

Abstract
Lecanemab is a humanized immunoglobulin G1 monoclonal antibody that selectively binds to soluble Aβ aggregate species, while demonstrating low affinity for Aβ monomer. This article describes the population pharmacokinetic (PK) and PK/pharmacodynamic (PD) analyses for amyloid plaques, as measured using positron emission tomography (PET), and biomarkers of amyloid pathology as evidenced by Aβ42/40 ratio and plasma p‐tau181 following i.v. administration of lecanemab in subjects with early Alzheimer's disease. Lecanemab PKs were well‐characterized with a two‐compartment model with first‐order elimination. Final PK model contained covariate effects of anti‐drug antibody positive status, sex, body weight, and albumin on clearance. The time course of amyloid PET standard uptake ratio (SUVr), plasma Aβ42/40 ratio, and p‐tau181 were described using indirect response models with lecanemab exposure as a maximum effect function stimulating the reduction of SUVr, and as a linear function increasing Aβ42/40 ratio and decreasing p‐tau181 formation rates. PK/PD simulations show that 10 mg/kg biweekly dosing results in larger and faster decrease in SUVr and p‐tau181 and increase in Aβ42/40 ratio as compared to 10 mg/kg monthly dose. Furthermore, the PK/PD simulations showed that after treatment discontinuation the brain amyloid re‐accumulation to baseline levels is slow with a recovery half‐life of ~4 years, whereas plasma Aβ42/40 ratio and p‐tau181 return to baseline levels faster than amyloid. Given the relationship between changes in amyloid PET SUVr and soluble biomarkers, the developed PK/PD models can be used to inform lecanemab dose regimens in future clinical studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Reductions in amyloid plaques assessed by amyloid positron emission tomography (PET) is a well‐established imaging biomarker to visualize brain amyloid accumulation for Alzheimer's disease (AD). Changes in plasma biomarkers, such as Aβ42/40 ratio and p‐tau181, are potential surrogate markers for amyloid reduction as measured by amyloid PET.
WHAT QUESTION DID THIS STUDY ADDRESS?
This article describes serum lecanemab population pharmacokinetic (PK) and PK/pharmacodynamic (PD) relationships for amyloid PET standard uptake value ratio, and two plasma biomarkers (Aβ42/40 ratio and p‐tau181) in patients with early AD.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
PKs of lecanemab were well‐characterized by a linear, two‐compartment model and identified covariates were consistent with other monoclonal antibodies. The PK/PD model captured a slow re‐accumulation of brain amyloid and improved our understanding of how lecanemab impacts important surrogate markers for amyloid reduction.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The developed PK and PK/PD models provide insights in the effect of lecanemab dosing on the extent of brain amyloid removal and plasma biomarkers of amyloid and tau pathology.
INTRODUCTION
Alzheimer's disease (AD) is a progressive, neurodegenerative disorder of unknown etiology and the most common form of dementia among older people. Risk factors for AD are increasing age, genetics, and family history. 1 Whereas several genes increase the risk of AD, the ε4 allele of the apolipoprotein E (APOE4) gene is the strongest known genetic risk factor. 2 , 3 AD is defined biologically by the presence of two abnormal protein deposits: extracellular deposits of brain amyloid plaques (comprising β‐amyloid [Aβ] peptides) and neurofibrillary tangles (comprising abnormal tau protein). Biomarker, 4 clinicopathological, 5 and cohort 6 studies indicate that the disease process commences 10 to 20 years before the clinical onset of symptoms. The “amyloid cascade” hypothesis postulates that neurodegenerative processes in AD are driven by accumulation of aggregated Aβ species from an imbalance between Aβ production and Aβ clearance in the brain. 7
Biological classification of AD involves biomarker evidence of AD pathology, 8 , 9 such as confirmation of Aβ pathology by use of amyloid positron emission tomography (PET), or cerebrospinal fluid (CSF) measurements of Aβ. It is understood that biofluid biomarkers, such as CSF and plasma Aβ and p‐tau, become abnormal before imaging biomarkers, suggesting that biofluid biomarkers are more sensitive, early indicators of the pathophysiological changes that become manifest in imaging. 10 , 11 The plasma Aβ42/40 ratio has been shown to correlate with amyloid plaque load, where a low Aβ42/40 ratio (relative to those without brain amyloid pathology) indicates elevated (positive) brain amyloid. 12 , 13 , 14 Phosphorylated‐tau181 (p‐tau181) is a biomarker of tau pathophysiology 8 that correlates with tau pathology in the brain. It was reported that Aβ could induce tau phosphorylation and toxicity in cholinergic neurons, indicative of a relationship between these biomarkers. 15 Further, biomarker clinical research indicated that plasma p‐tau181 has been significantly correlated with brain amyloid. 16 Fluid biomarkers, such as plasma Aβ42/40 ratio and p‐tau181, may be a more sensitive and convenient method of tracking disease progression than imaging biomarkers.
Lecanemab (BAN2401) is a humanized immunoglobulin G1 monoclonal antibody (mAb) that selectively binds to soluble Aβ aggregate species (oligomers and protofibrils), while demonstrating low affinity for Aβ monomer. 17 , 18 , 19 , 20 , 21 , 22 The antibody was safe and well‐tolerated in two phase I studies, with dose proportional exposure. 23 In a phase II study, lecanemab treatment led to a dose‐dependent reduction in brain amyloid, a slowing of clinical decline across a number of outcome measures, and directionally consistent biomarker changes at 18 months in subjects with early AD (mild cognitive impairment [MCI] due to AD or mild AD dementia). 24
Here, we report population PK and PK/PD analyses of lecanemab in subjects with early AD correlating lecanemab exposure with amyloid PET standard uptake value ratio (SUVr), and plasma Aβ42/40 ratio and p‐tau181.
METHODS
Study design and treatments
The phase II study 201 core phase was a double‐blind, parallel‐group, placebo‐controlled, multicenter study utilizing a dose‐finding response adaptive randomization design to evaluate the safety, tolerability, and efficacy of lecanemab in subjects with MCI due to AD or with mild AD dementia. Study 201 core randomized 856 subjects across six treatment groups: placebo, 2.5 mg/kg biweekly, 5.0 mg/kg monthly, 5.0 mg/kg biweekly, 10 mg/kg monthly, or 10 mg/kg biweekly for 18 months. The open‐label extension (OLE) phase of study 201 was initiated following the core study to allow subjects to receive open‐label lecanemab 10 mg/kg biweekly for up to 60 months (5 years). For all subjects, there was a gap of 9–59 months (average 24 months) without treatment between the last dose in the core study and the first dose in the OLE phase (hereafter called the “gap period”). An overview of the study design is shown in Figure S1.
The pharmacokinetic (PK) analysis was performed on pooled data from subjects receiving lecanemab who participated in two phase 1 studies (studies 101 23 and 104) and study 201 core and OLE. The PK/pharmacodynamic (PD) analyses for SUVr and plasma Aβ42/40 ratio and p‐tau181 were performed on data from subjects with early AD (mild AD dementia and MCI due to AD) receiving either lecanemab or placebo who participated in study 201 core and OLE.
