

Abstract
Joubert syndrome (JS), a well-established ciliopathy, is characterized by the distinctive molar tooth sign on brain MRI, ataxia, and neurodevelopmental features. Other manifestations can include polydactyly, accessory frenula, renal or liver disease. Here we report individuals meeting criteria for JS with de novo heterozygous variants in SLC30A7 (Chr1p21.2). The first individual is a female with history of unilateral postaxial polydactyly, classic molar tooth sign on MRI, macrocephaly, ataxia, ocular motor apraxia, neurodevelopmental delay (NDD), and precocious puberty. Exome sequencing (ES), detected a de novo heterozygous missense variant in SLC30A7: NM_133496.5: c.407T>C, (p.Val136Ala). The second individual had bilateral postaxial polydactyly, molar tooth sign, macrocephaly, developmental delay, and an extra oral frenulum. A de novo deletion-insertion variant in SLC30A7, c.490_491delinsAG (p.His164Ser) was found. Both de novo variants affect highly conserved residues. Variants were not identified in known Joubert genes for either case. SLC30A7 has not yet been associated with a human phenotype. The SLC30 family of zinc transporters, like SLC30A7, permit cellular efflux of zinc, and although it is expressed in the brain its functions remain unknown. Published data from proteomic studies support SLC30A7 interaction with TCTN3, another protein associated with JS. The potential involvement of such genes in primary cilia suggest a role in Sonic Hedgehog signaling. SLC30A7 is a candidate JS-associated gene. Future work could be directed toward further characterization of SLC30A7 variants and understanding its function.
Keywords: SLC30A7, Joubert syndrome, ciliopathies, molar tooth sign, zinc transporter
INTRODUCTION
Joubert syndrome (JS), is a condition characterized by a distinctive development of the cerebellum and brainstem, appearing as the molar tooth sign on brain MRI and is a well-established ciliopathy (Bachmann-Gagescu et al., 2020; Parisi, 2019; Romani et al., 2013). Typical clinical features include ataxia, ocular motor apraxia, and developmental delay while other manifestations are variably present, such as retinal dystrophy, nephronophthisis, and liver disease (Bachmann-Gagescu et al., 2020). JS is part of an expanding group of primary ciliopathies with new gene candidates being identified using sequencing and phenotyping (Parisi, 2019). A molecular diagnosis of JS can be established in ~62-85% of individuals with clinical diagnosis of JS with the remaining cases molecularly unsolved. Moreover, the veracity and accuracy of a JS clinical diagnosis is not secure based on clinical signs and symptoms alone bringing into question the recurrence risk counseling of parents of a sporadic case (Bachmann-Gagescu et al., 2015; Shaheen et al., 2016; Weghe et al., 2021).
Here we report a candidate novel gene association with JS in two individuals with de novo rare heterozygous variant alleles in SLC30A7 (Chr1p21.2) and features of JS including physical and brain MRI findings. SLC30A7 is an SLC30 transporter family member, which is expressed in the brain and other tissues (Brion et al., 2021; Huang & Tepaamorndech, 2013). Rare de novo variants in the SLC30A7 in patients with JS, suggest its involvement in ciliopathies.
METHODS
Patient consent was obtained for both families reported. Research activities were approved by the institutional review board (IRB#: H-29697). Whole exome sequencing (ES) was performed as a trio (proband with both biological parents). Variants were evaluated using the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015) and the Sequence Variant Interpretation Working Group (SVI WG) recommendations (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/).
RESULTS
CLINICAL REPORT
Patient 1
A 14-year-old Latino female was born at term to a G1P1 19-year-old mother by emergency C-section after prolonged labor. Birth weight: 3.8 kg (80th centile, on CDC girls 0-36 months growth chart). She was noted to have post-axial polydactyly. At the age of 6 months, she was noted to have trouble tracking objects and was hypotonic. She sat independently at 12-18 months and started walking at 2 years of age. She said her first words at around 2 years of age. There was no history of abnormal respiratory pattern. She had an unsteady gait, fell frequently and was diagnosed with ataxia. Her history was also significant for ocular motor apraxia, rotary nystagmus with normal fundoscopy and macrocephaly (OFC 57cm at 7 years of age, >98th centile or Z-score >2.05). By age 7 she had premature adrenarche and true precocious puberty with advanced bone age (chronological age 7 years 9 months and bone age of 11 years) treated with leuprolide for 2 years. By age 14, she could read, write paragraphs and was learning multiplication and division. She had good balance for functional mobility and was not falling but had difficulty with higher level balance skills.
