, respectively.
Acknowledgements
This manuscript is dedicated to the memory of Robert Hanau. This work was primarily supported by US Department of Agriculture (USDA)–Cooperative State Research, Education and Extension Service (CSREES) grant 2007-35600-17829 (L.J.V., M.B.D., S.R. and M.R.T.), the Max Planck Society (R.J.O.) and Deutsche Forschungsgemeinschaft (SPP1212) grant OC 104/1-3 (R.J.O.). Other funding sources included USDA-CSREES grant 2009-34457-20125 (L.J.V.), the University of Kentucky College of Agriculture Research Office (L.J.V.), Ministerio de Ciencia e Innovación (MICINN) of Spain grants AGL2008-03177/AGR and AGL2011-29446/AGR (M.R.T.) and the Programme for Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry (P.G. and K. Shirasu). This is manuscript number 12-12-011, published with the approval of the Director of the University of Kentucky Agricultural Experiment Station. See the Supplementary Note for a full list of acknowledgments.
Supplementary Note
Life-style transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses
Inventory of membrane transporters in C. higginsianum and C. graminicola and their orthology (XLSX 137 kb)
Inventory of key secondary metabolism enzymes in C. higginsianum and C. graminicola and their orthology (XLSX 67 kb)
Inventory of secondary metabolism gene clusters in C. higginsianum and C. graminicola (XLSX 92 kb)
Genome-wide expression analysis of C. higginsianum genes measured across four developmental stages using RNA-Seq (XLSX 5061 kb)
Genome-wide expression analysis of C. graminicola genes measured across three developmental stages using RNA-Seq (XLSX 1954 kb)
Author Contributions
L.J.V., R.J.O., M.R.T., L.-J.M., P.S.-L. and M.B.D. initiated the project. R.J.O. and L.J.V. coordinated the project. M.R.T., E.V.L.v.T. and L.-J.M. coordinated bioinformatics. L.-J.M., B.W.B., C.A.B., U.D., Y.T., R.R., E.V.L.v.T., K. Stüber, H.T., J.K., L.J.V., A.D.L., J.P.D., J.A.S., C.L.S. and S.R. performed DNA and expressed sequence tag sequencing. D.H., S.Y., L.-J.M., C.A.B., E.V.L.v.T., K.N. and K. Stüber performed sequence assembly. S.Z., D.C.S., J.Z., D.H., L.-J.M., J.A.R., M.P. and S.G.A. performed optical map and synteny analyses. Q.Z., Z.C., L.A.-B., N.S., L.-J.M., E.V.L.v.T., S.G.A., N.M., M.R.T. and B.K. performed gene predictions and managed the data. E.A.B., M.R.T., S.A.S., L.E., M.F.T., R.J.O., L.J.V., K. Stüber, E.V.L.v.T., J.K. and H.C.v.d.D. manually curated gene models. U.D., M.R.T., Y.-H.L. and G.R. performed phylogeny and evolution analyses. M.R.T. and G.R. performed comparative genomics and MCL analyses. S.G.A., M.L.F., M.R.T. and E.V.L.v.T. analyzed repeats. S.H., E.V.L.v.T., J.K., H.C.v.d.D., B.K., J.A., C.B. and R.J.O. performed and analyzed C. higginsianum RNA-Seq. E.A.B., M.F.T., S.G.A., L.J.V. and M.K. performed and analysed C. graminicola RNA-Seq. L.M.V. and C.K. annotated transporters. L.J.V., M.F.T., M.-H.L., Y.K., D.G.P., R.H.P., C.L.S., S.H., R.J.O., M.R.T. and S.G.A. annotated secondary metabolism genes. B.H., L.E., H.T. and M.-H.L. annotated CAZymes and chitin-binding proteins. M.R.T., S.A.S. and S.H. annotated secretomes and effectors. P.G. and K. Shirasu annotated phytohormone genes. M.R.T. and R.J.O. annotated proteases. Y.-H.L. and J.C. annotated transcription factors and P450 proteins. J.A.C., F.T. and U.R.S. annotated mating-type genes. J.K. and I.W. performed reporter assays. D.P., N.A. and I.M. performed pH and ammonia assays. S.H., U.N., E.V.L.v.T., R.J.H., J.K., L.J.V., S.G.A., U.D., M.R.T., L.-J.M., E.A.B. and R.J.O. prepared tables, figures and text. R.J.O. wrote and edited the paper with input from L.J.V., L.-J.M., M.R.T., P.S.-L., R.P., S.H., J.K., M.-H.L., L.E., E.V.L.v.T., E.A.B. and S.G.A.
Accession codes
Primary accessions
Gene Expression Omnibus
Competing interests
The authors declare no competing financial interests.
Footnotes
Richard J O'Connell and Michael R Thon: These authors contributed equally to this work.
Contributor Information
Richard J O'Connell, Email: oconnel@mpipz.mpg.de.
Lisa J Vaillancourt, Email: vaillan@uky.edu.