Study designs and treatment regimens across these studies are detailed in Tables S1‐S10. All studies were approved by relevant institutional review boards/ethics committees and conducted in accordance with International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and all applicable local Good Clinical Practice (GCP) guidelines, including the Declaration of Helsinki.
Bioanalytical assays and amyloid PET assessment
Serum concentrations of lecanemab were measured by validated immunoprecipitation—liquid chromatography—tandem mass spectrometry (IP/LC–MS/MS) methods using antihuman IgG antibody to precipitate lecanemab from a serum sample. For study 101, a validated enzyme‐linked immunosorbent assay was used for the measurement of serum concentrations of lecanemab. Lecanemab anti‐drug antibody (ADA) and neutralizing ADA (NAb) were measured in serum using validated immunoassays.
The amyloid PET SUVr normalized to whole cerebellum mask, measured using 18F florbetapir as a PET ligand, was used as a measure of brain amyloid levels. Amyloid negativity was determined as amyloid PET SUVr less than 1.17, which is equivalent to amyloid PET visual read in this study. 25
Plasma concentrations of Aβ42 and Aβ40 were measured using the IP/LC–MS/MS technology platform (PrecivityAD assay, C2N), and the ratio of plasma Aβ42/40 was calculated from the output. Plasma concentrations of p‐tau181 were measured using commercially validated single molecule array (Simoa) assay developed by Quanterix.
Modeling software
Population PK and PK/PD analyses were conducted using the first‐order conditional estimation with interaction method as implemented in the NONMEM software system (version 7.4.3; ICON Development Solutions) aided by Perl‐speaks‐NONMEM (version 4.9.0) or PDx‐Pop (version 5.2; ICON Development Solutions). R software (version 4.0.3) was used for the preprocessing and postprocessing of NONMEM, output creation of diagnostic plots, and graphical visualization. Simulations were performed using the R package mrgsolve (0.10.7). The final NONMEM model codes are provided in Supplementary Texts S1‐S4.
Population PK analysis
The population PK model utilized in the current analysis was developed previously using data from studies 101, 104, and 201 core. 26 Lecanemab PK was described by a two‐compartment linear model parameterized for the clearance (CL), the volumes of distribution of the central (V 1) and peripheral (V 2) compartments and intercompartmental clearance. The final PK model contained the covariate effects of sex, body weight, and albumin on CL, sex, and body weight on V 1 and Japanese race on V 2. This model was updated following pooling additional data from subjects receiving lecanemab in study 201 OLE. The effect of formulation from manufacturing process B, used in the OLE (detailed in Tables S1‐S10), was evaluated as a covariate on CL as well by assessing its bioavailability (F) relative to the process used in study 201 core and earlier studies. Additionally, the effect of ADA status as a categorical time‐variant covariate and ADA titer as a continuous time‐variant covariate on CL were also evaluated. In the categorical covariate analysis, both ADA negative conclusive and ADA negative inconclusive were assigned as ADA negative because no systematic exposure difference was observed between the two categories. 27 All PK observations with missing ADA status were assumed to be ADA negative.
The impact of significant covariates in the final PK model on the lecanemab exposures (area under the curve [AUC] and maximum concentration [C max]) was evaluated using a forest plot analysis. Lecanemab exposures at steady‐state with extreme values (5th and 95th percentiles) of each individual covariate were simulated and compared with a reference exposure derived based on population typical covariate values. The median and 95% confidence intervals (CIs) of individual covariate effects were generated based on 1000 simulations using final estimates of typical values and the variance–covariance matrix from the final PK model.
The final population PK model was used to derive the post hoc estimates of individual PK parameters that were used in the subsequent PK/PD analyses for amyloid PET SUVr and plasma biomarkers.
PK/PD analysis for amyloid PET SUVr and plasma Aβ42/40 ratio and p‐tau181
PK/PD models were built in a sequential manner and the structures and equations of the models are shown in Figure 1.
FIGURE 1.
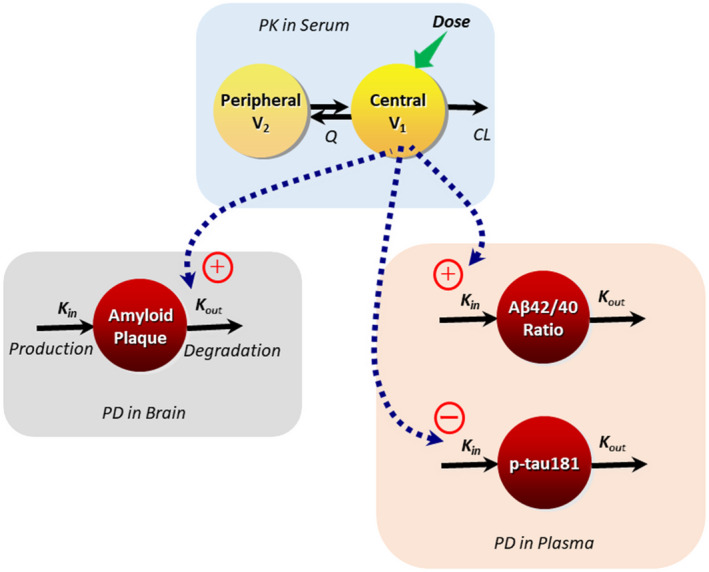
Schematic of the population PK and PK/PD models for lecanemab. CL, clearance; K in, zero‐order rate constant for production of biomarker; K out, first‐order rate constant of degradation of biomarker; PD, pharmacodynamic; PK, pharmacokinetic; Q, intercompartmental clearance; V 1, central volume of distribution; V 2, peripheral volume of distribution. The equation for standard uptake ratio (SUVr) PK/PD model is presented below: Estimated parameters included baseline SUVr, the zero‐order production rate constant of amyloid plaque (K in), maximum exposure effect (E max), and lecanemab concentration resulting in half of the maximum drug effect (EC50), where K out = K in/baseline. The equations for Aβ42/40 ratio and p‐tau181 models are presented below: For both Aβ42/40 ratio and p‐tau181 estimated parameters included baseline, first order degradation rate constant of biomarker (K out) and slope for exposure effect, where K in = K out * baseline.
The relationship between serum lecanemab concentration and the amyloid PET SUVr reduction time course was well‐described by an indirect response model with lecanemab concentration in the central compartment at the time of SUVr measurement, as a maximum effect (E max) function, acting to increase the rate of plaque removal.
For plasma Aβ42/40 ratio and p‐tau181, absolute measurements over time were correlated with PK model‐predicted lecanemab concentration in the central compartment at the time of the assessment by an indirect response model with exposure as a linear function increasing Aβ42/40 ratio or decreasing p‐tau181.
In each PK/PD analysis, all subjects receiving lecanemab with serum PK information or receiving placebo in study 201 core and who had baseline and at least one postdose assessment were included. Subjects treated with lecanemab 10 mg/kg biweekly in OLE and who had OLE baseline and postdose assessments were also included.
