

Abstract
The mammalian distal nephron is a target of highly effective antihypertensive drugs. Genetic variants that alter its transport activity are also inherited causes of high or low blood pressure, clearly establishing its central role in human blood pressure regulation. Much has been learned during the past 25 years about salt transport along this nephron segment, spurred by the cloning of major transport proteins and by the discovery of disease-causing genetic variants. There is increasing recognition of substantial cellular and segmental heterogeneity along this segment, with electroneutral sodium transport dominating more proximal segments and electrogenic sodium transport dominating more distally. Coupled with recent insights into factors that modulate transport along these segments, we now understand one important mechanism by which dietary potassium intake influences sodium excretion and blood pressure. This has solved the aldosterone paradox, by demonstrating how aldosterone can be kaliuretic when plasma potassium is elevated and anti-natriuretic, when extracellular fluid volume is low. It has also become clear, however, that aldosterone itself only stimulates a portion of the mineralocorticoid receptors along this segment, with the others being activated by glucocorticoid hormones instead. These recent insights provide an increasingly clear picture of how this short nephron segment contributes to blood pressure homeostasis and provide important implications for hypertension prevention and treatment.
Introduction
The moniker mammalian distal nephron, an imprecise but practical term, is commonly used to describe kidney tubule segments that include the distal convoluted tubule, the connecting tubule, and the initial portion of the collecting duct. Originally defined anatomically and later studied physiologically, this segment has most recently been interrogated for patterns of protein and transcript expression. Yet, the new information must be integrated with established anatomical and physiological knowledge and not viewed in isolation, to gain a full understanding of distal nephron structure and function 1. As the distal nephron is both a target of highly effective antihypertensive drugs and a site at which genetic variants disrupt blood pressure homeostasis, its contributions to human hypertension are clearly established. Here, we will provide a contemporary discussion of its structure, function, and modes of regulation. While this segment has often been thought to be synonymous with the aldosterone-sensitive distal nephron, we will also review recent data suggesting that only a fraction of mineralocorticoid receptors along this segment is sensitive to aldosterone.
Anatomy and molecular characteristics
The distal convoluted tubule (DCT), which arises during development from the mesenchymal mesenchyme, arises a short distance beyond the macula densa region of the thick ascending limb, and is identified by an increase in cell height; DCT cells have apically-oriented nuclei, extensive basolateral membrane infoldings, lateral interdigitations, and abundant palisading mitochondria. The connecting tubule (CNT) lies distal to the DCT. Connecting tubules of juxtamedullary nephrons form arcade structures, with several connecting tubules joining together, whereas connecting tubules of superficial nephrons are uninterrupted. The CNT leads into the collecting duct (CD), a branched structure that derives from the ureteric bud. In all mammalian species, the transition from thick ascending limb (TAL) to DCT is abrupt. In rabbit, the junctions between DCT and CNT, and CNT and CD, are also abrupt, while in rodents and humans, these transitions are gradual.
Fluid entering the DCT is dilute, with a concentration of NaCl below that of extracellular fluid (ECF). The DCT reabsorbs NaCl against its electrochemical gradient and is also water-impermeable. The CNT and CD also reabsorb Na+ actively, but in this case, the transport is primarily electrogenic and coupled functionally to the secretion of K+ and H+. Along these segments, chloride is reabsorbed transcellularly by intercalated cells and across paracellular tight junctions.
DCT cells and principal-like cells of the CNT and CD all have abundant Na+/K+-ATPase along their basolateral surface, which maintains intracellular sodium concentration low and potassium concentration high2. Owing to basolateral potassium conductance, the inside of the cell is electrically negative relative to the outside; the voltage across the epithelium is near zero along the DCT, becoming more lumen negative along CNT and CD3.
Knepper and colleagues recently proposed transcriptomic ‘Gold Standard’ markers for cell types along the nephron1. Along the distal nephron, apical sodium and water transport proteins dominate these markers. The predominant Na+ entry pathway along the DCT is the electroneutral NaCl cotransporter (NCC, encoded by SLC12A3), the expression of which is limited to the DCT1. Principal-like cells of the CNT and CD share many features, including apical Na+ channels (ENaC, a trimeric channel encoded by SCNN1 α, β, and γ) and water channels (aquaporin-2, encoded by AQP2), at least in a subpopulation. Additionally, the potassium channel ROMK (encoded by KCNJ1) is expressed all the way from the TAL to the CD 4, although ROMK activity cannot be detected at the apical membrane of DCT1 cells5. The CNT is also differentiated from the CD both anatomically and because of its high expression of calcium transport proteins, including the calcium channel, TRPV5, and the Na/Ca exchanger (encoded by Slc8a1) 6,7.
In humans, rats, and mice, transitional segments along the distal nephron, have been identified. The first molecular segmentation defined a DCT1 and DCT2 (also called ‘early’ and ‘late’ distal convoluted tubules) 7. Both subsegments express NCC, but the DCT2 also expresses the epithelial sodium channel (ENaC), the sodium-calcium exchanger (NCX) and calbindin-D28K, proteins typically observed along the CNT1,8. Welling and colleagues noted that the CNT itself could be divided into a portion that lacks AQP2 (they named it CNT1) and a portion that expresses it (CNT2) 4, a finding more recently supported by fate-mapping studies 9. Recently, Yang and colleagues proposed that the DCT2 and CNT1 be designated as CNTe (for early CNT) and the CNT2 as CNTas (for CNT aldosterone-sensitive), based on ENaC activity responsiveness to aldosterone 10–13. According to this nomenclature, part of the CNTe expresses NCC and the other part does not. While new results are creating a richer picture of the distal nephron, they can lead to confusion. Figure 1A compares the traditional and alternative naming schema. Because NCC has been used as the anchor marker for DCT1, we will use the terms DCT2, CNT1, and CNT2 in this paper.
Figure 1.

(A) Expression sites for key transporters and regulatory factors along the distal nephron. Traditional and alternative naming conventions of distal nephron segments are shown (alternative convention proposed by Yang et al.18). TAL, thick ascending limb; DCT, distal convoluted tubule; CNT, connecting tubule; CNTe, early CNT; CNTas, aldosterone sensitive CNT; CCD, cortical collecting duct; NKCC2, Na+:K+:Cl- cotransporter NKCC; NCC, Na+:Cl- cotransporter; γENaC, Epithelial Na+ channel ENaC, γ subunit; AQP2: aquaporin 2; 11HSD2: 11-beta-hydroxysteroid dehydrogenase type 2; MR, mineralocorticoid receptor. (B) Changes in NCC activity affect the structure of the distal nephron. Increased activity of NCC is associated with DCT growth and deceased CNT volume, whereas decreased NCC activity is associated with reduced DCT volume and CNT/CD growth.22,31,67,69 (C) DCT model showing NCC regulation by extracellular potassium, ([K+]ec, see text for details). Adrenal zona glomerulosa model showing [K+]ec also directly stimulates aldosterone secretion. (D) Hormones that increase intracellular [cAMP] stimulate NCC activity through signaling pathways that depend on PKA activation. Active PKA phosphorylates WNK4 in sites that promote kinase activation. In addition, PKA phosphorylates the PP1 regulatory subunit I1, a phosphorylation event that allows I1 to repress PP1 activity towards WNK4 and perhaps towards SPAK/OSR1 and NCC.
Along the CNT and CD, intercalated cells are interspersed between the major cell type. Intercalated cells are typically categorized as A, B, and non-A non-B cells. Lineage tracing suggests that intercalated cells may arise from a progenitor cell that also develops into principal-like cells9; recent transcriptomic data have suggested that some cells expressing both principal and intercalated cell genes represent a transitional cell type14. Although intercalated cells are most abundant along the CNT and CD, they also may be found along portions of the DCT2. In addition to modulating acid/base balance, intercalated cells are now recognized as playing an important role in chloride reabsorption and ECF volume 15.
Transport
Fluid entering the DCT has a NaCl concentration of approximately 40–60 mM, regardless of salt intake16,17. Approximately 65% of the delivered Na+ load is reabsorbed along the DCT and CNT, with approximately two thirds of that occurring along the DCT18. Along this segment, Na+ and Cl- are reabsorbed electroneutrally, with reabsorption by DCT1 mediated overwhelmingly by NCC19.
DCT cells do not express AQP220, so tubule fluid dilution continues along this segment. Little potassium is secreted along the DCT121; in contrast, potassium secretion is robust along DCT2 and CNT22. Importantly, although mineralocorticoid receptors are expressed along the DCT, the enzyme 11-β-hydroxysteroid dehydrogenase 2, which enables aldosterone to stimulate ENaC, is expressed either at very low levels 23, or not at all (Figure 1A)24.
Sodium reabsorption along the CNT is primarily electrogenic, generating a transepithelial voltage oriented with the lumen negative, relative the blood. The major salt transport mechanisms found along the CNT also extend into the CD. Although ENaC appears to be structurally identical along CNT and CD, many channels in more proximal segments are constitutively active, whereas channels in more distal segments depend on stimulation by aldosterone to be active 11,12,25.
