

Abstract
Background and Objectives
Pyridoxine-dependent epilepsy (PDE-ALDH7A1) is a developmental epileptic encephalopathy characterized by seizure improvement after pyridoxine supplementation. Adjunct lysine reduction therapies (LRTs) reduce the accumulation of putative neurotoxic metabolites with the goal to improve developmental outcomes. Our objective was to examine the association between treatment with LRTs and cognitive outcomes.
Methods
Participants were recruited from within the International Registry for Patients with Pyridoxine-Dependent Epilepsy from August 2014 through March 2021. The primary outcome was standardized developmental test scores associated with overall cognitive ability. The relationship between test scores and treatment was analyzed with multivariable linear regression using a mixed-effects model. A priori, we hypothesized that treatment in early infancy with pyridoxine and LRTs would result in a normal developmental outcome. A subanalysis was performed to evaluate the association between cognitive outcome and LRTs initiated in the first 6 months of life.
Results
A total of 112 test scores from 60 participants were available. On average, treatment with pyridoxine and LRTs was associated with a nonsignificant increase of 6.9 points (95% CI −2.7 to 16.5) on developmental testing compared with treatment with pyridoxine alone. For the subanalysis, a total of 14 developmental testing scores were available from 8 participants. On average, treatment with pyridoxine and LRTs in the first 6 months of life was associated with a significant increase of 21.9 points (95% CI 1.7–42.0) on developmental testing.
Discussion
Pyridoxine and LRTs at any age was associated with mild improvement in developmental testing, and treatment in early infancy was associated with a clinically significant increase in developmental test scores. These results provide insight into the mechanism of intellectual and developmental disability in PDE-ALDH7A1 and emphasize the importance of treatment in early infancy with both pyridoxine and LRTs.
Classification of Evidence
This study provides Class IV evidence that in PDE-ALDH7A1, pyridoxine and LRTs compared with pyridoxine alone is not significantly associated with overall higher developmental testing scores, but treatment in the first 6 months of life is associated with significantly higher developmental testing scores.
Pyridoxine-dependent epilepsy due to a deficiency of α-aminoadipic semialdehyde (α-AASA) dehydrogenase (PDE-ALDH7A1) is a developmental epileptic encephalopathy characterized by a clinical response to pyridoxine. Pharmacologic doses of pyridoxine have remained central to the treatment of this disease for more than 60 years.1 Historically, the withdrawal of pyridoxine and subsequent recurrence of seizures documented dependence on chronic pyridoxine supplementation for seizure control,2-6 and several reports describe patients who died before initiation of pyridoxine treatment, emphasizing the critical role of pyridoxine in the treatment of this disorder.7-9
However, pyridoxine supplementation alone is insufficient to achieve a normal developmental outcome. Even when seizure control is achieved, most patients experience intellectual disability or developmental disability (ID/DD).10,11 This is highlighted by patients who commenced pyridoxine antenatally, often without postnatal evidence of seizures, who were still reported to experience ID/DD.12 The deficiency of α-AASA dehydrogenase results in the accumulation of several metabolites including pipecolic acid,13 α-AASA,14 Δ1-piperideine-6-carboxylate,15 6-oxo-pipecolate,16 and 2S,6R-oxopropylpiperidine-2-carboxylic acid.17 These metabolites aid in confirming the diagnosis of PDE-ALDH7A1, and the chronic accumulation of 1 or more of these metabolites may contribute to the ID/DD pervasive in this disease.18
Lysine reduction therapies (LRTs) aim to reduce the accumulation of these putative neurotoxic metabolites to improve developmental outcomes. These treatment strategies include a lysine-restricted diet,19 pharmacologic doses of arginine, which acts as a competitive inhibitor of lysine transport,20,21 and a combination of these 2 therapeutic strategies and pyridoxine referred to as triple therapy.22 To date, 13 observational studies have reported the benefit of LRT,8,19,20,22-31 and both pyridoxine and LRT are recommended for most patients with PDE-ALDH7A1.32 Yet not all patients have benefited from the addition of LRT, highlighting the need for further evaluation of these adjunct treatments. In this study, we described the association between treatment with LRT and developmental outcomes in observational study of research participants with PDE-ALDH7A1. Our primary research question was whether treatment with pyridoxine and LRT associated with improved developmental test scores.
Methods
Study Design
This is a multicenter, observational cohort study of participants with PDE-ALDH7A1. The Strengthening the Reporting of Observational Studies in Epidemiology statement for reporting observational studies was used for reporting these results.33
Standard Protocol Approvals, Registrations, and Patient Consents
Participants were recruited from the International Registry for Patients with Pyridoxine-Dependent Epilepsy (PDE registry, pdeonline.org). The PDE registry was initially approved by the University of British Columbia Children's and Women's Research Ethics Board (H14-01832), and local approval was obtained by all sites where required. Participants were recruited from August 2014 through March 2021, and ambispective clinical data were collected for each participant.
Individuals of any age with a confirmed diagnosis of PDE-ALDH7A1 were eligible for enrollment. The diagnosis of PDE-ALDH7A1 was defined as the elevation of α-AASA or biallelic pathogenic variants in ALDH7A1. Additional inclusion criteria included at least 1 developmental assessment. Exclusion criteria included other causes of pyridoxine-responsive seizures and comorbid findings that may have severely affected a participant's development. Following informed consent, each participant's clinical, biochemical, and genetic data were collected using Research Electronic Data Capture software.34 The timing of follow-up visits were based on published recommendations18,35 and consensus guidelines32 for the management of PDE-ALDH7A1.