On physical examination she had a friendly demeanor, broad forehead and slightly downturned corners of the mouth at rest (Figure 1A and B). A brain MRI at 14 years of age showed classical molar tooth sign (Figure 1B and C). Workup included normal liver transaminases, blood counts, and serum zinc level. On abdominal ultrasound the kidneys and spleen were normal, but the liver was mildly echogenic and at upper limits of normal size. Genetic evaluation included a normal chromosomal microarray (CMA). Clinical exome sequencing (cES), (Mendelsohn et al., 2020) detected a de novo heterozygous missense variant in NM_133496.5 (SLC30A7): c.407T>C, (p.Val136Ala) annotated according the GRCh37/hg19 (Figure 2A). SLC30A7 maps to chromosome 1p21.2. The variant allele and de novo status was verified using family trio DNA and Sanger dideoxynucleotide sequencing. This variant allele changes a highly conserved residue and is predicted to be deleterious (MutationTaster, SIFT, CADD score 27.8, and REVEL score 0.59) (Kircher et al., 2014).
Figure 1.
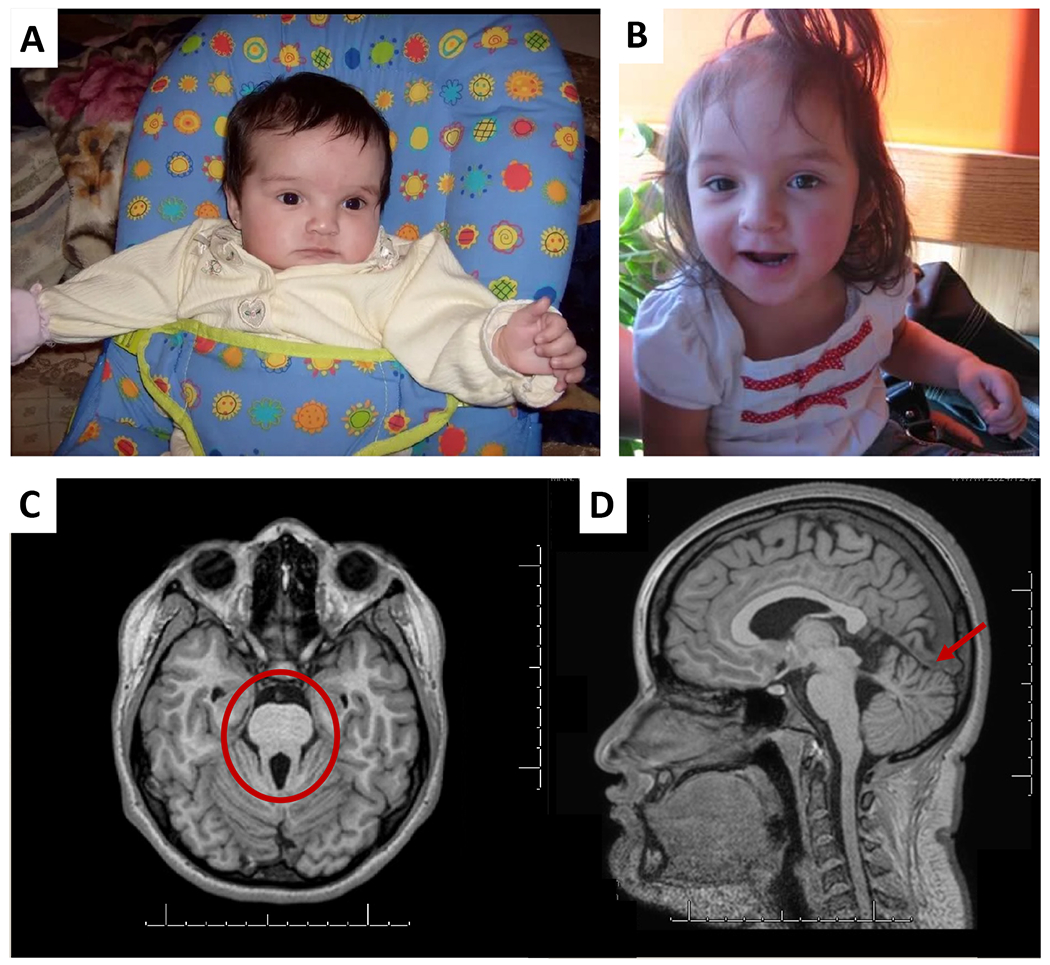
Upper panel. Patient 1 A) At age 4 months. Note the left postaxial polydactyly, prominent forehead and widely spaced eyes. B) At 3 years and 2 months. High eyebrows everted lower lip and thin upper lip. Lower panel: Brain MRI for patient 1 at 14 years of age A) T1- weighted image on axial view through the cerebellum and brainstem showing the Molar Tooth Sign (circled). B) Mid-sagittal view showing appearance of the superior portion of the cerebellar vermis with focal hypoplasia more on the left side of the superior vermis (arrow).
