

Abstract
Marzeptacog alfa (activated) (MarzAA) is an activated recombinant human FVII (rFVIIa) variant developed as subcutaneous (s.c.) administration for the treatment or prevention of bleeding episodes in patients with hemophilia A (HA) or hemophilia B (HB) with inhibitors and other rare bleeding disorders. Population pharmacokinetic (PK) modeling was applied for dose selection for a pivotal phase III clinical trial evaluating s.c. MarzAA for episodic treatment of spontaneous or traumatic bleeding episodes. The population PK model used MarzAA intravenous and s.c. data from previously completed clinical trials in patients with HA/HB with or without inhibitors. Based on the model, clinical trial simulations were performed to predict MarzAA exposure after different dosing regimens. The exposure target was identified using an exposure‐matching strategy with a wild‐type rFVIIa but adjusting for the difference in potency between the two compounds. Simulations demonstrated a sufficient absorption rate and prolonged exposure following a single 60 μg/kg dose leading to 51% and 70% of the population reaching levels above the target after 3 and 6 h, respectively. According to the phase III protocol, if a second dose was required after 3 h because of a lack of efficacy, 90% of the population was observed to be above target 6 h after the initial dose. The model‐informed drug development approach integrated information from several trials and guided dose selection in the pivotal phase III clinical trial for episodic treatment of an acute bleeding event in individuals with HA or HB with inhibitors without the execution of a phase II trial for that indication.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Novel subcutaneously administered hemostatic drugs for acute bleeding events are needed to enhance treatment outcomes and prevent bleeding‐related sequelae and morbidity. There is currently no approved subcutaneous treatment available for the episodic treatment of acute bleeding.
WHAT QUESTION DID THIS STUDY ADDRESS?
Selection of dose was supported for a phase III clinical trial by characterizing the population pharmacokinetics in plasma following intravenous and subcutaneous administration, enabling clinical trial simulations based on pharmacokinetic information to meet a predefined target.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The model provides a pharmacokinetic characterization of activated recombinant FVII and scientific rationale for dose selection in a pivotal phase III trial treating subjects with inhibitor‐complicated hemophilia for acute bleeding events. The model bridges proof‐of‐concept data from a phase II trial focused on prophylaxis to a trial targeting acute bleeding events.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Our framework demonstrates how clinical development of drugs for rare diseases may benefit from using information throughout development even when data are obtained under varying conditions, for example, preventive versus episodic treatment.
INTRODUCTION
Marzeptacog alfa (activated) (MarzAA) is a novel activated recombinant FVII (rFVIIa) variant differing from wild‐type (wt) rFVIIa by an increase in catalytic activity for factor X activation, increased bioavailability (F), prolonged half‐life, and therapeutic activity following subcutaneous (s.c.) administration. 1 Currently available therapies for patients on prophylaxis but who experience acute bleedings are subject to a delay in administration and a potential lack of adherence 2 evolving from barriers that arise with intravenous (i.v.) formulations, 3 leading to the accumulation of blood in joints or other areas while awaiting treatment. The s.c. formulations provide an easier‐to‐administer alternative and remove the need for home nursing or travel to a clinic for an i.v. injection. 2 Effective treatment of episodic bleeding within 2 h is essential and recommended by the World Federation of Hemophilia for patient care, as promptly stopping bleeding events as well as reducing or eliminating the presence of rebleeds is important for the prevention of hemorrhage‐induced joint damage. 4 Furthermore, in a public meeting held by the US Food and Drug Administration (FDA) on hemophilia A (HA) and B (HB), von Willebrand disease, and other inherited bleeding disorders, the participants expressed that s.c. administration together with therapies that were longer acting and safe with zero risk of inhibitor formation were desired product attributes. 5 In contrast to prophylaxis treatment, there is currently no s.c. rFVIIa treatment options approved for the treatment of acute bleeding events.