Covariates were selected based on clinical relevance and biological plausibility and tested using a stepwise approach. The covariates tested were sex, APOE4 carrier status, AD diagnosis (MCI or mild AD), ADA, and NAb at subject level. In the SUVr analysis, covariates were tested on baseline, first‐order rate constants for the production of an effect (K in), E max, and half‐maximal effective concentration (EC50), age, and weight on baseline and E max. For plasma Aβ42/40 ratio and p‐tau181 models, covariates were tested on baseline, first‐order rate constants for the removal of an effect (K out) and slope for exposure effect, in addition to the effect of age and weight on baseline and slope and observed baseline on slope. Univariate analysis was performed first for the effect of each covariate and all significant covariates at a significant level of α = 0.01 were pooled in a full multivariate model which was followed by backward elimination at a significant level of α = 0.001 to verify that the retained covariates were relevant in the final model.
PK and PK/PD model evaluation
Final population PK and PK/PD models were evaluated for performance using goodness‐of‐fit plots, simulated prediction‐corrected visual predictive checks (pcVPC), 28 and nonparametric bootstrapping. 29 , 30 For estimates of precision, asymptotic relative standard errors (RSEs), and nonparametric bootstrap 95% CIs were obtained for each model parameter.
Model‐based PK/PD simulations
Model‐based simulations were performed to explore the effect of lecanemab dosing regimens of 10 mg/kg biweekly or 10 mg/kg monthly for a treatment duration of 18 months on SUVr and Aβ42/40 ratio and plasma p‐tau181, accounting for the uncertainty of PD parameter estimates. Uncertainty was assessed by sampling 1000 sets of PD parameters, using final estimates of typical values and the variance–covariance matrix from final PK/PD models. Simulations were also performed to explore the potential impact of the treatment discontinuation on SUVr and plasma biomarkers after 18 months of continuous treatment at 10 mg/kg biweekly.
In addition, based on the 1000 model‐predicted individual profiles for SUVr, considering interindividual variabilities of PD parameters, the percentage of subjects achieving amyloid negativity for SUVr less than 1.17 at 18 months following continuous treatment with lecanemab at 10 mg/kg biweekly or monthly were derived.
RESULTS
Analyses population
The PK dataset included 9027 serum lecanemab observations, from 725 subjects, of which 661 (7.3%) were from study 101, there were 371 (4.1%) from study 104, and 7995 (88.6%) from study 201 core and OLE. The SUVr PK/PD dataset included 1213 observations from 374 subjects from study 201 core and OLE. There was a total of 2021 observations from 562 subjects in the plasma p‐tau181 dataset and a total of 1254 observations from 284 subjects in the plasma Aβ42/40 ratio dataset, both from study 201 core and OLE.
Summaries of subject baseline demographics and covariates for the population PK and PK/PD datasets are presented in Table S2.
Population PK analysis
Lecanemab PK full profiles from two phase I studies (studies 101 and 104) and sparse data from study 201 core were well‐described by a two‐compartment model with covariate effects previously developed, 26 and this model was used as a base model in updating the model after adding data from study 201 OLE. In the presence of known covariates, the effects of ADA on CL were newly evaluated. Effect of ADA status on CL was identified as significant covariate and included in the final PK model. Additionally, the relative F of the formulation (process B vs. process A) as an additional PK parameter was identified to be significant (Table S3).
The final population PK parameters for the final covariate model are presented in Table 1. All model parameters were estimated with good precision (%RSE ≤24.4%).
TABLE 1.
Population PK parameters and bootstrap CIs for the final lecanemab covariate model
| Parameter (units) | NONMEM | Bootstrap | |
|---|---|---|---|
| Estimate | %RSE | Median (95% CI) | |
| PK parameters | |||
| CL (L/h) | 0.0181 | 2.55 | 0.0181 (0.0175–0.0188) |
| V 1 (L) | 3.22 | 1.18 | 3.22 (3.15–3.28) |
| Q (L/h) | 0.0349 | 8.02 | 0.0349 (0.0294–0.0396) |
| V 2 (L) | 2.19 | 7.21 | 2.20 (2.00–2.40) |
| F for process B | 0.998 | 4.07 | 0.999 (0.950–1.06) |
| Covariate effects | |||
| Weight ~ CL (exponent) | 0.403 | 9.73 | 0.393 (0.217–0.495) |
| Albumin ~ CL (exponent) | −0.243 | 17.2 | −0.237 (−0.405 to −0.0771) |
| Females ~ CL (ratio) | 0.792 | 3.43 | 0.790 (0.735–0.825) |
| ADA positive ~ CL (ratio to ADA negative) | 1.09 | 0.586 | 1.09 (1.05–1.12) |
| Weight ~ V 1 (exponent) | 0.606 | 7.52 | 0.603 (0.548–0.663) |
| Females ~ V 1 (ratio) | 0.893 | 1.75 | 0.893 (0.870–0.919) |
| Japanese race ~ V 2 (ratio) | 0.455 | 24.4 | 0.450 (0.338–0.583) |
| Interindividual variability (CV%) | |||
| CL | 38.9 | 6.69 | 38.9 (36.7–40.4) |
| V 1 | 14.0 | 8.38 | 14.0 (12.7–15.1) |
| V 2 | 99.5 | 7.91 | 99.8 (84.4–109) |
| F | 34.2 | 17.7 | 34.1 (28.7–37.4) |
| Residual variability (CV%) | |||
| Proportional: study 101 | 14.0 | 3.49 | 14.0 (12.6–15.1) |
| Proportional: study 104 | 19.7 | 4.66 | 19.6 (16.9–22.1) |
| Proportional: study 201 | 30.3 | 0.803 | 30.2 (28.7–31.2) |
Abbreviations: %RSE, percent relative standard error of the estimate = SE/parameter estimate × 100; ADA, anti‐drug antibody; CI, confidence interval; CL, clearance; CV%, square root of variance × 100; F, relative bioavailability; PK, pharmacokinetic; Q, intercompartment clearance; V 1, central volume of distribution; V 2, peripheral volume of distribution.
.
.
.
.
ADA, 0 (ADA negative) or 1 (positive); ALB, albumin; BW, body weight; SEX, 0 (male) or 1 (female); JPN, 0 (non‐Japanese) or 1 (Japanese); FORM, 0 (Process A) or 1 (Process B).
Eta shrinkage (%): ETA_CL, 9.96%; ETA_V 1, 30.5%, ETA_V 2, 31.7%, ETA_F1, 63.2%.
The magnitude of significant covariates in the final PK model (Table 1 ) on predicted steady‐state PK exposures (AUC and C max) after 10 mg/kg biweekly is illustrated in the forest plot (Figure 2). As depicted, none of the covariates have any clinically meaningful effect on exposure to lecanemab to warrant any dose adjustment, as CIs of all covariates either fell within or overlapped the reference 0.8–1.25 interval.
FIGURE 2.