Na+ reabsorption in the distal convoluted tubule
NCC activity is regulated by a diverse set of signals that not only modulate Na+ homeostasis, but also impact K+ secretion in downstream segments 26,27. Here, we will first describe how rare mendelian diseases have identified important transporters and their regulation. Then, we will discuss more common acquired diseases of these nephron segments.
Genetic disease of the DCT
Gitelman syndrome (GS), caused by loss-of function genetic variants in the SLC12A3 gene (encoding NCC), results in urinary salt wasting, hypochloremia, hypokalemia, metabolic alkalosis, hypomagnesemia, and hypocalciuria 28. Altered function of proteins that act in concert with NCC to mediate Na+:Cl- reabsorption in the DCT also produce (GS)-like tubulopathies. Basolateral Cl- exit from DCT cells occur through the Cl- channel ClC-Kb (ClC-K2 in mice) 29. Basolateral membrane K+ conductance is mediated by heterotetrameric K+ channels, Kir4.1 and Kir5.1 (encoded by KCNJ10 and KCNJ16, respectively),30–32 which modulate the basolateral membrane voltage of the DCT, which indirectly affects Cl- exit though ClC-Kb. In addition, Kir4.1/Kir51 activity is also important for the basolateral recycling of K+ that enters the cell through the Na+:K+ ATPase. Thus, disruptive genetic variants in CLCNKB and KCNJ10 negatively affect Na+:Cl- reabsorption in the DCT and produce Gitelman-like electrolyte disturbances 29,32–36. More recently, genetic variants affecting other genes have been shown to produce Gitelman-like phenotypes in humans. These genes include KCNJ16 and the mitochondrial genes encoding for mitochondrial transfer RNAs MT-TI and MT-TF. For a recent review see 37.
Activation of NCC occurs in another genetic disease known as Familial Hyperkalemic Hypertension (FHHt) (also known as Gordon syndrome or Pseudohypoaldosteronism type 2) that presents with a phenotype opposite to GS: hypertension, hyperchloremia, hyperkalemia, metabolic acidosis, and hypercalciuria 38. Genetic variants in four different genes can give rise to FHHt and the characterization of the role played by the encoded proteins has elucidated a previously-unrecognized mechanism by which NCC is regulated 38–41.
The With No lysine (K) (WNK) kinases are atypically large kinases that phosphorylate and promote activation of two paralogous kinases called STE20-Proline Alanine rich Kinase (SPAK, STK39) and the Oxidative Stress Responsive kinase 1 (OSR1, gene symbol OXSR1) 42–44. Both kinases can directly phosphorylate NCC in vitro, but SPAK is primarily responsible in vivo45,46. The WNK kinases are degraded by a Cullin-Ring-Ligase (CRL) complex that includes Cullin 3 (CUL3).47–49 The complex, via kelch-like protein 3 (KLHL3), interacts with WNK kinase acidic motifs to promote WNK ubiquitylation and proteasomal degradation. Thus, FHHt-causative genetic variants that affect WNK4 and WNK1 in the acidic motifs lead to WNK accumulation in the DCT, where KLHL3 expression is enriched, increasing SPAK and NCC phosphorylation.
Interestingly, variants affecting the acidic domain of WNK1 likely produce overexpression of the short, kidney-specific and kinase-deficient form of WNK1 (known as KS-WNK1) because KS-WNK1 is especially sensitive to KLHL3-targeted degradation41,50 and L-WNK1 transcript levels are barely measurable in the DCT 51,52. In vitro, KS-WNK1 co-expression with WNK4 promotes WNK4’s phosphorylation and NCC activation 53.
A second type of FHHt-causative variants in WNK1 are large deletions within intron 1 that produce the ectopic expression of L-WNK1 in the DCT 38,54. Ectopic expression of kinase-active L-WNK1 in the DCT stimulates NCC directly. This view is supported by the observation that WNK4 knockout mice lack NCC activity, yet convert to an FHHt phenotype when crossed with mice that express L-WNK1 ectopically 55.
Finally, genetic variants in KLHL3 and CUL3 also lead to FHHt 47,49,50. Variants in CUL3 are particularly interesting because they affect the splicing of exon 9. Studies have suggested that this mutated Cullin3 protein loses its activity to mediate WNK kinase degradation, but also promotes degradation of the substrate adaptor, KLHL3, thus decreasing the amount of KLHL3, but increasing the amount of WNK456. Only one FHHt-causative mutation in CUL3 has been reported that does not affect splicing of exon 9. This variant lacks amino acid residues 474–477, a deletion that appears to reduce CUL3 levels due to autoubiquitylation and promote the formation of KLHL3 complexes that are unable to ubiquitylate WNKs 57.
Despite the absence of net K+ transport in the DCT, the most notable feature resulting from disrupted DCT function is altered K+ balance. For instance, the milder form of FHHt, due to variants in the acidic motif in WNK1, presents with hyperkalemia and normal blood pressure41. Changes in NCC activity have been shown to indirectly affect K+ secretion by the contiguous K+ secretory segments (see next section). Interestingly, relatively recent data suggest that this effect is at least in part due to nephron remodeling events that occur in the distal nephron probably triggered by altered Na+ delivery to the CNT/CD and altered Na+ transport in the DCT 27,58. A decrease in NCC activity reduces the DCT volume and promotes growth of the CNT/CCD, whereas an increase in NCC activity is associated with DCT growth and decreased CNT/CCD volume 17,27,59–61 (Figure 1B). Thus, the observation that NCC phosphorylation and activity is exquisitely modulated in response to altered K+ intake and altered plasma [K+] concentrations62–65 led investigators to propose that the DCT serves as a K+ switch to regulate urinary K+ excretion65.
Potassium regulation of NaCl transport in the DCT
Our understanding of the molecular mechanisms regulating NCC has greatly increased in the recent years. The currently accepted model proposes that Kir4.1/Kir5.1 channels at the basolateral membrane function as the K+ sensor by facilitating changes in the basolateral membrane voltage of DCT cells in response to changes in extracellular K+ 32,65 (Figure 1C). These changes alter the driving force for Cl- movement across the membrane through ClC-Kb channels, altering intracellular [Cl-] 66,67. The activity of WNK kinases has been shown to be sensitive to changes in intracellular [Cl-] 68–70. Interestingly, WNK4 is inhibited at lower [Cl-] levels than other WNK kinases 69,70 making it able to modulate NCC, given the low intracellular chloride concentration of DCT cells67. Of note, Kir4.1/Kir5.1 activity in the basolateral membrane of DCT is also affected by dietary K+ intake,32 further affecting NCC activity. Note that [K+] is also sensed by adrenal zona glomerulosa cells to regulate aldosterone secretion (Figure 1C).
Other mechanisms contribute to NCC modulation by K+. For instance, dietary K+ deprivation enhances KLHL3 phosphorylation71, which impairs KLHL3-WNK interaction72. This phosphorylation site in KLHL3 is homologous to sites in the regulatory N- and C-terminal domains of WNK4 and can be phosphorylated in vitro by the same kinases (PKC and PKA) 73. Phosphorylation of these sites in WNK4 has been reported to increase in mice on low K+ diet 74. In vitro, these phosphorylation events have been shown to promote kinase activation, and thus, this may constitute an additional mechanism to promote maximal WNK4 activation. Activity of WNK kinases has also been shown to be directly affected by changes in K+ concentration75. Finally, phosphatases contributing to NCC dephosphorylation may be activated in conditions of high plasma [K+], as suggested by the observation that in mice harboring a Cl--insensitive WNK4 mutant, dephosphorylation of NCC is observed when mice are placed on high K+ diet for four days 76. Thus, the inhibition of WNK4 by high intracellular Cl- and the activation of phosphatases may act in concert to suppress NCC activity.
Other regulators of NCC activity
It has long been known that NCC activity is modulated by dietary NaCl intake 17,62,77. For many years, NCC was believed to be activated by aldosterone directly; yet three groups provided strong evidence that aldosterone is not a dominant factor activating NCC 78–80, and instead that plasma [K+] and AngII are responsible. pNCC increases in adrenalectomized rats infused with angiotensin II (AngII) suggested that this hormone may be the signal that promotes activation of NCC in response to low NaCl diet 81. Expression of WNK4 in the DCT was shown to be critical for AngII-induced activation of NCC77. Evidence of direct modulation by AngII on DCT primary cells has been published, 82 although AngII effects on [K+] may also contribute 83. Further research is needed to determine whether AngII can directly promote NCC activation in DCT cells
Finally, other reported regulators of NCC activity include arginine vasopressin (AVP), extracellular Ca2+, β-adrenergic agonists, insulin, and sex hormones 84. AVP and β-adrenergic agonists activate signaling pathways that increase intracellular [cAMP] and promote PKA activation (Figure 1D). Ex vivo activation of PKA in kidney slices increases NCC phosphorylation85. Phosphorylation by PKA of the PP1 regulatory subunit I1, an event that promotes PP1 inhibition, participates in NCC modulation by cAMP-stimulating hormones 85. PKA-mediated phosphorylation of WNK4-RRXS sites may also be important for WNK4 activation under these conditions. Indeed, AVP infusion promotes WNK4 RRXS phosphorylation and WNK4-dependent activation 86. Extracellular Ca2+ has been proposed to act through a similar pathway involving phosphorylation of RRXS sites in WNK4 and KLHL3. Activation of the Ca2+ sensing receptor (CaSR) in DCT cells was proposed to mediate this effect through the activation of an endogenous PKC isoform87. Another possible mechanism may involve modulation of Kir4.1/Kir5.1 by CaSR88.