Figure 2.
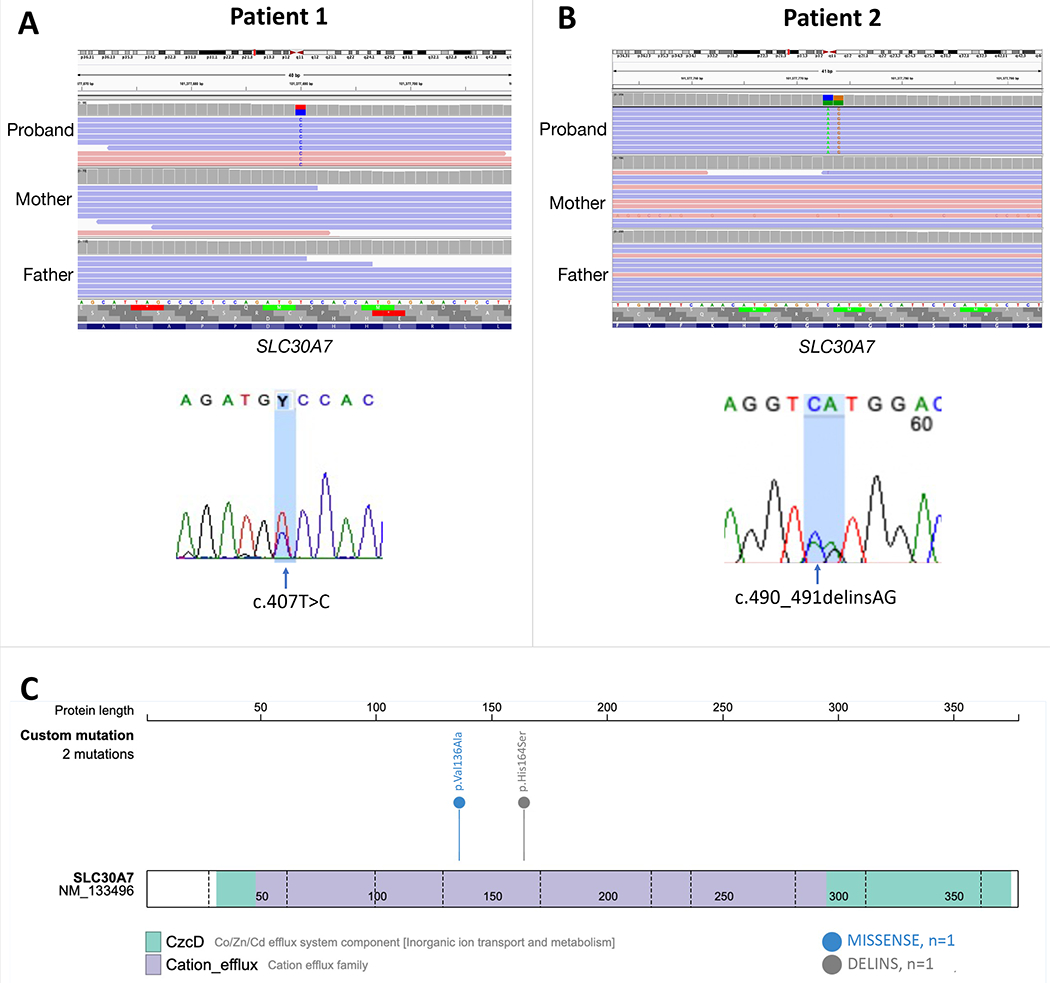
A) Top: Integrated Genome Viewer of the BAM file showing the de novo SLC30A7 variant from patient 1 and its absence in parents. Bottom: Chromatogram confirming the heterozygous mutation in SLC30A7: chr1:101377690, NM_133496.5: c.407T>C, (p.Val136Ala). B) Top: Integrated Genome Viewer of the BAM file showing the de novo SLC30A7 variant from patient 2 and its absence in parents. Bottom: Chromatogram confirming the heterozygous mutation in SLC30A7: chr1:101377773-101377774, NM_133496.5: c.490_491delinsAG, (p.His164Ser). C) Schematic of the SLC30A7 protein and the location of the mutations in each specific domain.