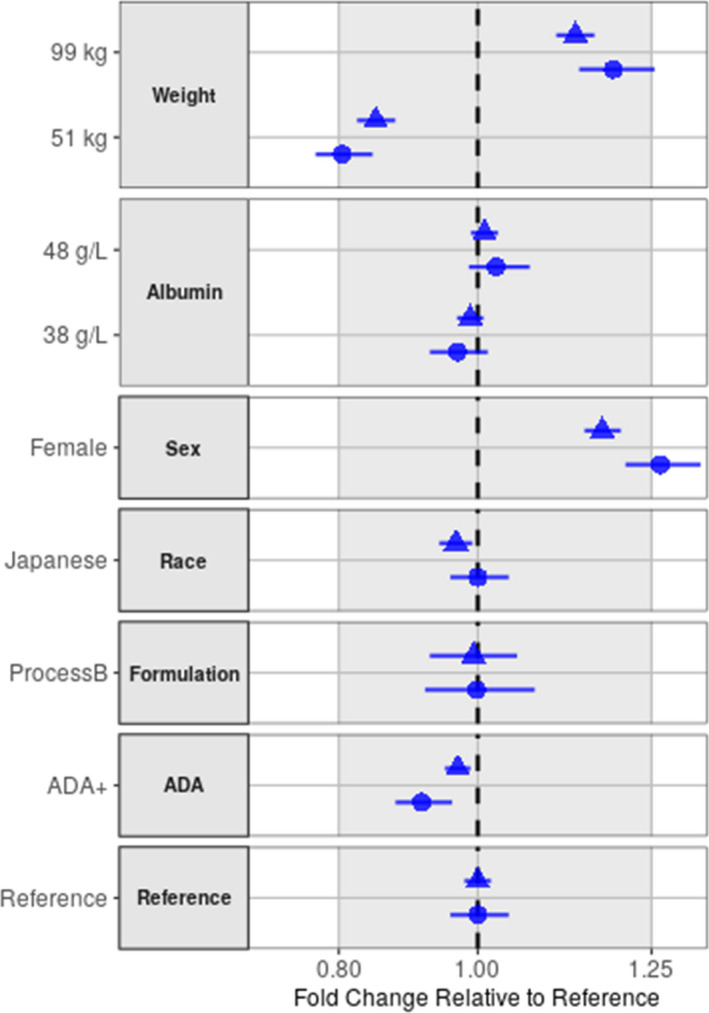
Effect of covariates on lecanemab AUC and C max at steady‐state after 10 mg/kg biweekly. Covariate effects are expressed as lecanemab exposures at steady‐state relative to a reference subject. Body weight and albumin test categories (51 and 99 kg for body weight, 38 and 48 g/L for albumin) represent the 5% and 95% percentiles of PK analysis set, respectively. Plot displays relative change in a parameter with uncertainty as 90% CIs for each covariate relative to the reference 73.4 kg male, non‐Japanese subject who was administered process A formulation and is ADA negative. Filled circle and triangle: median of AUC and C max, horizontal line: 90% CI, gray area: acceptance interval (0.80–1.25), vertical dashed line: reference. ADA, anti‐drug antibody; AUC, area under the curve; CI, confidence interval; Cmax, maximum concentration; PK, pharmacokinetic.
PK/PD analysis for amyloid PET SUVr
Steps taken in the development of the base PK/PD model for SUVr are summarized in Table S4. Based on these results, the base model selected for subsequent covariate analysis used an E max function for exposure effect with interindividual variability (IIV) on baseline and E max only with a covariance and proportional residual variability. Although IIV could not be estimated for K in and EC50, the effect of categorical covariates on these parameters were examined for exploratory purposes.
From the univariate analysis, only the effects of age on both E max and baseline SUVr and of APOE4 carrier status on baseline SUVr were identified as significant covariates (Table S5). Following multivariate analysis with backward elimination, only the effects of APOE4 carrier on baseline and age on E max remained significant and retained in the final PK/PD model (Table S6).
The population PK/PD parameters for the final model for SUVr are presented in Table 2. All key model parameters were estimated with good precision (%RSE < 21%).
TABLE 2.
Population PD parameters and bootstrap CIs for the final PK/PD model for Amyloid PET SUVr
| Parameter | NONMEM | Bootstrap | |
|---|---|---|---|
| Estimate | %RSE | Median (95% CI) | |
| PD parameters | |||
| Baseline SUVr | 1.34 | 0.873 | 1.34 (1.31–1.38) |
| K in (1/year) | 0.232 | 11.1 | 0.232 (0.174–0.277) |
| E max | 1.54 | 11.8 | 1.56 (1.10–2.01) |
| EC50 (μg/ml) | 75.0 | 19.6 | 77.1 (38.5–121) |
| Covariate | |||
| APOE4 carrier ~ baseline | 1.04 | 1.04 | 1.04 (1.02–1.07) |
| Age ~ E max (exponent) | 1.58 | 20.9 | 1.60 (0.972–2.07) |
| Interindividual variability (CV%) | |||
| Baseline | 10.9 | 8.12 | 11.0 (9.79–11.9) |
| Correlation baseline ~ E max (R) | 0.669 | 11.5 | 0.674 (0.556–0.713) |
| E max | 50.3 | 12.0 | 50.8 (42.1–59.7) |
| Residual variability (CV%) | |||
| Proportional | 5.01 | 2.75 | 5.02 (4.48–5.59) |
Abbreviations: %RSE, percent relative standard error of the estimate = SE/parameter estimate × 100; CI, confidence interval; CV%, square root of variance × 100; EC50, lecanemab concentration at which 50% of maximum drug effect is achieved; E max, maximum drug effect; K in, rate of production; PD, pharmacodynamic; PET, positron emission tomography; PK, pharmacokinetic; SUVr, standard uptake ratio.
.
AGE = age; APOE = 0 (APOE4 carrier) or 1 (non‐carrier).
Eta shrinkage (%): ETA_Baseline = 6.94%, ETA_E max = 27.7%.
Covariates identified in the final SUVr model indicate that APOE4 carrier subjects have higher baseline SUVr, and older subjects have higher maximum plaque removal (E max) by lecanemab (Table 2). In addition, the final PK/PD model for SUVr could estimate the covariance between baseline SUVr and E max, and its correlation coefficient was 0.608. This indicates that subjects with higher baseline SUVr have greater SUVr reduction, which is consistent with the observed trend in study 201 core shown in Figure S4.
The magnitude of significant covariates in the final model on SUVr change from baseline at 18 months after 10 mg/kg biweekly is illustrated in the forest plot (Figure S5), which displays relative change in SUVr from baseline at 18 months with uncertainty as 90% CI for each covariate relative to the reference subjects who is 72‐years‐old and APOE4 non‐carrier. Older age subjects, such as 84 years (95th percentile of analysis set), and younger age subjects, such as 57‐years‐old (5th percentile), had 24% higher and 29% lower SUVr reduction, respectively, than a reference 72‐year‐old subject.
PK/PD analysis for plasma Aβ42/40 ratio
Steps taken in the development of the base PK/PD model for Aβ42/40 ratio are summarized in Table S7. Based on these results, the base model used a linear function to describe the effect of exposure on slope, with exponential IIV on baseline and slope and proportional residual variability. No covariate effects were retained in the final model for Aβ42/40 ratio.
The final population PK/PD parameters for the final model for Aβ42/40 ratio are presented in Table 3.
TABLE 3.