Acquired disease of the DCT
Loss of function
While GS is typically defined as an inherited disease, a similar phenotype, ‘acquired GS’, is most commonly associated with Sjogren syndrome, systemic sclerosis, or systemic lupus erythematosus 89–91. The pathogenesis involves a secondary loss of NCC function as a result of interstitial nephritis, and perhaps to antibodies against the NCC protein itself 92. Interestingly, a few cases of acquired GS have been reported to occur in patients heterozygous for GS variants in SLC12A3. As the GS mutant allele frequency is estimated to be 1:100 worldwide, and more common in Asia, such a ‘two-hit’ effect should be considered in appropriate situations. As hypokalemia from Sjogren syndrome is also associated with distal renal tubular acidosis, overlap with GS may complicate the diagnosis.
Gain of function
Clinical features of FHHt can also be acquired. Diabetes mellitus can cause the syndrome of hyporeninemic hypoaldosteronism, which presents with hyperkalemia and hypertension. Additionally, there are a number of drug-induced disease processes, which enhance NCC activity. The best studied results from calcineurin inhibiting drugs, such as tacrolimus, which impair inactivation of NCC and perhaps NKCC2 93,94.
Na+ reabsorption in the CNT/CD
Sodium reabsorption through principal-like cells of the CNT and CD is electrogenic, mediated by the coupled activity of basolateral Na+/K+ ATPase and the apical Na+ channel ENaC; this generates a lumen-negative transepithelial voltage that drives K+ secretion and paracellular Cl- reabsorption. K+ secretion is mainly mediated by the apical ROMK channel expressed in these cells 4,13. This molecular machinery is also present in the late DCT (DCT2) where significant K+ secretion also takes place. The mineralocorticoid receptor (MR) is the master regulator of this Na+/K+ exchange system.
Genetic disease of the CNT/CD
In humans, loss of function variants in any of the three genes encoding ENaC subunits (α, β, or γ) cause autosomal recessive Pseudohypoaldosteronism type I (PHAI), characterized by severe salt wasting, hyperkalemia, and metabolic acidosis with high aldosterone levels 95. Interestingly, loss of function variants in the MR also cause an autosomal dominant form of PHAI, with a similar, although less severe, presentation that often fades during development 95. Similar phenotypes are observed in mice in which targeted deletion of any of the ENaC subunits or deletion of the MR is induced in adulthood specifically in the renal tubule, indicating that ENaC disfunction in the renal tubule has a profound effect on renal Na+ reabsorption and K+ secretion. Levels of cleaved α and γ ENaC (their active form) in the kidneys of kidney-specific MR knockout mice are greatly decreased 78, and amiloride-sensitive currents are significantly blunted in the DCT2 and completely abolished in the late CNT of these mice 10,12.
Aberrant activation of the MR leads to the mirror image phenotype in mice and humans. For instance, the gain of function mutation reported in humans, S810L 96, which affects the ligand binding domain of the receptor, allows aberrant activation by steroids that lack 21-hydroxyl groups, like progesterone and the classic MR antagonist spironolactone. Early onset hypertension with suppressed renin activity and aldosterone levels are observed in affected patients. Hypertension is exacerbated during pregnancy when circulating progesterone levels increase. A tendency towards lower plasma K+ levels among carriers was also reported. As suggested by the binding characteristics noted above, spironolactone is contraindicated in these patients.
Aberrant activation of MR by steroids other than aldosterone also occurs in the apparent mineralocorticoid excess (AME) syndrome caused by loss-of function variants in the gene encoding 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2). 11β-HSD2 catalyzes the conversion of cortisol to cortisone in aldosterone-responsive cells. Absence of 11β-HSD2 activity in an MR-expressing cell allows cortisol to activate MR (cortisol circulates at much higher concentrations than aldosterone and shows similar potency for MR activation). Patients present early-onset hypertension, hypokalemia, and metabolic alkalosis with suppressed renin and aldosterone levels in plasma. The signs and symptoms respond to treatment with MR antagonists (MRA)97.
The same phenotype is observed in patients with Liddle syndrome, which results from gain-of-function variants in any of the three ENaC subunits 98–101. In contrast to AME, Liddle syndrome does not respond to MRAs, but responds to the ENaC blocking drug, amiloride 102. Most cases are due to genetic variants in β or γ ENaC that result in truncated subunits lacking a portion of the intracellular C-terminus 98,99. In some patients, missense mutations are observed that affect a PPPxY motif present in the C-terminus of βENaC 103–105. In vitro, this motif is key in regulating channel activity, as it mediates the interaction with the WW domain of the ubiquitin ligase Nedd4–2 responsible for channel`s ubiquitylation that regulates its surface expression 106,107.
Aldosterone induces apical translocation of ENaC108 and a link between Nedd4–2-mediated regulation of ENaC surface expression and aldosterone was described when it was shown that the Serum and Glucocorticoid-induced Kinase 1 (SGK1), whose transcription is rapidly induced by aldosterone, can phosphorylate Nedd4–2 and prevent its interaction with ENaC in vitro 109–111 (Figure 2). This was later supported by the observation that kidney-specific, inducible SGK1 knockout mice have decreased renal levels of phosphorylated Nedd4–2, decreased levels of cleaved α and γ ENaC, and decreased total γ and β ENaC, despite increased circulating aldosterone 112. Urinary Na+ and K+ wasting were reported in these mice when on low Na+ diet 112, and hyperkalemia was observed under high K+ diet 113. However, in mice with global constitutive SGK1 deletion, direct measurement of amiloride sensitive currents showed equal increases during high K+ diet in CCDs and during aldosterone infusion in CNTs of these mice 114,115. Thus, SGK1 is central in the regulation of ENaC trafficking, but other mechanisms must also be involved.
Figure 2.

Signaling pathways implicated in ENaC regulation. The mineralocorticoid receptor (MR) plays a central role in ENaC regulation. However, MR-independent pathways also participate in the regulation of ENaC in response to hormones/signals different to aldosterone. α and γ ENaC proteolytic processing is reduced in MR and SGK1 knockout mice, suggesting that aldosterone can modulate these processes. However, the molecular mechanism underlying this regulation are still obscure. In addition, aldosterone-independent mechanisms can influence ENaC proteolytic processing (not depicted). For instance, serine proteases that are abnormally filtered in patients with nephrotic syndrome can cleave and activate ENaC. AT1, angiotensin II (AngII) receptor type 1; V2, arginine vasopressin (AVP) receptor type 2.
Regulation of Na+ reabsorption in the CNT/CD by extracellular signals
Aldosterone
In addition to regulation of SGK1 transcription, other effects of aldosterone in molecular players of principal-like cells have been reported that affect Na+ reabsorption and K+ secretion (Figure 2). For instance, aldosterone enhances the expression of the α1 and β1 subunits of the Na+:K+ ATPase 116. It also increases transcription of the αENaC subunit by direct binding of MR to a glucocorticoid response element in the genés promoter, and relieves transcriptional repression of αENaC by a protein complex formed by transcription factors Dot1a, AF9, and AF17 117.
ENaC is a unique ion channel because it is activated by proteolytic cleavage of its α and γ subunits. This form of regulation evolved as vertebrates migrated from aqueous to terrestrial habitats, suggesting an important physiological role 118. Both α and γ ENaC contain two polybasic cleavage sites within their extracellular domains that appear to have evolved independently 118. These cleavage events greatly increase the channel’s open probability by removing “Gating Relief of Inhibition by Proteolysis” (GRIP) domains involved in the channel’s inhibition by extracellular Na+ 119–121. Maximal open probability is only achieved when all sites are cleaved 122. Both sites on α are cleaved by furin, an event believed to occur in the Golgi apparatus, before the channel traffics to the plasma membrane. In contrast, the γ subunit is only cleaved on its more proximal site by furin. The distal site instead can be cleaved in vitro by a number of proteases. For instance, co-expression of Channel Activating Proteases (CAP) 1– 3 with ENaC in X. laevis oocytes promotes channel cleavage and increases ENaC-dependent Na+ uptake. Deletion of CAP1 (also known as prostasin) in the mouse’s lung reduces ENaC-mediated alveolar Na+ clearance. The protease responsible modulation of ENaC activity in the kidney in vivo has yet to be identified 123,124.