Variables and Data Sources
A standardized developmental assessment with a focus on mental and cognitive scores was used as the primary outcome measure. For this analysis, full-scale intelligence quotient (as determined by Der Hamburg-Wechsler-intelligenztest für Kinder, Wechsler Adult Intelligence Scale, Wechsler Intelligence Scale for Children, Wechsler Preschool & Primary Scale of Intelligence, Woodcock-Johnson Tests of Cognitive Abilities), mental developmental index (Bayley Scales of Infant & Toddler development), SON-intelligence quotient (Snijders-Oomen Nonverbal Intelligence Test), and mental processing composite intelligence quotient (Kaufman Assessment Battery for Children) were treated as equivalent outcome measures. These 8 developmental tests were scaled so that the mean and standard deviation in the general population is 100 and 15, respectively. Treatment during developmental testing was categorized based on treatment with either pyridoxine monotherapy or treatment with pyridoxine + LRT. LRT was defined as the use of a lysine-restricted diet, arginine supplementation, or a combination of these 2 therapeutic strategies.32
Most of the data were collected from the participants' medical records. Demographic data (sex assigned at birth, race, and ethnicity), gestational age, diagnostic data (α-AASA levels, genotype), and initial presentation (age at first seizure and age at treatment with pyridoxine) were recorded during enrollment. Treatment data included the use of pyridoxine monotherapy or pyridoxine + LRT. For all participants, the age at the start of pyridoxine and LRT were recorded. Developmental testing data were additionally collected. For each standardized developmental assessment, the age and treatment during testing were recorded.
Investigators also provided a subjective assessment of whether a participant had an abnormal development at last visit, such as global developmental delay, speech or language delay, gross or fine motor delay, communication delay, or difficulties with activities of daily life. Investigators were instructed to use formal developmental assessments, clinical evaluations, and third-party informants for these assessments.
Covariates and Confounders
The age at first seizure was considered a potential covariate. Late-onset of seizures, defined as seizures starting after 2 months of age, has been associated with a relatively good clinical outcome.26 The onset of seizures may also be a proxy for α-AASA dehydrogenase activity resulting in decreased α-AASA accumulation compared with those participants with a complete absence of enzyme activity. Therefore, genotype was not considered as a covariate because this may also be associated with residual enzyme activity. Other potential confounders included sex, gestational age, age at pyridoxine supplementation, delay of pyridoxine supplementation, and age at developmental testing. The delay of pyridoxine treatment was defined as the time between seizure onset and consistent pyridoxine treatment.
Statistical Analysis
Statistical analyses were performed with R software version 3.6.3.36 Continuous variables were summarized as either mean and SD or median and interquartile range (IQR). Categorical variables were summarized as the number and percentage of all participants.
The relationship between developmental outcomes and treatment modality was analyzed with multivariable linear regression using a mixed-effects model. Univariate analysis was performed to explore potential covariates as predictors for the developmental outcome, and any potential covariate with a significance of ≤0.05 was included in the multivariable model. Treatment modality was a dichotomous variable defined as either treatment with pyridoxine monotherapy or pyridoxine + LRT during developmental testing. Gestational age (full-term or <37 weeks gestation) and onset of seizures (≤2 months of age or late onset) were also treated as dichotomous variables. The delay of pyridoxine supplementation (in days) and age at developmental testing (years) were treated as continuous variables. The interaction between treatment modality and length of treatment modality was tested.
A linear mixed-effects analysis of the relationship between treatment with pyridoxine + LRT and developmental test results was performed using the statistical software R and the lme4 package.37 The length of treatment (as an interaction term with treatment), gestational age, the age at initial onset of seizures, the delay in treatment with pyridoxine from the onset of seizures, and the age of the participants during developmental testing were added as fixed effects. Several participants had multiple developmental test results. As a result, the intercept for participants was entered as a random effect. Visual inspection of the residual plots did not reveal any obvious deviations from homoscedasticity or normality.
A priori, we also hypothesized that treatment in early infancy with pyridoxine + LRT would result in a normal developmental outcome. A subanalysis of the data was performed by categorizing each participant as either treated with pyridoxine + LRT in the first 6 months of life or other treatment modality. This age cutoff was based on observational data, which suggested that participants treated with LRT in the first 6 months of life had optimal neurologic outcomes.25
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Participants
Initial inclusion criteria were met by 103 participants (eTable 1, links.lww.com/WNL/C281). Three participants were excluded because of a history of prepartum hypoxic brain injury, a history of prematurity resulting in mild spastic diplegia and motor delay, and a history of prematurity and neonatal sepsis, respectively. Of the 100 remaining participants, 99 had biallelic variants identified in ALDH7A1. Participants in whom a variant of uncertain significance was identified also had an elevation of pathognomonic biomarkers, which supported the diagnosis of PDE-ALDH7A1. In the remaining participant, urine α-AASA was extremely elevated, and disorders of sulfite accumulation were excluded.
