


Abstract
Trimethylguanosine synthase 1 (TGS1) is a highly conserved enzyme that converts the 5′-monomethylguanosine cap of small nuclear RNAs (snRNAs) to a trimethylguanosine cap. Here, we show that loss of TGS1 in Caenorhabditis elegans, Drosophila melanogaster and Danio rerio results in neurological phenotypes similar to those caused by survival motor neuron (SMN) deficiency. Importantly, expression of human TGS1 ameliorates the SMN-dependent neurological phenotypes in both flies and worms, revealing that TGS1 can partly counteract the effects of SMN deficiency. TGS1 loss in HeLa cells leads to the accumulation of immature U2 and U4atac snRNAs with long 3′ tails that are often uridylated. snRNAs with defective 3′ terminations also accumulate in Drosophila Tgs1 mutants. Consistent with defective snRNA maturation, TGS1 and SMN mutant cells also exhibit partially overlapping transcriptome alterations that include aberrantly spliced and readthrough transcripts. Together, these results identify a neuroprotective function for TGS1 and reinforce the view that defective snRNA maturation affects neuronal viability and function.
INTRODUCTION
The correct processing of small non-coding RNAs is essential to prevent severe neurodegenerative conditions (1). Small nuclear RNAs (snRNAs) are components of the spliceosomal small nuclear ribonuclear proteins (snRNPs). Defects in the snRNP assembly pathway have been observed in spinal muscular atrophy (SMA), a neurological disease caused by deficiency of the survival motor neuron (SMN) protein (2). Another neurodegenerative disorder, Pontocerebellar hypoplasia type 7 (PCH7), is caused by mutations in the TOE1 deadenylase (Target Of EGR1) that prevents accumulation of 3′-end-extended pre-snRNAs (3).
The Sm class snRNAs include components of both the major (U1, U2, U4 and U5) and minor (U11, U12, U4atac and U5) spliceosome. These snRNAs are transcribed by RNA polymerase II (Pol II) and the formation of their 3′ ends is co-transcriptionally controlled (4,5). Nascent U1 and U2 snRNA transcripts are subject to cleavage by the Integrator complex, resulting in precursors carrying extended 3′ ends that are trimmed after export to the cytoplasm (6–8).
All snRNAs transcribed by Pol II acquire an m7-monomethylguanosine cap (m7G/MMG cap) at their 5′ end. Immediately after transcription, the monomethylated snRNA precursors bind the cap-binding complex (CBC) and are exported to the cytoplasm (9,10), where they bind the Gemin5 subunit of the SMN complex (11) that mediates their assembly with the seven Sm proteins (Sm core) to form snRNPs (12). The snRNA precursors associated with Gemin5 have extended 3′ tails that can be oligoadenylated or uridylated (11). The U2 snRNA precursors have 3′ extended tails including 12 genomic-encoded nucleotides, which are trimmed by the TOE1 deadenylase (3).
Following interaction with SMN and the Sm core, the MMG cap of snRNAs is converted to a 2,2,7-trimethylguanosine cap (henceforth abbreviated to TMG cap) by trimethylguanosine synthase 1 (TGS1) (13), which physically interacts with SmB and SMN (14). Re-entry of the snRNPs into the nucleus is mediated by both the TMG cap and the Sm core (15,16). In somatic cells of Xenopus and mammals, the Sm signal is both necessary and sufficient for nuclear import, while the TMG cap accelerates the rate of import (17–19). Once in the nucleus, the snRNPs concentrate in the Cajal bodies (CBs) and undergo further maturation steps (20).
3′-end processing of snRNAs is thought to occur after TMG cap formation (21), but whether TGS1 and cap hypermethylation have roles in subsequent steps of snRNP processing is unknown. This is a relevant biomedical issue, as abnormal accumulations of 3′-end-extended snRNA precursors have been observed in PCH7 patients (3) and in cells deficient for SMN (11). To elucidate the role of TGS1 in snRNA maturation and its functional relationships with SMN, here we explored the phenotypic consequences of TGS1 loss in human cells and in the Drosophila, Caenorhabditis elegans and Danio rerio model organisms. Collectively, our results indicate that TGS1 plays a conserved role in 3′ processing of snRNAs and acts as a neuroprotective factor that can counteract the phenotypic consequences of SMN deficiency.
MATERIALS AND METHODS
Caenorhabditis elegans strains
Nematodes were grown and handled following standard procedures, under uncrowded conditions, at 20°C, on NGM (Nematode Growth Medium) agar plates seeded with Escherichia coli strain OP50 (22). Wild-type animals used in this work were C. elegans variety Bristol, strain N2. The transgenic strains used in this work were: NA1330 gbIs4 [GBF109 punc-25::cesmn-1(RNAi) (200 ng/μl); GB301 pchs-2::GFP (10 ng/μl)] III; NA1617 gbEx593 [GBF109 punc-25::cesmn-1(RNAi) (10 ng/μl); GB301 pchs-2::GFP (10 ng/μl)]; NA1541 gbEx568 [GBF321 punc-25::cekal-1(RNAi) (10 ng/μl); GB301 pchs-2::GFP (10 ng/μl)]; NA1252 gbEx504 [GBF322 punc-119::dsRED (20 ng/μl); pelt-2::RFP (30 ng/μl)] (23); and EG1285 oxIs12 [punc-47::GFP; lin-15(+)]. EG2185 and N2 were provided by the Caenorhabditis Genetics Center (CGC), funded by the NIH Office of Research Infrastructure Programs (P40 OD010440); Is[punc-47::RFP] was kindly provided by K. Shen (Stanford University, USA). A complete description of the transgenes obtained in this work is reported in Table 1.
Table 1.
List of the C. elegans transgenes
| Strain | Transgene name | Transgene description | Notes |
|---|---|---|---|
| NA1539 | gbEx579 | cetgs-1 knock-down in D-type MNs | |
| NA1972 | gbEx647 | GB335 punc-119::hTGS1 (20 ng/μl); podr-1::RFP (30 ng/μl) | hTGS1 overexpression in all neurons |
| NA1981 | gbEx648 | GB338 punc-119::cetgs-1 (20 ng/μl); podr-1::RFP (30 ng/μl); gbls4 III | ceTGS-1 overexpression in all neurons with smn-1 knock-down in D-type MNs |
| NA1533 | gbEx576 | GBF329 pcetgs-1a::GFP (50 ng/μl) | GFP expression under cetgs-1 isoform a promoter |
| NA1535 | gbEx577 | GBF330 pcetgs-1b::GFP (50 ng/μl) | GFP expression under cetgs-1 isoform b promoter |
Transgenic C. elegans strains
The constructs GBF329 (pcetgs-1a::GFP) and GBF330 (pcetgs-1b::GFP) for analyzing the expression of cetgs-1 isoforms were created by polymerase chain reaction (PCR) fusion (24) of two DNA fragments: the presumptive promoters of cetgs-1 and the green fluorescent protein (GFP) sequence. The two promoters of cetgs-1 isoforms (a and b) have been chosen upstream of the ATG of the two isoforms, to span 360 bp (cetgs-1a) and 960 bp (cetgs-1b), respectively. The putative regulatory regions were amplified by PCR using as template wild-type genomic DNA. The GFP, followed by the 3′-untranslated region (UTR) of the unc-54 gene, was amplified from plasmid pPD95.75, kindly provided by A. Fire (Stanford University, CA, USA). The rescue plasmids (GB335 punc-119:: hTGS1 and GB338 punc-119::cetgs-1) for pan-neuronal expression were created by cloning the hTGS1 or cetgs-1 coding sequence (CDS) into the plasmid pBY103, kindly provided by O. Hobert (Columbia University, NY, USA), which contains the pan-neuronal promoter punc-119. hTGS1 and cetgs-1 CDS were amplified by PCR from cDNA libraries and cloned in the BamHI site of pBY103. D-type motor neuron (MN)-specific RNA interference (RNAi) transgenic lines were obtained as previously described (23,25). To obtain the specific knock-down of cetgs-1, we used a short form of the unc-25/GAD promoter (180 bp), which is specifically expressed from embryonic to adult stages only in the 19 D-type MNs in the ventral cord and not in other GABAergic neurons (RME, AVL, DVB and RIS) (23,26). For gene silencing, we amplified, from genomic DNA, the same exon-rich region that was used for the RNAi plasmid library prepared for RNAi by feeding experiments (27). Exon-rich regions were amplified in two separate PCRs to obtain the sense and antisense fragments. The unc-25 promoter was amplified using specific primers. The promoter was subsequently fused to each orientation of the target gene by PCR fusion using internal primers. All primers are available upon request. Germline transformation was accomplished as described (28) by injecting into the gonad of adult animals a DNA mixture containing a transgenic construct together with a phenotypic marker for selection of transgenic progeny. As co-injection marker we used podr-1::RFP kindly provided by C. Bargmann, The Rockfeller University, NY, USA [red fluorescent protein (RFP) expression in AWC and AWB neurons] at 30 ng/μl and GB301 pchs-2::GFP (GFP expression in the glandular pharyngeal g1 and g2 cells, and in m3 and m4 myoepithelial cells) (29) at 10 ng/μl. At least two independent transgenic lines were examined for each experiment. Genetic crosses were made to transfer transgenes to the appropriate genetic background. In all cases, the presence of the desired transgenes was verified by phenotyping. Two independent clones with the same genotype were examined after each cross, and the mean of the two clones has been reported in the Results. The punc-25 promoter, used for neuron-specific silencing of cetgs-1 and cesmn-1, is expressed in 19 neurons among a total of 959 somatic cells and ∼2000 germ cells in each animal (30), so it is estimated that only 0.6% of total cells have been silenced for cetgs-1 and cesmn-1 in each animal (23). Thus, changes in gene expression compared with the wild type cannot be appreciated in whole animals. In contrast, cetgs-1 overexpression is driven in all 302 neurons thanks to the pan-neuronal promoter punc-119 and through the formation of extrachromosomal arrays, which carry a variable number of copies (between 80 and 300 of the gene of interest (31).
Caenorhabditis elegans behavioral assay
Well-fed, young adult animals were used for backward movement assay (32) to test D-type MN function. The assay was performed blind on NGM plates, 6 cm in diameter, seeded with bacteria. Using an eyelash, the animal was touched first on the tail to induce a forward movement and then on the head to test for backward movement. A defective movement was scored when animals were unable to fully move backward. For each dataset, the percentage of animals with normal locomotion among the total number of tested animals was calculated. One-way analysis of variance (ANOVA) test was used for statistical analysis.
Caenorhabditis elegans microscopy analysis
Animals were immobilized in 0.01% tetramisole hydrochloride (Sigma-Aldrich) on 4% agar pads and visualized using Zeiss Axioskop or Leica DM6B microscopes. All microscopes were equipped with epifluorescence and differential interference contrast (DIC) Nomarski optics, and images were collected with Leica Hamamatsu C11440 digital cameras. Confocal images have been collected using a DMi8 confocal microscope.
Zebrafish experiments
Zebrafish procedures were approved by the local animal protection committee LANUV NRW; reference 84-02.04.2012.A251. Experiments for caudal primary MN (CaP-MN) analysis were performed in the wild-type TL/EK (Tupfel long fin/Ekkwill) line.
Zebrafish injection
Control (non-targeting) and tgs1 morpholino oligonucleotides (MOs) were purchased from (GeneTools, LLC) using the tgs1 XM_003197865.5 sequence as reference. MO sequences are reported in Supplementary Table S11. For TGS1 mRNA injection, human TGS1 cDNA (NM_001317902) was cloned into an N-terminal flag-pCS2 + mRNA expression vector. In vitro transcription of 5′-capped mRNAs was performed using the mMACHINE SP6 Transcription Kit (Ambion) following the manufacturer's protocol and as previously described in (33,34). Embryos from TL/EK wild-type crossings were used to visualize the CaP-MN phenotype. Embryos were injected with the respective dose of MOs or mRNA in an aqueous solution containing 0.05% PhenolRed and 0.05% Rhodamine-Dextran (Sigma-Aldrich). At 6–7 h after injection, embryos were sorted according to homogeneity of the rhodamine fluorescence signal.
Semi-quantitative RT–PCR of zebrafish samples
RNA isolation from zebrafish larvae [∼34 h post-fertilization (hpf)] was performed following the procedure described in (35). RNA was extracted using the RNeasy kit (Qiagen) and concentrations were determined by the RiboGreen method (Life Technologies). A 600 ng aliquot of RNA was reverse transcribed to cDNA with the Quantitect Reverse Transcription Kit (Qiagen). The reverse transcription–PCR (RT–PCR) experiments were performed as previously described in (36). Multiplex analyses were optimized at 30 cycles to amplify both spliced and unspliced tgs1 transcripts. In addition to the multiplex analyses, single analyses were performed in the linear phase (22–26 cycles) to ensure reliable quantitative measurements and normalization against endogenous control (ef1a).
We observed a clear dose-dependent increase of unspliced transcripts with increasing morpholino concentration. Primers were designed using Primer-BLAST and purchased from Integrated DNA technologies (IDT). Primers sequences (Drer-tgs1 and Drer ef1α) are reported in Supplementary Table S11. Amplification products from semi-quantitative RT–PCR were gel-purified using the QIAquick gel extraction kit (Qiagen) and Sanger sequenced. Densitometric analyses were performed with ImageLab 5.2.1 (BioRAD).
Immunostaining of zebrafish caudal primary motor neurons
Zebrafish larvae immunostaining was performed as previously described in (33,34). Briefly, ∼34 hpf zebrafish were manually dechorionated, fixed in 4% paraformaldehyde in phosphate-buffered saline (PFA-PBS) and permeabilized by proteinase K digestion of whole larvae. To visualize CaP-MNs, fish larvae were blocked in blocking solution [PBS, 0.1% Tween (PBST) + 5% fetal calf serum (FCS), 2% bovine serum albumin (BSA) and 1% dimethylsulfoxide (DMSO)] and incubated overnight at 4°C with mouse α-Znp1 (Synaptotagmin) antibody diluted 1:150 in blocking solution. After five washes of 1 h with washing solution (PBST + 1% FCS + 1% BSA), the secondary mouse α-AlexaFluor488 antibody was diluted 1:250 in blocking solution and incubated at 4°C overnight. Following five washes of 20 min each, stained fish were stored in 80% glycerol in PBS at 4°C.