Extensive evidence shows that proteolytic processing of both α and γ subunits can be induced by aldosterone 125,126. For instance, ENaC cleavage is strikingly reduced in mice lacking mineralocorticoid receptors along the nephron 78, and substantially reduced in mice deficient in aldosterone 127,128. Despite initial evidence suggesting that CAP1 renal expression was induced by aldosterone in mice and humans 129, more recent evidence has failed to support this 130. In the kidney, CAP1 is mainly expressed in proximal tubule cells that are not responsive to aldosterone. Very recently, it was shown that deleting CAP1 in the kidney does not lead to salt-wasting or affect channel cleavage 131. Some investigators have suggested that tissue kallikrein may be an important aldosterone-induced protease, but tissue kallikrein knockout mice do not recapitulate the MR-deficient phenotype 132–134.
ENaC cleavage that is independent of aldosterone may play an pathophysiological role in the sodium retention that occurs in nephrotic syndrome. Several serine proteases enter the filtrate when the glomerulus is disrupted, and these appear to cleave and activate ENaC, either directly or indirectly 135. Trials of ENaC inhibition for nephrotic patients are currently recruiting (ClinicalTrials.gov NCT05079789).
The site of action of aldosterone
For many years the focus on ENaC and ROMK-mediated Na+ and K+ transport was placed on the CD, given the accessibility of this segment for in vitro microperfusion. As suggested by Meneton and coworkers, however, more recent evidence has demonstrated that the DCT2 and CNT have much greater Na+ and K+ transport capacities than the CD 25. Electrophysiological recordings of ENaC and ROMK currents in two different anatomical regions show that, under basal dietary conditions, the DCT2 has significantly higher ENaC activity than the late CNT2/CCD (Figure 3)10–13. This agrees with the observation that, in the DCT2/CNT1, apical localization of the channel is observed under basal conditions, whereas in the late CNT2/CCD, a more diffuse cytoplasmic signal is observed 13,136. In the DCT2/CNT1, there is little increase in ENaC-mediated currents in response to low salt diet 11,12 or high K+ diet 12,13, and this activity is not reduced in aldosterone synthase knockout mice 10, suggesting that ENaC apical translocation is not regulated by aldosterone here. This led Yang and colleagues to propose the term CNTe (early CNT) to refer to the aldosterone-unresponsive segment (Figure 1). In contrast, in the late CNT segment (and CD), basal amiloride-sensitive currents are small, but are greatly increased by low salt or high K+ diet 11–13; they are absent in aldosterone synthase knockout mouse11. Thus, this segment was termed CNTas (aldosterone sensitive CNT) by Yang et al.
Figure 3.
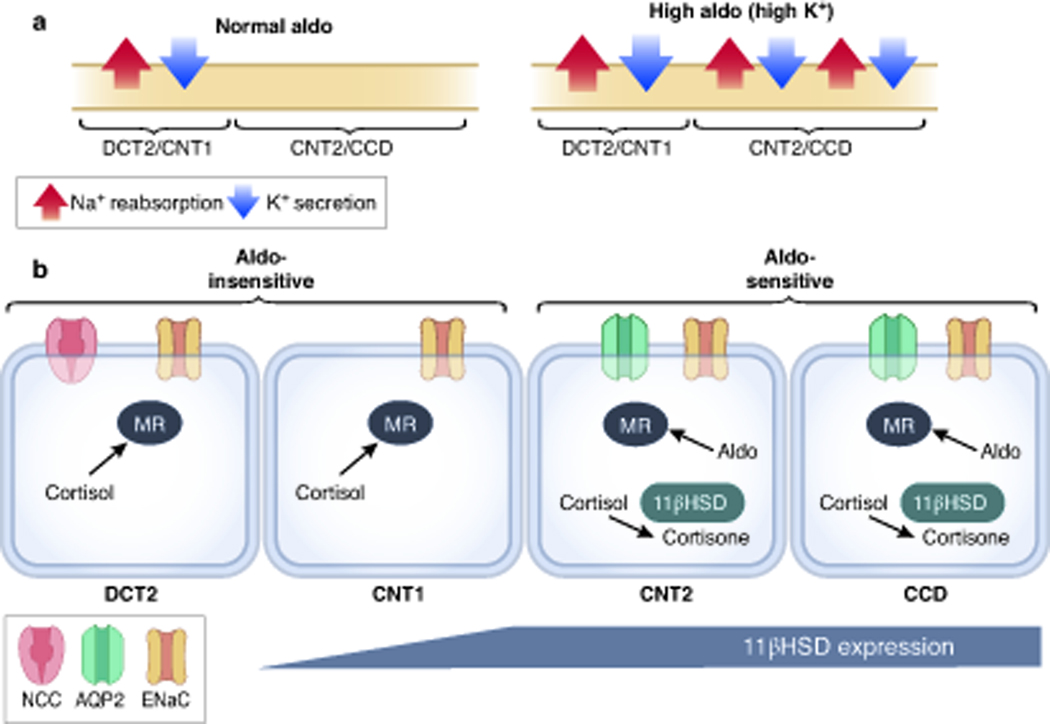
The site of action of aldosterone. (A) Under “normal” dietary Na+ and K+ intake, electrogenic Na+ reabsorption and K+ secretion mainly take place in the DCT2/CNT1 segments. Only under conditions of high circulating aldosterone, significant Na+ and K+ transport is observed in more distal segments (CNT2/CCD). (B) In the DCT2/CNT1, MR-dependent ENaC stimulation is mainly cortisol dependent (due to absence of 11βHSD). In contrast, in the CNT2 and CCD segments MR activation is aldosterone dependent.
Interestingly, in comparison with the observed effects of aldosterone deficiency on ENaC activity, two independent groups reported a significant decrease in ENaC activity in the DCT2/CNT1 of MR knockout mice, suggesting that MR regulation in this segment is aldosterone-independent 10,12. Given the absence of 11β-HSD expression in the DCT2 (Figures 1 and 3) 10,24, it was proposed that activation of MR by cortisol (corticosterone in rodents) is responsible for this constitutive ENaC activity. Recently, Maeoka and colleagues showed that MR nuclear localization was not affected along the DCT2 by depletion of aldosterone, whereas it was reduced along the CCD. They also showed that the MRA eplerenone induced natriuresis in mice lacking aldosterone and transformed their mild phenotype to one resembling that observed with MR deletion. This provides strong evidence supporting the physiologically relevant occupation of MR by glucocorticoids 127.
This analysis has largely resolved the ‘aldosterone paradox’, the observation that aldosterone primarily retains sodium, when the ECF volume is low, and causes kaliuresis, when the plasma [K+] is elevated. As shown in Figure 3A, NCC is inhibited when plasma [K+] is elevated, displacing a portion of the sodium reabsorption from the DCT to the CNT, where aldosterone (secreted in response to high plasma [K+]) stimulates potassium secretion. In contrast, when ECF volume depletion increases aldosterone, both NCC (AngII) and ENaC (Aldosterone) are active. In this situation, however, sodium-potassium exchange is limited by sodium reabsorption upstream.
MR-independent mechanisms for ENaC regulation
Multiple studies have pointed out a role of angiotensin II (AngII) in ENaC regulation 137–140. Wu and colleagues showed that AngII infusion for 3 days increased ENaC currents more prominently in the DCT2/CNT1 than in the CCDs12. Losartan treatment (an AngII receptor blocker) reduced amiloride-sensitive Na+ currents in the DCT2/CNT1, but not in the CCD. Interestingly, in mice with inducible deletion of MR in the kidney, in which amiloride-sensitive Na+ currents in the DCT2/CNT1 were still present, losartan further decreased these amiloride-sensitive currents, suggesting that AngII can modulate ENaC activity in an MR-independent manner. Supporting this, in these kidney-specific MR knockout mice, AngII infusion augmented ENaC currents in their DCT2/CNT1, increased expression of ENaC subunits, augmented benzamil-induced natriuresis, increased basal urinary K+ excretion, and corrected hyperkalemia. Also consistent with this concept, it was previously shown that aldosterone synthase knockout mice, which maintain Na+ and K+ balance in the absence of aldosterone, developed severe hyperkalemia when treated with losartan and redistributed ENaC from the apical membrane to the cytoplasm in the DCT2/CNT 141.
Although recent transcriptomic studies have suggested that levels of the AngII receptor transcript in the CNT and CCD are very low 51, urinary concentrating ability was low in two different principal cell-specific AT1a knockout models 142,143, suggesting an action in the CD. In addition, in AngII-infused mice, full-length and cleaved αENaC levels were lower in principal cell-specific knockout mice than in their wild type littermates, correlating with partial reduction in AngII-induced hypertension 143.