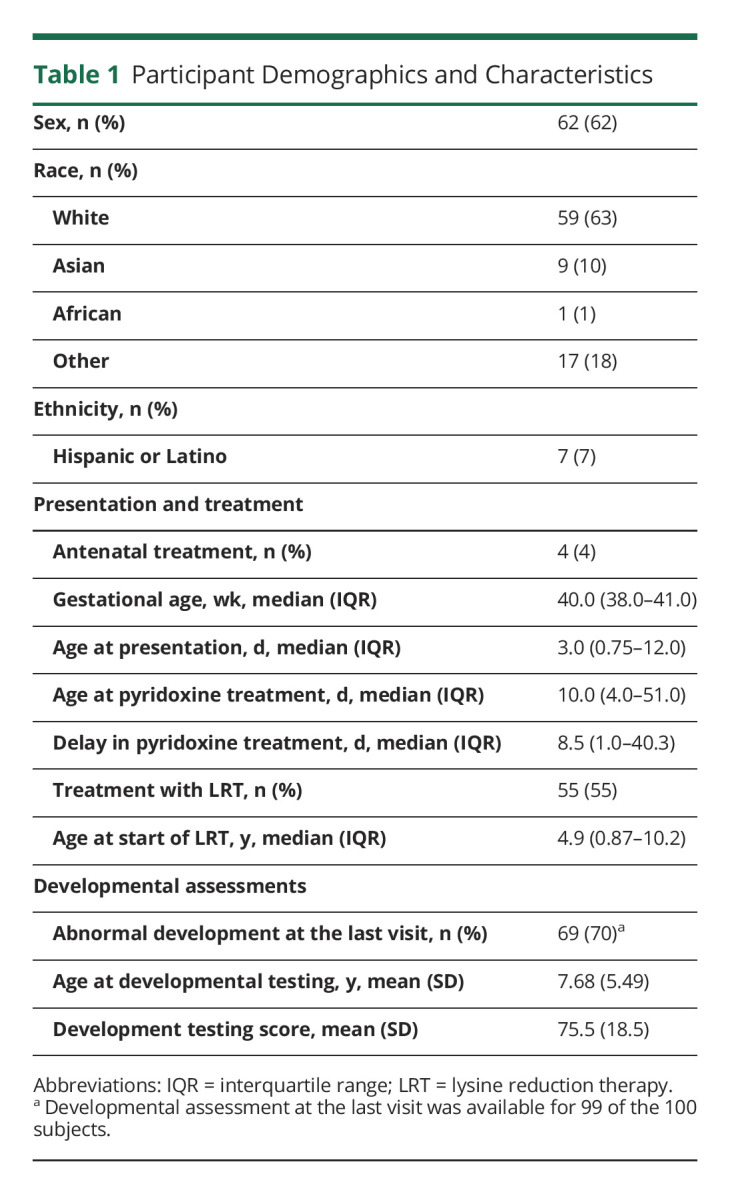
Most of the participants (63%) were White and not of Hispanic or Latino ethnicity (Table 1). Most participants presented with seizures in the neonatal or early infantile period, and 13% presented with seizures after 2 months of age. The delay from onset of seizures to treatment with pyridoxine ranged from 0 day to 10.5 years. Most participants (70.4%) were described as having developmental delay at their most recent visit, and the average developmental testing score was 75.5 (SD 18.5). These results are comparable with those of previous publications describing developmental outcomes in this disorder.10,11 Notably, 55% of all participants had been treated with adjunct LRT during at least 1 study visit, likely representing the increasing use of this therapy.
Table 1.
Participant Demographics and Characteristics
Primary Outcome LRT and Developmental Testing Results
A total of 112 standardized developmental testing scores were available from 60 participants (eTable 2, links.lww.com/WNL/C281). Fifty (44.6%) of the standardized developmental testing scores were obtained when participants were treated with pyridoxine monotherapy. When treated with pyridoxine monotherapy, the mean standardized developmental score was 74.9 (95% CI 69.9–79.9) compared with a mean of 76.0 (95% CI 71.3–80.7) (p = 0.8) when treated with pyridoxine + LRT.
Univariate model estimates were obtained for all possible covariates and confounders. The age at onset of seizures and delay of pyridoxine treatment were probable covariates and were significant in the univariate analysis. Gestational age at birth and age of the individual when developmental testing was performed were also significant in the univariate analysis, and all 4 of these variables were kept in the final model. Participants were not assigned a given treatment or treated for a fixed amount of time before developmental testing. The length of treatment is the amount of time (in years) that an individual was on a given treatment modality, and the duration of treatment during developmental testing ranged from 0.1 to 37.0 years. Available preliminary data suggest that the benefit of LRT would not be realized immediately. Therefore, the length of treatment was an interactive value with treatment modality.