For imaging of CaP-MN, fish were laterally embedded in 2% low melting agarose (LMA) microslides under a binocular microscope. Slides were analyzed with the fluorescence microscope AxioImagerM2 (Zeiss). The first 10 motor axons posterior to the yolk sac were considered for quantification. Based on overall appearance, CaP-MN axons were classified as: normal, truncation (shortened axonal projection) or atrophy (absent axonal projection). Based on terminal branching, axons were classified as normal, mild (branching ventral from the midline), medium (2–3 or more branches at ventral or midline) or severe (>3 branches ventral or dorsal from the midline). Brigthfield images of zebrafish larvae were acquired with a Leica S8AP0 binocular attached to an AxioCam ERc5s camera (Zeiss).
Drosophila strains and transgenic constructs
The UAS-Smn RNAi construct [P{TRiP.HMC03832}attP40 (UAS-SmnRNAi)] and the nsyb-GAL4 driver were obtained from the Bloomington Stock Center. The deficiency that removes the Smn gene (the SmnX7 allele) is a gift from Dr Artavanis-Tsakonas (37). The UAS-GFP-dTgs1 strain carries the pPGW-Tgs1 construct, generated by cloning the dTgs1 CDS into the pPGW destination vector (stock number 1077, Drosophila Genomics Resource Center, supported by NIH grant 2P40OD010949), using Gateway technology (Thermo Fisher Scientific); the UAS-mst construct (UAS-CTRL) encoding the Misato protein is ppGW-GFP-Mst (38). The UAS-hTGS1 construct carrying human TGS1 carries the full-length human TGS1 gene cloned into the pUAST-attB vector (39). Transgenic flies were obtained by injecting the constructs into either the w1118 or the y1 v1; P{CaryP}attP40 (2L, 25C6) strains. All embryo injections were carried out by BestGene (Chino Hills, CA, USA). The dTgs1R1 mutant alleles were generated by CRISPR/Cas9 [clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated peptide 4] genome editing as described in (40). The dTgs1CB0 mutant allele is the P{PTT-GB}moiCB02140 insertion (41,42). The Oregon-R strain was used as a wild-type control. All flies were reared according to standard procedures at 25°C. Lethal mutations were balanced over either TM6B, Hu, Tb or CyO-TbA, Cy, Tb (43). All genetic markers and special chromosomes are described in detail in FlyBase (http://www.flybase.org).
Cell culture, transfections, generation of TGS1- and SMN-CRISPR HeLa cell lines and transductions
HeLa S3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin at 37°C, in 5% CO2. Lipofectamine 2000 (Life Technologies) was used for all cDNA transfection experiments. TGS1 or SMN CRISPR cells were generated by transfection of HeLa cells with pSpCas9-2A-GFP (PX458) plasmid (44) encoding 3× FLAG Cas9-T2A–GFP and guide RNAs to the TGS1 locus (TGS1-1, AGAGAAACATTTCCGCCACG; TGS1-2, TGTCAGAGCGTATCTTCAGC) (45) or the SMN locus (SMN-1, CACCCCACTTACTATCATGC). GFP-positive cells were single-cell sorted into 96-well plates by fluorescence-activated cell sorting (FACS), and clones carrying mutations affecting TGS1 expression were screened by immunoblotting using anti-TGS1 or anti-SMN antibody, respectively.
To generate TGS1 mutant cells stably expressing FLAG-TGS1 (TGS1 rescued cells), we transfected 293T cells with the pCDH-CMV-PURO-3×FLAG-TGS1 construct (FLAG-TGS1) and packaging constructs; 48 h later, viral supernatant was collected and concentrated using Retro-X (Clontech). HeLa TGS1 mutant clones were transduced in the presence of 5 μg/ml polybrene and selected in 2 μg/ml puromycin.
For TGS1 knockdown, UMUC 3 cells (46) were cultured in Eagle’s minimum essential medium (EMEM)–Eagle’s balanced salt solution (EBSS) supplemented with 2 mM glutamine, 0.1 mM non-essential amino acids, 10% FBS, 1.5 g/l sodium bicarbonate, 1 mM sodium pyruvate, and treated for up to 10 days (every 72 h) with 50 nM SMARTpool: siGENOME TGS1 siRNA or ON-TARGET plus Non-targeting siRNA using Dharmafect I (Dharmacon). The PAPD5 KO and TCAB1 K1 cell lines are described in (47) and (48). Cumulative population doubling values were determined over a period of 30 days. A total of 1.5 × 104 cells/well were seeded in 6-well plates in duplicates; cells were counted after 72 h.
Western blotting
For western blotting, 10–50 μg of protein extracts were separated by sodium dodecylsulfate—polyacrylamide gel electrophoresis (SDS–PAGE), transferred onto a polyvinylidene fluoride (PVDF) membrane (GE Healthcare, RPN303F) and blotted according to standard procedures. Milk (5%) in PBST was used for all blocking and antibody incubation steps; PBST was used for all washes. Detection was performed with the SuperSignal™ West Pico Chemiluminescent Substrate (Thermo); images were acquired with Chemidoc (Biorad) and analyzed using the QuantityOne image analysis software (Biorad), or with the Odyssey infrared imaging system (LiCoR). We used the following primary antibodies: FLAG mouse monoclonal (clone M2), Sigma, F1804, 1:1000; PIMT/TGS1, Bethyl A300-814A (lot 1), 1:1500; β-tubulin, mouse monoclonal (clone D-10) Santa Cruz, SC-5274; SMN, mouse monoclonal antibody (clone 2B1), 05-1532 Sigma-Aldrich, 1:1000; Coilin, mouse monoclonal [IH10], abcam, (ab87913); CBP20, rabbit NCBP2, Bethyl laboratory, A302-553A; and GAPDH, rabbit, CST, D16H11. Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit 1:5000 (GE Health Care).
Immunofluorescence staining
Immunofluorescence experiments were carried out on cells grown on coverslips. Cells were fixed with 4% PFA and permeabilized with 0.5% Triton X-100 in PBS. Coverslips were incubated with primary antibodies in 3% BSA for 1 h at room temperature: monoclonal anti-SMN (05-1532 Sigma-Aldrich clone 2B1, 1:1000); rabbit anti-Coilin (H300 Santa Cruz SC-32860, 200 μg/ml); rabbit anti-TCAB1 ((49), 25 ng/ml). Coverslips were washed three times with PBS and incubated with secondary Alexa Fluor-conjugated antibodies (Jackson Immunoresearch). Coverslips were washed three times in PBS, counterstained in a 300 nM 4′,6-diamidino-2-phenylindole (DAPI) solution and mounted in ProLong Gold Anti-fade Mountant. Images were captured on a Leica wide-field fluorescence microscope and processed using Leica LAS AF and Photoshop.
RNA immunoprecipitation (RIP) of TMG-capped RNA
TRIzol-purified RNA was treated with DNase and subjected to immunoprecipitation with the R1131 anti-TMG cap-specific antibody (50). A 50 μl aliquot of protein G beads was washed with PBS and blocked with 20 μg of tRNA and 20 μg of BSA, then washed with NT2 buffer [50 mM Tris–HCl pH 7.5; 150 mM NaCl; 1 mM MgCl2; 0.05% Nonidet P40 (NP40); 1 mM dithiothreitol (DTT); 100 U/ml RNasin; 400 μM vanadyl ribonucleoside complexes (VRCs)] and coupled overnight to either 20 μl of anti-TMG cap antibody or 1 μg of IgG in 250 μl of NT2 buffer. Beads were then briefly washed with NT2 buffer and incubated with TRIzol-purified RNA in 250 μl of NT2 buffer for 2 h at 4°C, while rotating. Beads were then washed six times for 2 min while rotating with NT2 buffer, and precipitated RNA was TRIzol extracted. Both TMG-immunopurified and input RNAs were then subjected to reverse transcription and real-time quantitative PCR (RT-qPCR; see below).
Purification of yeast U1 and U2 snRNAs as spike-in controls for TMG-IPs
To extract total yeast RNA, 1 ml of suspension culture of Saccharomyces cerevisiae (YPD base + 1% glucose) was pelleted and resuspended in 500 μl of TRIzol, together with 100 μl of TRIzol-pre-washed glass beads (425–600 μm, Sigma G-8772). The mixture was shaken vigorously for at least 15 min on a vortexer to ensure efficient lysis and thereby optimal yield. RNA was extracted with Direct-Zol (Zymo R2050) including the DNase step, eluted in ddH2O, snap-frozen and stored at –80°C. Direct-Zol was performed according to the instructions except that an extra 2 min top speed centrifugation was added to get rid of residuals from the column before the elution step.
To prepare RNA molecular weight marker for yeast U1 (SNR19, 568 nt) or yeast U2 (LSR1, 1175 nt), 20 μl in vitro transcription (IVT) reactions were carried out (HiScribe T7 kit, NEB E2040). IVT DNA templates were generated by PCRs, in which two randomly selected amplicons of 568 or 1175 nt were amplified from the vector px458 (Addgene #48138) using forward primers that begin with the T7 promoter sequence 5′-AATACGACTCACTATAG-3′. The IVT-RNA markers were purified by Direct-Zol.
For gel purification of yeast U1 and U2, 10 μg of total yeast RNA was resolved in a 3.5% urea–PAGE gel (Hoefer Vertical Unit) and stained with SYBR Green II dye (Sigma, S9305, used at 1:10 000). RNA species co-migrating with IVT RNA markers (568 nt for yeast U1 and 1175 nt for yeast U2) were excised and eluted from the gel according to (51) in Gel Elution buffer (20 mM Tris–HCl pH 7.5, 250 mM sodium acetate, 1 mM EDTA, 0.25% SDS) with end-to-end mixing overnight. Gel eluates were finally purified by Direct-Zol. The presence and enrichment for yeast U1 or yeast U2 from the corresponding gel slices were confirmed by RT-qPCR.
TMG immunoprecipitationn (TMG-IP) from whole-cell extracts
HeLa cells cultured in a 15 cm dish were trypsinized, washed with PBS and resuspended in RBS-250 buffer [250 mM NaCl, 10 mM Tris–HCl pH 7.4, 2.5 mM MgCl2, 0.1% NP40, with freshly added protease inhibitors (Sigma P8340) and RNase inhibitor (Lucigen 302812) as 1000× stock]. Cells were lysed with a water bath sonicator (Q-Sonica Q800) with 70% energy output three times for 30 s each. Cell lysates were clarified by centrifugation at 13 000 × g at 4°C for 20 min. The supernatant was quantitated by Bradford protein assay (BioRad 5000006) before snap-freezing, and stored at –80°C. Yeast purified U1/U2 snRNAs were spiked into 200 μg of whole-cell RSB extracts, with 10% of such input set aside and stored at 4°C. Protein A–Sepharose beads (Invitrogen 101041) were pre-washed by PBS and blocked in PBS/1% BSA (w/v) solution (Jackson ImmunoResearch 001-000-162). For each immunoprecipitation, a 10 μl bed volume of beads or 20 μl of slurry were coupled with 4 μg of anti-TMG antibodies (EMD Millipore, MABE302, clone K121) or control IgGs for 2 h and then combined with the input extract for 2 h rotation at 4°C. The immunoprecipitated complex was then washed and eluted with TRIzol, in parallel with the ‘Input’, followed by the Direct-Zol procedure including the DNase I step and subjected to reverse transcription and RT-qPCR (see below).
RT-qPCR
Total RNA was extracted with TRIzol reagent (Ambion), treated with Ambion™ DNase I (RNase-free) and extracted with phenol/chloroform. The integrity of RNA samples was evaluated by gel electrophoresis. A 1 μg aliquot of intact RNA (with a 28S:18S rRNA ratio = 2:1) was reverse transcribed with the RevertAid H Minus Reverse Transcriptase kit (Thermo Scientific, EP0451). Real-time PCRs were performed with the Brilliant II SYBR® Green QPCR Master Mix (Agilent, 600828). The relative quantification in gene expression was carried out using the 2−ΔΔCt method (52). Using this method, we obtained the fold changes in gene expression normalized to the 5.8S gene (the amplification efficiencies were not significantly different for the target and reference among all samples). A total of three experiments were performed for three biological replicates and the significance was assessed by one- or two-way ANOVA.
For RT-qPCR of RNA immunoprecipitated with the R1131 antibody, samples were processed as above; fold change was calculated by normalizing each RIP sample to the relative input.
In RIP-qPCR analysis, the values reported in the histogram bars represent the fold enrichment of RNA in each immunoprecipitation relative to TMG-IP from control samples (expressed as a percentage of input) and were calculated as follows:
fold enrichment of RNA in the immunoprecipitate, relative to control immunoprecipitate = 2(–ΔΔCt [RIPtest sample/ RIPCTR]), where
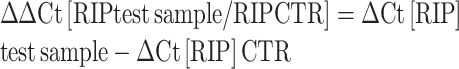 |
and
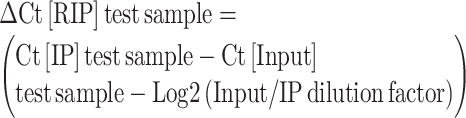 |
For RT-qPCR of RNA immunoprecipitated with the K121 antibody, the RNA eluates were quantitated by one-step RNA-to-Ct qRT-PCR (Brilliant III SYBR green mix, Agilent 600886) containing MMLV reverse transcriptase and ROX reference dye. Reactions (5 μl) were performed in duplicate in a 384-well plate, using the QuantStudio 6 Pro qPCR system (Applied Biosystems).
The Ct or Cq value for each RNA target in TMG-IPs was normalized to the corresponding Inputs using the 2–ΔCt method. These ‘% of input’ values of human RNA targets were then normalized with those of yeast U1 or yeast U2 (the spike-in controls), enabling direct comparison across TMG-IPs from different extracts. Three independent experiments were performed, and the resulting data were analyzed and graphed with Prism software. For C. elegans experiments, worm pellets were flash-frozen three times before RNA extraction in TRI Reagent® (Merck, Darmstadt, Germany), according to the manufacturer's protocol. RNA was treated with DNA-free™ DNA Removal (Invitrogen™). Reverse transcription was performed with SuperScript Reverse Transcriptase (Invitrogen™) and real-time PCRs were performed using the Power SYBR Green PCR Master Mix (Applied Biosystems) on the 7900HT Fast Real Time PCR System (Applied Biosystems), following standard procedures. cetgs-1 expression levels were normalized to the average level of the housekeeping gene ceact-1. Each experimental assay was performed in triplicate in three independent experiments Primers are listed in Supplementary Table S11.