Another MR-independent mechanism for ENaC regulation involves the rapamycin-insensitive mTOR complex 2 (mTORC2) (Figure 2). The mTOR kinase can catalyze the phosphorylation of SGK1 kinase within a hydrophobic C-terminal motif (S422), promoting its activation 144. In support of a role for this pathway in physiological ENaC activation, non-selective mTOR1 and 2 inhibition, but not mTORC1-specific inhibition with rapamycin, prevented SGK1 phosphorylation in mice and caused substantial natriuresis without kaliuresis; inhibition also decreased the ENaC-dependent transepithelial voltage in isolated perfused tubules, and reduced amiloride-sensitive ENaC activity, but not ROMK activity in rat CDs145. Conflicting observations, however, were made by Grahammer and coworkers, who showed that mice lacking mTORC2 activity in the distal nephron had normal amiloride-sensitive Na+ currents in split-open CNT/CCD, but had almost completely abolished apical K+ currents (ROMK activity) 146. These mice had also significantly reduced levels of pSGK1, but were able to achieve Na+ balance when on low Na+ diet. On a high K+ diet, however, they developed severe hyperkalemia despite markedly elevated plasma aldosterone. It seems likely that both ENaC-dependent and ROMK-dependent effects of mTORC2 contribute.
More recently, Sorensen et al. showed that amiloride-sensitive Na+ currents in isolated mouse CCDs and cultured mpkCCD cells can be directly stimulated by increases in basolateral [K+]147. These increases correlated with an increase in SGK1-S422 phosphorylation and were prevented with an SGK1 inhibitor, suggesting they were SGK1-dependent. They were also prevented with an mTOR inhibitor and were dependent on WNK1 presence. Thus, it was proposed that direct sensing of changes in basolateral [K+] by principal-like cells modulates K+ secretion through a pathway that involves SGK1-dependent activation of ENaC by mTORC2, and in which WNK1 appears to be also involved.
Finally, multiple studies have shown that Na+ reabsorption in the CCD can be modulated by arginine vasopressin (AVP). AVP has been shown to stimulate both Na+:K+ ATPase and ENaC activity in this segment 2. Patch clamp studies in microdissected, split-open CCDs have shown that stimulation with AVP or cAMP analogs increases amiloride-sensitive Na+ currents within minutes by increasing the open probability of the channel and the number of channels in the cell surface 148–150. Based on in vitro experiments, it was proposed that AVP increases ENaC surface expression by stimulating the PKA-mediated phosphorylation of Nedd4–2, which prevents Nedd4–2-ENaC interaction 151 (Figure 2). Although in vivo confirmation of this pathway is lacking, it would constitute an additional MR and SGK1- independent mechanism for ENaC modulation. This is consistent with the observation that AVP levels increase in adrenalectomized mice and appear to contribute to maintenance of ENaC activity 152. Stimulation of ENaC by AVP was proposed to be relevant for maximum urinary concentration capacity, because benzamil treatment in mice drinking a hypertonic solution reduced urine osmolarity and exacerbated hypernatremia 153. More recently, Exchange Proteins Activated by cAMP (EPACs) were proposed to play a role in ENaC regulation 154. Epac1−/−, Epac2−/−, and double knockout mice displayed delayed anti-natriuresis when placed on low salt diet, as well as reduced ENaC activity. Thus, this may constitute an alternative pathway for cAMP-mediated regulation of the channel.
Concluding remarks
We have provided an up-to-date landscape of the structure of the DCT and CNT and of mechanisms governing sodium reabsorption, thus affecting sodium balance and long-term blood pressure regulation. Despite the vast knowledge we have gained about the physiology of this nephron segment, this field of research continues to be very active as relevant molecular players continue to be described and tools are developed that allow us to generate new knowledge and question established knowledge.
ACKNOWLEDGEMETS
This work was supported by the grants from Conacyt Mexico No. 101720 to MCB, and from NIH DK51496, DK054983 and U54TR001628 to DHE. DHE was also supported by I01BX002228 from the Department of Veterans Affairs to DHE, and a Leducq Foundation Transatlantic Network of Excellence 17CVD05.
Footnotes
Disclosure
Nothing to disclose.
Editor’s Note
The review of Drs. Castaneda-Bueno and Ellison extends our series on hypertension. Here the authors discuss insights from physiology and genetic variants which have been instrumental to better understand how the distal tubule contributes to blood pressure regulation and how this can be used for preventive and therapeutic actions.
References
- 1.Chen L, Clark JZ, Nelson JW, Kaissling B, Ellison DH, Knepper MA. Renal-tubule epithelial cell nomenclature for single-cell rna-sequencing studies. J Am Soc Nephrol. 2019;30(8):1358–1364. doi: 10.1681/ASN.2019040415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Féraille E, Doucet a. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev. 2001;81(1):345–418. [DOI] [PubMed] [Google Scholar]
- 3.Reilly RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80(1):277–313. [DOI] [PubMed] [Google Scholar]
- 4.Wade JB, Fang L, Coleman RA, et al. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Physiol. 2011;300(6):F1385–F1393. doi: 10.1152/ajprenal.00592.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan XP, Gu L, Xiao Y, et al. Norepinephrine-Induced Stimulation of Kir4.1/Kir5.1 Is Required for the Activation of NaCl Transporter in Distal Convoluted Tubule. Hypertension. 2019;73(1):112–120. doi: 10.1161/HYPERTENSIONAHA.118.11621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loffing J, Loffing-Cueni D, Valderrabano V, et al. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol - Ren Physiol. 2001;281(6):F1021–F1027. doi: 10.1152/ajprenal.0085.2001 [DOI] [PubMed] [Google Scholar]
- 7.Obermuller N, Bernstein P, Velazquez H, et al. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol - Ren Fluid Electrolyte Physiol. 1995;269(6 38–6). doi: 10.1152/ajprenal.1995.269.6.f900 [DOI] [PubMed] [Google Scholar]
- 8.Ransick A, Lindström NO, Liu J, et al. Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev Cell. 2019;51(3):399–413.e7. doi: 10.1016/j.devcel.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao C, Chen L, Chen E, Tsilosani A, Xia Y, Zhang W. Generation of Distal Renal Segments Involves a Unique Population of Aqp2 + Progenitor Cells. J Am Soc Nephrol. 2021;32(12):3035–3049. doi: 10.1681/ASN.2021030399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nesterov V, Bertog M, Canonica J, et al. Critical role of the mineralocorticoid receptor in aldosterone-dependent and aldosterone-independent regulation of ENaC in the distal nephron. Am J Physiol - Ren Physiol. 2021;321(3):F257–F268. doi: 10.1152/ajprenal.00139.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol - Ren Physiol. 2012;303(9):1289–1299. doi: 10.1152/ajprenal.00247.2012 [DOI] [PubMed] [Google Scholar]
- 12.Wu P, Gao ZX, Zhang DD, et al. Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice. J Am Heart Assoc. 2020;9(7):e014996. doi: 10.1161/JAHA.119.014996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Xu Y, Gravotta D, Frindt G, Weinstein AM, Palmer LG. Enac and romk channels in the connecting tubule regulate renal k+ secretion. J Gen Physiol. 2021;153(8):1–16. doi: 10.1085/jgp.202112902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J, Shrestha R, Qiu C, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science (80- ). 2018;360(6390):758–763. doi: 10.1126/science.aar2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wall SM. The role of pendrin in blood pressure regulation. Am J Physiol - Ren Physiol. 2016;310(3):F193–F203. doi: 10.1152/ajprenal.00400.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Physiol. 2005;289(4):F699–F720. doi: 10.1152/ajprenal.00043.2005 [DOI] [PubMed] [Google Scholar]
- 17.Ellison DH, Velazquez H, Wright FS. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest. 1989;83(1):113–126. doi: 10.1172/JCI113847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinstein AM. A mathematical model of the rat nephron: K+-induced natriuresis. Am J Physiol Renal Physiol. 2017;312:F925–F950. doi: 10.1152/ajprenal.00505.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellison DH, Velázquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol. 1987;253(3 Pt 2):F546–54. http://www.ncbi.nlm.nih.gov/pubmed/3631283. [DOI] [PubMed] [Google Scholar]
- 20.Coleman RA, Wu DC, Liu J, Wade JB. Expression of aquaporins in the renal connecting tubule. Am J Physiol - Ren Physiol. 2000;279(5 48–5):874–883. doi: 10.1152/ajprenal.2000.279.5.f874 [DOI] [PubMed] [Google Scholar]