References
- 1.Crous PW, Gams W, Stalpers JA, Robert V, Stegehuis G. MycoBank: an online initiative to launch mycology into the 21st century. Stud. Mycol. 2004;50:19–22. [Google Scholar]
- 2.Perfect SE, Hughes HB, O'Connell RJ, Green JR. Colletotrichum—a model genus for studies on pathology and fungal-plant interactions. Fungal Genet. Biol. 1999;27:186–198. doi: 10.1006/fgbi.1999.1143. [DOI] [PubMed] [Google Scholar]
- 3.Huser A, Takahara H, Schmalenbach W, O'Connell R. Discovery of pathogenicity genes in the crucifer anthracnose fungus Colletotrichum higginsianum, using random insertional mutagenesis. Mol. Plant Microbe Interact. 2009;22:143–156. doi: 10.1094/MPMI-22-2-0143. [DOI] [PubMed] [Google Scholar]
- 4.Narusaka M, et al. RRS1 and RPS4 provide a dual resistance-gene system against fungal and bacterial pathogens. Plant J. 2009;60:218–226. doi: 10.1111/j.1365-313X.2009.03949.x. [DOI] [PubMed] [Google Scholar]
- 5.O'Connell R, et al. A novel Arabidopsis-Colletotrichum pathosystem for the molecular dissection of plant-fungal interactions. Mol. Plant Microbe Interact. 2004;17:272–282. doi: 10.1094/MPMI.2004.17.3.272. [DOI] [PubMed] [Google Scholar]
- 6.Frey TJ, Weldekidan T, Colbert T, Wolters PJCC, Hawk JA. Fitness evaluation of Rcg1, a locus that confers resistance to Colletotrichum graminicola (Ces.) G.W. Wils. Using Near-Isogenic Maize Hybrids. Crop Sci. 2011;51:1551–1563. doi: 10.2135/cropsci2010.10.0613. [DOI] [Google Scholar]
- 7.Mims CW, Vaillancourt LJ. Ultrastructural characterization of infection and colonization of maize leaves by Colletotrichum graminicola, and by a C. graminicola pathogenicity mutant. Phytopathology. 2002;92:803–812. doi: 10.1094/PHYTO.2002.92.7.803. [DOI] [PubMed] [Google Scholar]
- 8.Rouxel T, et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat. Commun. 2011;2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaw SM, Chang CC, Chen HL, Li WH. Dating the monocot-dicot divergence and the origin of core Eudicots using whole chloroplast genomes. J. Mol. Evol. 2004;58:424–441. doi: 10.1007/s00239-003-2564-9. [DOI] [PubMed] [Google Scholar]
- 10.Amselem J, et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011;7:e1002230. doi: 10.1371/journal.pgen.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh BC, Choi WC, Park S, Kim YO, Oh TK. Biochemical properties and substrate specificities of alkaline and histidine acid phytases. Appl. Microbiol. Biotechnol. 2004;63:362–372. doi: 10.1007/s00253-003-1345-0. [DOI] [PubMed] [Google Scholar]
- 13.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Brink J, de Vries RP. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011;91:1477–1492. doi: 10.1007/s00253-011-3473-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King BC, et al. Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol. Biofuels. 2011;4:4. doi: 10.1186/1754-6834-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinlan RJ, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. USA. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beeson WT, Phillips CM, Cate JH, Marletta MA. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 2012;134:890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 18.Vogel J. Unique aspects of the grass cell wall. Curr. Opin. Plant Biol. 2008;11:301–307. doi: 10.1016/j.pbi.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Stergiopoulos I, de Wit PJGM. Fungal effector proteins. Annu. Rev. Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 20.Ma L-J, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farman ML. Telomeres in the rice blast fungus Magnaporthe oryzae: the world of the end as we know it. FEMS Microbiol. Lett. 2007;273:125–132. doi: 10.1111/j.1574-6968.2007.00812.x. [DOI] [PubMed] [Google Scholar]
- 22.Kämper J, et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101. doi: 10.1038/nature05248. [DOI] [PubMed] [Google Scholar]
- 23.Crouch JA, Beirn LA. Anthracnose of cereals and grasses. Fungal Divers. 2009;39:19–44. [Google Scholar]
- 24.Collemare J, Billard A, Bohnert HU, Lebrun MH. Biosynthesis of secondary metabolites in the rice blast fungus Magnaporthe grisea: the role of hybrid PKS-NRPS in pathogenicity. Mycol. Res. 2008;112:207–215. doi: 10.1016/j.mycres.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Spanu PD, et al. Genome expansion and gene loss in powdery mildew fungi reveal functional trade-offs in extreme parasitism. Science. 2010;330:1543–1546. doi: 10.1126/science.1194573. [DOI] [PubMed] [Google Scholar]
- 26.Khaldi N, et al. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collemare, J. & Lebrun, M.-H. Fungal secondary metabolites: ancient toxins and novel effectors in plant-microbe interactions. in Effectors in Plant-Microbe Interactions (eds. Martin, F. & Kamoun, S.) 379–402 (Wiley-Blackwell, Oxford, 2011).