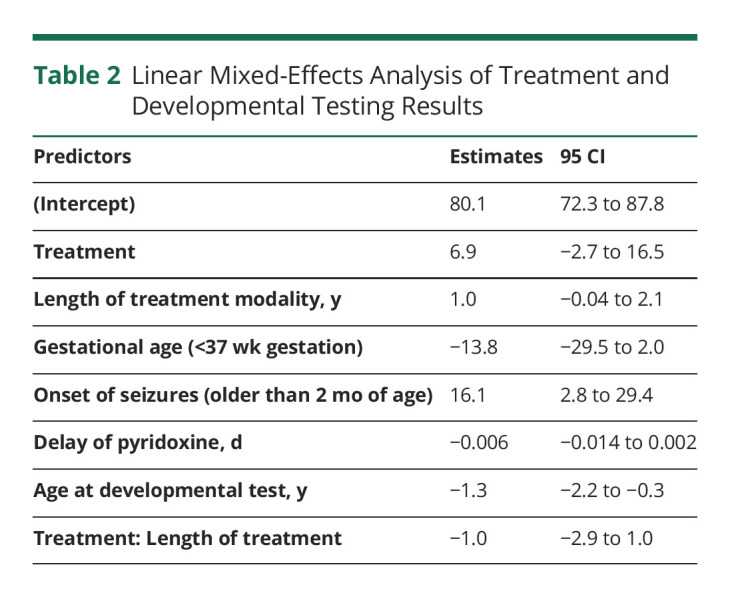
Results from the linear mixed-effects model are summarized in Table 2. The interactions of the variables and the intercept trended in biologically plausible fashions (i.e., decreasing when an individual is born premature and increasing with late-onset seizures). On average, the use of pyridoxine + LRT was associated with an increase of 6.9 points on developmental testing (95% CI −2.7 to 16.5) (p = 0.2).
Table 2.
Linear Mixed-Effects Analysis of Treatment and Developmental Testing Results
Analysis of LRT Started in Neonatal Period or Early Infancy
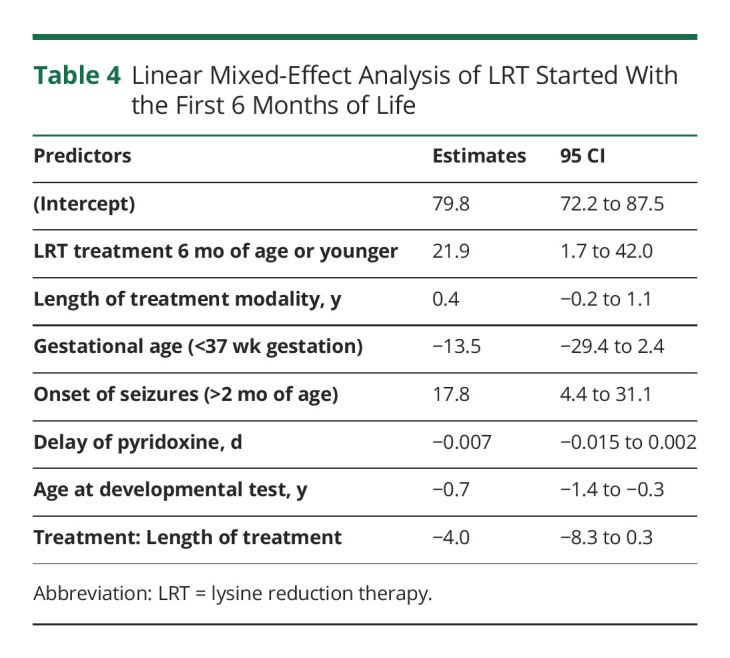
We had hypothesized treatment with LRT should be started in early infancy for individuals to have a normal developmental outcome. A total of 14 developmental testing scores were available from 8 participants who were treated with pyridoxine + LRT in the first 6 months of life (Table 3). The mean developmental test score in the early treated group was 87.3 (95% CI 79.5–95.0) compared with a mean of 73.8 (95% CI 73.8–77.5) (p < 0.01) in the rest of the cohort (Figure 1). After adjusting for the previously described confounders and covariates, treatment with LRT in the first 6 months of life was also evaluated in the linear mixed-effects model noted earlier. On average, treatment with pyridoxine + LRT was associated with an increase of 21.9 points (95% CI 1.7–42.0, p = 0.04) on developmental testing compared with treatment with pyridoxine alone or treatment with pyridoxine + LRT initiated after 6 months of life (Table 4).
Table 3.
Participants Treated With Lysine Reduction Therapy in the First 6 Months of Life

Figure 1. Developmental Outcomes Based on Timing of Treatment.

Raincloud plot of developmental testing scores based on treatment with pyridoxine or pyridoxine and lysine reduction therapies (LRTs) started after 6 months of age compared with treatment with pyridoxine and LRT in the first 6 months of life. The normality of the data was confirmed by the Shapiro-Wilk test, and p value is based on an independent t test.
Table 4.
Linear Mixed-Effect Analysis of LRT Started With the First 6 Months of Life
Classification of Evidence
This study provides Class IV evidence that in PDE-ALDH7A1, pyridoxine and lysine reduction therapies compared with pyridoxine alone is not significantly associated with overall higher developmental testing scores, but treatment in the first 6 months of life is associated with significantly higher developmental testing scores.
Discussion
PDE-ALDH7A1 is a readily treatable disorder, and pharmacologic doses of pyridoxine dramatically improve the outcome for control of otherwise intractable epileptic seizures. Despite adequate seizure control, most patients continue to experience ID/DD. LRT aims to reduce the accumulation of putative neurotoxic metabolites with the goal of improving developmental outcomes.32 In this study, we reported an international observational cohort study of research participants with PDE-ALDH7A1, which describes the association between treatment with LRT and objective developmental outcomes.