Cell fractionation and northern blotting
Nuclear extracts (NE) and cytosolic extracts (S100) were obtained using the Dignam protocol as shown in (53). For northern blotting, total RNA is extracted by resuspending pelleted cells with TRIzol reagent. Nuclear and cytoplasmic RNAs were extracted from 4 μg of NE and 12 μg of S100, respectively with a 5× volume of TRIzol. Precipitated RNAs were dried and resuspended sequentially in 5 μl of ddH2O, and then with 5 μl of RNA loading dye, containing 0.1× TBE, 50 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol, 93% formamide. Boiled and rapidly chilled RNAs were fractionated in a 18 × 24 cm 6% urea–PAGE (19:1) gel, running at 45 V for > 20 h until the bromophenol blue run-off. The gel was then transferred to a Hybond-N nitrocellulose membrane under 400 mA for 2 h in the cold room. The post-transfer gel was stained by ethidium bromide (EtBr) for rRNAs as loading control. UV-cross-linked membrane was blocked and hybridized in ULTRAhyb, in a 42°C oven overnight, with 32P-end-labeled oligonucleotide probes (sequence listed in Supplementary Table S11). The washed membrane was exposed to a phosphor-imager screen, scanned by a Typhoon scanner and quantitated with the TotalLab software.
Single molecule FISH (Yn situ)
The single molecule fluorescence in situ hybridization (FISH) protocol was carried out according to the protocol described in (54). Oligonucleotides used as Yn situ probes for U1 and U2 snRNAs are listed in Supplementary Table S11.
Nascent RNAend-Seq and steady-state 3′ RACE-Seq
For the isolation of nascent RNA from HeLa cells, cells were pulsed for 1 h with 250 μM 4-thiouridine (4sU) or for 4 h with 50 μM 4sU. For chase experiments, 4sU was removed, the cells were washed in PBS and 2.5 mM uridine-containing medium was added. Cells were harvested and cell pellets resuspended in TRIzol. RNA was extracted using the standard TRIzol protocol. A 100 μg aliquot of RNA was biotinylated with Biotin-HPDP (Pierce 21341) at 0.5 mg/ml in 40% demethylformamide (DMF) and 10 mM Tris pH 7.4, 1 mM EDTA, for 1.5 h at room temperature. In vitro transcribed luciferase RNA transcribed in the presence of 4sU was spiked into the biotinylation mixture for a final concentration of 0.01 ng/μl. RNA was extracted using isopropanol/ethanol precipitation. Biotinylated RNA was immunoprecipitated from total RNA using the mMacs Streptavidin Kit (Miltenyi 130-074-101). The depleted fraction was recovered using isopropanol/ethanol precipitation. Biotinylated RNA was released from beads using 100 mM DTT and cleaned using an RNeasy MinElute Kit (QIAGEN 74204). Half of the eluted RNA or 600 ng of depleted RNA was ligated to 5 μM of 5′ adenylated, 3′ blocked adaptor (Universal miRNA cloning linker, NEB S1315S) with 250 U of T4 RNA ligase-truncated KQ (NEB M0373S), 25% polyethylene glycol (PEG) 8000, and 1 μl of RnaseOUT (ThermoFisher 10777019) in a 20 μl reaction at 25°C for 16 h. Ligated RNA was cleaned up with RNA clean and concentrator columns (Clontech 740955.50) and DNase treatment. cDNA was synthesized with universal primer and SuperScript III (ThermoFisher 18080093). Amplification was carried out with Phusion (New England Biosystems M0530) and primer sets universal/specific for the RNA of interest. PCR products were directly run on an 8% PAGE gel and visualized with SYBR Gold (ThermoFisher S-11494), or subject to AMPure XP beads (Beckman Coulter A63881) for PCR clean-up and library preparation. Libraries were prepped using a Kapa Hyperprep Kit (Kapa KK8504), quantified with Qubit and Bioanalyzer, and run on Illumina miSeq at the Stanford Functional Genomics Facility. Reads were paired using the fastq-join tool at Galaxy (http://usegalaxy.org). Reads were binned into the various forms of each specific snRNA using custom Python scripts (https://github.com/cmroake/NAR_TGS1), and the number of reads in each bin was normalized to total snRNA reads. Primer sequences can be found in Supplementary Table S11. Data have been deposited at SRA with BioProject ID PRJNA628085.
Transcriptome library preparation and RNA-sequencing
Total RNA was extracted with TRIzol reagent (Ambion), treated with Ambion™ DNase I (RNase-free) and extracted with phenol/chloroform. RNA samples were quantified and quality-tested by Agilent 2100 Bioanalyzer RNA assay (Agilent Technologies, Santa Clara, CA, USA). The TruSeq Stranded mRNA kit (Illumina, San Diego, CA, USA) was used for library preparation following the manufacturer's instructions (library type: fr-firstrand). Final libraries were checked with both Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and Agilent Bioanalyzer DNA assay. Libraries were then prepared for sequencing and sequenced on paired-end 150 bp mode on NovaSeq 6000 (Illumina, San Diego, CA, USA).
The number of reads (in millions) produced for each sample is listed in Table 2.
Table 2.
Number of Illumina sequencing reads
| Sample name | Reads (M) |
|---|---|
| ID1232_1–1A-CTR1 | 311.29 |
| ID1232_2–1B-CTR2 | 310.44 |
| ID1232_3–2A-TGS1 M1 | 299.96 |
| ID1232_4–3A-TGS1 M2 | 314.74 |
| ID1232_5–4A-SMN C1 | 358.39 |
| ID1232_6–5A-SMN C2 | 272.79 |
Primary bioinformatic analysis includes the following steps.
Basecalling and demultiplexing. Processing raw data for both format conversion and demultiplexing by Bcl2Fastq 2.20 version of the Illumina pipeline (https://support.illumina.com/content/dam/illuminasupport/documents/documentation/software_documentation/bcl2fastq/bcl2fastq2-v2-20-software-guide15051736-03.pdf).
Adapter masking. Adapter sequences are masked with Cutadapt v1.11 from raw fastq data using the following parameters: –anywhere (on both adapter sequences) –overlap 5 –times 2 –minimum-length 35 –mask-adapter (http://dx.doi.org/10.14806/ej.17.1.200).
Folder ‘raw_reads’ contains files with raw reads (R1, first read sequence; R2, second read sequence) and a multiqc_report.html file, which aggregates results from primary bioinformatic analysis into a single report file with parameters that give insight into overall processing and sequencing quality.
Transcript reconstruction and analysis
Paired-end Illumina sequencing was performed on RNA isolated from TGS1 mutant clones (M1 and M2), SMN mutant clones (C1 and C2) and parental HeLa cells (CTRL, two replicates) at a depth of 160 million reads/sample. Mutant samples were clustered based on both transcript and gene expression profiles (Supplementary Figure S7). We mapped reads to the human reference genome (Gencode annotation v33) using STAR v2.7.1a (55) and ran Scallop (56) on the resulting BAM files to find all novel transcripts in the data. We combined the Scallop-detected novel transcripts with the reference transcripts and used Kallisto (57) to quantify transcript expression levels. Differentially expressed novel and annotated transcripts were found using Sleuth v0.30.0 (57) (Wald test, false discovery rate-adjusted P-value < 0.05, beta value > 2). We compared the 3′ side of each novel transcript with its closest annotated transcript to determine whether the novel transcript has a shorter, extended or the same 3′ side as compared with the annotated transcript. To investigate the extent of global splicing changes induced by TGS1 and SMN knockouts, we performed differential splicing analysis. We extracted alternative splicing events from the GTF file containing all novel and annotated transcripts and then used SUPPA2 (58) to find significantly changed alternative splicing events. We considered all seven major categories of alternative splicing events and found the percentage of splicing inclusion (PSI) for each extracted splicing event. A differentially spliced event between mutant and control samples should have a difference in PSI of at least 0.1 and an adjusted P-value < 0.05.
Methodological approach used for quantification of intron retention (IR) levels (Figure 7D)
Figure 7.

Mutations in TGS1 and SMN cause global changes in RNA expression and splicing. (A, B) Heatmaps depicting the expression levels (TPM values estimated by Kallisto) for differentially expressed (DE) transcripts in TGS1 M1 and TGS1 M2 (A), or SMN C1 and SMN C2 (B) mutant cells, compared with parental HeLa cells (CTR). Transcripts are classified into five types: annotated transcripts per GENCODE (orange); novel transcripts with extended 3′ (magenta) or shortened 3′ (blue); novel transcripts with annotated identical 3′ (pink); and other (intergenic and opposite-strand) novel transcripts (purple). Transcripts within each group are ranked by unsupervised clustering. Total transcript number for each group and the DE status for each transcript (either up- or down-regulated in mutant cells) are annotated by the green/red sidebar to the right (see also Supplementary Table S1). (C) Venn diagram from data in (A) and (B), showing the significant number of shared DE transcript isoforms (both annotated and unannotated) between TGS1 and SMN mutant cells. See also Supplementary Table S1. (D) Methodological approach used for quantification of intron retention (IR) levels (see also the Materials and Methods). (E, F) Scatter plots showing for each IR event between mutant (TGS1 M1 and M2 or SMN C1 and C2) and CTR cells the differential PSI (dPSI) against the differential expression level (dTPM) of the major transcript (intron exclusion). The significant IR events are color-coded in the plots, with significantly up-regulated events colored in orange and significantly down-regulated in teal.
We performed a transcriptome-wide analysis of the expression levels for the major transcript isoforms (i.e. the transcript without the intron) in the IR events. IR is quantified by the PSI, which is computed as the percentage of reads mapping to the intron inclusion isoform, relative to total reads mapping to both intron inclusion and intron exclusion isoforms. Differential expression of the spliced isoforms (intron exclusion) was computed as the difference in TPM (or transcript per million) values (dTPM) between mutant and control cells (plotted on the x-axis in Figure 7E, F). The expression of the major transcripts in most of the significant IR events does not change considerably (small difference in TPM) between control and mutant samples (Figure 7E, F). As an independent pipeline for detecting differentially expressed transcripts, we also used a combination of Salmon (59) for transcript quantification and DESeq2 for differential analysis. Statistical analyses on individual transcripts were made with the test implemented in the software.
Nanopore sequencing and analyses
Data generation
Total RNA was extracted with TRIzol reagent (Ambion), treated with Ambion™ DNase I (RNase-free) and extracted with phenol/chloroform. All samples have been checked for RIN score > 8 by Agilent 2100 Bioanalyzer RNA assay (Agilent Technologies). The same extracted RNA was used both for Illumina and Nanopore library preparation. Nanopore libraries were generated according to the PCR-cDNA barcoding (SQK-PCB109) protocol and sequenced on FLO-MIN106 flow cells for 48–72 h with –180 mV starting voltage (see Supplementary Figure S9).
Basecalling and mapping
Samples were sequenced using Oxford Nanopore MinION on FLO-MIN106 flow cells. Primary data acquisition was performed by MinKNOW. Supplementary Figure S9 reports information about each run. Basecalling and demultiplexing of Fast5 files obtained by sequencing were performed using Guppy v4.2.2. Reads with Qscore > 7 were mapped to the hg38 genome using Minimap2 v2.17 (60) with parameters -ax splice.
Differential splicing analysis
To assemble and quantify transcripts from Nanopore reads, we used FLAIR v1.4 (61). BAM files obtained with Minimap2 were converted into BED12 files using the bam2Bed12.py utility included in FLAIR. After this, splice junctions of reads were corrected using the FLAIR correct module employing both the GENCODE v33 GTF file (gencode.v33.chr_patch_hapl_scaff.annotation.gtf) (62) and short Illumina reads of the corresponding samples. BED12 files were then filtered to maintain only reads with at least one splice junction. Filtered BED12 files from all samples were used to assemble the transcriptome using the FLAIR collapse module, providing the GENCODE GTF file to annotate the results and requiring at least 10 supporting reads to identify novel transcripts. Transcripts from both the GENCODE and FLAIR GTF files were quantified using the FLAIR quantify module. The TPM values were thus calculated and provided to SUPPA2 to identify significantly changed splicing events, following the same procedure used for Illumina short reads.
Analysis of transcript readthrough
To study readthrough transcription in wild-type and mutant samples, we first assigned multi-exonic corrected reads to their corresponding genes starting from the GTF of assembled transcripts obtained by running another run of FLAIR collapse, with the minimum number of supporting reads set to 3 to identify all the possible splicing junctions for each gene. Each read was assigned to the gene with which it shared the highest number of splicing junctions. For the identification of the last exon of each gene, we first retained only those transcripts from the reference GTF that have an average TPM of > 1 in at least one condition (wild type and TGS1 mutant in wild type versus TGS1 comparison, wild type and SMN mutant in wild type versus SMN comparison); then, for each gene, we selected the last exon of the transcript with the most distal termination site. BEDTools v2.29.1 intersect module (63) was employed to select, among the reads assigned to each gene, those mapping to the last exon. Such reads, each one representing a different transcript, were classified as readthrough if their last mapping position was > 10 nt downstream of the termination of the last exon. Differential readthrough analysis was performed by comparing each mutant sample against the pooled WT sample, obtained by combining the reads of both WT samples. For each last exon having at least 10 reads in both WT and mutant samples under analysis, the proportion of readthrough reads in both samples was compared by performing a Fisher's exact test. In this way, we identified differential readthrough events (DREs) as those having a P-value < 0.05. DREs in which the proportion of readthrough reads was higher in the mutant sample and those in which such a proportion was higher in the control samples were defined as ‘Preferential Readthrough in Mutant’ and ‘Preferential Readthrough in CTR’ events, respectively. The reads mapping to exons undergoing ‘Preferential Readthrough in Mutant’ events were used to determine the association between readthrough and differential splicing. Readthrough reads from mutant samples were classified as aberrant if their splice junction chains were not found among the non-readthrough reads from the pooled wild-type sample. The same was done for the non-readthrough reads from mutant samples. The proportions of aberrant reads among readthrough and non-readthrough reads were compared using the χ2 test. To identify the alternative splicing events carried by aberrant reads, we generated several GTF files, each one containing an aberrant read and the non-readthrough reads from the pooled wild-type sample, and provided such files to the SUPPA2 generateEvents module.
To identify fusion transcripts of adjacent genes, we focused on readthrough reads with one or more exons after the termination site and searched for overlaps with downstream genes, defined as those genes for which the first splicing junction is located after the last splicing junction of the gene to which the readthrough reads were assigned. We also evaluated whether at least one splice junction of the overlapping readthrough reads coincided with a splice junction of the downstream gene.
Phylogenetic alignment
TGS1 homologs were identified by searching in the UniProt knowledgebase (https://www.uniprot.org/). The amino acid alignment was built using Geneious prime employing Clustal Omega. To build the phylogenetic tree, pairwise distances were calculated based on the multiple sequence alignment using the Jukes–Cantor distance model. The tree was built using the UPGMA method.