Two phase I trials with MarzAA (NCT01439971 6 [i.v. dosing], NCT04072237 [i.v. and s.c. dosing]) and one phase II (NCT03407651 [i.v. and s.c. dosing]) trial have been conducted. Intravenously administered MarzAA was shown to be tolerated without serious adverse events at all dose levels 6 and exhibited a desirable biomarker response. 6 However, a desirable biomarker response in phase I does not in general imply clinical efficacy in phase II and phase III trials. From an efficacy perspective, the phase II trial 1 studied s.c. daily prophylactic administration in 10 subjects with inhibitors. 1 Subjects enrolled had an annual bleeding rate (ABR) ≥12 and started receiving s.c. MarzAA at 30 μg/kg/day for 50 consecutive days. If bleeding occurred before day 50, the subject was dose escalated to a daily dose of 60 μg/kg s.c. administration of MarzAA for another 50 consecutive days. The null hypothesis was that ABR equal to 12 versus the alternative hypothesis of ABR <12. If the true ABR for MarzAA is ≤6 with a one‐tailed 2.5% significance level, the power was 99% using a one‐sample Poisson test of the null hypothesis even if only six subjects provided data for analysis. 1 Nine subjects completed the trial, and all subjects demonstrated a clinically and statistically significant effect according to the primary outcome measure, which was a reduction of the baseline ABR from an average baseline of 19.8 to 1.6 (95% confidence interval [CI], 15.2–21.1; p = 0.009) while on treatment. A statistically and clinically meaningful reduction in the proportion of days (secondary outcome measure) with bleeding was observed from a pretreatment level of 12.3% to 0.8% while on treatment (95% CI, 7.5%–15.6%; p = 0.009). Two subjects who experienced breakthrough bleeding events at a dose of 30 μg/kg daily were escalated to 60 μg/kg daily, after which they did not experience any additional bleeding events during the subsequent 50 days. 1 After 517 exposure days, only five mild or moderate injection site reactions were reported, and no antidrug antibodies (ADAs) were observed. The study indicated that MarzAA had an acceptable safety profile and significantly reduced bleed frequency and duration. As the rarity of the disease introduces challenges in finding eligible patients, a dose‐selection strategy was targeted that incorporated preclinical potency difference in relation to NovoSeven® (Novo Nordisk Inc) together with an exposure‐matching approach. This strategy was endorsed by regulators and enabled acceleration of the clinical development program to target the unmet medical need.
The work presented here supported dose selection in a pivotal phase III clinical trial for the treatment of bleeding events in subjects with HA/HB with inhibitors, without prior execution of a phase II trial for stoppage of a bleed. All available pharmacokinetic (PK) information from the clinical development program were pooled, and a population PK model was developed to conduct clinical trial simulations with the objective of guiding dose selection in HA/HB patients based on PK information. The work bridges information through preclinical and clinical phases of drug development and demonstrates an example of how PK modeling and simulation helped to support dose selection for a pivotal phase III trial in a rare disease where traditional drug development programs are challenging.
METHODS
Study design and patients
MarzAA concentrations versus time data were obtained from previously completed trials in adults (NCT01439971, NCT04072237, and NCT03407651). In NCT01439971 (Table 1), a single i.v. dose of MarzAA was administered at five different dose levels—0.5, 4.5, 9, 18, and 30 μg/kg—to subjects with severe HA/HB with inhibitors. Samples were taken predose and at 1, 3, 6, 9, 12, 24, and 48 h postdose. Drug administration occurred in the nonbleeding state. The 0.5‐μg/kg dose level was excluded from analysis as only one subject received that dose. NCT04072237 included a single i.v. dose followed by ascending s.c. doses of MarzAA in adult subjects with HA/HB, with or without inhibitors. The study included nine different stages representing different single‐dose escalations followed by multiple dosing. The dose levels were 18 (i.v.), 30, 45, 60, 60 (2 × 30 at the same time at two anatomically different locations), 90, and 120 μg/kg for Stages 1–7, 60 μg/kg two times 3 h apart in Stage 8, and 60 μg/kg three times 3 h apart in Stage 9. Samples were obtained at predose and then 5 min and 1, 2, 6, 9, 12, and 24 h after the i.v. dose in Stage 1 and at predose followed by 0.5, 1, 1.5, 2, 6, 9, 24, and 48 h postdose in Stages 2–7 and at predose followed by 2, 3, 4, 5, 6, 7, 8, 9, 12, 24, 48, and 72 h after the dose for Stages 8 and 9. NCT03407651 included a single i.v. dose (18 μg/kg) with samples taken at 5 min followed by 0.5, 1, 3, 6, 9, 12, and 24 h after the dose. The subjects were then administered a single s.c. dose (30 μg/kg) 24 h after the first dose, and samples were obtained at predose followed by 3, 5, 7, 9, 12, 24, 30, and 48 h after the s.c. dose. In the second part of the study, MarzAA was administered daily for 50 consecutive days (s.c., 30 μg/kg), and samples were collected at predose and 7 h after the dose on Days 1, 3, 5, 7, 14, 21, 28, and 50 and the end of the study. If a subject experienced a bleeding event while on 30 μg/kg, then the dose was escalated to 60 μg/kg and then the patient was followed for another 50 consecutive days after the dose escalation. As other activated factor VII (FVIIa)‐bypassing therapies that have shown body weight–dependent PK 7 and have been dosed clinically per killigram of body weight, 8 MarzAA was also dosed per body weight in all trials. In total, 46 male subjects were included across these three trials, including a total of 1225 observations (Figure 1). The median and 10th–90th percentile body weights were 75 kg and 60–94 kg, respectively. The minimum and maximum body weights in the data set were 43 and 120 kg. The median and 10th–90th percentile age were 32 years and 23–52 years, and the minimum and maximum age were 18 and 62 years, respectively. Number of observations per dose level and trial and number of subjects per dose group are listed in Table 1. A total of 19 observations, which comprised 1.6% of all samples obtained, were below the limit of quantification (BLOQ) and were excluded from the analysis. An additional three samples were excluded because of either missing sampling time information or for being obtained during unscheduled visits.