In 60 participants, 112 standardized developmental test results, such as full-scale intelligence quotient, were reported. Fifty-five percentage of developmental tests were performed when individuals were treated with pyridoxine + LRT. There was not a significant difference in the test results of those treated with pyridoxine + LRT compared with the results of those participants treated with pyridoxine supplementation alone. Of note, 15 participants (25%) had developmental testing performed while on pyridoxine monotherapy and subsequently on pyridoxine + LRT. This change in therapy was not unexpected because recommendations to treat patients with LRT were only recently published.32,35 Nine participants had an improved developmental test score on the subsequent developmental test after changing to pyridoxine + LRT. Two other participants had developmental test score improvements over time (eFigure 1, links.lww.com/WNL/C280). Some caution should be taken when interpreting this data. It remains possible that only those individuals who had a poor developmental trajectory were recommended to change therapy. Second, this data set does not control for critical variables such as the age at the start of LRT treatment and the length of LRT treatment.
Because a simple model did not account for the variability of developmental outcomes and multiple developmental test results were available for some participants, a linear mixed-effects model was chosen to evaluate the association of LRT on developmental test scores. After controlling for the length of treatment, prematurity, timing of seizure onset, delay of treatment with pyridoxine, and age of individuals at developmental testing, the use of pyridoxine + LRT was associated with a moderate increase in developmental testing scores with an average increase of 6.9 points on developmental testing (95% CI −2.7 to 16.5). This may be clinically important even if the results are not statistically significant.
These results support previous observational studies, which noted clinical improvement in some patients after the addition of LRT. While encouraging, the benefit of LRT in this study is rather modest and suggests that effect of LRT may not significantly affect the cognitive outcome in all patients. The recommendation for dietary treatment is not benign. In similar metabolic disorders, dietary treatment has been associated with personal and financial burdens on caregivers.38,39 Overtreatment can also lead to a protein deficiency.40 Although this risk seems to be relatively low,41,42 LRT should be closely monitored by a multidisciplinary team including a metabolic dietitian.32
One possible explanation for the modest effect of LRT on cognitive development is that treatment may need to be started early in life to avoid the sequelae from long-term exposure to neurotoxic metabolites. This is consistent with observational data in patients with PDE-ALDH7A125 and with other inherited metabolic disorders such as phenylketonuria43 and cystathionine β-synthase–deficient homocystinuria.44 All individuals treated with pyridoxine + LRT were included in this analysis irrespective of the age of the participant when LRT was initiated. The median age at the start of LRT was 4.9 years of life (IQR 0.87–10.2 years). Lysine restriction may have also been insufficiently controlled in some individuals because details concerning lysine intake and surrogate biomarkers of dietary lysine restriction were not evaluated.
We performed a subanalysis that suggested treatment with pyridoxine + LRT in the first 6 months of life is associated with a significantly higher developmental testing score. After controlling for the length of treatment, prematurity, timing of seizure onset, delay of treatment with pyridoxine, and age of individuals at developmental testing, treatment with pyridoxine + LRT in the first 6 months of life was associated with an increase of 21.9 points on developmental testing. These results emphasize the importance of an early diagnosis and early initiation of treatment with pyridoxine + LRT. This further strengthens the evidence for the 2021 international consensus guidelines in which the initiation of LRT is encouraged as soon as the diagnosis of PDE-ALDH7A1 is confirmed.32
These data also suggest that the delay in diagnosis and treatment may be a main contributor to the poor developmental outcome in this disease. In phenylketonuria, the paradigm condition for newborn screening, even a few weeks delay in dietary therapy can affect an individual's cognitive outcome.43 The potentially disastrous effects of diagnostic and therapeutic delays, in addition to the recent elucidation of stable biomarkers, makes PDE-ALDH7A1 a potential candidate for newborn screening.16,17,45
Previous observational studies noted that late-onset seizures may be associated with relatively good developmental outcomes.26 Indeed, our results confirmed that late-onset seizure is a significant predictor of developmental test results. In total, we reported 8 participants who had onset of seizures after 2 months of age with a mean developmental test score of 92.2 (95% CI 82.9–101.0). Although 6 of the 8 participants with late-onset seizures were also treated with pyridoxine + LRT, which limits the ability to discern the effect of late-onset seizures on developmental test scores alone.
It is important to reiterate that the developmental outcomes in patients with PDE-ALDH7A1 are not associated with seizure control. Several patients had been treated with pyridoxine antenatally and never had clinical or electroencephalographic evidence of seizures.46 Despite the absence of seizures, some of these patients had significant intellectual disability.12,47 It remains possible that late-onset seizures is the result of residual α-AASA dehydrogenase activity, which would also result in lower levels of neurotoxic metabolites. Given the association between late-onset seizures and relatively good developmental outcomes, patients who present with onset of seizures after 2 months of age may only require treatment with pyridoxine supplementation. Conversely, patients with attenuated phenotypes may benefit the most from early and aggressive therapy. Future studies are needed to investigate which patients would benefit from treatment with both LRT and pyridoxine.