Patient 2
Patient 2 is 9-year-old male delivered at 38 weeks via Cesarean section for failure to progress. He was born to a 26-year-old mother. Prenatal ultrasound and first trimester screening were normal. Genetics was consulted on day of life 2 because of dysmorphic features and failed newborn hearing screen. Growth percentiles at birth were: FOC (97.7th), length (80.2th), and weight (73.9th). Physical examination showed relative macrocephaly, low set ears, midfacial hypoplasia, bilateral upper extremity postaxial polydactyly, and 2/3 toe syndactyly. Brain MRI showed the molar tooth sign, mild decrease in supratentorial white matter volume, borderline thinning of the corpus callosum, mild prominence of the supratentorial ventricular system, and hypoplasia of the adenohypophysis. Renal ultrasound and sterol analysis were normal. Genetic testing including chromosome microarray analysis (CMA) and clinical ES were non-diagnostic. CMA array showed a region of at least 0.158 Mb copy number loss on the proximal long arm of chromosome (2q13) encompassing the juvenile nephronophthisis, NPHP1 gene.
Patient was re-evaluated by Genetics at age 7 months and a clinical diagnosis of JS was discussed. Deceleration of linear growth (3rd percentile), moderate hypotonia, global developmental delay, poor visual attentiveness, multiple superior and inferior gingival-labial frenula, a median groove of the upper lip, and micropenis were noted. Evaluation by Endocrinology at age 11 months for micropenis and poor linear growth showed normal thyroid function and hCG stimulation studies suggested normal testicular function. Intramuscular testosterone injections were given to improve penile length though patient was lost to follow up with Endocrinology. Evaluation by Ophthalmology at age 8 months showed subnormal vision. Short stature was recognized by age 2 years.
Patient was lost to follow up from age 7 months until age 9 years when a rare de novo deletion-insertion variant NM_133496.5: c.490_491delinsAG (p.His164Ser) was identified in research analysis within the gene SLC30A7 (Figure 2B) (Eldomery et al., 2017). History obtained at Genetics reevaluation at age 9 years and 5 months found that child’s early clinical course was notable for global developmental delay, chronic cough, aspiration, and recurrent respiratory infections in early childhood, which had improved with age. Autism spectrum disorder was diagnosed in early childhood and ABA therapy was instituted. He achieved the ability to walk between age 5 and 6 years and while ambulatory at age 9 years, his parents reported poor balance and lack of stamina. His parents also noted poor vision and poor balance in low-light environments, and a recent diagnosis of “night blindness”. At 9 years 5 months patient was non-verbal and communicated via a picture image system, constructing sentences such as “I am hungry”. Physical examination showed relative macrocephaly with OFC at 88%ile, short stature (Z= −2.31) and BMI at 96%ile. There were bilateral epicanthal folds, a dysconjugate gaze, subjectively poor visual focus, median groove of the upper lip, and superior and inferior gingival-labial frenula. The gait was wide based. Formal ophthalmologic examination and abdominal ultrasound were recommended yet not completed.
These rare variants in SLC30A7 in patients 1 and 2 are not present in the population data in gnomAD (v2.1.1, access date: January 10th, 2022). Val136 is in the second predicted extracellular domain of SLC30A7, and His164 is in the adjacent cytoplasmic domain (Figure 2C) (Kirschke & Huang, 2003). These residues are conserved across 100 vertebrate species, although the region is not globally depleted in missense variants (not in a missense depleted region, or showing subgenic intolerance) (Ge et al., 2016; Pérez-Palma et al., 2020; Silk et al., 2019). The variants in SLC30A7 associated with JS are potentially ultrarare in frequency (<1/10,000) but important in terms of potential phenotype (Hansen et al., 2019). Evaluation of several hundred families with JS who have undergone ES did not immediately reveal further de novo/suspicious variants in SLC30A7. Matching by gene for other investigators who have encountered SLC30A7 variants only revealed inherited variants of uncertain significance (VUS) in individuals with phenotypes not suggestive of JS (Sobreira et al., 2015). SLC30A7 variants observed as de novo variants in available autism/neurodevelopmental sequencing studies showed only intronic variants in SLC30A7 with a maximum CADD score of 13.5.