Statistics
Statistical analyses were performed in GraphPad Prism 6. Type of test, sample size, data representation and P-values are indicated in the figure legends. P ≤ 0.0001 is indicated as ****; P > 0.05 was considered not significant.
RESULTS
The C. elegans TGS1 ortholog is expressed in motor neurons and required for neuron survival and worm locomotion
To investigate the role of TGS1 in the nervous system and its relationships with SMN biology, we exploited the C. elegans model (23). We first analyzed the expression of the C. elegans ortholog of TGS1, T08G11.4 (Supplementary Figure S1) (64), hereafter referred to as cetgs-1, using a GFP reporter approach (24) in which the promoter regions of the two annotated cetgs-1 isoforms were fused to GFP (pcetgs-1::GFP a and b). Both isoforms are expressed in ventral cord MNs, 19 of which are D-type γ-aminobutyric acid (GABA) motor neurons (MNs), which are specifically marked by the expression of punc-47::RFP (Figure 1A) (65,66).
Figure 1.

Knockdown of cetgs-1 or cesmn-1 in D-type MNs results in similar phenotypes that are rescued by cetgs-1 and hTGS1 overexpression. (A) The promoters of the pcetgs-1a and pcetgs-1b isoforms drive the expression of GFP in MNs of the ventral cord. D-type (GABA) MNs express RFP under the control of the MN-specific promoter (punc-47). In merged images, the D-type MNs expressing both cetgs-1 (GFP)and punc-47(RFP) are marked by asterisks. The cells expressing only cetgs-1(GFP) are MNs other than D-type (arrowheads). Anterior is left and ventral is down in all images. Scale bar, 50 μm. (B) Knockdown of cetgs-1 or cesmn-1 in D-type MNs leads to similar locomotion defects. Wild-type (Ctr) and cekal-1-silenced animals are controls. The locomotion defect elicited by cesmn-1 silencing is rescued by cetgs-1 (driven by the pan-neuronal punc-119 promoter) but not by dsRED expression. Bars represent the percentage of animals with normal backward locomotion ± SEM, from at least two independent lines/clones. Numbers within bars are the animals tested. ****P < 0.0001 (one-way ANOVA). No statistically significant differences were observed between cesmn-1(RNAi) and cetgs-1(RNAi) (P = 0.41) or cesmn-1(RNAi) and cesmn-1(RNAi Is) (P = 0.50). (C) Transgenic worms expressing GFP [oxIs12 (punc-47::GFP)] show 19 D-type MNs of the ventral cord (Ctr). cetgs-1(RNAi) worms display fewer GFP-expressing MNs in the ventral cord compared with controls. This phenotype is partially rescued by hTGS1 expression. MNs are indicated by arrowheads; the RNAi construct is not expressed in the tail, where one GFP-positive cell is always detectable (asterisk); the heads (underlined) express the pchs-2::GFP injection marker in both cetgs-1(RNAi) and cetgs-1(RNAi); hTGS1 worms; the latter were also injected with the podr-1::RFP marker (arrow). Scale bar, 75 μm. (D) Quantification of ventral cord D-type MNs. Neuron loss caused by cetgs-1 knockdown is partially rescued by pan-neuronal expression of human TGS1 (hTGS1) but not of dsRED. Each bar represents the mean number of visible MNs from at least two independent lines/clones ± SEM. Numbers within the bars are the animals tested. ****P < 0.0001 (one-way ANOVA). (E) In oxIs12 [punc-47::GFP] transgenic animals, both MN cell bodies and axons are visible. In control (Ctr) and cekal-1(RNAi) animals, commissures appear as single, straight axons directed to the upper side. cetgs-1 knockdown worms exhibit commissures with extra branching and guidance defects (arrows). (F) Apoptotic autofluorescence signals (arrowheads) in dying MNs of cetgs-1(RNAi) and cesmn-1(RNAi Is) animals. (G) Quantification of dying MNs. Bars represent the average number of dying MNs in transgenic animals from at least two independent lines/clones ± SEM. Numbers within and next to bars are the animals tested. ****P < 0.0001 (one-way ANOVA). No statistically significant differences were observed by comparing: cesmn-1 (RNAi) and cetgs-1(RNAi) (P = 0.27) or cesmn-1(RNAi) and cesmn-1(RNAi Is) (P = 0.08). (H) RT-qPCR showing the overexpression of cetgs-1 in cesmn-1(RNA Is); cetgs-1 animals, driven by the pan-neuronal punc-119 promoter (>10-fold higher than in the wild type). The reduction in cetgs-1 expression could not be detected in cetgs-1(RNAi) animals, as silencing occurs in 19 neurons that represent just 0.6% of total cells. Data are from three biological replicates, are normalized to ceact-1 and are relative to wild-type animals (Ctr). ****P < 0.0001 (one-way ANOVA).
To selectively silence cetgs-1 in the D-type MNs, we generated a strain carrying the punc-25::cetgs-1(RNAi) transgene. This strain is viable, fertile and has a normal development. As a control, we used a transgenic RNAi line that targets cekal-1, a neurodevelopmental gene not expressed in the D-type MNs (67). We also exploited a cesmn-1(RNAi) line expressing an extrachromosomal RNAi construct (23). D-type MNs control C. elegans backward movement (68). A total of 86% of wild-type controls and 85% of cekal-1-silenced animals showed normal backward locomotion; this proportion was reduced to 40% and 29% in cetgs-1(RNAi) and cesmn-1(RNAi) worms, respectively (Figure 1B), indicating that both cetgs-1 and cesmn-1 are required for proper MN function and locomotor behavior.
We next asked whether the cetgs-1-dependent locomotion defect is due to an alteration in MN morphology and survival. We introduced in cetgs-1(RNAi) animals the oxIs12[punc-47::GFP] transgene that expresses GFP in D-type MNs, allowing their visualization in living animals. Wild-type or cekal-1-silenced worms invariably showed 19 D-type MNs, each extending a circumferential commissure to the dorsal side (Figure 1C–E) (68). In cetgs-1(RNAi) animals, the number of visible D-type MNs was on average seven per worm, while cesmn-1(RNAi) animals displayed only four neurons/worm, as previously reported (23) (Figure 1C, D). The D-type MNs of cetgs-1(RNAi) animals (n = 606 commissures) showed a defect in axonal morphology, consisting of extra branching and guidance defects (Figure 1E). This defect was present in 14% of the commissures of cetgs-1(RNAi) worms but was never observed in control (n = 586) or cekal-1(RNAi) (n = 410) animals (P < 0.0001; non-parametric z-test.). Remarkably, a similar defect was found in cesmn-1 knockdown worms (23).
Next, we expressed in D-type MNs either human TGS1 (punc-119::hTGS1, referred to as hTGS1) or the RFP dsRED that served as a control (punc-119::dsRED). hTGS1, but not dsRED, partially rescued the MN loss caused by cetgs-1(RNAi) (from seven to nine neurons/animal; Figure 1C, D), indicating that this phenotype is specifically caused by cetgs-1 silencing and suggesting that the TGS1 requirement for neuron survival is conserved from worms to humans.
Finally, we asked whether the loss of D-type MNs observed in cetgs-1(RNAi) worms is caused by cell death. Apoptotic MN death in cesmn-1 knocked down animals is revealed by the accumulation of an endogenous autofluorescent marker (23). Wild-type worms showed no dying MNs, while cekal-1(RNAi) worms showed an average of one dying MN per animal. In contrast, cetgs-1(RNAi) and cesmn-1(RNAi) worms displayed seven and eight dying MNs per worm, respectively (Figure 1F, G). Thus, loss of either cesmn-1 or cetgs-1 causes D-type MN death.
cetgs-1 overexpression mitigates the defects elicited by cesmn-1 depletion
To determine whether cetgs-1 interacts genetically with cesmn-1, we expressed cetgs-1 under the control of a pan-neuronal promoter (punc-119::cetgs-1, referred to as cetgs-1) in worms that also express an integrated cesmn-1 RNAi construct [cesmn-1(RNAi Is)] (23). The overexpression of cetgs-1 was confirmed by RT-qPCR (Figure 1H). A total of 62% of the cesmn-1(RNAi Is) worms showed abnormal locomotion. Expression of cetgs-1 in these worms lowered the proportion of animals with abnormal locomotion to 28% (Figure 1B), while dsRED expression did not change the frequency of worms with locomotory defects (68%, P = 0.9) (Figure 1B). In addition, while in cesmn-1(RNAi Is) or cesmn-1(RNAi Is); dsRED worms, we observed an average of nine dying MNs/worm, in the presence of the cetgs-1 or hTGS1 transgenes, the average number of dying MNs was slightly but significantly lowered to seven and eight, respectively (Figure 1F, G). Thus, cetgs-1 overexpression partially suppresses locomotion defects and neuronal death caused by cesmn-1 silencing.
Overexpression of dTgs1 ameliorates the Smn loss-of-function phenotype in Drosophila
We next assayed the effects of TGS1 overexpression in a fly SMA model. It has been reported that Drosophila Tgs1 (dTgs1) depletion affects motor behavior (40,69,70). We have previously shown that RNAi against Smn in neurons perturbs the circuits that control post-eclosion events, leading to defective wing expansion (71). A similar phenotype has been observed in hypomorphic dTgs1 mutants (40). We generated flies expressing a UAS-Smn RNAi construct in neurons, using the nsyb-GAL4 driver. A total of 16% of these Smn RNAi flies displayed unexpanded wings (Figure 2A, B), and this percentage was increased to 44% when RNAi was performed in flies carrying only one copy of Smn+ (ΔSmn/+; Figure 2B). We then examined flies co-expressing UAS-Smn-RNAi and UAS-dTgs1, both driven by nsyb-GAL4. As control, we used flies carrying a UAS-CTRL construct encoding the unrelated Mst protein (38). An increase in the dosage of dTgs1, but not of Mst, significantly lowered the frequency of flies with unexpanded wings (from 16% to 9%) (Figure 2B). A human TGS1 transgene (UAS-hTGS1), which fully rescues the lethality associated with null dTgs1 mutations (40), decreased the proportion of flies with unexpanded wings from 16% to 10% in UAS-Smn-RNAi; Smn+/Smn+ flies, and from 44% to 30% in UAS-Smn-RNAi flies bearing a single copy of Smn (Figure 2B). These results indicate that dTgs1 overexpression partially suppresses the defects caused by Smn deficiency in at least a subset of fly neurons, and that human TGS1 can substitute for the neuronal function of its fly ortholog.
Figure 2.

Tgs1 overexpression ameliorates the wing expansion defects caused by neuronal knockdown of Smn in Drosophila. (A) Examples of flies showing wing expansion failure upon nsyb-GAL4-driven (neur>) Smn RNAi. (B) Frequencies of the defective wing expansion phenotype in flies co-expressing a UAS-Smn RNAi (Smn RNAi) construct and the indicated UAS constructs driven by nsyb-GAL4 (neur>). UAS dTgs1 encodes GFP-tagged dTgs1; UAS hTGS1 encodes GFP-tagged human TGS1; UAS CTR is a control construct expressing the unrelated Mst protein. ΔSmn/+ flies are heterozygous for a deficiency of the Smn locus. Error bars: ± SEM. Numbers within and next to bars are the animals tested. P-values: one-way ANOVA with Tukey's multiple comparisons test.
TGS1 down-regulation in zebrafish causes motor axon defects
To determine the consequences of TGS1 loss in a vertebrate model, we exploited zebrafish. To down-regulate tgs1, we injected larvae with two antisense MOs: a translation-blocking MO (tgs1 ATG-MO) and a splicing-blocking MO (tgs1 Sp-MO) (Supplementary Figure S2A, B). Each MO was injected at an optimized dosage and caused little or no effect on the overall larval morphology and development (Supplementary Figure S2C).
Injection of 2 ng of tgs1 ATG-MO resulted in defects in CaP-MN, including axonal truncations and increased terminal branching. A total of 10% of the CaP-MNs exhibited truncated axonal projections and 25% showed increased terminal branching (Figure 3A, B). Negligible CaP-MN defects were observed in uninjected larvae and in larvae injected with a non-targeting MO (Figure 3A, B). A 1.5 ng aliquot of tgs1 Sp-MO injection led to 15% of CaP-MNs with axonal truncations and 35% with increased terminal branching (Figure 3B). Notably, these results recapitulate the MN defects observed in smn morphants (36,72,73).
Figure 3.

Tgs1 down-regulation in zebrafish leads to defects in CaP-MNs. (A) Lateral views of whole-mount embryos immunostained with a synaptotagmin antibody (Znp1) that labels the CaP-MNs. Embryos were untreated or injected with the indicated MOs. Injection of either tgs1 ATG-MO or tgs1 Sp-MO results in truncated or absent motor axons (solid arrowheads) and terminally branched axons (open arrowheads). Scale bar, 25 μm. See also Supplementary Figure S2. (B) Based on overall appearance, CaP-MNs were classified as: normal, short (truncated axonal projection) or absent (axonal atrophy). Based on terminal branching, axons were classified as normal, mild (branching ventral from midline), intermediate (2–3 or more branches at ventral or midline) or severe (>3 branches ventral or dorsal from the midline). Zebrafish larvae injected with 1.5 ng of tgs1 Sp-MO or 2 ng tgs1 ATG-MO displayed CaP-MN defects compared with control (non-targeting) MOs and uninjected fish. Results are percentages from three independent experiments (n = axons analysed; N = animals tested. ****P < 0.0001 χ2 test); n.s. not significant. See also Supplementary Figure S2D.
We also attempted to perform rescue experiments by co-injecting FLAG-tagged human TGS1 mRNA and the tgs1 Sp-MO. Co-injection of tgs1 Sp-MO and 100 pg of FLAG-hTGS1 mRNA resulted in a significant rescue of the neurological phenotype (Supplementary Figure S2D). However, injection in embryos of FLAG-hTGS1 mRNA (100–400 pg) caused developmental defects in larvae (Supplementary Figure S2C), preventing a firm conclusion about the rescue capacity of human TGS1. Collectively, these results show that the TGS1 function is required for correct MN development in zebrafish.