TABLE 1.
Number of observations per dose level and trial and subjects per dose group
| Dose level (μg/kg) | Total subjects | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4.5 a | 9 a | 18 a | 30 a , b , c | 45 b | 60 b , c | 90 b | 120 b | ||
| NCT01439971 | 72 | 72 | 72 | 72 | – | – | – | – | 288 |
| NCT04072237 | – | – | 63 | 74 | 68 | 344 | 67 | 67 | 683 |
| NCT03407651 | – | – | 81 | 150 | – | 23 | – | – | 254 |
| Total observations | 72 | 72 | 216 | 296 | 68 | 367 | 67 | 67 | 1225 |
| Total subjects | 6 | 6 | 26 | 23 | 8 | 12 | 8 | 8 | 46 |
Abbreviations: i.v., intravenous; s.c., subcutaneous.
The i.v. dose levels.
The s.c. dose levels.
Multiple s.c. dosing.
FIGURE 1.
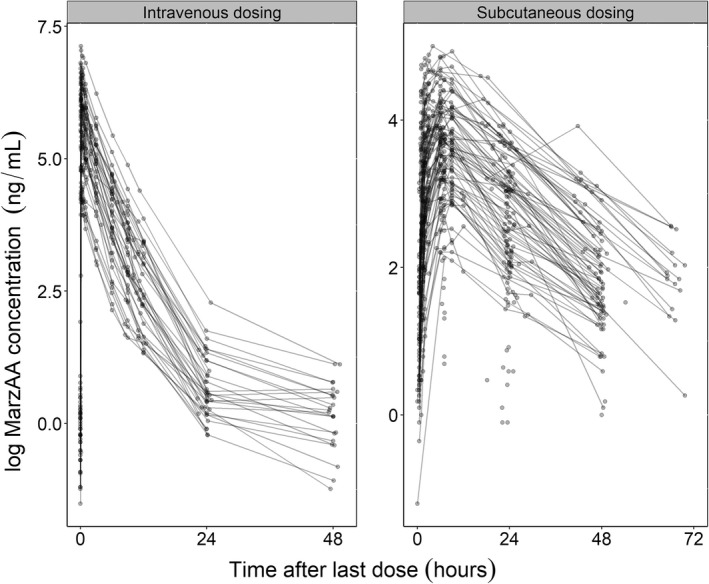
Circles illustrate observed concentrations for all dose groups on a log scale after i.v. and s.c. dosing. The black lines connect observations in an individual. MarzAA, marzeptacog alfa (activated).
Drug quantification
All details around drug quantification can be found in the supplementary materials (Data S2).
Model building
The overall model building was divided into steps as data became available. Briefly, the model was first developed to describe only i.v. data to capture the PK of MarzAA independent of absorption processes. One‐, two‐ and three‐compartment disposition models were explored. Interindividual variability (IIV) was tested in all parameters. Different residual error models were tested. Covariance was tested in appropriate model parameters. To account for baseline levels of endogenous FVIIa, the baseline values were modeled as additive to the concentrations of FVIIa. The individual prediction of FVIIa concentrations (IPRED) was expressed as:
in which BASE is endogenous FVIIa levels in a typical patient, and IIVbase is the IIV in BASE introduced exponentially (exp). A is the amount in the central compartment, and V c is the volume of distribution of the central compartment. Allometric scaling was employed in all clearance (CL) and volume (V) terms with fixed exponents of 0.75 and 1 as in the example of CL and V c. 9
where TVCL and TVVc are typical CL and V of distribution values in the central compartment, respectively. Scaling was done using 70 kg of body weight. Subsequently, to study potential nonlinear trends, Michaelis–Menten elimination and nonlinear V of distribution were explored.
The i.v.‐related parameters were then fixed, and s.c. data were included to describe absorption. Zero‐order, first‐order, and a combination were evaluated to describe absorption into the central compartment from a depot compartment (D). A zero‐order input into the D was explored. Models with and without an absorption lag‐time were explored. IIV was evaluated in all absorption parameters. Interoccasional variability (IOV) was explored for all PK parameters as s.c. data were obtained at more than one occasion per individual. Subsequently, all PK parameters were simultaneously estimated given both i.v. and s.c. data followed by a backward exclusion of all variability parameters and nonlinearities. Subsequently, covariances were tested between IIV parameters.
After the evaluation of IIV and IOV in all parameters, the assessment of covariate effects with different relationships on all parameters were performed using the stepwise covariate modeling approach. 10 Age was tested on all parameters, whereas body weight was tested on absorption parameters and baseline FVIIa as all CL and V terms already included body weight–based allometric scaling. No ADAs were detected related to MarzAA, and as such they were not included in the covariate search. Statistical significance was tested (p = 0.05) for the forward search, followed by a stricter criterion (p = 0.01) in the backward deletion. A covariate effect was considered clinically significant if the effect on maximum concentration (C max) was a >30% change in a subject with the 10th and 90th percentiles of age in the data set compared with the median age. C max was used for the evaluation of clinical significance as it was considered the seconday PK parameter that is believed to be most correlated with the probability of stopping acute bleeding. Only statistically and clinically significant covariates were included in the final population PK model.