Population PD parameters and bootstrap CIs for the final PK/PD models for plasma Aβ42/40 ratio and p‐tau181
| Parameter | NONMEM | Bootstrap | |
|---|---|---|---|
| Estimate | %RSE | Median (95% CI) | |
| Aβ42/40 ratio | |||
| PD parameters | |||
| Baseline plasma Aβ42/40 ratio | 0.0842 | 4.28 | 0.0843 (0.0834–0.0852) |
| K out (1/year) | 0.367 | 1.97 | 0.368 (0.255–0.521) |
| Slope (1/μg/ml) | 0.00155 | 9.32 | 0.00155 (0.00110–0.00213) |
| Interindividual variability | |||
| Baseline (CV%) | 6.78 | 16.1 | 6.73 (4.58–8.76) |
| Slope (CV%) | 44.1 | 11.1 | 44.3 (21.4–58.9) |
| Residual variability | |||
| Proportional (CV%) | 6.41 | 2.82 | 6.42 (6.03–6.82) |
| p‐tau181 | |||
| PD parameters | |||
| Baseline plasma p‐tau181 | 4.06 | 1.61 | 4.06 (3.97–4.14) |
| K out (1/year) | 0.468 | 20.7 | 0.502 (0.183–0.934) |
| Slope (1/μg/ml) | 0.00313 | 15.6 | 0.00328 (0.00178–0.00596) |
| Covariate effects | |||
| Weight ~ baseline (exponent) | −0.300 | 24.2 | −0.304 (−0.439 – −0.211) |
| Interindividual variability | |||
| Baseline (CV%) | 35.1 | 5.63 | 35.1 (33.0–37.4) |
| Slope (SD) | 0.00151 | 50.2 | 0.00162 (0.000662–0.00280) |
| Residual variability | |||
| Proportional (CV%) | 19.4 | 2.39 | 19.4 (18.3–20.6) |
Abbreviations: %RSE = percent relative standard error of the estimate = SE/parameter estimate × 100; CI = confidence interval; CV% = Square root of variance × 100; K out = rate constant of degradation; PD, pharmacodynamic; PK, pharmacokinetic.
p‐tau181: .
BW = body weight.
Aβ42/40 ratio ‐ Eta shrinkage (%): ETA_Baseline = 11.9%, ETA_Slope = 63.1%.
p‐tau181 ‐ Eta shrinkage (%): ETA_Baseline = 4.65%, ETA_Slope = 68.4%.
PK/PD analysis for plasma p‐tau181
Steps taken in the development of the base PK/PD model for p‐tau181 are summarized in Table S8. Based on these results, the base model selected for subsequent covariate analysis was with linear function for exposure effect with proportional IIV on baseline, additive IIV on slope for exposure effect, and proportional residual variability. Although IIV could not be estimated for K out the effect of covariates on this parameter were examined for exploratory purposes.
From the univariate analysis, the effects of ADA positive status, diagnosis, and weight on p‐tau181 baseline were identified as significant covariates (Table S9). Only the effect of body weight on baseline remained significant and retained in the final PK/PD model for p‐tau181 following backward elimination (Table S10).
The final population PK/PD parameters for the final covariate model for p‐tau181 are presented in Table 3. A 50 kg subject (5th percentile of dataset) and a 96 kg subject (95th percentile of dataset) had 11.7% higher and 8.2% lower baseline plasma p‐tau181, respectively, than a reference 72 kg subject. The magnitude of body weight effect on plasma p‐tau181 change from baseline at 18 months after 10 mg/kg biweekly is illustrated in the forest plot (Figure S6), however, there was no impact of body weight on plasma p‐tau181 change from baseline.
PK and PK/PD model evaluation
For PK model and PK/PD models for PET SUVr, Aβ42/40 ratio and p‐tau181, goodness‐of‐fit plots did not indicate model deficiencies (Figures S2 and S7‐S9).
Results of pcVPC for the final PK model, stratified by study, are presented in Figure S3. Lecanemab concentration time course for studies 201 and 104 has been reasonably well‐defined by the final PK model with good predictive performance. The pcVPC plot for study 101 indicated a trend to overprediction for timepoints for terminal elimination phase, however, the reason of overprediction in study 101 is unknown.
Prediction‐corrected observed data from subjects receiving placebo and lecanemab 10 mg/kg biweekly in the core study, and simulated data are plotted for PK/PD models for PET SUVr, p‐tau181, and Aβ42/40 ratio in Figure 3. The pcVPC plots indicated good agreement between simulated and observed data, indicating that the SUVr, Aβ42/40 ratio, and p‐tau181 time courses have been reasonably well‐defined by the final model with good predictive performance. The pcVPC plots of subjects receiving lecanemab treatment except for 10 mg/kg biweekly are shown in Figures S10‐S12.
FIGURE 3.
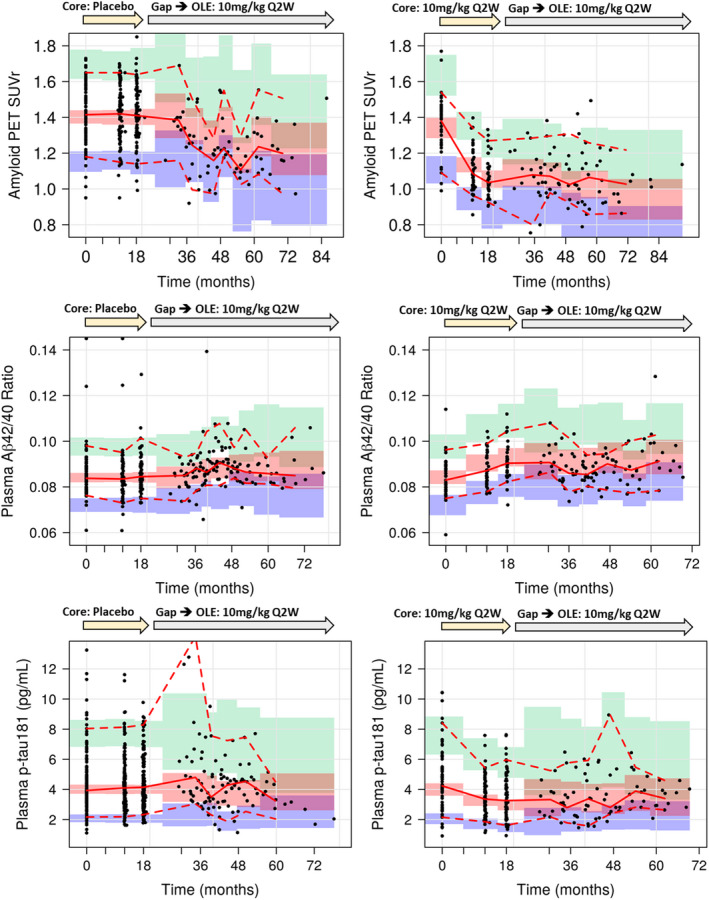
Prediction‐corrected visual predictive check plots for PK/PD models for PET SUVr and plasma Aβ42/40 ratio and p‐tau181 (left: core placebo, right: core 10 mg/kg biweekly). Black solid line: predicted median, black dashed line: predicted 5th and 95th percentiles, red solid line: observed median, red dashed line: observed 5th and 95th percentiles, green area: 95% CI for predicted 95th percentile, red area: 95% CI for predicted median, blue area: 95% CI for predicted 5th percentile. CI, confidence interval; OLE, open‐label extension; PD, pharmacodynamic; PET, positron emission tomography; PK, pharmacokinetic; SUVr, standard uptake ratio.