The observational nature of this study is a limitation because participants were not assigned to an intervention nor treated for a standard amount of time before developmental testing. Not all participants underwent the same developmental testing battery. There may also be a selection bias for which participants underwent developmental testing because individuals who were assessed to be developmentally appropriate may be less likely to undergo formal developmental testing. However, this approach did provide valuable insights into treatment outcomes with use of available data. We also recognize that standardized developmental testing is only one measure of evaluating a child's development and does not fully describe the effect of this disorder on an individual's quality of life or the benefit of therapy. Previously, studies demonstrated the potential benefit of adjunct LRT in other domains, such as in motor, verbal, and behavioral skills.8,19,20,22,24,30,31 Future studies focused on various domains of development, including patient-reported outcome measures, would allow for improved guidance on the use of adjunct LRT.
The results of early treatment are compelling, although only 8 participants were treated with pyridoxine + LRT in the first 6 months of life. It is also important to note the average age of developmental testing in this cohort was significantly younger (3.6 years of age, 95% CI 2.9–4.3) compared with that of the rest of the participants (8.3 years of age, 95% CI 7.2–9.4). It remains possible that the developmental outcomes in this cohort may wane with age. Future studies are needed to confirm the effect of neonatal treatment with pyridoxine + LRT on cognitive development.
Several patients have also been reported with abnormal cerebral imaging and varying degrees of neurologic sequelae.48-50 The effect of these findings on developmental outcomes is currently unclear. While age at seizure onset and treatment initiation was available, the information on recurrent episodes of status epilepticus and burden of seizures before and after treatment initiation was not available; the latter variable could also influence developmental outcomes. Although these results suggest treatment with LRT in early infancy may ameliorate the developmental delay in this disease, the developmental testing results remain 1 standard deviation below the general population. Taken together, these data suggest that LRT is an important treatment to improve clinical outcomes although other treatment strategies are needed to further improve the developmental outcome in this disorder.
This report supports the association between treatment with LRT and improved developmental outcomes. On average, the use of LRT was associated with a nonsignificant increase of 6.9 points on developmental testing. A significant increase in developmental testing results was associated when treatment with LRT was initiated in early infancy with an average increase of 21.9 points on developmental testing. These results highlight the importance of an early diagnosis and initiation of treatment with both pyridoxine and LRT. PDE-ALDH7A1 joins other neurotoxic biochemical genetic conditions such as phenylketonuria and cystathionine β-synthase–deficient homocystinuria where early initiation of a dietary intervention has a well-established effect on neurodevelopmental outcome.
Acknowledgment
The authors are grateful to the patients and families for participating in this study, the clinicians and dietitians for their clinical care, and the laboratory specialists for diagnostics and monitoring.
Glossary
- α-AASA
α-aminoadipic semialdehyde
- ID/DD
intellectual disability or developmental disability
- IQR
interquartile range
- LRT
lysine reduction therapy
- PDE
pyridoxine-dependent epilepsy
Appendix 1. Authors
Appendix 2. Coinvestigators

Footnotes
Editorial, page 1025
Class of Evidence: NPub.org/coe
Study Funding
This project has received funding from the European Union's Horizon 2020 research and innovation program under the EJP RD COFUND-EJP No. 825575. This project/publication is supported in part by NIH/NCATS Colorado CTSA Grant No. UL1 TR002535.
Disclosure
L.A. Tseng reports an honorarium for a lecture from Nutricia, and A.M.J. van Wegberg reports a research grant from Nutricia and travel grants from Nutricia and Vitaflo. Both Nutricia and Vitaflo produce medical formulas for the treatment of PDE-ALDH7A1. All other authors report no relevant disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Hunt AD, Stokes J, McCrory WW, Stroud HH. Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics. 1954;13(2):140-145. [PubMed] [Google Scholar]
- 2.Gül-Mert G, İncecik F, Hergüner MÖ, Ceylaner S, Altunbaşak Ş. Pyridoxine-dependent epilepsy in two Turkish patients in Turkey and review of the literature. Turk J Pediatr. 2015;57(4):394-397. [PubMed] [Google Scholar]
- 3.Nam SH, Kwon M-J, Lee J, et al. Clinical and genetic analysis of three Korean children with pyridoxine-dependent epilepsy. Ann Clin Lab Sci. 2012;42(1):65-72. [PubMed] [Google Scholar]