Model evaluation
All details around model evaluation can be found in the supplementary materials (Data S2).
Clinical trial simulations
The final population PK model was used for clinical trial simulations to inform the dosing regimen in a planned pivotal phase III trial. MarzAA concentration versus time data were simulated in 1000 virtual subjects with rich sampling following 60, 90, and 120 μg/kg with different dosing frequencies: single dose (t = 0 h), two doses (t = 0 and 3 h), three doses (t = 0, 3, and 6 h), and following 60 μg/kg (t = 0 and 6 h and t = 0, 6, and 12 h). As dosing was based on body weight (micrograms of MarzAA per killigrams of body weight), body weights were simulated for each of the 1000 virtual subjects from a truncated normal distribution using the mean and standard deviation from the body weights in the analysis data set. The lower and upper boundaries of the distribution were set to 40 kg and 105 kg, respectively.
Median and 80% prediction interval (PI) of secondary PK parameters, area under the concentration versus time curve from 0 to 24 h (AUC0–24h), maximum concentration over 24 h (Cmax 24h), minimal concentration over 24 h (Cmin24h), and time to maximal concentrations over 24 h (Tmax 24h) were calculated from the simulated concentration versus time profiles. The percentage of the population above target at different timepoints and the time required to reach therapeutic target concentrations (median and 80% PI) were derived. The target range in HA/HB patients was defined as 24–120 ng/ml, which is equivalent to 6–30 IU/ml wt‐rFVIIa, assuming that MarzAA is at least fivefold more potent than wt‐rFVIIa in vivo. The assumption was derived from preclinical experiments that showed a higher potency of 5–10 fold 11 , 12 , 13 when compared with NovoSeven®.
Ethics
The studies were conducted in accordance with the International Council for Harmonization Good Clinical Practice guidelines and with national and local laws and regulations. The study protocols and all amendments were reviewed and approved by the institutional review board or independent ethics committee at each participating study site. All subjects or their legally authorized representative were required to provide written informed consent prior to participation in each study, and withdrawal from the studies was accepted at any time.
RESULTS
Model development
A two‐compartment model with IIV in CL, V, and baseline FVIIa described the i.v. data well and performed better compared with one‐ or three‐compartment models with IIV in the same parameters. Michaelis–Menten elimination of FVIIa was statistically significant (p < 0.05; change in objective function value [ΔOFV] = −8.4). Nonlinear V of distribution, evaluated both as the change in rate and extent with dose, was not supported by the data.
The second step incorporated data after s.c. administration of MarzAA. Exclusion of Michaelis–Menten elimination gave an increase in objective function value (OFV) by 8.9 points and was therefore kept in the model. A change to a three‐compartmental model gave a ΔOFV = −4.3 but produced high uncertainty in additional intercompartmental CL (relative standard error [RSE] = 165%) and V of distribution (RSE = 82%). No notable changes were observed in structural and random parameters associated with the distribution and elimination compared with when modeling i.v. data only. A zero‐order release into the D followed by first‐order absorption into the central compartment gave a drop in OFV by 145 points. A direct zero‐order uptake into the central compartment followed by an addition of a lag‐time parameter were evaluated, but goodness‐of‐fit plots were in favor of the former absorption model. IIV was supported in the zero‐order release into the absorption but was excluded as the addition of IOV followed by backward exclusion ruled out the IIV parameter in favor of IOV only. Lastly, more s.c. data were obtained, and the evaluation of additional variability parameters were performed but produced no additions when simultaneously estimating all parameters. In a final step, Michaelis–Menten kinetics were excluded from the model, and OFV increased by 4.3 points but produced no changes in visual predictive checks (VPCs) and was therefore excluded from the model. In addition, covariance between IIV of CL and V was included in the model to account for high correlation (Table S1). The automated covariate search resulted in age as a statistically significant covariate on the absorption constant (kA) (p < 0.01), predicting lower absorption with increase in age. Following a dose of 60 μg/kg in a typical patient, C max was found to increase by 18% in a 23‐year‐old subject compared with a 32‐year‐old subject (median age in the data set) and decrease by 22% in a 52‐year‐old subject compared with a 32‐year‐old subject. The choice of 23 and 52 years of age to demonstrate the effect was because they correspond to the 10th and 90th percentiles of age in the data set. As such, age was not considered clinically significant and was not included in the final model.
The final population PK model was a two‐compartment disposition model with zero‐order input to a D and first‐order absorption from the D into the central systemic compartment. IIV parameters were supported for CL, V, baseline FVIIa, F, and k A, whereas IOV was supported for a duration of zero‐order input of FVIIa into the D, F, and V c. Covariance between the IIV of CL and V was supported by the data, and the residual error model was proportional. All final parameter values and their associated uncertainties can be found in Table S1. The model described the data well as seen in the prediction‐corrected VPCs and dose‐stratified VPCs (Figures S1 and S2). The NONMEM code for the final model is given in the supplementary materials (Data S1).