- 28.Voegele RT, Mendgen KW. Nutrient uptake in rust fungi: how sweet is parasitic life? Euphytica. 2011;179:41–55. doi: 10.1007/s10681-011-0358-5. [DOI] [Google Scholar]
- 29.Catanzariti A-M, Dodds PN, Lawrence GJ, Ayliffe MA, Ellis JG. Haustorially expressed secreted proteins from flax rust are highly enriched for avirulence elicitors. Plant Cell. 2006;18:243–256. doi: 10.1105/tpc.105.035980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peñalva MA, Arst HN. Regulation of gene expression by ambient pH in filamentous fungi and yeasts. Microbiol. Mol. Biol. Rev. 2002;66:426–446. doi: 10.1128/MMBR.66.3.426-446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alkan N, Fluhr R, Sherman A, Prusky D. Role of ammonia secretion and pH modulation on pathogenicity of Colletotrichum coccodes on tomato fruit. Mol. Plant Microbe Interact. 2008;21:1058–1066. doi: 10.1094/MPMI-21-8-1058. [DOI] [PubMed] [Google Scholar]
- 32.Kleemann J, et al. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen. Colletotrichum higginsianum. PLoS Pathog. 2012;8:e1002643. doi: 10.1371/journal.ppat.1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank B. Ueber einige neue und weniger bekannte Pflanzenkrankheiten. Ber. Deutsch. Bot. Gesell. 1883;1:29–34. [Google Scholar]
- 34.Wang H, Xu Z, Gao L, Hao B. A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol. Biol. 2009;9:195. doi: 10.1186/1471-2148-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–W312. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haas BJ, Zeng Q, Pearson MD, Cuomo CA, Wortman JR. Approaches to fungal genome annotation. Mycology. 2011;2:118–141. doi: 10.1080/21501203.2011.606851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parra G, Blanco E, Guigó R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borodovsky M, McIninch J. GeneMark: parallel gene recognition for both DNA strands. Comput. Chem. 1993;17:123–133. doi: 10.1016/0097-8485(93)85004-V. [DOI] [Google Scholar]
- 39.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thon MR, Nuckles EM, Vaillancourt LJ. Restriction enzyme-mediated integration used to produce pathogenicity mutants of Colletotrichum graminicola. Mol. Plant Microbe Interact. 2000;13:1356–1365. doi: 10.1094/MPMI.2000.13.12.1356. [DOI] [PubMed] [Google Scholar]
- 41.Zhou S, et al. Validation of rice genome sequence by optical mapping. BMC Genomics. 2007;8:278. doi: 10.1186/1471-2164-8-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou S, et al. A single molecule scaffold for the maize genome. PLoS Genet. 2009;5:e1000711. doi: 10.1371/journal.pgen.1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engels R, et al. Combo: a whole genome comparative browser. Bioinformatics. 2006;22:1782–1783. doi: 10.1093/bioinformatics/btl193. [DOI] [PubMed] [Google Scholar]
- 45.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horton P, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38:D227–D233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahara H, Dolf A, Endl E, O'Connell R. Flow cytometric purification of Colletotrichum higginsianum biotrophic hyphae from Arabidopsis leaves for stage-specific transcriptome analysis. Plant J. 2009;59:672–683. doi: 10.1111/j.1365-313X.2009.03896.x. [DOI] [PubMed] [Google Scholar]
- 49.Kleemann J, Takahara H, Stüber K, O'Connell R. Identification of soluble secreted proteins from appressoria of Colletotrichum higginsianum by analysis of expressed sequence tags. Microbiology. 2008;154:1204–1217. doi: 10.1099/mic.0.2007/014944-0. [DOI] [PubMed] [Google Scholar]
- 50.Kankanala P, Czymmek K, Valent B. Roles for rice membrane dynamics and plasmodesmata during biotrophic invasion by the blast fungus. Plant Cell. 2007;19:706–724. doi: 10.1105/tpc.106.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metz RP, Kwak H-I, Gustafson T, Laffin B, Porter WW. Differential transcriptional regulation by mouse single-minded 2s. J. Biol. Chem. 2006;281:10839–10848. doi: 10.1074/jbc.M508858200. [DOI] [PubMed] [Google Scholar]
- 52.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 56.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 58.Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- 59.Lücking R, Huhndorf S, Pfister DH, Plata ER, Lumbsch HT. Fungi evolved right on track. Mycologia. 2009;101:810–822. doi: 10.3852/09-016. [DOI] [PubMed] [Google Scholar]
- 60.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 61.Szewczyk E, et al. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 2006;1:3111–3120. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- 62.Ushimaru T, et al. Development of an efficient gene targeting system in Colletotrichum higginsianum using a non-homologous end-joining mutant and Agrobacterium tumefaciens–mediated gene transfer. Mol. Genet. Genomics. 2010;284:357–371. doi: 10.1007/s00438-010-0572-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.