TGS1 deficiency impairs 3′ processing of snRNAs in human cells
Given the similarity between the phenotypes elicited by TGS1 and SMN depletion, we asked whether TGS1 loss affects snRNP biogenesis (74). To address this, we used two HeLa clones carrying CRISPR/Cas9-induced mutations in TGS1 (TGS1 M1 and M2) (45), which decrease TGS1 expression to < 10% of the normal level but do not affect the expression of the SMN protein (Figure 4A; Supplementary Figure S3C, D). We have previously shown that both mutant lines exhibit a rather diffuse distribution of the CB marker Coilin (45) and fail to accumulate SMN in CBs, in agreement with previous studies (75). This defect is rescued by the expression of FLAG-TGS1 (TGS1 M1R cells, Figure 4A; Supplementary Figure S3A, B). To ascertain whether TGS1 deficiency affects snRNA expression, we performed RT-qPCR on total RNA from control cells, TGS1 mutant cells and TGS1 mutant cells bearing a FLAG-TGS1 rescue construct. There were no significant differences in the abundance of different snRNAs, with minor changes probably reflecting clonal variability (Figure 4B).
Figure 4.

TGS1 deficiency affects maturation of U2 snRNAs in human cells. (A) Western blotting (WB) showing TGS1 expression in two independent CRISPR clones of HeLa cells (M1, M2). CTR is the parental cell line; TGS1 M1R and CTR R are a TGS1 M1 and a parental line stably expressing FLAG-TGS1. Tubulin is a loading control. The abundance of the SMN protein is not affected by mutations in TGS1 (see also Supplementary Figures S3C, D and S6B). (B) RT-qPCR showing the abundances of the indicated snRNAs in TGS1 M1, TGS1 M2 (collectively indicated as TGS1 M) and TGS1 M1R cells. Data from three independent experiments are relative to parental HeLa cells (set to 1) and normalized to 5.8S rRNA. The U3 snoRNA (U3) and the uncapped U6 and U6atac snRNAs are controls. No statistically significant differences were observed between TGS1 M and TGS1 M1R cells (error bars: ± SEM; P-values: all > 0.05; two-way ANOVA with Sidak's multiple comparisons test). (C) Total RNA from control (CTR), TGS1 M1 and TGS1 M1R cells was precipitated with the R1131 anti-TMG CAP antibody (50,76,77) (TMG IP) or control IgGs (IgG IP). Histogram bars represent the fold enrichment of the indicated transcripts in RIP eluates (relative to input), determined by RT-qPCR and normalized to TMG IP from control cells (expressed as the percentage of input). IgG IP bar: values of IgG IPs for the three cell types were pooled into a single bar. MMG-capped GAPDH mRNA and the uncapped 5.8S rRNA are negative controls. Data are from 3–5 independent experiments. P-value: two-way ANOVA with Sidak's multiple comparisons test. See also Supplementary Figure S4A and B. (D) RIP was performed with the R1131 anti-TMG antibody (TMG) or control IgG on nuclear extracts from control (CTR), TGS1 M1 and TGS1 M1R cells. Membranes were probed for U3 snoRNA, U2 and U1 snRNAs; the 5S rRNA, which lacks a TMG cap, is a negative control. Histogram bars represent the quantification of the U2, U1 and U3 RNAs, by densitometric analysis. Data are from three biological replicates. Note the presence of U2 snRNA precursors (arrow) in nuclear extracts from TGS1 M1 cells, which display reduced hypermethylation of U2 snRNA and U3 snoRNA, but not of U1 snRNA. (E) Representative northern blots showing U2 snRNA precursors (arrow) in nuclear and cytoplasmic fractions. RNA was purified from nuclear and cytoplasmic extracts from: TCAB1 K1 HeLa mutant cells [this cell line, described in (48) has reduced CBs and was used as a control]; CTR, parental HeLa cells; TGS1 M1 and TGS1 M1R cells. Membranes were probed for the U1, U2, U4atac, U5 snRNA and the U3 snoRNA. 5S rRNA and tRNA-Arg were used as loading controls. EtBr rRNA staining is a loading control. Western blots with Coilin, CBP20, alpha-tubulin and GAPDH were used as loading controls.
Next, to determine the effects of TGS1 deficiency on cap hypermethylation of different snRNAs, we performed RIP on total RNA, with the anti-TMG antibody R1131 that specifically recognizes the TMG cap (50,76,77). RT-qPCR on RNA precipitated with the TMG antibody showed that the proportion of TMG-capped U2, U12 and U4atac snRNAs, but not U1 snRNA, was more abundant in control cells than in TGS1 mutant cells (Figure 4C). The U2 snRNA was the most affected by TGS1 deficiency, as TMG-capped U2 snRNA molecules in TGS1 M1 cells were reduced 4-fold compared with the control. TMG-capped U4atac and U12 snRNA molecules were reduced by 50% in TGS1 M1 cells. In contrast, the abundance of TMG-capped U1 snRNA was not reduced in TGS1 M1 cells. This observation was confirmed by an independent set of RIP experiments, performed with the anti-TMG antibody K121 (77) and in which gel-purified yeast U1 or U2 snRNAs were added as spike-in TMG-capped RNAs to control for immunoprecipitation efficiency (Supplementary Figure S4A, B). As a positive control for these experiments, we used telomerase RNA (hTR). The abundance of TMG-capped hTR was reduced by 50% in TGS1 M1 cells (Figure 4C), consistent with our previous work showing that TGS1 hypermethylates hTR and down-regulates its abundance (45). The GAPDH and 5.8S RNAs were used as negative controls as GAPDH mRNA has an MMG cap that is not recognized by the anti-TMG antibody, and the 5.8S rRNA is normally uncapped. As expected, these RNAs bound very poorly to the anti-TMG antibody and were precipitated in similarly low proportions from both TGS1 M1 and control cells (Figure 4C). RIP with the anti TMG antibody, performed on nuclear RNA fractions and followed by northern blotting, confirmed that, compared with control and TGS1 M1R cells, TGS1 M1 cells exhibit a reduction in the TMG-capped U2 snRNA and U3 snoRNA, but not in the U1 snRNA (Figure 4D). This experiment also showed that nuclear extracts of TGS1 M1 cells are enriched in slower migrating U2 species (Figure 4D), which are likely to correspond to U2 precursors carrying genome-encoded extensions at their 3′ ends (11,21).
Next, we analyzed by NB the distribution of the extended snRNA species in nuclear and cytoplasmic fractions of both control and TGS1 mutant cells. Extended RNA precursors were detected for U2 snRNAs but not for U1, U3, U4atac and U5 snRNAs. U2 snRNA precursors were clearly enriched in both nuclear and cytoplasmic RNA of TGS1 M1 and M2 cells, compared with TGS1-proficient cells (Figure 4E; Supplementary Figure S5A, B). Nuclear or cytoplasmic accumulations of extended U2 precursors were not enriched in cells carrying mutations in TCAB1 (48) that disrupt CB stability (Figure 4E). Immunostaining of TGS1 M1 cells with antibodies against the TMG cap and the SmB protein showed a strong reduction in the speckled nuclear distribution of snRNPs compared with control or TGS1 M1R cells (Supplementary Figure S5C). In addition, after immunostaining with these antibodies, TGS1 M1 nuclei showed a diffuse cytoplasmic halo that was not observed in control or TGS1 M1R cells, suggesting that TGS1 deficiency leads to retention of some snRNPs in the cytoplasm (Supplementary Figure S5C). Consistent with this finding, single-molecule FISH detected cytoplasmic retention of U2 species in TGS1 M1 cells but not in TGS1-proficient cells (Supplementary Figure S5D). In contrast, FISH analysis showed that the nuclear localization of U1 snRNA is unaffected by TGS1 deficiency (Supplementary Figure S5D). These results indicate that TGS1 deficiency causes aberrant accumulation of unprocessed U2 precursors that might interfere with spliceosome activity, as occurs in yeast (78).
We characterized the 3′ ends of the pre-U2 molecules in total RNA by 3′ rapid amplification of cDNA ends (RACE) and next-generation sequencing as previously described (47,79). In control cells, 90% of the reads mapped to the mature forms of U2, most of which contained 188 nt plus a 3′ adenosine added post-transcriptionally (≤189 nt), in agreement with previous results (3,80). The remaining 10% of the U2 molecules had extended tails including genome-encoded nucleotides at their 3′ ends (Figure 5A, B). TGS1 mutants displayed a 4-fold higher proportion of 3′-extended U2 molecules (41%) compared with control (10%), with an average tail length of 12 nt (Figure 5A, B). In TGS1 M1R cells, the abundance of the extended U2 molecules was reduced from 41% to 20%, showing rescue of the mutant phenotype (Figure 5A). 3′ RACE and sequencing performed on nuclear and cytoplasmic RNA confirmed the data obtained by northern blotting (Figures 4D, E and 5C, D). In the nuclear RNA fractions, the abundance of the U2 extended species was 18% in TGS1 mutant cells, 7% in control cells and 8% in TGS1 M1R cells (Figure 5C). In the cytoplasmic fractions, the extended U2 molecules were 52% in TGS1 mutant cells, 13% in control cells and 20% in TGS1 M1R cells (Figure 5C). TGS1 mutant cells showed no variations in the proportion of mature U2 molecules carrying a 3′-untemplated adenosine compared with controls; in both cases, the 189/188 ratio was ∼80%. Importantly, the finding that the nuclei of TGS1 mutant cells contain more extended U2 molecules than control nuclei (Figures 4D, E and 5C) suggests that a fraction of these unprocessed species completes the cytoplasmic assembly pathway and is imported in the nucleus.
Figure 5.

TGS1 loss leads to an accumulation of 3′-extended U2 and U4atac snRNAs in human cells. (A) 3′ RACE and sequencing on total RNA showing that TGS1 mutant HeLa cells accumulate more U2-extended snRNA molecules than either parental (CTR) or TGS1 M1R cells. UMUC3 cells treated with TGS1 siRNAs (siTGS1) also accumulate more U2-extended snRNAs than cells exposed to non-targeting siRNAs (siSCR). Nascent and steady-state RNA fractions were purified from control HeLa cells (see the Materials and Methods). Extended U2 molecules carry extra 3′ sequences of templated and/or untemplated nucleotides. Average reads per sample > 78 000 ± 41 000. Data are mean values of two independent clones + SEM. P-values: two-sided Student's t-test. (B) Plots showing the percentage of the different sequence reads of U2 snRNAs in the same HeLa cell samples as in (A). Numbers on the x-axis: additional nucleotides beyond the 189 nt of the mature form. Position 0 includes mature U2 snRNA species of 188 nt or 188 nt plus a post-transcriptionally added A. y-axis: percentage of total reads. Blue, genomic-templated nucleotides; green, untemplated adenosine; orange, untemplated uridine. (C) Characterization of U2 snRNA molecules from nuclear and cytoplasmic fractions from TGS1 M1, TGS1 M2, TGS1 M1R and control (CTR) cells. (D) Tail length and composition of U2 snRNAs in the samples shown in (C). y-axis: percentage of total reads. (E) Percentages of U2 molecules with tails ending with post-transcriptionally added uridines in the HeLa cell RNA samples described in (A). Total reads per sample > 75 000. (F) Percentages of 3′-extended U4atac snRNAs in the RNA samples described in (A). Average reads per sample > 106 000 ± 21 000 (Nascent: >11 000). (G) Tail lengths and composition of the 3′ ends of U4atac snRNAs in TGS1 M1, TGS1 M2, TGS1 M1R and control (CTR) cells. Numbers on the x-axis: additional nucleotides beyond the 130 nt of mature U4atac (position 0). y-axis: percentage of total reads. (H) Percentages of U4atac snRNA molecules with extended tails ending with untemplated uridines. Total reads per sample > 75 000.
To assess whether TGS1 deficiency affects processing of the U2 precursors in a different cell type, we knocked down TGS1 by RNAi in UMUC3 cells, which showed a strong reduction in the TGS1 protein and defective CBs (Supplementary Figure S3E, F). 3′ RACE and sequencing on total RNA revealed that in TGS1-RNAi cells the proportion of extended molecules is 3-fold higher than in cells treated with control siRNA (Figure 5A).
To confirm that the extended species are precursors of U2 molecules, we performed metabolic labeling of newly transcribed RNA with 4-thiouridine for 4 h in control HeLa cells, and affinity-purified this nascent RNA population with thiol-specific biotinylation and streptavidin-coated magnetic beads (47). Unlabeled RNAs (steady-state RNA) recovered after this treatment are enriched in mature RNA species, while labeled RNAs are enriched in short-lived RNA precursors. A comparison of the sequence profiles showed that extended U2 molecules are 5-fold more abundant in nascent RNA samples (59% of total U2 reads) than in steady-state RNA (12%; Figure 5A, B), indicating that the extended U2 species are immature U2 precursors, not yet processed at their 3′ ends.
In total RNA from control cells, a small fraction of the U2 reads are extended molecules carrying non-templated adenosines (0.2%) or uridines (0.6%) added post-transcriptionally at their 3′ ends. The acquisition of an extra A or U is specific for the extended molecules with a mean tail length of 12 nucleotides. In TGS1 mutant cells, the fraction of extended U2 molecules incorporating extra uridines is six times and twice higher than in control and TGS1 M1R-rescued cells, respectively (Figure 5B, E). A 5-fold increase in uridylated molecules is also observed in TGS1 RNAi UMUC3 cells compared with cells treated with non-targeting double-stranded RNA (dsRNA) (Figure 5E). These data suggest that TGS1 affects not only 3′ trimming but also the profile of post-transcriptional modifications on the U2 precursors. Interestingly, oligo(U)-extended molecules are 5-fold more abundant in nascent RNA than in mature RNA, indicating that these modifications represent an intermediate step in the 3′ processing of U2 precursors (Figure 5E).
We also analyzed the 3′ sequences of the U1 and U4atac snRNAs. In both control and TGS1 mutant cells, 98% of U1 snRNA molecules were 164 nt long and < 1% of these molecules carried untemplated adenosine or uridine residues at their ends, indicating that in the TGS1 mutant clones, neither cap hypermethylation of U1 snRNAs nor their biogenesis is affected (Supplementary Figure S4C). In contrast, TGS1 mutant cells were enriched in U4atac-extended forms, showing a mean tail length of 10 nt; these forms were 19% of total reads in TGS1 mutants, 6% in control cells and 9% in rescued cells (Figure 5F, G). Moreover, in TGS1 mutant cells, 3% of U4atac molecules incorporated additional uridines, compared with 0.25% in control cells and 0.7% in rescued cells (Figure 5H).
Collectively, these results indicate that TGS1 depletion results in defective hypermethylation of some snRNA species, which largely correlates with the accumulation of extended U2 and U4atac snRNA molecules. These snRNAs differ in the type and proportion of 3′ end extensions, most probably reflecting diverse dynamics in their 3′ end processing.