DISCUSSION
The molar tooth sign, clinical findings of JS and rare, predicted-deleterious, de novo SLC30A7 variants in two patients with classic features of JS suggest this gene as a strong candidate for the JS phenotype. JS is a developmental disorder with marked genetic heterogeneity that exhibits predominately autosomal recessive, and rarely X-linked inheritance. The clinical and genomic data we present support that heterozygous SLC30A7 variants are implicated in JS. Patients 1 and 2 had three diagnostic criteria for JS: Molar tooth sign on brain MRI, hypotonia in infancy with later ataxia, and developmental delays (Bachmann-Gagescu et al., 2020; Parisi, 2019). Polydactyly was also a key clinical feature observed in both individuals, and the accessory frenula reminiscent of oral-facial-digital sub-type of JS was observed in patient 2. Despite the facts that ACMG guidelines, and the SVI WG recommendations, are typically applied for well-established known disease associations, we attempted to assess the impact of our variants in our candidate gene. The SLC30A7 c.407T>C and the SLC30A7 c.490_491delinsAG meet the criteria for being likely pathogenic by applying the following rules: PS2_strong (de novo), PS4_supporting, PM2_supporting (absent from gnomAD) and PP3 (SLC30A7 c.407T>C is damaging by in silico predictions). Of note, since the in-silico prediction algorithms are developed to assess missense alterations, we opted not to apply the PP3 rule for the c.490_491delinsAG variant. The PS4_supporting rule was potentially justified based on the specificity of the clinical phenotype of Joubert Syndrome and the absence of the variants from the gnomAD database (Richards et al., 2015). Other candidates identified on cES were deemed unlikely candidates. Additionally, the sequencing did not find candidate variants in other genes currently associated with JS. While a variety of variants (non-coding, structural) can be missed by exome sequencing, these variants are infrequent causes, so the rare de novo SLC30A7 variants remain the highest candidates in these patients after in depth review. Alternatively, these variants could be chance occurrences in individuals with JS.
The pipeline used at the time of data reanalysis (Eldomery et al., 2017) called the deletion-insertion variant c.490_491delinsAG (p.His164Ser) as two independent calls c.490C>A:p.H164N and c.491A>G:p.H164R, which apparently can be misinterpreted as a two missense substitution in the cis configuration (Figure 2C). The de novo mutation (DNM) finder algorithm (https://github.com/BCM-Lupskilab/DNM-Finder) uncovered the de novo nature of these alterations that were subsequently confirmed with Sanger sequencing. According to the Sequence Variant Nomenclature recommendations (https://varnomen.hgvs.org/), these two alterations should be merged and called deletion-insertion to ensure the appropriate annotation with public databases (e.g., ClinVar).
The zinc transporter (ZnT)/SLC30 subfamily of the cation diffusion facilitator family, including SLC30A7, permit cellular efflux of zinc. How this mechanism or other SLC30A7 functions may lead to potential ciliopathies or disruption of forebrain development remains unknown. Further cases will be needed to add to the findings presented here.
Overlapping JS features in diverse ciliopathies reflect a shared molecular pathophysiology involving the primary cilia. Previous published centrosome-cilium interface mapping through biotinylation with TCTN3 as a bait protein show interaction with SLC30A7 linking it to a known JS-associated protein. Tectonic family member 3 (TCTN3), which complexes with TCTN1-2 in the ciliary transition zone is implicated in Sonic Hedgehog (SHH) signaling and their disruption leads to JS and other ciliopathies (Reiter & Skarnes, 2006). Indeed, JS associated proteins localize to multiple subregions of the cilium, but the transition zone, where TCTN3 and SLC30A7 interact, is a proposed hotspot for ciliopathy related proteins (Shaheen et al., 2016).
The family of SLC30 transporters is involved in zinc homeostasis in the body, by exporting zinc to the extracellular space or sequestering this trace element into the intracellular compartments if levels rise (Huang & Tepaamorndech, 2013). Zinc transporters, including ZNT7 are abundantly expressed in the mouse cerebellum suggesting a significant role of zinc homeostasis in this organ (Hui-Ling et al., 2009). Null mutation of Slc30a7 in mice results in a lean phenotype that cannot be corrected by oral zinc (Huang & Tepaamorndech, 2013). Yet, loss of function of other ZnT paralogs, has been established in other neurological conditions like epileptic encephalopathies, and Alzheimer disease (Boone et al., 2011; Duan et al., 2021; Marafi et al., 2021).