For PK and PK/PD models, the bootstrap medians were concordant with the population predicted values, indicating that each of the final model was valid and stable and produced well‐estimated parameters (Tables 1, 2, 3).
DISCUSSION
Lecanemab PK full profiles from two phase I studies and sparse data from study 201 core and OLE were well‐described by a two‐compartment model with linear elimination from the central compartment. Although the PKs of mAbs generally show the nonlinear process by target‐mediated drug disposition (TMDD), 31 the TMDD was not observed for lecanemab because its antigens (Aβ oligomers, protofibrils, and plaques) are dominantly located in the brain. Clearance and volume of distribution estimates were consistent with typical values for a mAb. 32 As a comparison with other mAbs for AD, the terminal half‐life of lecanemab for the typical patient estimated from PK model is ~9.5 days, which is similar with donanemab (10 days) 33 but shorter than aducanumab (24.8 days) 34 and gantenerumab (22 days). 35
Lecanemab CL and V 1 increased with increasing body weight with exponents of 0.403 and 0.606, respectively, and both parameters were slightly lower in women compared to men. These findings are consistent with results for other mAbs. 36 The rationale for the difference in PKs of mAbs between men and women is unclear, however, there may be differences in the lymph flow rate and expression of the Fc receptor. 37 Lecanemab CL was found to decline with increasing albumin levels. The neonatal Fc receptor (FcRn) facilitates IgG and albumin homeostasis by recycling them across cell membranes back to the central circulatory system. 38 Thus, a higher albumin concentration could be an indicator of an increased number of FcRn and related reduced lecanemab elimination. However, none of the significant covariates had a clinically relevant effect on steady‐state exposure to lecanemab in terms of both C max and AUC. Age and APOE4 carrier status, which are risk factors for AD, were not found to significantly affect lecanemab PKs. 26
In study 201 core, lecanemab treatment removing amyloid plaques in the brain was associated with the increase of Aβ42 and decrease of p‐tau181 in CSF. 24 Because plasma Aβ42/40 ratio and p‐tau181 have been reported to reflect the changes in CSF Aβ42 and p‐tau181, increase of plasma Aβ42/40 ratio, and decrease of plasma p‐tau181 by lecanemab treatment are possibly reflecting the changes of CSF Aβ42 and CSF p‐tau181. 10 , 39 , 40 Observed amyloid PET SUVr and plasma p‐tau181 and Aβ42/40 ratio profiles showed a dose and time‐dependent change across all doses versus placebo in study 201 core (Figures 3 and S10‐S12). During the gap period, in which all subjects who participated in the OLE were off treatment for an average of ~2 years (9–59 months), amyloid plaque levels re‐accumulated slowly, and p‐tau181 and Aβ42/40 ratio slowly increased and decreased, respectively. The relationships between exposure to lecanemab and SUVr, p‐tau181, and Aβ42/40 ratio time course were well‐described by indirect response models. Based on our PK/PD analysis for SUVr the population estimate of K out is 0.173/year and thus the time for amyloid re‐accumulation to reach 50% recovery of baseline level (recovery half‐life) is estimated to be ~4 years. Kandadi Muralidharan et al. reported the estimate of K out for SUVr PK/PD model based on data from aducanumab clinical studies was 0.745/year, 34 therefore the recovery half‐life for amyloid re‐accumulation is estimated to be about 1 year, which is shorter than that from our analysis for lecanemab. In our previous SUVr PK/PD for lecanemab with data from study 201 core only, without off treatment data, the value of K out was estimated to be 0.757/year, 26 which is similar to that reported for aducanumab. These findings underscore the importance of off‐treatment data following amyloid reduction in determining amyloid re‐accumulation rates, where the model suggests slow re‐accumulation of amyloid consistent with the natural history of amyloid accumulation in AD, based on data from our clinical study that includes a relatively long period off treatment following amyloid reduction.
The recovery half‐life of plasma Aβ42/40 ratio level to baseline level prior to initiating lecanemab treatment is estimated to be ~1.9 years, which is comparable to that for plasma p‐tau181 (~1.5 years) but shorter than that for SUVr. This result suggests that changes of plasma Aβ42/40 and p‐tau181 may be reflecting the early dynamic aggregation process of soluble Aβ aggregates in the brain, in contrast to slower accumulation of insoluble Aβ fibrils into amyloid plaques assessed by PET.
Covariates identified in the final PK/PD model for SUVr indicated that APOE4 carriers have slightly higher baseline SUVr than non‐carriers, and older subjects have higher maximum plaque removal (E max). Although the effect of APOE4 on baseline SUVr was small, the effect was retained in the model based on the significant improvement in objective function value and the clinical interest. Greater amounts of Aβ burden in APOE4 carriers than in non‐carriers were reported in some studies. 41 , 42 The small effect of APOE4 on baseline SUVr identified in our analysis will be further evaluated based on additional data in future study. Although the mechanism of observed higher E max in older subjects is unclear, it is also reported in the PK/PD model for aducanumab, 34 which posited that greater exposure of the drug into the brain due to the functional decline of blood–brain barrier in older subjects may have contributed to the finding. Neither ADA nor neutralizing antibodies were significant covariates in PK/PD model for SUVr and PK/PD models for plasma biomarkers.
The dose‐dependency in the decrease in SUVr and p‐tau181 and increase in Aβ42/40 ratio are illustrated in Figure 4, which shows time profiles for 10 mg/kg biweekly and 10 mg/kg monthly following 18 months of treatment. The profiles show that 10 mg/kg biweekly results in a faster and larger decrease in both SUVr and p‐tau181 and a faster and larger increase in Aβ42/40 ratio. The percentage of subjects achieving amyloid negativity for SUVr (<1.17) 25 following 10 mg/kg biweekly for 18 months is predicted to be 64.6%, higher than corresponding values following 10 mg/kg monthly (36.7%). Additionally, simulations (Figure 5) showed that when treatment at 10 mg/kg biweekly is continued beyond 18 months SUVr keeps declining further, whereas once treatment is discontinued after 18 months SUVr starts to increase slowly, and it takes over 15 years for amyloid to re‐accumulate to baseline levels prior to treatment initiation with lecanemab. For plasma Aβ42/40 ratio and p‐tau181, simulation showed that it takes ~6–8 years for these biomarkers to reach the plateau by continuous lecanemab dosing or to return to baseline levels after the treatment discontinuation.
FIGURE 4.