Tgs1 controls 3′ processing of snRNAs in Drosophila
To ask whether the role of TGS1 in snRNA 3′ processing is conserved in flies, we exploited dTgs1R1, a null CRISPR/Cas9-induced allele, which in homozygosity causes death in late second instar larvae (40). We also used dTgs1R1/dTgs1CB0 heterozygous flies that die as third instar larvae. The lethality of both mutants is rescued by ubiquitous expression of either a dTgs1 or a human TGS1 transgene (40,42).
We characterized the 3′ ends of U2 snRNAs by 3′ RACE and RNA sequencing on total RNA from wild-type larvae (CTR), dTgs1R1 homozygous larvae and dTgs1R1/dTgs1CB0 larvae; as control, we also examined mutant larvae constitutively expressing dTgs1. dTgs1 mutant larvae showed an increase in 3′-extended U2 species compared with either wild-type larvae or larvae bearing the rescue construct (Figure 6A). A total of 95% of these extended U2 species displayed one additional uridine at the 3′ end (Figure 6B). The remaining 5% of the extended molecules included species containing longer tails incorporating both genome-templated and untemplated A or U nucleotides. Thus, TGS1 is required for proper 3′ processing of U2 snRNAs also in flies.
Figure 6.

3′-Extended U2 snRNA molecules accumulate in Drosophila dTgs1 mutants. (A) Percentage of extended U2 snRNA molecules determined by 3′ RACE and sequencing on RNA from equally aged second or third instar mutant larvae, rescued mutant larvae (bearing a dTgs1 construct) and wild-type larvae (CTR). The most abundant population of U2 snRNAs is 192 nt long. Extended U2 snRNAs are longer than 192 nt and incorporate templated or untemplated nucleotides at their 3′ ends. Total reads > 2900. (B) Tail length and composition of the 3′ ends of U2 snRNAs for the second and third instar larvae described in (A). Numbers on the x-axis: additional nucleotides beyond the mature form of 192 nt (position 0). y-axis: percentage of total reads.
In contrast, an analysis of the U1 snRNAs showed that they are substantially unaffected by dTgs1 mutations, consistent with the results obtained with human cells. In mutant and wild-type larvae, extended species represented ∼1.5% and 0.5% of total U1 RNAs, respectively.
Aberrant transcripts accumulate in both TGS1 and SMN mutant cells
Studies conducted in different systems demonstrated that SMN deficiency causes profound perturbations in the transcriptome (81–86). To explore the consequences of TGS1 loss on mRNA splicing and gene expression, we performed deep sequencing on total RNA extracted from TGS1 mutant HeLa cells, and from two independent SMN mutant clones derived from the same HeLa cell line used to generate TGS1 mutant cells. These lines, designated as SMN C1 and SMN C2, carry CRISPR-induced mutations in the sixth exon of the gene and produce mutated versions of the SMN protein, which are expressed at reduced levels, compared with full-length SMN (Supplementary Figure S6A, B). These lines have normal amounts of the TGS1 protein (Supplementary Figure S6B) and do not exhibit significant changes in viability, compared with the HeLa parental cell line (CTR) and TGS1 M1 mutant cells (Supplementary Figure S6C). SMN C1 has a small deletion in the SMN proline-rich domain and SMN C2 carries frameshift mutations (see Supplementary Figure S6A). The SMN proteins encoded by the SMN C2 cells have a lower molecular weight compared with the wild-type protein. The SMN C1 and SMN C2 mutant cells are defective in CB formation and exhibit fewer SMN-enriched CBs compared with control cells (Supplementary Figure S6D, E).
Paired-ended Illumina sequencing (160 million reads/sample) was performed on RNA isolated from TGS1 mutant clones (M1 and M2), SMN mutant clones (C1 and C2) and parental HeLa cells (CTR). Independent mutant clones and controls were clustered based on both transcript and gene expression profiles (Supplementary Figure S7A–C). We used Kallisto (87) and Sleuth (57) for transcript quantification and differential analysis, respectively (see the Materials and Methods). TGS1 and SMN mutant cells showed a differential expression of both annotated and unannotated transcripts compared with controls [reconstructed by Scallop (56); Figure 7A, B; Supplementary Table S1]. Our analysis revealed significant changes in the expression levels of 3084 and 351 transcript isoforms in TGS1 and SMN mutant cells, respectively (Figure 7A, B; Supplementary Table S1). We found changes in the expression levels of many unannotated transcripts (1080/3084 for TGS1 and 158/351 for SMN mutants); 48 of these novel isoforms were found to be affected in both mutant cell types (Figure 7C; Supplementary Table S1). The number of differentially expressed transcripts was higher in TGS1 mutant cells than in SMN mutant cells (Figure 7A–C), probably reflecting the higher degree of functional inactivation of TGS1 compared with SMN (Figure 4A; Supplementary Figure S6A, B).
The numbers of annotated and unannotated transcripts that were found to be differentially expressed in both SMN and TGS1 mutant cells were significantly greater than would be expected by chance (P-value < 10e-22, hypergeometric test, 110 400 total annotated transcripts, 28 100 total unannotated transcripts with computed P-value by Sleuth) (Figure 7C; Supplementary Table S1; see the Materials and Methods). Notably, 71% and 76% of the differentially expressed unannotated transcripts found in TGS1 and SMN mutant cells, respectively, showed an increased number of reads for intergenic regions compared with controls (Figure 7A, B; Supplementary Table S1). These 3′-elongated transcripts extended beyond the 3′ end of the gene and sometimes included the coding region of the next gene, probably reflecting an altered transcription termination. Most of the differentially expressed transcripts were also identified using an independent analysis workflow (56,59) (Supplementary Tables S3–S5). The mutant cells also exhibited changes in splicing behavior (Supplementary Figure S8A), with intron retention (IR) being the most frequent defect in both TGS1 (3.5% and 4.5% of annotated and unannotated IR events, respectively) and SMN mutants (1.2% and 2.1% of annotated and unannotated IR events, respectively); Figure 7D–F; Supplementary Figure S8A, B and Supplementary Table S2.
Long-read Oxford Nanopore sequencing was performed on the six RNA samples, described above, to gain additional information on the transcripts that accumulate in TGS1 and SMN mutant cells, and to assess the association of splicing defects and 3′ extensions on the same transcript isoform. In total, 72 million reads were generated, pre-processed and mapped to the human genome. The FLAIR pipeline (61) was employed for splicing junction correction, yielding 24.7 million corrected multi-exonic reads that were used for transcriptome reconstruction (see the Materials and Methods and Supplementary Figure S9). A toal of 55 815 transcripts, 41 565 of which were not present in the reference annotation, were identified and quantified using FLAIR; differential splicing between mutant and control samples was assessed using SUPPA2 (Figure 8A–C; Supplementary Figure S10A). The number of shared annotated and unannotated differential splicing events between SMN and TGS1 mutant cells is greater than expected by chance (Figure 8D). Furthermore, there is a significant overlap between the differential splicing events identified by Illumina and Nanopore sequencing (Figure 8E; Supplementary Table S8), providing further support for the reproducibility of our findings on the global splicing defects caused by TGS1 and SMN depletion. Multi-exonic reads intersecting the last exon of each gene were used to identify loci in which the proportion of readthrough transcripts was significantly different in TGS1 and SMN mutant samples compared with control (Figure 8F; Supplementary Table S9). Interestingly, in both comparisons, most of the differential readthrough events consist of a higher proportion of readthrough reads in the mutant, indicating a defective termination leading to the production of longer transcripts. Many of these longer transcripts extended into the downstream gene. For the genes showing such preferential readthrough in mutant samples, SUPPA-2 was used to determine the number of readthrough and non-readthrough reads harboring an alternative spicing event in the mutant samples. These analyses (Figure 8G; Supplementary Figure S10B; Supplementary Table S10) indicate that in both TGS1 and SMN mutant cells, readthrough reads have a higher probability of also containing an alternative splicing event compared with non-readthrough reads.
Figure 8.

Analysis of the aberrant transcripts that accumulate in TGS1 and SMN mutant cells by Oxford Nanopore sequencing. (A) Methodological approach used for transcript quantification and evaluation of transcript differential splicing. (B,C) Scatter plots showing, for each AS event between mutant (TGS1 M1, M2 and SMN C1, C2) and parental HeLa cells (CTR), the differential PSI (dPSI) against the differential expression level (dTPM) of the major transcript (that does not contain the AS event). The significant differential splicing events are color-coded in the plots, with significantly included events colored in orange and significantly excluded events in teal. Non-significant events are in gray (see also Supplementary Figure S10A for a classification of the AS events and Supplementary Table S6). The arrows indicate up- or down-regulation of AS inclusion transcripts (pink) and AS exclusion transcripts (green), as depicted in (A), respectively. (D) Venn diagrams showing the number of annotated and novel differential splicing events found in TGS1 M1, M2 and SMN C1, C2 mutant cells by Nanopore sequencing. The statistical significance of the intersection was determined via Fisher's exact test. See also Supplementary Table S7. (E) Venn diagram showing the number of the differential splicing events identified by Illumina and Nanopore analysis in TGS1 and SMN mutant cells. The statistical significance of the intersection was determined via Fisher's exact test. See also Supplementary Table S8. (F) Dot plots showing differential readthrough events in mutant cells (orange dots) versus control cells (gray dots). For each readthrough event, the value of the log2(fold change) (mutant versus control) is reported on the x-axis. Statistical significance of the fold enrichment for each event was determined by Fisher's exact test (P-values on the y-axis; P < 0.05 was considered significant). The numbers of genes with ‘Preferential Readthrough in Mutant’ (orange) or ‘Preferential Readthrough in CTR’ (gray) are reported below the graphs. See also Supplementary Table S9; and the Materials and Methods. (G) Bar plot showing that, in mutant samples, the percentage of reads that carry an aberrant splicing event is higher among readthrough reads compared with non-readthrough reads. Only reads mapping to the last exons undergoing ‘Preferential Readthrough in Mutant’ events were used for this analysis. Aberrant splicing was assessed by comparing reads from mutant samples against non-readthrough reads from control samples. The significance of the difference in the proportions of aberrant splicing reads between readthrough and non-readthrough reads was assessed using the χ2 test. See also Supplementary Table S10.
To confirm the results obtained using high-throughput data analysis, we performed targeted validation for four transcripts by RT-PCR or RT-qPCR on RNA samples from CTR, TGS1M and TGS1M1R cells. Amplification products consistent with the predicted novel transcript isoforms were enriched in the two TGS1 mutant clones compared with both the parental cell line and TGS1 M1R cells (Supplementary Figures S11 and S12). Two of these validated novel transcripts were found to be enriched in TGS1 mutant clones also by Nanopore analysis and are representative examples of readthrough transcripts that extend into the downstream gene (Supplementary Figure S11B, E).
DISCUSSION
We have shown that the highly conserved TGS1 hypermethylase localizes to MNs in C. elegans, and that its deficiency results in morphological and functional abnormalities in neurons from both C. elegans and D. rerio. Consistent with these defects, TGS1 deficiency impairs locomotion in worms and wing expansion in flies, which is governed by bursicon-expressing neurons (88). Previous work has also shown that mutations in Drosophila Tgs1 cause abnormal larval locomotion (40,69,70). The phenotypes elicited by TGS1 inhibition in flies, worm and zebrafish are highly reminiscent of those caused by loss of function of the SMN gene. Importantly, we showed that TGS1 overexpression ameliorates the neurological phenotypes elicited by the impairment of the SMN function in both fly and worm model systems. These findings suggest commonalities in the molecular and cellular effects induced by either TGS1 or SMN loss of function.
Our investigation on the consequences of TGS1 loss in snRNA biogenesis uncovers the involvement of this hypermethylase in 3′-end processing. The HeLa cell model was instrumental to compare the molecular consequences of SMN and TGS1 loss in the same isogenic background. Additionally, we demonstrate that 3′-extended mRNA transcripts accumulate in both TGS1 and SMN mutant cells. Lastly, our analyses of different animal models provide robust evidence that TGS1 together with SMN plays a conserved, important role in neuronal function.
Several studies have linked neurodegeneration to defective snRNA processing, snRNP biogenesis and/or pre-mRNA splicing (3,74,85,89,90). It has been reported that SMN-depleted cells exhibit an enrichment of 3′-unprocessed snRNA precursors resulting from their impaired assembly into snRNPs (11). In a mouse model of SMA, MNs display a much greater reduction in the snRNPs levels relative to unaffected spinal cells (91), and their selective death is the result of converging mechanisms of p53 activation driven by dysregulation of distinct mRNA splicing pathways (92–94). It has been also reported that mutations in a single gene of the U2 multicopy cluster (Rnu2-8) result in defective splicing and cause ataxia and neurodegeneration in mice (89). Unprocessed snRNAs accumulate in cells from patients affected by PCH7, which is also characterized by MN loss (3). Interestingly, we show that TGS1 mutant cells accumulate extended snRNA species with post-transcriptionally added adenosines or uridines similar to cells from PCH7 patients (3,95,96).
Our 3′ RACE and sequencing experiments show that TGS1 loss results in the accumulation of untrimmed 3′-extended U2 snRNA molecules, but has no effect on U1 snRNA in both human cells and Drosophila lethal mutants. TGS1-deficient human cells also accumulate untrimmed U4atac snRNAs. Interestingly, viable hypomorphic Drosophila Tgs1 mutants that cause male sterility exhibit a reduction in TMG-capped U1 and U2 snRNAs in testes and a concomitant accumulation of long precursors for most snRNAs (97). Collectively, these results indicate that TGS1-mediated cap hypermethylation affects 3′-end processing of snRNAs.
Immature snRNA precursors produced by different snRNA genes differ in length (11,21,97–100). In human cells deficient for either TGS1 (this study) or TOE1 (3,96), the tails of the extended molecules generated by different U snRNA subclasses vary in length, number of templated nucleotides and number/type of untemplated residues. We found that in TGS1-deficient human cells and flies, the U2 snRNAs acquire different 3′-extended structures. While the majority of extended human U2 snRNAs have templated tails with or without an untemplated U residue, Drosophila U2 molecules preferentially gain untemplated uridines. Uridylation is thought to recognize misprocessed and/or low-quality snRNPs and target them for degradation (96,101–103). The variation in the 3′ ends of snRNAs may reflect differences in promoter sequences (104), gene bodies or 3′ regions, which are crucial determinants for the cleavage and maturation of the snRNA precursor transcripts (105). The different susceptibilities of individual snRNAs to loss of maturation factors might also depend on the specificity of these factors in 3′-end processing and on their residual levels. It is possible that TGS1 hypermethylates different snRNAs with different efficiencies and that our experimental conditions did not allow detection of differences in the hypermethylation level of U1 snRNA.