Evidence suggests a direct mechanistic link between zinc homeostasis and Hedgehog (Hh) signaling. Zinc binds directly to the Hint domain of the Hh protein inhibiting its auto processing (N-S acyl shift and cholesteroylation) inhibiting the generation of Hh ligand and Hh signaling. Thus, zinc dyshomeostasis disrupts Hh signaling and may lead to pathogenesis, however the mechanism of Hh ligand disruption in disease is poorly understood. Existing research also supports the role of zinc in oligodendrogenesis, neurogenesis, neuronal differentiation, and white matter growth. Structural central nervous system abnormalities may also be associated with defects in zinc transport (Brion et al., 2021), although whether this is relevant in JS requires further study.
Discovery of additional individuals with pathogenic variants in SLC30A7 will strengthen the evidence regarding this specific gene and JS association. Future work could be directed toward understanding how SLC30A7 dysfunction might cause JS and potentially other ciliopathies.
Acknowledgments
This study was supported in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG; UM1 HG006542); NHGRI GREGoR (U01 HG011758); U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and Muscular Dystrophy Association (MDA) (512848 to J.R.L.).
Financial Support
This study was supported in part by the U.S. National Institutes of Health (NIH) National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHLBI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542); NHGRI Baylor College of Medicine Genomic Research to Elucidate the Genetics of Rare (BCM-GREGoR, U01 HG011758); National Institute of Neurological Disorders and Stroke (NINDS, R35NS105078 to J.R.L.); and the Muscular Dystrophy Association (MDA, 512848 to J.R.L.); NHGRI University of Washington Mendelian Genomics Research Center (UW-MGRC, U01 HG011744, NICHD R01 HD100730 to D.D)
Footnotes
Disclosure
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories; JRL is a member of the Scientific Advisory Board of BG. Other authors have no potential conflicts to report.
REFERENCES
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, Roak BJO, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, Lacy N De, Day DO, Alswaid A,A,RR, Lingappa L, Lourenço C, Martorell L, Ozyürek H, … Doherty D (2015). Joubert syndrome : a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Gene, 52, 514–522. 10.1136/jmedgenet-2015-103087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Gagescu Ruxandra., Dempsey JC, Bulgheroni S, Chen ML, D’Arrigo S, Glass IA, Heller T, Héon E, Hildebrandt F, Joshi N, Knutzen D, Kroes HY, Mack SH, Nuovo S, Parisi MA, Snow J, Summers AC, Symons JM, Zein WM, … Doherty D (2020). Healthcare recommendations for Joubert syndrome. American Journal of Medical Genetics, Part A, 182(1), 229–249. 10.1002/ajmg.a.61399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Liu P, Zhang F, Carvalho CMB, Towne CF, Batish SD, & Lupski JR (2011). Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genetics in Medicine, 13(6), 582–592. 10.1097/GIM.0b013e3182106775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion LP, Heyne R, & Lair CS (2021). Role of zinc in neonatal growth and brain growth: review and scoping review. Pediatric Research, 89(7), 1627–1640. 10.1038/s41390-020-01181-z [DOI] [PubMed] [Google Scholar]
- Duan R, Saadi NW, Grochowski CM, Bhadila G, Faridoun A, Mitani T, Du H, Fatih JM, Jhangiani SN, Akdemir ZC, Gibbs RA, Pehlivan D, Posey JE, Marafi D, & Lupski JR (2021). A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy. American Journal of Medical Genetics, Part A, 185(7), 1972–1980. 10.1002/ajmg.a.62192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, Küry S, Mercier S, Lessel D, Denecke J, Wiszniewski W, Penney S, Liu P, Bi W, Lalani SR, Schaaf CP, Wangler MF, Bacino CA, Lewis RA, … Lupski JR (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Medicine, 9(1), 1–15. 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, Waisfisz Q, Weiss MM, Hendriks Y, Stuurman KE, Nelson SF, Grody WW, Lee H, Kwok PY, & Shieh JTC (2016). Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. Npj Genomic Medicine, 1(August). 10.1038/npjgenmed.