- 4.Russell KE, Mulligan SR, Mallory LA. Diagnosis of pyridoxine-dependent seizures in a nineteen-year-old patient. Pediatr Neurol. 2012;47:141-143. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Yang X, Wu Y, et al. Clinical diagnosis, treatment, and ALDH7A1 mutations in pyridoxine-dependent epilepsy in three Chinese infants. PLoS One. 2014;9(3):e92803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben Younes T, Kraoua I, Benrhouma H, et al. Pyridoxine-dependent epilepsy: a novel mutation in a Tunisian child. Arch Pediatr. 2017;24(3):241-243. [DOI] [PubMed] [Google Scholar]
- 7.Baumgart A, Spiczak Svon, Verhoeven-Duif NM, et al. Atypical vitamin B6 deficiency: a rare cause of unexplained neonatal and infantile epilepsies. J Child Neurol. 2014;29(5):704-707. [DOI] [PubMed] [Google Scholar]
- 8.Toldo I, Bonardi CM, Bettella E, et al. Brain malformations associated to Aldh7a1 gene mutations: report of a novel homozygous mutation and literature review. Eur J Paediatr Neurol. 2018;22(6):1042-1053. [DOI] [PubMed] [Google Scholar]
- 9.Marguet F, Barakizou H, Tebani A, et al. Pyridoxine-dependent epilepsy: report on three families with neuropathology. Metab Brain Dis. 2016;31(6):1435-1443. [DOI] [PubMed] [Google Scholar]
- 10.Basura GJ, Hagland SP, Wiltse AM, Gospe SM. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. 2009;168(6):697-704. [DOI] [PubMed] [Google Scholar]
- 11.Bok LA, Halbertsma FJ, Houterman S, et al. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2012;54(9):849-854. [DOI] [PubMed] [Google Scholar]
- 12.Rankin PM, Harrison S, Chong WK, Boyd S, Aylett SE. Pyridoxine-dependent seizures: a family phenotype that leads to severe cognitive deficits, regardless of treatment regime. Dev Med Child Neurol. 2007;49(4):300-305. [DOI] [PubMed] [Google Scholar]
- 13.Plecko B, Stöckler-Ipsiroglu S, Paschke E, Erwa W, Struys EA, Jakobs C. Pipecolic acid elevation in plasma and cerebrospinal fluid of two patients with pyridoxine-dependent epilepsy. Ann Neurol. 2000;48(1):121-125. [PubMed] [Google Scholar]
- 14.Struys EA, Jakobs C. Alpha-aminoadipic semialdehyde is the biomarker for pyridoxine dependent epilepsy caused by alpha-aminoadipic semialdehyde dehydrogenase deficiency. Mol Genet Metab. 2007;91(4):405. [DOI] [PubMed] [Google Scholar]
- 15.Struys EA, Bok LA, Emal D, Houterman S, Willemsen MA, Jakobs C. The measurement of urinary Δ1-piperideine-6-carboxylate, the alter ego of α-aminoadipic semialdehyde, in Antiquitin deficiency. J Inherit Metab Dis. 2012;35(5):909-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wempe MF, Kumar A, Kumar V, et al. Identification of a novel biomarker for pyridoxine-dependent epilepsy: implications for newborn screening. J Inherit Metab Dis. 2019;42(3):565-574. [DOI] [PubMed] [Google Scholar]
- 17.Engelke UF, van Outersterp RE, Merx J, et al. Untargeted metabolomics and infrared ion spectroscopy identify biomarkers for pyridoxine-dependent epilepsy. J Clin Invest. 2021;131(15):148272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stockler S, Plecko B, Gospe SM, et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104(1-2):48-60. [DOI] [PubMed] [Google Scholar]
- 19.van Karnebeek CDM, Hartmann H, Jaggumantri S, et al. Lysine restricted diet for pyridoxine-dependent epilepsy: first evidence and future trials. Mol Genet Metab. 2012;107(3):335-344. [DOI] [PubMed] [Google Scholar]
- 20.Mercimek-Mahmutoglu S, Cordeiro D, Cruz V, et al. Novel therapy for pyridoxine dependent epilepsy due to ALDH7A1 genetic defect: L-arginine supplementation alternative to lysine-restricted diet. Eur J Paediatr Neurol. 2014;18(6):741-746. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt Z, Murthy G, Ennis M, Stockler-Ipsiroglu S, Elango R. Impact of enteral arginine supplementation on lysine metabolism in humans: a proof-of-concept for lysine-related inborn errors of metabolism. J Inherit Metab Dis. 2020;43(5):952-959. [DOI] [PubMed] [Google Scholar]
- 22.Coughlin CR, van Karnebeek CDM, Al-Hertani W, et al. Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: neurodevelopmental outcome. Mol Genet Metab. 2015;116(1-2):35-43. [DOI] [PubMed] [Google Scholar]
- 23.Mahajnah M, Corderio D, Austin V, et al. A Prospective case study of the safety and efficacy of lysine-restricted diet and arginine supplementation therapy in a patient with pyridoxine-dependent epilepsy caused by mutations in ALDH7A1. Pediatr Neurol. 2016;60:60-65. [DOI] [PubMed] [Google Scholar]
- 24.Yuzyuk T, Thomas A, Viau K, et al. Effect of dietary lysine restriction and arginine supplementation in two patients with pyridoxine-dependent epilepsy. Mol Genet Metab. 2016;118(3):167-172. [DOI] [PubMed] [Google Scholar]
- 25.Al Teneiji A, Bruun TUJ, Cordeiro D, et al. Phenotype, biochemical features, genotype and treatment outcome of pyridoxine-dependent epilepsy. Metab Brain Dis. 2017;32(2):443-451. [DOI] [PubMed] [Google Scholar]
- 26.de Rooy RLP, Halbertsma FJ, Struijs EA, et al. Pyridoxine dependent epilepsy: is late onset a predictor for favorable outcome? Eur J Paediatr Neurol. 2018;22(4):662-666. [DOI] [PubMed] [Google Scholar]