There is also evidence suggesting that the U1 snRNAs behave differently from the other U snRNA subclasses. Recent work has revealed that 3′-end processing of some snRNAs is a biphasic process that takes place both before and after the assembly with the SMN complex (96). Human U1 has a peculiar Sm core assembly pathway and differs from the other snRNAs in the affinity for SMN-Gemin5 (11,106). These features suggest a specific regulation of the biogenesis of the U1 snRNP, which is the most abundant snRNP, playing roles in both splicing and poly(A) site selection (telescripting) (107). Thus, the finding that the U1 snRNAs are not affected by loss of TGS1 might depend on several factors, including the specificity of the TGS1 enzymatic activity and the peculiar maturation pathway of the U1 snRNPs. We cannot rule out the possibility that in our hypomorphic mutants, a minimal residual TGS1 activity, that is sufficient to sustain cell viability, is enough to provide the modification of U1 snRNA.
TGS1 deficiency results in extensive alterations in the human transcriptome, including changes in the efficiency of intron removal and accumulation of transcripts with 3′ extensions spanning intergenic regions, and often incorporating exons of adjacent genes. Similar alterations are also observed in the transcriptome of SMN mutant cells. Long-read Oxford Nanopore Technology (ONT) sequencing confirmed that TGS1- and SMN-deficient cells accumulate mRNAs that carry both splicing and 3′-end cleavage defects. Many of these are chimeric transcripts, in which the exon of a gene is fused to exons of the downstream gene. Recent work has suggested that SMN directly mediates proper transcription termination by favoring Pol II release (108), and SMN deficiency has been linked to accumulation of R-loops and DNA damage (83,109). Accumulation of aberrant readthrough transcripts is consistent with these results and may contribute to the neurodegenerative defects observed in TGS1 and SMN mutant models.
Previous work has shown that U2 snRNP alterations caused by deficiency of the SF3B1 protein result in splicing defects and transcription proceeding beyond the canonical polyadenylation [poly(A)] sites, leading to severe pathological consequences (61,110). In addition, it has been shown that U1 snRNPs prevent premature transcription termination before the last exon by inhibiting 3′-end cleavage at cryptic, early polyadenylation sites (107). The results of our long-read ONT sequencing of transcripts from TGS1- and SMN-deficient cells support the hypothesis that at least a fraction of the aberrant readthrough mRNAs observed in TGS1 and SMN mutant cells are caused by a primary defect in splicing, and consolidate the notion that reduced efficiency of co-transcriptional splicing impairs correct transcription termination (111,112). However, given that the TMG cap contributes to nuclear import of snRNPs (16–20), it cannot be excluded that inefficient import of monomethylated snRNA precursors could decrease the levels of functional snRNPs, with possible repercussions on the efficiency of termination. Our observations are also compatible with the possibility that TGS1 cooperates with SMN in transcription termination and that its loss directly contributes to the formation of long readthrough transcripts.
The characterization of transcriptional defects in TGS1- and SMN-deficient cells was carried out on steady-state poly(A) transcripts and therefore the occurrence of non-polyadenylated and unstable species that are subjected to clearance by the RNA surveillance machineries may have been overlooked. It has been reported that inverted Alu repeats promote the expression of circular RNAs from the SMN locus (113,114). In our sequencing datasets we did not find aberrant transcripts arising from the SMN loci; however, it is possible that transcriptional readthrough of the SMN gene might enhance the expression of SMN circular RNAs (113). Further analyses with methods that allow the isolation of non-polyadenylated transcripts as in (115) and (116) and by long-read sequencing of nascent RNA will probably expand the repertoire of RNAs affected by TGS1 and SMN depletion.
Collectively, our data support a model where TGS1 and SMN have closely related functions in preventing neurodegeneration (Figure 9A, B). TGS1 plays roles in snRNA 3′ maturation and its loss affects the accuracy of both pre-mRNA splicing and transcription termination. SMN promotes assembly of snRNA into snRNPs and is essential for splicing and efficient transcription termination (85,108). Thus, loss of either protein leads to substantial transcriptome alterations. These alterations may be neurotoxic by directly affecting specific transcripts important for neuronal survival and function, a scenario supported by increasing experimental evidence (92–94,117). In addition, reduced efficiency of splicing and transcription termination may be a source of transcriptional stress that results in genome-wide accumulation of R-loops and DNA damage, which may also affect neuronal survival (118). Future studies on ultra-fine characterization of aberrant mRNA molecules and their effects in neurons are needed to support this possibility, which has strong relevance not only for SMA but also for other neurodegenerative disorders. Importantly, we found that TGS1 overexpression can partially rescue the neurological phenotypes caused by SMN depletion in the fly and worm models. Moreover, previous work has shown that TGS1 directly interacts with the SMN protein in human cells (14), and that Drosophila Tgs1 physically associates with most components of the SMN complex in vivo (40). Thus, the physical interaction between TGS1 and SMN and the similar phenotypes elicited by their deficiency suggest a functional interaction between these proteins. We hypothesize that TGS1 overexpression in SMN-depleted cells might enhance snRNP biogenesis and possibly improve the fidelity of splicing and transcription termination, functionally compensating at least in part for defects caused by SMN deficiency (Figure 9).
Figure 9.

SMN and TGS1 protect against neurodegeneration through both common and specific routes. (A) Roles of SMN and TGS1 in snRNA biogenesis. The dashed arrow line indicates that the role of TGS1 in 3′-end processing could be indirect. (B) A model for the roles of TGS1 and SMN in prevention of neurodegeneration. The effects of TGS1 or SMN deficiency on snRNA biogenesis and general transcription suggest that these proteins play interconnected roles in prevention of neurodegeneration.
Recent remarkable advances in the field have led to the approval of disease-modifying therapies for SMA patients, which include SMN replacement via gene therapy as well as increased production of full-length SMN by splicing modulation with antisense oligonucleotides or small molecules (119–122). These treatments are most effective in improving the SMA phenotypes when administered to pre-symptomatic patients (123,124). However, an expansion of SMA therapeutic options that could complement SMN-inducing treatments is especially desirable for those patients displaying more limited clinical benefit due to delayed intervention or other variables (123). The development of SMN-independent strategies will require a more profound knowledge of disease-relevant downstream pathways (119,120,124–129). Studies in model organisms have led to the identification of factors, whose modulation can alter the pathogenic consequences of SMN deficiency (73,81,93,94,130). This study expands the list of these factors by showing that TGS1 overexpression can compensate, at least in part, for SMN loss in animal models of neurodegeneration.
DATA AVAILABILITY
The custom Python scripts used for the analysis of 3′ RACE data are available at https://github.com/cmroake/NAR_TGS1.
The custom R scripts used for the analysis of transcriptome data produced via Illumina sequencing are available at https://github.com/roozbehdn/TGS1_SMN.
The RNAend-Seq data discussed in this publication have been deposited with links to BioProject accession number PRJNA628085 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
The Illumina and Nanopore RNA-Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (131) and are accessible through GEO Series accession numbers GSE189413 and GSE189138, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Bazzicalupo for critical reading of the manuscript, G. Zampi and F. Sola for technical support, S. Schneider for support in zebrafish procedures, and P. De Piante for data analysis. For strains and plasmids, we thank A. Fire, K. Shen, O. Hobert, C. Bargmann and the Caenorhabditis Genetics Center (CGC), funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Reinhard Lührmann for sharing the R1131 TMG antibody.
Contributor Information
Lu Chen, Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biochemistry, Stanford University School of Medicine, Stanford, CA 94305, USA; Cancer Signaling and Epigenetics Program and Cancer Epigenetics Institute, Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia, PA 19111, USA.
Caitlin M Roake, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biochemistry, Stanford University School of Medicine, Stanford, CA 94305, USA.
Paolo Maccallini, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Francesca Bavasso, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Roozbeh Dehghannasiri, Department of Biochemistry, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA.
Pamela Santonicola, Institute of Biosciences and BioResources, IBBR, CNR, Naples, Italy.
Natalia Mendoza-Ferreira, Institute of Human Genetics, Center for Molecular Medicine Cologne, Institute for Genetics, University of Cologne, 50931 Cologne, Germany.
Livia Scatolini, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Ludovico Rizzuti, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Alessandro Esposito, Institute of Genetics and Biophysics, IGB-ABT, CNR, Naples, Italy.
Ivan Gallotta, Institute of Genetics and Biophysics, IGB-ABT, CNR, Naples, Italy.
Sofia Francia, IFOM-The FIRC Institute of Molecular Oncology, Milan, Italy; Istituto di Genetica Molecolare, CNR-Consiglio Nazionale delle Ricerche, Pavia, Italy.
Stefano Cacchione, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Alessandra Galati, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Valeria Palumbo, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
Marie A Kobin, Cancer Signaling and Epigenetics Program and Cancer Epigenetics Institute, Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia, PA 19111, USA.
Gian Gaetano Tartaglia, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy; Center for Life Nano- & Neuro-Science, Fondazione Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy; Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy.
Alessio Colantoni, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy; Center for Life Nano- & Neuro-Science, Fondazione Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy; Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy.
Gabriele Proietti, Center for Life Nano- & Neuro-Science, Fondazione Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy; Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy.
Yunming Wu, Cancer Signaling and Epigenetics Program and Cancer Epigenetics Institute, Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia, PA 19111, USA; Department of Biology, Howard Hughes Medical Institute, Stanford University, Stanford, CA 94305, USA.
Matthias Hammerschmidt, Institute for Zoology, Developmental Biology, University of Cologne, 50674 Cologne, Germany.
Cristiano De Pittà, Department of Biology, University of Padova, Padua, Italy.
Gabriele Sales, Department of Biology, University of Padova, Padua, Italy.
Julia Salzman, Department of Biochemistry, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA.
Livio Pellizzoni, Center for Motor Neuron Biology and Disease, Columbia University, NY 10032, USA; Department of Pathology and Cell Biology, Columbia University, NY 10032, USA; Department of Neurology, Columbia University, NY 10032, USA.
Brunhilde Wirth, Institute of Human Genetics, Center for Molecular Medicine Cologne, Institute for Genetics, University of Cologne, 50931 Cologne, Germany; Center for Rare Diseases, University Hospital of Cologne, University of Cologne, 50931 Cologne, Germany.
Elia Di Schiavi, Institute of Biosciences and BioResources, IBBR, CNR, Naples, Italy; Institute of Genetics and Biophysics, IGB-ABT, CNR, Naples, Italy.
Maurizio Gatti, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy; Istituto di Biologia e Patologia Molecolari (IBPM) del CNR, Rome, Italy.
Steven E Artandi, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biochemistry, Stanford University School of Medicine, Stanford, CA 94305, USA.