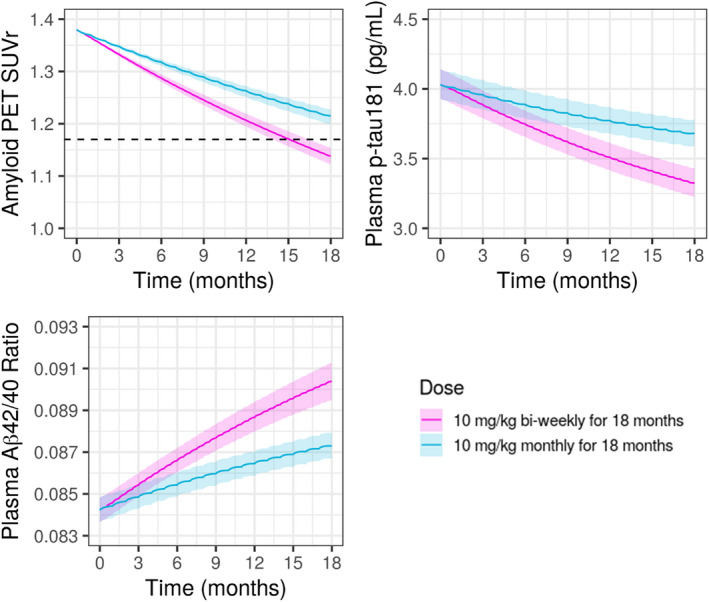
Model‐predicted SUVr and plasma Aβ42/40 ratio and p‐tau181 following 18 months treatment with lecanemab at 10 mg/kg biweekly or 10 mg/kg monthly. Black dashed line in SUVr plot represents SUVr = 1.17, indicating amyloid negative line. For SUVr, baseline SUVr = 1.38 (median of Study 201) was assumed. Pink solid line and shaded area: predicted median and 95% CI for 10 mg/kg biweekly, blue solid line and shaded area: predicted median and 95% CI for 10 mg/kg monthly. CI, confidence interval; PET, positron emission tomography; SUVr, standard uptake ratio.
FIGURE 5.
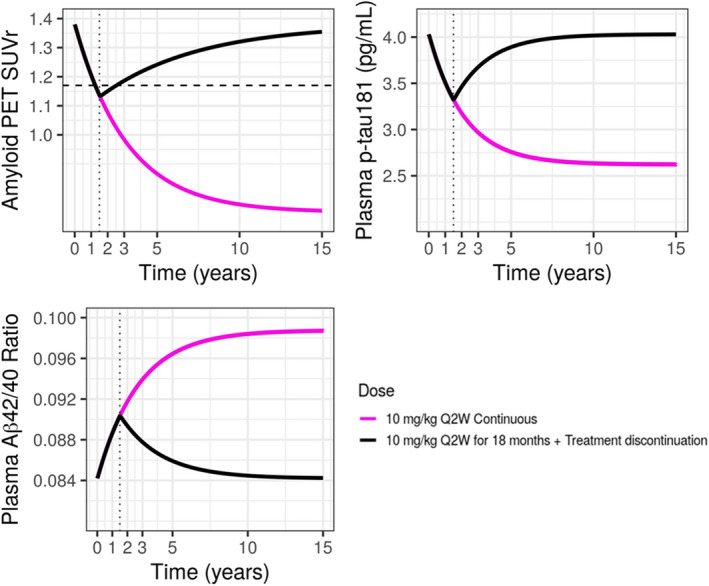
Model‐predicted SUVr and plasma Aβ42/40 ratio and p‐tau181 following continuous 10 mg/kg biweekly with or without treatment discontinuation. Black horizontal dashed line in SUVr plot represents SUVr = 1.17, indicating amyloid negative line. Black vertical dotted lines represent time = 1.5 years (18 months). Pink solid line: continuous 10 mg/kg biweekly for 15 years, black solid line: 10 mg/kg biweekly for 18 months followed by treatment discontinuation. PET, positron emission tomography; SUVr, standard uptake ratio.
In conclusion, PK of lecanemab was well‐characterized in patients with AD by linear, two‐compartment models and detected covariates were consistent with other monoclonal antibodies. Model‐based PK/PD simulations demonstrated the effectiveness of lecanemab 10 mg/kg biweekly dosing on amyloid plaque in the brain as the therapeutic dosing regimen in patients with early AD, a finding consistent with effects observed in biofluid biomarkers as evidenced by an increase in plasma Aβ42/40 ratio and a decrease in plasma p‐tau181. Furthermore, developed PK/PD models inform the relationships between lecanemab exposure and brain amyloid or plasma biomarkers, and describe changes of these biomarkers following discontinuation of lecanemab treatment, capturing the dynamics of neurodegenerative processes associated with early AD.
AUTHOR CONTRIBUTIONS
All authors designed the research. S.H., O.T., and Z.H. performed the research. S.H. and Z.H. analyzed the data and wrote the manuscript.
Funding information
The study was funded by Eisai Co., Ltd.
CONFLICT OF INTEREST
S.H., O.T., A.K., and S.Y. are employees of Eisai Co., Ltd. S.R., I.L., C.S., and L.R. are employees of Eisai Inc. Z.H. is an employee of Eisai Ltd.
Supporting information
Table S1‐S10
Figures S1‐S3
Figures S4‐S9
Figures S10‐S12
Appendix S1‐S4
ACKNOWLEDGMENTS
The authors would like to thank all of the subjects who enrolled in the studies as well as their family, caregivers, and friends who supported them. These analyses would not be possible without all of the hard work and contributions from the dedicated investigators in the studies. The sponsor was involved in the study design and collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this publication.