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AW, Murugan M, Li H, Khayat MM, Wang L, Rosenfeld J, Andrews BK, Jhangiani SN, Coban Akdemir ZH, Sedlazeck FJ, Ashley-Koch AE, Liu P, Muzny DM, Allori A, Angrist M, Ashley P, Bidegain M, Boyd B, Chambers E, … Gibbs RA (2019). A Genocentric Approach to Discovery of Mendelian Disorders. American Journal of Human Genetics, 105(5), 974–986. 10.1016/j.ajhg.2019.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, & Tepaamorndech S (2013). The SLC30 family of zinc transporters-A review of current understanding of their biological and pathophysiological roles. Molecular Aspects of Medicine, 34(2–3), 548–560. 10.1016/j.mam.2012.05.008 [DOI] [PubMed] [Google Scholar]
- Hui-Ling G, Wan-Yu F, Xiao-Ling L, He X, Liping H, & Zhan-You W (2009). Golgi apparatus localization of ZNT7 in the mouse cerebellum. Histol Histopathol, 24(5), 567–572. 10.14670/HH-24.567 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschke CP, & Huang L (2003). ZnT7, a novel mammalian zinc transporter, accumulates zinc in the Golgi apparatus. Journal of Biological Chemistry, 278(6), 4096–4102. 10.1074/jbc.M207644200 [DOI] [PubMed] [Google Scholar]
- Marafi D, Fatih JM, Kaiyrzhanov R, Ferla MP, & Gijavanekar C (2021). Biallelic variants in SLC38A3 encoding a glutamine transporter cause epileptic encephalopathy. Brain. 10.1093/brain/awab369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn BA, Beleford DT, Abu-El-Haija A, Alsaleh NS, Rahbeeni Z, Martin PM, Rego S, Huang A, Capodanno G, Shieh JT, Van Ziffle J, Risch N, Alkuraya FS, & Slavotinek AM (2020). A novel truncating variant in ring finger protein 113A (RNF113A) confirms the association of this gene with X-linked trichothiodystrophy. American Journal of Medical Genetics, Part A, 182(3), 513–520. 10.1002/ajmg.a.61450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA (2019). The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Translational Science of Rare Diseases, 4(1–2), 25–49. 10.3233/trd-190041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Palma E, May P, Iqbal S, Niestroj LM, Du J, Heyne HO, Castrillon JA, O’Donnell-Luria A, Nürnberg P, Palotie A, Daly M, & Lal D (2020). Identification of pathogenic variant enriched regions across genes and gene families. Genome Research, 30(1), 62–71. 10.1101/gr.252601.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, & Skarnes WC (2006). Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes and Development, 20(1), 22–27. 10.1101/gad.1363606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, & Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani M, Micalizzi A, & Valente EM (2013). Joubert syndrome: Congenital cerebellar ataxia with the molar tooth. The Lancet Neurology, 12(9), 894–905. 10.1016/S1474-4422(13)70136-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, Al Hashem A, Derar N, Alsharif H, Aldahmesh MA, Alazami AM, Hashem M, Ibrahim N, Abdulwahab FM, Sonbul R, Alkuraya H, Alnemer M, Al Tala S, Al-Husain M, … Alkuraya FS (2016). Characterizing the morbid genome of ciliopathies. Genome Biology, 17(1), 1–11. 10.1186/s13059-016-1099-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk M, Petrovski S, & Ascher DB (2019). MTR-Viewer: identifying regions within genes under purifying selection. Nucleic Acids Research, 47(W1), W121–W126. 10.1093/nar/gkz457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. Oct;36(10):928–30. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Pierce-Hoffman E, Cummings BB, Alföldi J, Francioli LC, Gauthier LD, Hill AJ, O’Donnell-Luria AH, Armean IM, Banks E, Bergelson L, Cibulskis K, Collins RL, Connolly KM, Covarrubias M, Daly MJ, Donnelly S, Farjoun Y, Ferriera S, … MacArthur DG (2020). Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nature Communications, 11(1), 1–13. 10.1038/s41467-019-12438-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weghe J. C. Van De, Giordano JL, Mathijssen IB, Mojarrad M, Lugtenberg D, Miller CV, Dempsey JC, Sadat M, Mohajeri A, Leeuwen E Van, Pajkrt E, Klaver CCW, & Houlden H (2021). TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes. HGG Adv, 2(1), 1–14. 10.1016/j.xhgg.2020.100016.TMEM218 [DOI] [PMC free article] [PubMed] [Google Scholar]