Clinical trial simulations
The median and 80% PI of the secondary PK parameters for each of the different regimens are shown in Table 2. The percentage of the population (n = 1000) above the target for the different regimens is presented in Figure 2. All explored regimens achieved C max within the desired target range (24 to 120 ng/ml), as seen in Table 2. The median (80% PI) times to target in HA/HB patients were 2.2 h (1.1–5.0 h), 1.6 h (0.9–3.5 h), and 1.3 h (0.7–2.7 h), respectively, after a single dose of 60, 90, or 120 μg/kg. The corresponding percentage of patients above the lower end of the target was 51%, 74%, and 86% after 3 h and 70%, 87%, or 94% after 6 h following a single 60, 90, or 120 μg/kg dose (Figure 2). The second dose given 3 h after the first resulted in 90%, 96%, and 98% of the patients being above target at 6 h after 60, 90, or 120 μg/kg and 81%, 94%, or 98% at 24 h after 60, 90, or 120 μg/kg, respectively (Figure 2). If a third dose was given to the virtual population another 3 h later, that is, 6 h after the initial dose, the percentage of population above target was 96%, 99%, or 100% for the 60, 90, or 120 μg/kg dose levels, respectively (Figure 2).
TABLE 2.
Predicted secondary PK parameters following different phase III regimens in simulated HA/HB subjects using the final population PK model
| Regimen | Median Cmax24h (80% PI) ng/ml | Median Tmax24h (80% PI) h | Median Cmin24h (80% PI) ng/ml | Median AUC0–24h (80% PI) h · ng/ml |
|---|---|---|---|---|
| 60 μg/kg q.d. a | 34.5 (17.6–68.5) | 7.8 (5.3–11.1) | 19.3 (10.7–35.2) | 604 (310–1190) |
| 90 μg/kg q.d. a | 51.1 (26.1–102.1) | 7.8 (5.3–11.1) | 28.3 (15.6–52.2) | 905.3 (465.0–1785.2) |
| 120 μg/kg q.d. a | 67.7 (34.6–135.7) | 7.8 (5.3–11.1) | 37.4 (20.5–69.3) | 1207.1 (619.9–2380.3) |
| 60 μg/kg b.i.d. b | 68.7 (35.1–125.1) | 9.5 (7.1–13.0) | 40.4 (21.0–70.6) | 1166 (602–2081) |
| 60 μg/kg t.i.d. c | 106.1 (51.3–201.1) | 11.6 (8.7–14.8) | 66.7 (34.9–122.9) | 1715 (852–3210) |
| 60 μg/kg b.i.d. d | 67.2 (33.5–123.8) | 11.9 (9.0–15.5) | 43.5 (22.8–75.4) | 1098 (565–1981) |
| 60 μg/kg t.i.d. e | 100.1 (48.2–186.7) | 16.6 (12.1–20.1) | 77.0 (39.5–138.8) | 1483 (738–2782) |
| 90 μg/kg b.i.d. b | 102.3 (51.8–187.2) | 9.4 (7.1–13.0) | 59.9 (31.0–105.4) | 1749.3 (903.3–3120.9) |
| 120 μg/kg b.i.d. b | 136.1 (68.3–249.3) | 9.5 (7.1–13.0) | 79.6 (40.8–140.1) | 2332.4 (1204.5–4161.1) |
| 90 μg/kg t.i.d. c | 158.6 (76.4–301.0) | 11.6 (8.6–14.9) | 99.4 (51.7–183.7) | 2573.0 (1278.6–4815.0) |
| 120 t.i.d. c | 211.2 (101.4–401.1) | 11.6 (8.6–14.8) | 132.1 (68.7–244.6) | 3430.7 (1704.8–6419.9) |
Abbreviations: AUC0–24h, area under the concentration versus time curve from 0 to 24 h; b.i.d., twice daily; Cmax24h, maximum concentration over 24 h; Cmin24h, minimal concentration over 24 h; HA, hemophilia A; HB, hemophilia B; PI, prediction interval; PK, pharmacokinetic; q.d., once daily; t.i.d., thrice daily; Tmax24h, time to maximal concentrations over 24 h.
Dosing at t = 0 h.
Dosing at t = 0 and 3 h.
Dosing at t = 0, 3, and 6 h.
Dosing at t = 0 and 6 h.
Dosing at t = 0, 6, and 12 h.
FIGURE 2.