- 21.Ellison DH, Velazquez H, Wright FS. Unidirectional potassium fluxes in renal distal tubule: Effects of chloride and barium. Am J Physiol - Ren Fluid Electrolyte Physiol. 1986;250(5 (19/5)). doi: 10.1152/ajprenal.1986.250.5.f885 [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Xu Y, Gravotta D, Frindt G, Weinstein AM, Palmer LG. ENaC and ROMK channels in the connecting tubule regulate renal K + secretion. 2021;153(8):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bostanjoglo M, Reeves WB, Reilly RF, et al. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9(8):1347–1358. http://www.ncbi.nlm.nih.gov/pubmed/9697656. [DOI] [PubMed] [Google Scholar]
- 24.Hunter RW, Ivy JR, Flatman PW, et al. Hypertrophy in the Distal Convoluted Tubule of an 11 -Hydroxysteroid Dehydrogenase Type 2 Knockout Model. J Am Soc Nephrol. 2015;26(7):1537–1548. doi: 10.1681/ASN.2013060634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol. 2004;287(4):F593–601. doi: 10.1152/ajprenal.00454.2003 [DOI] [PubMed] [Google Scholar]
- 26.Hunter RW, Craigie E, Homer NZM, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. AJP Ren Physiol. 2014;306(4). doi: 10.1152/ajprenal.00339.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively Active SPAK Causes Hyperkalemia by Activating NCC and Remodeling Distal Tubules. J Am Soc Nephrol. 2017;28(9):2597–2606. doi: 10.1681/ASN.2016090948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz DN, Simon DB, Nelson-Williams C, et al. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension. 2001;37(6):1458–1464. doi: 10.1161/01.HYP.37.6.1458 [DOI] [PubMed] [Google Scholar]
- 29.Hennings JC, Andrini O, Picard N, et al. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J Am Soc Nephrol. 2017;28(1):209–217. doi: 10.1681/ASN.2016010085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH. Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol. 2019;30(8):1425–1438. doi: 10.1681/ASN.2019010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su X-T, Wang W-H. The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am J Physiol - Ren Physiol. 2016;311(1):F12–F15. doi: 10.1152/ajprenal.00112.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang MX, Cuevas CA, Su XT, et al. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018;93(4):893–902. doi: 10.1016/j.kint.2017.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Wang L, Zhang J, et al. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci U S A. 2014;111(32):11864–11869. doi: 10.1073/pnas.1411705111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grill A, Schießl IM, Gess B, Fremter K, Hammer A, Castrop H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol. 2016;218(3):198–211. doi: 10.1111/apha.12755 [DOI] [PubMed] [Google Scholar]
- 36.Zelikovic I, Szargel R, Hawash A, et al. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63(1):24–32. doi: 10.1046/j.1523-1755.2003.00730.x [DOI] [PubMed] [Google Scholar]
- 37.Schlingmann KP, de Baaij JHF. The genetic spectrum of Gitelman(-like) syndromes. Curr Opin Nephrol Hypertens. 2022;31(5):508–515. doi: 10.1097/MNH.0000000000000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson FH, Disse-Nicodèm S, Choate KA, et al. Human Hypertension Caused by Mutations in WNK Kinases. Science (80- ). 2001;293(5532):1107–1112. doi: 10.1126/science.1062844 [DOI] [PubMed] [Google Scholar]
- 39.Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis-Dit-Picard H, Barc J, Trujillano D, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44(4):456–460. doi: 10.1038/ng.2218 [DOI] [PubMed] [Google Scholar]
- 41.Louis-Dit-Picard H, Kouranti I, Rafael C, et al. Mutation affecting the conserved acidic WNK1 motif causes inherited hyperkalemic hyperchloremic acidosis. J Clin Invest. 2020;130(12):6379–6394. doi: 10.1172/JCI94171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gamba G. WNK lies upstream of kinases involved in regulation of ion transporters. Biochem J. 2005;391(Pt 1):e1–3. doi: 10.1042/BJ20051345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391(1):17–24. doi: 10.1042/BJ20051180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moriguchi T, Urushiyama S, Hisamoto N, et al. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005;280(52):42685–42693. doi: 10.1074/jbc.M510042200 [DOI] [PubMed] [Google Scholar]
- 45.Ferdaus MZ, Barber KW, López-Cayuqueo KI, et al. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol. 2016;594(17):4945–4966. doi: 10.1113/JP272311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin S-H, Yu I-S, Jiang S-T, et al. Impaired phosphorylation of Na+-K+−2Cl- cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci. 2011;108(42):17538–17543. doi: 10.1073/pnas.1107452108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci. 2013;110(19):7838–7843. doi: 10.1073/pnas.1304592110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohta A, Schumacher F, Mehellou Y, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J. 2013;451:111–122. doi: 10.1042/bj20121903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakabayashi M, Mori T, Isobe K, et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013;3(3):858–868. doi: 10.1016/j.celrep.2013.02.024 [DOI] [PubMed] [Google Scholar]
- 50.Ostrosky-Frid M, Chávez-Canales M, Zhang J, et al. Role of KLHL3 and dietary K + in regulating KS-WNK1 expression. Am J Physiol Physiol. 2021;320(5):F734–F747. doi: 10.1152/ajprenal.00575.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, Chou C, Knepper MA. A Comprehensive Map of mRNAs and Their Isoforms across All 14 Renal Tubule Segments of Mouse. J Am Soc Nephrol. 2021;32(4):897–912. doi: 10.1681/ASN.2020101406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vidal-Petiot E, Cheval L, Faugeroux J, et al. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One. 2012;7(5):1–9. doi: 10.1371/journal.pone.0037751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Argaiz ER, Chavez-Canales M, Ostrosky-Frid M, et al. Kidney-specific WNK1 isoform (KS-WNK1) is a potent activator of WNK4 and NCC. Am J Physiol Physiol. May 2018. doi: 10.1152/ajprenal.00145.2018 [DOI] [PMC free article] [PubMed]
- 54.Vidal-Petiot E, Elvira-Matelot E, Mutig K, et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci U S A. 2013;110(35):14366–14371. doi: 10.1073/pnas.1304230110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chávez-Canales M, Zhang C, Soukaseum C, et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension. 2014;64(5):1047–1053. doi: 10.1161/HYPERTENSIONAHA.114.04036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cornelius RJ, Yang CL, Ellison DH. Hypertension-causing cullin 3 mutations disrupt COP9 signalosome binding. Am J Physiol - Ren Physiol. 2020;318(1):F204–F208. doi: 10.1152/ajprenal.00497.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chatrathi HE, Collins JC, Wolfe LA, et al. Novel CUL3 Variant Causing Familial Hyperkalemic Hypertension Impairs Regulation and Function of Ubiquitin Ligase Activity. Hypertension. 2022;79(1):60–75. doi: 10.1161/HYPERTENSIONAHA.121.17624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunter RW, Craigie E, Homer NZM, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol - Ren Physiol. 2014;306(4):F457–F467. doi: 10.1152/ajprenal.00339.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanton BA, Kaissling B. Adaptation of distal tubule and collecting duct to increased Na delivery. II. Na+ and K+ transport. Am J Physiol Physiol. 1988;255(6):F1269–F1275. doi: 10.1152/ajprenal.1988.255.6.F1269 [DOI] [PubMed] [Google Scholar]
- 60.Loffing J, Loffing-Cueni D, Hegyi I, et al. Thiazide treatment of rats provokes apoptosis in distal tubule cells. Kidney Int. 1996;50(4):1180–1190. doi: 10.1038/ki.1996.426 [DOI] [PubMed] [Google Scholar]
- 61.Schnoz C, Carrel M, Loffing J. Loss of sodium chloride co-transporter impairs the outgrowth of the renal distal convoluted tubule during renal development. Nephrol Dial Transplant. 2020;35(3):422–432. doi: 10.1093/ndt/gfz172 [DOI] [PubMed] [Google Scholar]
- 62.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol - Ren Physiol. 2009;297(3):F704–F712. doi: 10.1152/ajprenal.00030.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, et al. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol - Ren Physiol. 2014;306(12):F1507–F1519. doi: 10.1152/ajprenal.00255.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sorensen M V, Grossmann S, Roesinger M, et al. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83(5):811–824. doi: 10.1038/ki.2013.14 [DOI] [PubMed] [Google Scholar]