- 27.Navarro-Abia V, Soriano-Ramos M, Núñez-Enamorado N, et al. Hydrocephalus in pyridoxine-dependent epilepsy: new case and literature review. Brain Dev. 2018;40(4):348-352. [DOI] [PubMed] [Google Scholar]
- 28.Kava MP, Bryant L, Rowe P, Lewis B, Greed L, Balasubramaniam S. Beneficial outcome of early dietary lysine restriction as an adjunct to pyridoxine therapy in a child with pyridoxine dependant epilepsy due to Antiquitin deficiency. JIMD Rep. 2020;54(1):9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coci EG, Codutti L, Fink C, et al. Novel homozygous missense mutation in ALDH7A1 causes neonatal pyridoxine dependent epilepsy. Mol Cell Probes. 2017;32:18-23. [DOI] [PubMed] [Google Scholar]
- 30.Mercimek-Mahmutoglu S, Corderio D, Nagy L, et al. Lysine-restricted diet and mild cerebral serotonin deficiency in a patient with pyridoxine-dependent epilepsy caused by ALDH7A1 genetic defect. Mol Genet Metab Rep. 2014;1:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minet P, Sarret C, Miret A, Mention K, Benoist JF, Remerand G. Clinical and biochemical outcome of a patient with pyridoxine-dependent epilepsy treated by triple therapy (pyridoxine supplementation, lysine-restricted diet, and arginine supplementation). Acta Neurol Belg. 2021;121(6):1669-1675. [DOI] [PubMed] [Google Scholar]
- 32.Coughlin CR, Tseng LA, Abdenur JE, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2021;44(1):178-192. [DOI] [PubMed] [Google Scholar]
- 33.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP; STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453-1457. [DOI] [PubMed] [Google Scholar]
- 34.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Karnebeek CDM, Stockler-Ipsiroglu S, Jaggumantri S, et al. Lysine-restricted diet as adjunct therapy for pyridoxine-dependent epilepsy: the PDE consortium consensus recommendations. JIMD Rep. 2014;15:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Core Team. A language and environment for statistical computing [online]. 2013. Accessed April 21, 2021. R-project.org.
- 37.Bates D, Maechler M, Bolker B. lme4: linear mixed-effects models using S4 classes [online]. 2012. Accessed April 21, 2021. cran.r-project.org/web/packages/lme4/index.html.
- 38.MacDonald A, Gokmen-Ozel H, van Rijn M, Burgard P. The reality of dietary compliance in the management of phenylketonuria. J Inherit Metab Dis. 2010;33(6):665-670. [DOI] [PubMed] [Google Scholar]
- 39.MacDonald A, Smith TA, de Silva S, Alam V, van Loon JMT. The personal burden for caregivers of children with phenylketonuria: a cross-sectional study investigating time burden and costs in the UK. Mol Genet Metab Rep. 2016;9:1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard JV. Komrower lecture: treatment of inborn errors of metabolism: a review. J Inherit Metab Dis. 2006;29(2-3):275-278. [DOI] [PubMed] [Google Scholar]
- 41.Müller E, Kölker S. Reduction of lysine intake while avoiding malnutrition—major goals and major problems in dietary treatment of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27(6):903-910. [DOI] [PubMed] [Google Scholar]
- 42.Boy N, Haege G, Heringer J, et al. Low lysine diet in glutaric aciduria type I—effect on anthropometric and biochemical follow-up parameters. J Inherit Metab Dis. 2013;36(3):525-533. [DOI] [PubMed] [Google Scholar]
- 43.Smith I, Beasley MG, Ades AE. Intelligence and quality of dietary treatment in phenylketonuria. Arch Dis Child. 1990;65:472-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter JH, Wraith JE, White FJ, Bridge C, Till J. Strategies for the treatment of cystathionine beta-synthase deficiency: the experience of the Willink Biochemical Genetics Unit over the past 30 years. Eur J Pediatr. 1998;157(suppl 2):S71-S76. [DOI] [PubMed] [Google Scholar]
- 45.Jung S, Tran N-TB, Gospe SM, Hahn SH. Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS. Mol Genet Metab. 2013;110(3):237-240. [DOI] [PubMed] [Google Scholar]
- 46.Bok LA, Been JV, Struys EA, Jakobs C, Rijper EAM, Willemsen MA. Antenatal treatment in two Dutch families with pyridoxine-dependent seizures. Eur J Pediatr. 2010;169(3):297-303. [DOI] [PubMed] [Google Scholar]
- 47.Yeghiazaryan NS, Striano P, Spaccini L, et al. Long-term follow-up in two siblings with pyridoxine-dependent seizures associated with a novel ALDH7A1 mutation. Eur J Paediatr Neurol. 2011;15(6):547-550. [DOI] [PubMed] [Google Scholar]
- 48.Oesch G, Maga AM, Friedman SD, et al. Geometric morphometrics reveal altered corpus callosum shape in pyridoxine-dependent epilepsy. Neurology. 2018;91(1):e78-e86. [DOI] [PubMed] [Google Scholar]
- 49.Friedman SD, Ishak GE, Poliachik SL, et al. Callosal alterations in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2014;56(11):1106-1110. [DOI] [PubMed] [Google Scholar]
- 50.Strijker M, Tseng LA, van Avezaath LK, et al. Cognitive and neurological outcome of patients in the Dutch pyridoxine-dependent epilepsy (PDE-ALDH7A1) cohort, a cross-sectional study. Eur J Paediatr Neurol. 2021;33:112-120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.