Hayato S, Takenaka O, Sreerama Reddy SH, et al. Population pharmacokinetic‐pharmacodynamic analyses of amyloid positron emission tomography and plasma biomarkers for lecanemab in subjects with early Alzheimer's disease. CPT Pharmacometrics Syst Pharmacol. 2022;11:1578‐1591. doi: 10.1002/psp4.12862
REFERENCES
- 1. Alzheimer's Association . Alzheimer's disease facts and figures; 2021. Available from: https://www.alz.org/media/Documents/alzheimers‐facts‐and‐figures.pdf
- 2. Elias‐Sonnenschein LS, Viechtbauer W, Ramakers IH, et al. Predictive value of APOE‐ε4 allele for progression from MCI to AD‐type dementia: a meta‐analysis. J Neurol Neurosurg Psychiatry. 2011;82:1149‐1156. doi: 10.1136/jnnp.2010.231555 [DOI] [PubMed] [Google Scholar]
- 3. Mattsson N, Groot C, Jansen WJ, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer's disease. Alzheimers Dement. 2018;14:913‐924. doi: 10.1016/j.jalz.2018.02.009 [DOI] [PubMed] [Google Scholar]
- 4. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delacourte A, Sergeant N, Champain D, et al. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer's disease. Neurology. 2002;59:398‐407. doi: 10.1212/wnl.59.3.398 [DOI] [PubMed] [Google Scholar]
- 6. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64:492‐498. doi: 10.1002/ana.21509 [DOI] [PubMed] [Google Scholar]
- 7. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. doi: 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 8. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dubois B, Villain N, Frisoni GB, et al. Clinical diagnosis of Alzheimer's disease: recommendations of the international working group. Lancet Neurol. 2021;20:484‐496. doi: 10.1016/S1474-4422(21)00066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11:e11170. doi: 10.15252/emmm.201911170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hadjichrysanthou C, Evans S, Bajaj SL, et al. The dynamics of biomarkers across the clinical spectrum of Alzheimer's disease. Alzheimer's Res Therapy. 2020;12:74. doi: 10.1186/s13195-020-00636-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fandos N, Pérez‐Grijalba V, Pesini P, et al. Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimers Dement. 2017;8:179‐187. doi: 10.1016/j.dadm.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doecke JD, Pérez‐Grijalba V, Fandos N, et al. Total Aβ42/Aβ40 ratio in plasma predicts amyloid‐PET status, independent of clinical AD diagnosis. Neurology. 2020;94:1580‐1591. doi: 10.1212/WNL.0000000000009240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. West T, Kirmess KM, Meyer MR, et al. A blood‐based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener. 2021;16:30. doi: 10.1186/s13024-021-00451-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng W, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115:201‐211. doi: 10.1016/s0306-4522(02)00404-9 [DOI] [PubMed] [Google Scholar]
- 16. Shen XN, Huang YY, Chen SD, et al. Plasma phosphorylated‐tau181 as a predictive biomarker for Alzheimer's amyloid, tau and FDG PET status. Transl Psychiatry. 2021;11:585. doi: 10.1038/s41398-021-01709-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tucker S, Möller C, Tegerstedt K, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid‐β protofibrils in brain and cerebrospinal fluid of tg‐ArcSwe mice. J Alzheimers Dis. 2015;43:575‐588. doi: 10.3233/JAD-140741 [DOI] [PubMed] [Google Scholar]
- 18. Sehlin D, Hedlund M, Lord A, et al. Heavy‐chain complementarity‐determining regions determine conformation selectivity of anti‐Aβ antibodies. Neurodegener Dis. 2011;8:117‐123. doi: 10.1159/000316530 [DOI] [PubMed] [Google Scholar]
- 19. Sehlin D, Englund H, Simu B, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7:e32014. doi: 10.1371/journal.pone.0032014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Magnusson K, Sehlin D, Syvänen S, et al. Specific uptake of an amyloid‐β‐protofibril‐binding antibody‐tracer in AβPP transgenic mouse brain. J Alzheimers Dis. 2013;37:29‐40. doi: 10.3233/JAD-130029 [DOI] [PubMed] [Google Scholar]
- 21. Englund H, Sehlin D, Johansson AS, et al. Sensitive ELISA detection of amyloid‐beta protofibrils in biological samples. J Neurochem. 2007;103:334‐345. doi: 10.1111/j.1471-4159.2007.04759.x [DOI] [PubMed] [Google Scholar]
- 22. Lord A, Gumucio A, Englund H, et al. An amyloid‐beta protofibril‐selective antibody prevents amyloid formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;36:425‐434. doi: 10.1016/j.nbd.2009.08.007 [DOI] [PubMed] [Google Scholar]
- 23. Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401 ‐ a clinical study in Alzheimer's disease with a protofibril selective Aβ antibody. Alzheimer's Res Therapy. 2016;8:14. doi: 10.1186/s13195-016-0181-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double‐blind, phase 2b proof‐of‐concept clinical trial in early Alzheimer's disease with lecanemab, an anti‐Aβ protofibril antibody. Alzheimer's Res Therapy. 2021;13:80. doi: 10.1186/s13195-021-00813-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68:1404‐1411. doi: 10.1001/archneurol.2011.150 [DOI] [PubMed] [Google Scholar]
- 26. Schuck EL, Reyderman L, Hayato S, et al. Population pharmacokinetic/pharmacodynamic analyses of BAN2401 in patients with early Alzheimer's disease: correlation of BAN2401 exposure, PET standard uptake value ratio, and cognitive outcomes. Poster presented at Alzheimer's Association International Conference 2019. Volume 15, Issue 7S, Part 30. [Google Scholar]
- 27. Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides‐harmonized terminology and tactical recommendations. AAPS J. 2014;16:658‐673. doi: 10.1208/s12248-014-9599-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13:143‐151. doi: 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28:171‐192. doi: 10.1023/a:1011555016423 [DOI] [PubMed] [Google Scholar]
- 30. Efron B. Missing data, imputation, and the bootstrap. J Am Stat Assoc. 1994;89:463‐475. [Google Scholar]
- 31. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic‐pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol. 2009;5:803‐812. doi: 10.1517/17425250902992901 [DOI] [PubMed] [Google Scholar]
- 32. Keiser RJ, Huitema ADR, Schellens JHM, Bejinen JH. Clinical pharmacokinetics of therapeutic monocloncal antibodies. Clin Pharmacokinet. 2010;49:493‐507. doi: 10.2165/11531280-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 33. Lowe SL, Willis BA, Hawdon A, et al. Donanemab (LY3002813) dose‐escalation study in Alzheimer's disease. Alzheimers Dement. 2021;7:e12112. doi: 10.1002/trc2.12112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kandadi Muralidharan K, Tong X, Kowalski KG, et al. Population pharmacokinetics and standard uptake value ratio of aducanumab, an amyloid plaque‐removing agent, in patients with Alzheimer's disease. CPT Pharmacometr Syst Pharmacol. 2022;11:7‐19. doi: 10.1002/psp4.12728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Portron A, Jordan P, Draper K, et al. A phase 1 study to assess the effect of speed of injection on pain, tolerability, and pharmacokinetics after high‐volume subcutaneous administration of gantenerumab in healthy volunteers. Clin Ther. 2020;42:108‐120. doi: 10.1016/j.clinthera.2019.11.015 [DOI] [PubMed] [Google Scholar]
- 36. Bajaj G, Suryawanshi S, Roy A, Gupta M. Evaluation of covariate effects on pharmacokinetics of monoclonal antibodies in oncology. Br J Clin Pharmacol. 2019;85:2045‐2058. doi: 10.1111/bcp.13996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gill KL, Machavaram KK, Rose RH, Chetty M. Potential sources of inter‐subject variability in monoclonal antibody pharmacokinetics. Clin Pharmacokinet. 2016;55:789‐805. doi: 10.1007/s40262-015-0361-4 [DOI] [PubMed] [Google Scholar]
- 38. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin Immunol. 2007;122:146‐155. doi: 10.1016/j.clim.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura A, Kaneko N, Villemagne VL, et al. High‐performance plasma amyloid‐β biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. doi: 10.1038/nature25456 [DOI] [PubMed] [Google Scholar]
- 40. Schindler SE, Bollinger JG, Ovod V, et al. Highprecision plasma β‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647‐e1659. doi: 10.1212/WNL.0000000000008081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid‐β and tau accumulation in Alzheimer's disease. Alzheimer Res Therapy. 2020;12:140. doi: 10.1186/s13195-020-00710-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Resnick SM, Bilgel M, Moghekar A, et al. Changes in Aβ biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol Aging. 2015;36:2333‐2339. doi: 10.1016/j.neurobiolaging.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S10
Figures S1‐S3
Figures S4‐S9
Figures S10‐S12
Appendix S1‐S4