- 65.Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21(1):39–50. doi: 10.1016/j.cmet.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cuevas CA, Su X-T, Wang M-X, et al. Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J Am Soc Nephrol. 2017;28(6):1814–1825. doi: 10.1681/ASN.2016090935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su X, Klett NJ, Sharma A, et al. Distal convoluted tubule Cl − concentration is modulated via K + channels and transporters. Am J Physiol Physiol. 2020;319(3):F534–F540. doi: 10.1152/ajprenal.00284.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride Sensing by WNK1 Involves Inhibition of Autophosphorylation. Sci Signal. 2014;7(324):ra41–ra41. doi: 10.1126/scisignal.2005050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, et al. The Effect of WNK4 on the Na+-Cl- Cotransporter Is Modulated by Intracellular Chloride. J Am Soc Nephrol. 2015;26(8):1781–1786. doi: 10.1681/ASN.2014050470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang C-L, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2015;89(1):1–8. doi: 10.1038/ki.2015.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishizawa K, Xu N, Loffing J, et al. Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem Biophys Res Commun. 2016;480(4):745–751. doi: 10.1016/j.bbrc.2016.10.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shibata S, Arroyo JP, Castaneda-Bueno M, et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci. 2014;111(43):15556–15561. doi: 10.1073/pnas.1418342111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castañeda-Bueno M, Arroyo JP, Zhang J, et al. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc Natl Acad Sci. 2017;114(5):E879–E886. doi: 10.1073/pnas.1620315114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dubois E, Yuan X, Bennett D, et al. Ur Na R Oo F. Data Inf Manag. 2022:100001. doi: 10.1016/j.kint.2022.06.027 [DOI] [PMC free article] [PubMed]
- 75.Pleinis JM, Norrell L, Akella R, et al. WNKs are potassium-sensitive kinases. 2021. [DOI] [PMC free article] [PubMed]
- 76.Chen J-C, Lo Y-F, Lin Y-W, Lin S-H, Huang C-L, Cheng C-J. WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci. 2019;116(10):4502–4507. doi: 10.1073/pnas.1817220116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, et al. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci. 2012;109(20):7929–7934. doi: 10.1073/pnas.1200947109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Terker AS, Yarbrough B, Ferdaus MZ, et al. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol. 2016;27(8):2436–2445. doi: 10.1681/ASN.2015070815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, Loffing J. The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch Eur J Physiol. 2016;468(5):849–858. doi: 10.1007/s00424-016-1798-5 [DOI] [PubMed] [Google Scholar]
- 80.Canonica J, Sergi C, Maillard M, et al. Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch Eur J Physiol. 2016;468(5):895–908. doi: 10.1007/s00424-015-1785-2 [DOI] [PubMed] [Google Scholar]
- 81.van der Lubbe N, Lim CH, Fenton RA, et al. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79(1):66–76. doi: 10.1038/ki.2010.290 [DOI] [PubMed] [Google Scholar]
- 82.Markadieu N, San-Cristobal P, Nair AV., et al. A primary culture of distal convoluted tubules expressing functional thiazide-sensitive NaCl transport. Am J Physiol - Ren Physiol. 2012;303(6):F886–F892. doi: 10.1152/ajprenal.00114.2012 [DOI] [PubMed] [Google Scholar]
- 83.Veiras LC, Han J, Ralph DL, McDonough AA. Potassium Supplementation Prevents Sodium Chloride Cotransporter Stimulation during Angiotensin II Hypertension. Hypertension. 2016;68(4):904–912. doi: 10.1161/HYPERTENSIONAHA.116.07389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rojas-Vega L, Gamba G. Mini-review: regulation of the renal NaCl cotransporter by hormones. Am J Physiol Physiol. 2016;310(1):F10–F14. doi: 10.1152/ajprenal.00354.2015 [DOI] [PubMed] [Google Scholar]
- 85.Penton D, Moser S, Wengi A, et al. Protein phosphatase 1 inhibitor–1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J Am Soc Nephrol. 2019;30(5):737–750. doi: 10.1681/ASN.2018050540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carbajal-Contreras H, Murillo-de-Ozores AR, German M-A, Vazquez N, Gamba G, Castañeda-Bueno M. WNK4 is a transductor of V2 receptor signaling in the TAL and DCT. J Am Soc Nephrol. 2021;32:360. [Google Scholar]
- 87.Bazúa-Valenti S, Rojas-Vega L, Castañeda-Bueno M, et al. The Calcium-Sensing Receptor Increases Activity of the Renal NCC through the WNK4-SPAK Pathway. J Am Soc Nephrol. 2018;29(7):1838–1848. doi: 10.1681/ASN.2017111155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang C, Sindic A, Hill CE, et al. Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. Am J Physiol - Ren Physiol. 2007;292(3):1073–1081. doi: 10.1152/ajprenal.00269.2006 [DOI] [PubMed] [Google Scholar]
- 89.Masab M, Goyal A, Abrol S, Rangaswami J. Acquired Gitelman Syndrome Associated with Systemic Sclerosis. Cureus. 2019;11(1):10–12. doi: 10.7759/cureus.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gu X, Su Z, Chen M, Xu Y, Wang Y. Acquired Gitelman syndrome in a primary Sjögren syndrome patient with a SLC12A3 heterozygous mutation: A case report and literature review. Nephrology. 2017;22(8):652–655. doi: 10.1111/nep.13045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barathidasan GS, Krishnamurthy S, Karunakar P, et al. Systemic lupus erythematosus complicated by a Gitelman-like syndrome in an 8-year-old girl. CEN case reports. 2020;9(2):129–132. doi: 10.1007/s13730-019-00440-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim YK, Song HC, Kim W-Y, et al. Acquired Gitelman Syndrome in a Patient With Primary Sjögren Syndrome. Am J Kidney Dis. 2008;52(6):1163–1167. doi: 10.1053/j.ajkd.2008.07.025 [DOI] [PubMed] [Google Scholar]
- 93.Hoorn EJ, Walsh SB, McCormick JA, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17(10):1304–1309. doi: 10.1038/nm.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blankenstein KI, Borschewski A, Labes R, et al. Calcineurin inhibitor cyclosporine A activates renal NA-K-CL cotransporters via local and systemic mechanisms. Am J Physiol - Ren Physiol. 2017;312(3):F489–F501. doi: 10.1152/ajprenal.00575.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riepe FG. Clinical and Molecular Features of Type 1 Pseudohypoaldosteronism. Horm Res. 2009;72(1):1–9. doi: 10.1159/000224334 [DOI] [PubMed] [Google Scholar]
- 96.Geller DS, Farhi a, Pinkerton N, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;289(July):119–123. doi: 10.1126/science.289.5476.119 [DOI] [PubMed] [Google Scholar]
- 97.Razzaghy-Azar M, Yau M, Khattab A, New MI. Apparent mineralocorticoid excess and the long term treatment of genetic hypertension. J Steroid Biochem Mol Biol. 2017;165:145–150. doi: 10.1016/j.jsbmb.2016.02.014 [DOI] [PubMed] [Google Scholar]
- 98.Shimkets RA, Warnock DG, Bositis CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79(3):407–414. doi: 10.1016/0092-8674(94)90250-X [DOI] [PubMed] [Google Scholar]
- 99.Hansson JH, Nelson-Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76. 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 100.Salih M, Gautschi I, Van Bemmelen MX, et al. A missense mutation in the extracellular domain of aenac causes liddle syndrome. J Am Soc Nephrol. 2017;28(11):3291–3299. doi: 10.1681/ASN.2016111163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hiltunen TP, Hannila-Handelberg T, Petäjäniemi N, et al. Liddle’s syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel γ subunit. J Hypertens. 2002;20(12):2383–2390. doi: 10.1097/00004872-200212000-00017 [DOI] [PubMed] [Google Scholar]
- 102.Botero-Velez M, Curtis JJ, Warnock DG. Liddle’s Syndrome Revisited -- A Disorder of Sodium Reabsorption in the Distal Tubule. N Engl J Med. 1994;330(3):178–181. doi: 10.1056/NEJM199401203300305 [DOI] [PubMed] [Google Scholar]
- 103.Hansson JH, Schild L, Lu Y, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci. 1995;92(25):11495–11499. doi: 10.1073/pnas.92.25.11495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tamura H, Schild L, Enomoto N, et al. Liddle disease caused by a missense mutation of β subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97(7):1780–1784. doi: 10.1172/JCI118606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Furuhashi M, Kitamura K, Adachi M, et al. Liddle’s syndrome caused by a novel mutation in the proline-rich PY motif of the epithelial sodium channel β-subunit. J Clin Endocrinol Metab. 2005;90(1):340–344. doi: 10.1210/jc.2004-1027 [DOI] [PubMed] [Google Scholar]
- 106.Abriel H, Loffing J, Rebhun JF, et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest. 1999;103(5):667–673. doi: 10.1172/JCI5713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Staub O, Dho S, Henry P, et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996;15(10):2371–2380. doi: 10.1002/j.1460-2075.1996.tb00593.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loffing J, Zecevic M, Féraille E, et al. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol. 2001;280(4):F675–82. http://www.ncbi.nlm.nih.gov/pubmed/11249859. [DOI] [PubMed] [Google Scholar]
- 109.Chen S-y., Bhargava A, Mastroberardino L, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci. 1999;96(5):2514–2519. doi: 10.1073/pnas.96.5.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Náray-Fejes-Tóth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Tóth G. sgk Is an Aldosterone-induced Kinase in the Renal Collecting Duct. J Biol Chem. 1999;274(24):16973–16978. doi: 10.1074/jbc.274.24.16973 [DOI] [PubMed] [Google Scholar]
- 111.Debonneville C, Flores SY, Kamynina E, et al. Phosphorylation of Nedd4–2 by Sgk1 Regulates Epithelial Na+ Channel Cell Surface Expression. EMBO J. 2002;20(24):7052–7059. doi: 10.1093/emboj/20.24.7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Faresse N, Lagnaz D, Debonneville A, et al. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. AJP Ren Physiol. 2012;302(8):F977–F985. doi: 10.1152/ajprenal.00535.2011 [DOI] [PubMed] [Google Scholar]