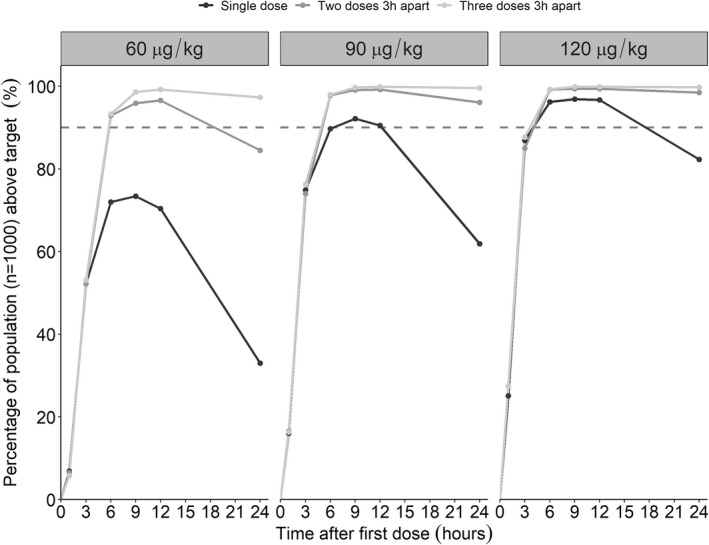
Percentage of population (n = 1000) above 24 ng/ml at different timepoints after dosing following different dose levels. The black lines illustrate a single dose, and the dark gray and gray lines illustrate repeated administration of total two or three doses, respectively. The dashed yellow line illustrates 90% of patients being above the target.
DISCUSSION
This population PK modeling and simulation work informed the dosing regimen in a phase III trial for development of the first s.c. treatment of episodic bleeding in HA/HB subjects with inhibitors. The model‐based approach enabled integration of data from different trials to be analyzed simultaneously, which resulted in the establishment of clinical trial simulations. Considering differences in potency between MarzAA and NovoSeven®, which has established therapeutic levels, together with evidence of MarzAA efficacy in a prophylaxis setting, the phase III clinical trial for stoppage of an acute bleed was supported without prior execution of a phase II trial. The simulations showed that all of the regimens explored in this work achieved C max levels above the desired target of 24 ng/ml (Table 2). The percentage of a virtual population (n = 1000) above the target 3 h after single doses of 60, 90, or 120 μg/kg were found to be 51%, 74%, and 86%, respectively. Moreover, 33%, 63%, and 78% stayed above target 24 h postdose. According to the protocol of the phase III trial and in a scenario of failure to reach hemostasis after 3 h, the first dose was suggested to be followed by a second dose. As the target was conservatively set with respect to potency, it is likely that stoppage of a bleeding event will occur in a vast majority of the subjects already at 3 h after the first dose. If not, the model‐based clinical trial simulations suggested that an additional dose would result in 90%, 96%, and 98% of the population being above target 3 h after the second dose (6 h after the first dose) following 60, 90, or 120 μg/kg, respectively. If hemostasis was not achieved 6 h after the first dose, a third and last dose was found to result in 98%, 100%, and 100% of the patients being above target 9 h after the first dose for 60, 90, and 120 μg/kg, respectively. As cost and injection volume are important factors to consider for dose selection, it is desirable to have as low dose and volume of injection as possible to reduce economic burden, administration barriers for the patient and increase utility of the product. Considering these factors, the model‐informed trial simulations were informative in guiding the phase III dose to ensure the lowest dose possible.
A crucial part of this work was the definition of the target range, which was derived based on an exposure‐matching strategy toward effective levels of wt‐rFVIIa (e.g., NovoSeven®). The approach accounted for potency differences seen with in vitro assays, for example, thrombin generation assay, as well as in preclinical animal models. Across the assays and animal models, MarzAA generally appeared 5‐ to 10‐fold more potent than the wt‐rFVIIa. To be conservative in terms of efficacy and because there is a considerable safety window (Good Laboratory Practice toxicology data on file), we chose to base the simulations on a scenario where MarzAA was only fivefold more potent than wt‐rFVIIa in humans. The target concentrations for wt‐FVIIa were derived based on the available literature. 14 For hemostasis, a target concentration of 6 IU/ml, inclusive of normal levels of FVIIa in hemophilia patients, has been shown to be adequate for effective hemostasis with rFVIIa treatment. 15 Continuous infusion has been used successfully and safely in hemophilia patients targeting a plasma level of 30 IU/ml. 16 Furthermore, significant variability in plasma concentrations has been observed in hemophilia patients receiving rFVIIa. 16 Therefore, a target range of at least 6–30 IU/ml or higher was planned, which is equivalent to 24–120 ng/ml MarzAA when taking the at least fivefold increase of potency into account. It is important to highlight that the target may vary for stoppage of a bleed that occurred following trauma or spontaneously. However, the design of the phase III trial was set to allow for additional doses with 3‐h intervals if hemostasis is not reached.