- 113.Al-Qusairi L, Basquin D, Roy A, et al. Renal tubular SGK1 deficiency causes impaired K + excretion via loss of regulation of NEDD4–2/WNK1 and ENaC. Am J Physiol - Ren Physiol. 2016;311(2):F330–F342. doi: 10.1152/ajprenal.00002.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang L, Frindt G, Lang F, Kuhl D, Vallon V, Palmer LG. SGK1-dependent ENaC processing and trafficking in mice with high dietary K intake and elevated aldosterone. Am J Physiol - Ren Physiol. 2017;312(1):F65–F76. doi: 10.1152/ajprenal.00257.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fejes-Tóth G, Frindt G, Náray-Fejes-Tóth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol - Ren Physiol. 2008;294(6):1298–1305. doi: 10.1152/ajprenal.00579.2007 [DOI] [PubMed] [Google Scholar]
- 116.Féraille E, Mordasini D, Gonin S, et al. Mechanism of control of Na,K-ATPase in principal cells of the mammalian collecting duct. Ann N Y Acad Sci. 2003;986:570–578. doi: 10.1111/j.1749-6632.2003.tb07255.x [DOI] [PubMed] [Google Scholar]
- 117.Zhang X, Zhou Q, Chen L, et al. Mineralocorticoid receptor antagonizes Dot1a-Af9 complex to increase αENaC transcription. Am J Physiol - Ren Physiol. 2013;305(10):1436–1444. doi: 10.1152/ajprenal.00202.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang XP, Balchak DM, Gentilcore C, Clark NL, Kashlan OB. Activation by cleavage of the epithelial Na+ channel α and γ subunits independently coevolved with the vertebrate terrestrial migration. Elife. 2022;11:1–20. doi: 10.7554/eLife.75796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kashlan OB, Blobner BM, Zuzek Z, Tolino M, Kleyman TR. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J Biol Chem. 2015;290(1):568–576. doi: 10.1074/jbc.M114.606152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife. 2018;7:1–23. doi: 10.7554/eLife.39340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noreng S, Posert R, Bharadwaj A, Houser A, Baconguis I. Molecular principles of assembly, activation, and inhibition in epithelial sodium channel. Elife. 2020;9:1–23. doi: 10.7554/eLife.59038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bruns JB, Carattino MD, Sheng S, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem. 2007;282(9):6153–6160. doi: 10.1074/jbc.M610636200 [DOI] [PubMed] [Google Scholar]
- 123.Anand D, Hummler E, Rickman O. ENaC activation by proteases. Acta Physiol. 2022;(March):1–13. doi: 10.1111/apha.13811 [DOI] [PMC free article] [PubMed]
- 124.Svenningsen P. Non-enzymatic function of prostasin and sodium balance. Acta Physiol. 2021;232(1):1–2. doi: 10.1111/apha.13649 [DOI] [PubMed] [Google Scholar]
- 125.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus Oocytes. J Gen Physiol. 2002;120(2):191–201. doi: 10.1085/jgp.20028598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389(6651):607–610. doi: 10.1038/39329 [DOI] [PubMed] [Google Scholar]
- 127.Maeoka Y, Su X-T, Wang W-H, et al. Mineralocorticoid Receptor Antagonists Cause Natriuresis in the Absence of Aldosterone. Hypertension. 2022;79(7):1423–1434. doi: 10.1161/HYPERTENSIONAHA.122.19159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang L, Frindt G, Xu Y, Uchida S, Palmer LG. Aldosterone-dependent and -independent regulation of Na+ and K+ excretion and ENaC in mouse kidneys. Am J Physiol - Ren Physiol. 2020;319(2):F323–F334. doi: 10.1152/ajprenal.00204.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Narikiyo T, Kitamura K, Adachi M, et al. Regulation of prostasin by aldosterone in the kidney. J Clin Invest. 2002;109(3):401–408. doi: 10.1172/JCI0213229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oxlund C, Kurt B, Schwarzensteiner I, et al. Albuminuria is associated with an increased prostasin in urine while aldosterone has no direct effect on urine and kidney tissue abundance of prostasin. Pflügers Arch - Eur J Physiol. 2017;469(5–6):655–667. doi: 10.1007/s00424-017-1938-6 [DOI] [PubMed] [Google Scholar]
- 131.Ehret E, Jäger Y, Sergi C, et al. Kidney-Specific CAP1 / Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway. 2022;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.El Moghrabi S, Houillier P, Picard N, et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci U S A. 2010;107(30):13526–13531. doi: 10.1073/pnas.0913070107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol - Ren Physiol. 2012;303(4):540–550. doi: 10.1152/ajprenal.00133.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Picard N, Eladari D, El Moghrabi S, et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem. 2008;283(8):4602–4611. doi: 10.1074/jbc.M705664200 [DOI] [PubMed] [Google Scholar]
- 135.Hinrichs GR, Jensen BL, Svenningsen P. Mechanisms of sodium retention in nephrotic syndrome. Curr Opin Nephrol Hypertens. 2020;29(2):207–212. doi: 10.1097/MNH.0000000000000578 [DOI] [PubMed] [Google Scholar]
- 136.Loffing J, Pietri L, Aregger F, et al. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am J Physiol Renal Physiol. 2000;279(2):F252–8. file://i/Chercheurs/Brochiero_Emmanuelle/Brochiero/RefManager/500/531.pdf%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/10919843. [DOI] [PubMed] [Google Scholar]
- 137.Peti-Peterdi J. Angiotensin II Directly Stimulates ENaC Activity in the Cortical Collecting Duct via AT1 Receptors. J Am Soc Nephrol. 2002;13(5):1131–1135. doi: 10.1097/01.ASN.0000013292.78621.FD [DOI] [PubMed] [Google Scholar]
- 138.Sun P, Yue P, Wang W-H. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. AJP Ren Physiol. 2012;302(6):F679–F687. doi: 10.1152/ajprenal.00368.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mamenko M, Zaika O, Ilatovskaya DV., Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na + channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287(1):660–671. doi: 10.1074/jbc.M111.298919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr Hypertens Rep. 2013;15(1):17–24. doi: 10.1007/s11906-012-0316-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Todkar A, Picard N, Loffing-Cueni D, et al. Mechanisms of Renal Control of Potassium Homeostasis in Complete Aldosterone Deficiency. J Am Soc Nephrol. 2015;26(2):425–438. doi: 10.1681/ASN.2013111156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stegbauer J, Gurley SB, Sparks MA, et al. AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol. 2011;22(12):2237–2246. doi: 10.1681/ASN.2010101095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen D, Stegbauer J, Sparks MA, et al. Impact of angiotensin type 1A receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension. 2016;67(6):1291–1297. doi: 10.1161/HYPERTENSIONAHA.115.06987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu M, Wang J, Jones KT, et al. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol. 2010;21(5):811–818. doi: 10.1681/ASN.2009111168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gleason CE, Frindt G, Cheng CJ, et al. MTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest. 2015;125(1):117–128. doi: 10.1172/JCI73935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grahammer F, Nesterov V, Ahmed A, et al. Mtorc2 critically regulates renal potassium handling. J Clin Invest. 2016;126(5):1773–1782. doi: 10.1172/JCI80304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sørensen MV, Saha B, Jensen IS, et al. Potassium acts through mTOR to regulate its own secretion. JCI Insight. 2019;4(11):1–17. doi: 10.1172/jci.insight.126910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol - Ren Physiol. 2009;297(5). doi: 10.1152/ajprenal.00371.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Frindt G, Palmer LG. Regulation of Na channels in the rat cortical collecting tubule: effects of cAMP and methyl donors. Am J Physiol Physiol. 1996;271(5):F1086–F1092. doi: 10.1152/ajprenal.1996.271.5.F1086 [DOI] [PubMed] [Google Scholar]
- 150.Frindt G, Silver RB, Windhager EE, Palmer LG. Feedback regulation of Na channels in rat CCT. III. Response to cAMP. Am J Physiol Physiol. 1995;268(3):F480–F489. doi: 10.1152/ajprenal.1995.268.3.F480 [DOI] [PubMed] [Google Scholar]
- 151.Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4–2. J Biol Chem. 2004;279(44):45753–45758. doi: 10.1074/jbc.M407858200 [DOI] [PubMed] [Google Scholar]
- 152.Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na + channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci U S A. 2012;109(25):10095–10100. doi: 10.1073/pnas.1201978109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mironova E, Chen Y, Pao AC, et al. Activation of ENaC by AVP contributes to the urinary concentrating mechanism and dilution of plasma. Am J Physiol - Ren Physiol. 2015;308(3):F237–F243. doi: 10.1152/ajprenal.00246.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tomilin VN, Pyrshev K, Stavniichuk A, et al. Epac1−/− and Epac2−/− mice exhibit deficient epithelial Na+ channel regulation and impaired urinary Na+ conservation. JCI Insight. 2022;7(3). doi: 10.1172/jci.insight.145653 [DOI] [PMC free article] [PubMed] [Google Scholar]