An important aspect in the treatment of hemophilia is compliance. Compliance is essential for the treatment of bleeding events and prevention of long‐term complications associated with delayed and/or missed treatment. As s.c. dosing is less invasive to the patient and avoids the need for home nursing or traveling to a clinic for i.v. injections, it is expected to improve compliance and enable faster dosing, which reduces time for destructive blood accumulation in the joints. The patients in the phase III trial were expected to perform self‐administration and record the time of administration, which might provide additional evidence that s.c. formulation can improve compliance. As repeated bleeding episodes of hemarthrosis may lead to arthropathy, 17 , 18 it is critical to quickly and effectively stop an episodic bleeding event as soon as possible after detecting it. It is hypothesized that because of the fast absorption rate and high specific activity (potency) of MarzAA, effective levels may be rapidly obtained and that the depot created by the s.c. administration will enable a protracted exposure profile; both of these properties, that is, fast absorption and long activity exposure, appear important for the episodic treatment of acute bleeding. This may hypothetically lead to a reduced amount of drug required to reach hemostasis and to prevent rebleeding, 3 which is an important hypothesis to study in the future. With a model built on clinical observations that characterize and handle processes governing absorption and elimination, the phase III clinical development of MarzAA for the episodic treatment of bleeding events was supported based on previously obtained PK and proof‐of‐concept data. Although data from several clinical trials were used, the sample size was still smaller compared with what is usually used in nonrare disease indications. Because of the rarity of the disease and an earlier proven efficacy in a different indication (prophylaxis), a registrational phase III trial was designed in communication with regulators and supported by the model‐informed drug development work presented herein. The results from the phase III trial indicated that s.c. MarzAA had the potential to treat bleeding episodes safely and effectively. 19 Although limited, the data might indicate the adequacy of the potency and PK bridging strategy presented in this work, which aided dose selection without previous exposure–response data for the indication. The results might also indicate that preclinical information on comparative potency between FVIIa products may be useful for informing the dose of new drugs.
Because of the accumulation of the PK data throughout the clinical program, the model was built in a sequential step, including interim analyses. The sequential nature of the model development allowed for several controls of components included in the model through retesting and backward exclusion. Extensive characterization of the variability and different structural phenomena as nonlinearities in PK was performed, and the final parameters were estimated with sufficient certainty (Table S1) and good descriptive properties (Figures S1 and S2). The first step of the model‐building process involved i.v. data with rich profiles from many dose levels, enabling characterization of the true PK without dependencies on absorption. The elimination was described with Michaelis–Menten kinetics supported by the i.v. data. In the second step, a significant proportion of s.c. data were added, providing more insight into the absorption process. A zero‐order input of FVIIa into a D from which FVIIa is absorbed with a first‐order absorption parameter described the absorption the best. IIV was supported for F (coefficient of variation [CV%] = 54) and k A (CV% = 34), and IOV was supported by the data for D. The IOV in D can depend on different yet known mechanisms that are related to administration and uptake. No changes were found in structural and random parameters associated with distribution and elimination, which may be attributed to the rich i.v. data used, informing these processes. Lastly, more s.c. data were obtained, and the model was challenged once again. No additional variability parameters were supported by the data when simultaneously estimating all parameters. The exclusion of Michaelis–Menten elimination did not impact the predictive performance of the model and produced a low increase in OFV (4.3 points) and was therefore excluded. Precision in model parameters were improved when excluding Michaelis–Menten elimination as uncertainty in maximum rate of elimination (V max) and concentration at half of V max (K m) was RSE% = 65 and RSE% = 68, respectively, compared with an RSE% = 7.5 for CL for the final PK model with linear elimination. A possible explanation is that most of the analyzed data were well below the estimated value of K m (1352 ng/ml) and in a range of linear elimination. Another aspect of importance when conducting PK analyses are handling of samples BLOQ. In this work, only 1.6% of the samples were below the detection limit, and as such, they were removed from the data set. For a higher degree of samples below the detection limit, alternative approaches should be considered. 20 Furthermore, allometry was applied with fixed exponents during model development. Estimating the exponents led to no change in OFV and high uncertainty and as such was not considered further. A likely reason for this outcome is that the data set covered a relatively narrow body weight range.
The selected dose of 60 μg/kg in the planned pivotal phase III clinical trial for episodic treatment of acute bleeding events was approved to be studied by regulatory agencies with support from the model‐based clinical trial simulations presented here. This work demonstrated how PK information through the clinical development program can be used to inform decision making in the absence of an established exposure–response relationship for a serious and rare indication such as HA/HB with inhibitors.
AUTHOR CONTRIBUTIONS
A.F., U.S.H.S., S.D., L.N., T.K., and G.E.B. wrote the manuscript. A.F., U.S.H.S., S.D., L.N., T.K., and G.E.B. designed the research. A.F., U.S.H.S., S.D., L.N., and T.K. performed the research. A.F., U.S.H.S., S.D., L.N., and T.K. analyzed the data.
FUNDING INFORMATION
This work was funded by Catalyst Biosciences.
CONFLICT OF INTEREST
S.D., L.N., G.E.B., and T.K. are employees and shareholders of Catalyst Biosciences. All other authors declared no competing interests for this work.
Supporting information
Figure S1.
Figure S2.
Table S1.
Data S1.
Data S2.
ACKNOWLEDGMENTS
The authors express gratitude to the individuals who participated in the clinical trials as well as the staff members who executed the trials.
Faraj A, Knudsen T, Desai S, Neuman L, Blouse GE, Simonsson USH. Phase III dose selection of marzeptacog alfa (activated) informed by population pharmacokinetic modeling: A novel hemostatic drug. CPT Pharmacometrics Syst Pharmacol. 2022;11:1628‐1637. doi: 10.1002/psp4.12872
REFERENCES
- 1. Mahlangu J, Levy H, Kosinova MV, et al. Subcutaneous engineered factor VIIa marzeptacog alfa (activated) in hemophilia with inhibitors: phase 2 trial of pharmacokinetics, pharmacodynamics, efficacy, and safety. Res Pract Thromb Haemost. 2021;5:e12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stoner KL, Harder H, Fallowfield LJ, Jenkins VA. Intravenous versus subcutaneous drug administration. Which do patients prefer? A systematic review. Patient. 2014;8:145‐153. doi: 10.1007/s40271-014-0075-y [DOI] [PubMed] [Google Scholar]
- 3. Saxena K. Barriers and perceived limitations to early treatment of hemophilia. J Blood Med. 2013;4:49‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carcao M, Goudemand J. Inhibitors in hemophilia: a primer. 5th ed. World Federation of Hemophilia. November 2018, No. 7. [Google Scholar]
- 5. FDA . Research, C. for D. E. and The Voice of the Patient: A Series of Reports from FDA's Patient‐Focused Drug Development Initiative. 2020. Accessed October 7, 2022. https://www.fda.gov/industry/prescription‐drug‐user‐fee‐amendments/fda‐led‐patient‐focused‐drug‐development‐pfdd‐public‐meetings
- 6. Gruppo RA, Malan D, Kapocsi J, et al. Phase 1, single‐dose escalating study of marzeptacog alfa (activated), a recombinant factor VIIa variant, in patients with severe hemophilia. J Thromb Haemost. 2018;16:1984‐1993. [DOI] [PubMed] [Google Scholar]
- 7. Klitgaard T, Nielsen TG. Overview of the human pharmacokinetics of recombinant activated factor VII. Br J Clin Pharmacol. 2008;65:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. FDA . Research, C. for B. E. and NovoSeven. 2022. Accessed October 7, 2022. https://www.fda.gov/vaccines‐blood‐biologics/blood‐blood‐products/novoseven
- 9. Anderson BJ, Holford NHG. Mechanism‐based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303‐332. [DOI] [PubMed] [Google Scholar]
- 10. Wählby U, Jonsson EN, Karlsson MO. Comparison of stepwise covariate model building strategies in population pharmacokinetic‐pharmacodynamic analysis. AAPS J. 2002;4:68‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meeks SL, Leissinger CA. The evolution of factor VIIa in the treatment of bleeding in haemophilia with inhibitors. Haemophilia. 2019;25:911‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Knudsen T, Kumar B, del Greco F, Neuman L, Levy H, Blouse GE. The combination of Marzeptacog alfa (activated) or Eptacog alfa (activated) with Emicizumab appears comparable As assessed by the thrombin generation test in hemophilia a plasma. Blood. 2019;134:1112.31558559 [Google Scholar]
- 13. Pharmacokinetics and pharmacodynamics of daily subcutaneously administered Marzeptacog alfa (activated) In hemophilia dogs (P076). Haemophilia. 2017;23:29‐140. [Google Scholar]
- 14. EMA NovoSeven . European Medicines Agency. 2018. Accessed October 7, 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/novoseven
- 15. Hedner U. Dosing and monitoring NovoSeven treatment. Haemostasis. 1996;26(Suppl 1):102‐108. [DOI] [PubMed] [Google Scholar]
- 16. Ludlam CA. The evidence behind inhibitor treatment with recombinant factor VIIa. Pathophysiol Haemost Thromb. 2002;32(Suppl 1):13‐18. [DOI] [PubMed] [Google Scholar]
- 17. Berntorp E, Shapiro AD. Modern haemophilia care. The Lancet. 2012;379:1447‐1456. [DOI] [PubMed] [Google Scholar]
- 18. Mannucci PM, Tuddenham EG. The Hemophilias — from Royal Genes to gene therapy. N Engl J Med. 2001;344:1773‐1779. [DOI] [PubMed] [Google Scholar]
- 19. ISTH Congress Abstracts . Crimson 1: A Phase 3 study to evaluate the efficacy and safety of subcutaneous marzeptocog alfa (activated) for on‐demand treatment of bleed events in subjects with hemophilia A or B, with inhibitors. Accessed October 7, 2022. https://abstracts.isth.org/abstract/crimson‐1‐a‐phase‐3‐study‐to‐evaluate‐the‐efficacy‐and‐safety‐of‐subcutaneous‐marzeptocog‐alfa‐activated‐for‐on‐demand‐treatment‐of‐bleed‐events‐in‐subjects‐with‐hemophilia‐a‐or‐b‐with‐inhibitors/
- 20. Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35:401‐421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Table S1.
Data S1.
Data S2.