Grazia D Raffa, Dipartimento di Biologia e Biotecnologie, Sapienza University of Rome, Rome, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by grants from Telethon GPP13147 (to G.D.R.) and GGP16203 (to E.D.S.), NIH AG056575 and CA197563 (to S.E.A.), NS102451 (to L.P.), AIRC IG 20528 (to M.G.), AIRC IG 26496 (to G.D.R.), Italian Ministry of Economy and Finance (FaReBio di Qualità) and Italian Ministry of Health RF 2009-1473235 (to E.D.S.), German Research Foundation [Wi 945/17-1 (project-ID 398410809); FOR2722 (project ID 384170921); Wi 945/19-1 (project-ID 417989143); KY96.1-2 (project-ID 269018619); SFB1451 (project-ID 431549029 – A01), and GRK1960 (project ID 233886668)], the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 956185 (SMABEYOND) and the Center for Molecular Medicine Cologne (project C18) (to B.W.). S.F. was supported by Fondazione Cariplo (grant 2014-1215) and by AriSLA (project DDRNA&ALS and DDR&ALS). A.C., G.P. and G.G.T are supported by European Research Council [RIBOMYLOME_309545 and ASTRA_855923] and the H2020 projects [IASIS_727658 and INFORE_825080]. L.C. was supported by a Stanford Cancer Institute 2018 Fellowship Award, and C.M.R. by an MSTP Training Grant GM007365 and a Gerald J. Lieberman Fellowship.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nussbacher J.K., Tabet R., Yeo G.W., Lagier-Tourenne C.. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron. 2019; 102:294–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wirth B., Karakaya M., Kye M.J., Mendoza-Ferreira N.. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu. Rev. Genomics Hum. Genet. 2020; 21:231–261. [DOI] [PubMed] [Google Scholar]
- 3. Lardelli R.M., Schaffer A.E., Eggens V.R., Zaki M.S., Grainger S., Sathe S., Van Nostrand E.L., Schlachetzki Z., Rosti B., Akizu N.et al.. Biallelic mutations in the 3′ exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing. Nat. Genet. 2017; 49:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Vegvar H.E., Lund E., Dahlberg J.E.. 3′ end formation of U1 snRNA precursors is coupled to transcription from snRNA promoters. Cell. 1986; 47:259–266. [DOI] [PubMed] [Google Scholar]
- 5. Hernandez N., Weiner A.M.. Formation of the 3′ end of U1 snRNA requires compatible snRNA promoter elements. Cell. 1986; 47:249–258. [DOI] [PubMed] [Google Scholar]
- 6. Egloff S., Zaborowska J., Laitem C., Kiss T., Murphy S.. Ser7 phosphorylation of the CTD recruits the RPAP2 ser5 phosphatase to snRNA genes. Mol. Cell. 2012; 45:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O’Reilly D., Kuznetsova O.V., Laitem C., Zaborowska J., Dienstbier M., Murphy S.. Human snRNA genes use polyadenylation factors to promote efficient transcription termination. Nucleic Acids Res. 2014; 42:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baillat D., Hakimi M.A., Naar A.M., Shilatifard A., Cooch N., Shiekhattar R.. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase iI. Cell. 2005; 123:265–276. [DOI] [PubMed] [Google Scholar]
- 9. Ohno M., Segref A., Bachi A., Wilm M., Mattaj I.W.. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell. 2000; 101:187–198. [DOI] [PubMed] [Google Scholar]
- 10. Kitao S., Segref A., Kast J., Wilm M., Mattaj I.W., Ohno M.. A compartmentalized phosphorylation/dephosphorylation system that regulates U snRNA export from the nucleus. Mol. Cell. Biol. 2008; 28:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yong J., Kasim M., Bachorik J.L., Wan L., Dreyfuss G.. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol. Cell. 2010; 38:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pellizzoni L., Yong J., Dreyfuss G.. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002; 298:1775–1779. [DOI] [PubMed] [Google Scholar]
- 13. Mouaikel J., Verheggen C., Bertrand E., Tazi J., Bordonne R.. Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell. 2002; 9:891–901. [DOI] [PubMed] [Google Scholar]
- 14. Mouaikel J., Narayanan U., Verheggen C., Matera A.G., Bertrand E., Tazi J., Bordonne R.. Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 2003; 4:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narayanan U., Achsel T., Luhrmann R., Matera A.G.. Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol. Cell. 2004; 16:223–234. [DOI] [PubMed] [Google Scholar]
- 16. Strasser A., Dickmanns A., Luhrmann R., Ficner R.. Structural basis for m3G-cap-mediated nuclear import of spliceosomal usnrnps by snurportin1. EMBO J. 2005; 24:2235–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fischer U., Heinrich J., van Zee K., Fanning E., Luhrmann R.. Nuclear transport of U1 snRNP in somatic cells: differences in signal requirement compared with Xenopus laevis oocytes. J. Cell Biol. 1994; 125:971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marshallsay C., Luhrmann R.. In vitro nuclear import of snRNPs: cytosolic factors mediate m3G-cap dependence of U1 and U2 snRNP transport. EMBO J. 1994; 13:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huber J., Cronshagen U., Kadokura M., Marshallsay C., Wada T., Sekine M., Luhrmann R.. Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure. EMBO J. 1998; 17:4114–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanek D. Cajal bodies and snRNPs—friends with benefits. RNA Biol. 2017; 14:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wieben E.D., Nenninger J.M., Pederson T.. Ribonucleoprotein organization of eukaryotic RNA. XXXII. U2 small nuclear RNA precursors and their accurate 3′ processing in vitro as ribonucleoprotein particles. J. Mol. Biol. 1985; 183:69–78. [DOI] [PubMed] [Google Scholar]
- 22. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallotta I., Mazzarella N., Donato A., Esposito A., Chaplin J.C., Castro S., Zampi G., Battaglia G.S., Hilliard M.A., Bazzicalupo P.et al.. Neuron-specific knock-down of SMN1 causes neuron degeneration and death through an apoptotic mechanism. Hum. Mol. Genet. 2016; 25:2564–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. BioTechniques. 2002; 32:728–730. [DOI] [PubMed] [Google Scholar]
- 25. Esposito G., Di Schiavi E., Bergamasco C., Bazzicalupo P.. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene. 2007; 395:170–176. [DOI] [PubMed] [Google Scholar]
- 26. Eastman C., Horvitz H.R., Jin Y.. Coordinated transcriptional regulation of the unc-25 glutamic acid decarboxylase and the unc-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J. Neurosci. 1999; 19:6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kamath R.S., Fraser A.G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M.et al.. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003; 421:231–237. [DOI] [PubMed] [Google Scholar]
- 28. Mello C.C., Kramer J.M., Stinchcomb D., Ambros V.. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991; 10:3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veronico P., Gray L.J., Jones J.T., Bazzicalupo P., Arbucci S., Cortese M.R., Di Vito M., De Giorgi C.. Nematode chitin synthases: gene structure, expression and function in Caenorhabditis elegans and the plant parasitic nematode Meloidogyne artiellia. Mol. Genet. Genomics. 2001; 266:28–34. [DOI] [PubMed] [Google Scholar]
- 30. Kimble J.E., White J.G.. On the control of germ cell development in Caenorhabditis elegans. Dev. Biol. 1981; 81:208–219. [DOI] [PubMed] [Google Scholar]
- 31. Stinchcomb D.T., Shaw J.E., Carr S.H., Hirsh D.. Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol. Cell. Biol. 1985; 5:3484–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McIntire S.L., Reimer R.J., Schuske K., Edwards R.H., Jorgensen E.M.. Identification and characterization of the vesicular GABA transporter. Nature. 1997; 389:870–876. [DOI] [PubMed] [Google Scholar]
- 33. Riessland M., Kaczmarek A., Schneider S., Swoboda K.J., Lohr H., Bradler C., Grysko V., Dimitriadi M., Hosseinibarkooie S., Torres-Benito L.et al.. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am. J. Hum. Genet. 2017; 100:297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendoza-Ferreira N., Coutelier M., Janzen E., Hosseinibarkooie S., Lohr H., Schneider S., Milbradt J., Karakaya M., Riessland M., Pichlo C.et al.. Biallelic CHP1 mutation causes human autosomal recessive ataxia by impairing NHE1 function. Neurol. Genet. 2018; 4:e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lan C.C., Tang R., Un San Leong I., Love D.R.. Quantitative real-time RT-PCR (qRT-PCR) of zebrafish transcripts: optimization of RNA extraction, quality control considerations, and data analysis. Cold Spring Harb. Protoc. 2009; 2009:pdb prot5314. [DOI] [PubMed] [Google Scholar]
- 36. Oprea G.E., Krober S., McWhorter M.L., Rossoll W., Muller S., Krawczak M., Bassell G.J., Beattie C.E., Wirth B.. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008; 320:524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang H.C., Dimlich D.N., Yokokura T., Mukherjee A., Kankel M.W., Sen A., Sridhar V., Fulga T.A., Hart A.C., Van Vactor D.et al.. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008; 3:e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palumbo V., Pellacani C., Heesom K.J., Rogala K.B., Deane C.M., Mottier-Pavie V., Gatti M., Bonaccorsi S., Wakefield J.G.. Misato controls mitotic microtubule generation by stabilizing the TCP-1 tubulin chaperone complex [corrected]. Curr. Biol. 2015; 25:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bischof J., Maeda R.K., Hediger M., Karch F., Basler K.. An optimized transgenesis system for drosophila using germ-line-specific phiC31 integrases. Proc. Natl Acad. Sci. USA. 2007; 104:3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maccallini P., Bavasso F., Scatolini L., Bucciarelli E., Noviello G., Lisi V., Palumbo V., D’Angeli S., Cacchione S., Cenci G.et al.. Intimate functional interactions between TGS1 and the SMN complex revealed by an analysis of the Drosophila eye development. PLoS Genet. 2020; 16:e1008815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buszczak M., Paterno S., Lighthouse D., Bachman J., Planck J., Owen S., Skora A.D., Nystul T.G., Ohlstein B., Allen A.et al.. The Carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007; 175:1505–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raffa G.D., Siriaco G., Cugusi S., Ciapponi L., Cenci G., Wojcik E., Gatti M.. The Drosophila modigliani (moi) gene encodes a HOAP-interacting protein required for telomere protection. Proc. Natl Acad. Sci. USA. 2009; 106:2271–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lattao R., Bonaccorsi S., Guan X., Wasserman S.A., Gatti M.. Tubby-tagged balancers for the Drosophila X and second chromosomes. Fly. 2011; 5:369–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen L., Roake C.M., Galati A., Bavasso F., Micheli E., Saggio I., Schoeftner S., Cacchione S., Gatti M., Artandi S.E.et al.. Loss of human TGS1 hypermethylase promotes increased telomerase RNA and telomere elongation. Cell Rep. 2020; 30:1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu L., Blackburn E.H.. Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol. Cell. 2007; 28:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roake C.M., Chen L., Chakravarthy A.L., Ferrell J.E., Raffa G.D., Artandi S.E.. Disruption of telomerase RNA maturation kinetics precipitates disease. Mol. Cell. 2019; 74:688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen L., Roake C.M., Freund A., Batista P.J., Tian S., Yin Y.A., Gajera C.R., Lin S., Lee B., Pech M.F.et al.. An activity switch in human telomerase based on RNA conformation and shaped by TCAB1. Cell. 2018; 174:218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venteicher A.S., Abreu E.B., Meng Z., McCann K.E., Terns R.M., Veenstra T.D., Terns M.P., Artandi S.E.. A human telomerase holoenzyme protein required for cajal body localization and telomere synthesis. Science. 2009; 323:644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luhrmann R., Appel B., Bringmann P., Rinke J., Reuter R., Rothe S., Bald R.. Isolation and characterization of rabbit anti-m3 2,2,7G antibodies. Nucleic Acids Res. 1982; 10:7103–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Petrov A., Wu T., Puglisi E.V., Puglisi J.D.. RNA purification by preparative polyacrylamide gel electrophoresis. Methods Enzymol. 2013; 530:315–330. [DOI] [PubMed] [Google Scholar]
- 52. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta delta C(T)) method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 53. Chen L., Ooi S.K., Conaway R.C., Conaway J.W.. Generation and purification of human INO80 chromatin remodeling complexes and subcomplexes. J. Vis. Exp. 2014; e51720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu Y., Xu W., Ma L., Yu Z., Wang Y., Yu C.R.. Robust and sensitive in situ RNA detection using Yn-situ. Cell Rep. Methods. 2022; 2:100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shao M., Kingsford C.. Accurate assembly of transcripts through phase-preserving graph decomposition. Nat. Biotechnol. 2017; 35:1167–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L.. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017; 14:687–690. [DOI] [PubMed] [Google Scholar]
- 58. Trincado J.L., Entizne J.C., Hysenaj G., Singh B., Skalic M., Elliott D.J., Eyras E.. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018; 19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tang A.D., Soulette C.M., van Baren M.J., Hart K., Hrabeta-Robinson E., Wu C.J., Brooks A.N.. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020; 11:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shaye D.D., Greenwald I.. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011; 6:e20085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spencer W.C., Zeller G., Watson J.D., Henz S.R., Watkins K.L., McWhirter R.D., Petersen S., Sreedharan V.T., Widmer C., Jo J.et al.. A spatial and temporal map of C. elegans gene expression. Genome Res. 2011; 21:325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaletsky R., Lakhina V., Arey R., Williams A., Landis J., Ashraf J., Murphy C.T.. The C. elegans adult neuronal IIS/FOXO transcriptome reveals adult phenotype regulators. Nature. 2016; 529:92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rugarli E.I., Di Schiavi E., Hilliard M.A., Arbucci S., Ghezzi C., Facciolli A., Coppola G., Ballabio A., Bazzicalupo P.. The kallmann syndrome gene homolog in C. elegans is involved in epidermal morphogenesis and neurite branching. Development. 2002; 129:1283–1294. [DOI] [PubMed] [Google Scholar]
- 68. McIntire S.L., Jorgensen E., Kaplan J., Horvitz H.R.. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993; 364:337–341. [DOI] [PubMed] [Google Scholar]
- 69. Komonyi O., Schauer T., Papai G., Deak P., Boros I.M.. A product of the bicistronic Drosophila melanogaster gene CG31241, which also encodes a trimethylguanosine synthase, plays a role in telomere protection. J. Cell Sci. 2009; 122:769–774. [DOI] [PubMed] [Google Scholar]
- 70. Borg R.M., Fenech Salerno B., Vassallo N., Bordonne R., Cauchi R.J.. Disruption of snRNP biogenesis factors tgs1 and pICln induces phenotypes that mirror aspects of SMN-Gemins complex perturbation in Drosophila, providing new insights into spinal muscular atrophy. Neurobiol. Dis. 2016; 94:245–258. [DOI] [PubMed] [Google Scholar]
- 71. Di Giorgio M.L., Esposito A., Maccallini P., Micheli E., Bavasso F., Gallotta I., Vernì F., Feiguin F., Cacchione S., McCabe B.D.et al.. WDR79/TCAB1 plays a conserved role in the control of locomotion and ameliorates phenotypic defects in SMA models. Neurobiol. Dis. 2017; 105:42–50. [DOI] [PubMed] [Google Scholar]
- 72. McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E.. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003; 162:919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hosseinibarkooie S., Peters M., Torres-Benito L., Rastetter R.H., Hupperich K., Hoffmann A., Mendoza-Ferreira N., Kaczmarek A., Janzen E., Milbradt J.et al.. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 2016; 99:647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tisdale S., Pellizzoni L.. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015; 35:8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lemm I., Girard C., Kuhn A.N., Watkins N.J., Schneider M., Bordonne R., Luhrmann R.. Ongoing U snRNP biogenesis is required for the integrity of Cajal bodies. Mol. Biol. Cell. 2006; 17:3221–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tycowski K.T., Aab A., Steitz J.A.. Guide RNAs with 5′ caps and novel box C/D snoRNA-like domains for modification of snRNAs in metazoa. Curr. Biol. 2004; 14:1985–1995. [DOI] [PubMed] [Google Scholar]
- 77. Jia D., Cai L., He H., Skogerbo G., Li T., Aftab M.N., Chen R.. Systematic identification of non-coding RNA 2,2,7-trimethylguanosine cap structures in Caenorhabditis elegans. BMC Mol. Biol. 2007; 8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Becker D., Hirsch A.G., Bender L., Lingner T., Salinas G., Krebber H.. Nuclear pre-snRNA export is an essential quality assurance mechanism for functional spliceosomes. Cell Rep. 2019; 27:3199–3214. [DOI] [PubMed] [Google Scholar]
- 79. Goldfarb K.C., Cech T.R.. 3′ terminal diversity of MRP RNA and other human noncoding RNAs revealed by deep sequencing. BMC Mol. Biol. 2013; 14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cho H.D., Tomita K., Suzuki T., Weiner A.M.. U2 small nuclear RNA is a substrate for the CCA-adding enzyme (tRNA nucleotidyltransferase). J. Biol. Chem. 2002; 277:3447–3455. [DOI] [PubMed] [Google Scholar]
- 81. Rizzo F., Nizzardo M., Vashisht S., Molteni E., Melzi V., Taiana M., Salani S., Santonicola P., Di Schiavi E., Bucchia M.et al.. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain. 2019; 142:276–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Doktor T.K., Hua Y., Andersen H.S., Broner S., Liu Y.H., Wieckowska A., Dembic M., Bruun G.H., Krainer A.R., Andresen B.S.. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 2017; 45:395–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jangi M., Fleet C., Cullen P., Gupta S.V., Mekhoubad S., Chiao E., Allaire N., Bennett C.F., Rigo F., Krainer A.R.et al.. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl Acad. Sci. USA. 2017; 114:E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Huo Q., Kayikci M., Odermatt P., Meyer K., Michels O., Saxena S., Ule J., Schumperli D.. Splicing changes in SMA mouse motoneurons and SMN-depleted neuroblastoma cells: evidence for involvement of splicing regulatory proteins. RNA Biol. 2014; 11:1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G.. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008; 133:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D.et al.. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012; 151:440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 88. Peabody N.C., Diao F., Luan H., Wang H., Dewey E.M., Honegger H.W., White B.H.. Bursicon functions within the Drosophila CNS to modulate wing expansion behavior, hormone secretion, and cell death. J. Neurosci. 2008; 28:14379–14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jia Y., Mu J.C., Ackerman S.L.. Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration. Cell. 2012; 148:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tisdale S., Lotti F., Saieva L., Van Meerbeke J.P., Crawford T.O., Sumner C.J., Mentis G.Z., Pellizzoni L.. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep. 2013; 5:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ruggiu M., McGovern V.L., Lotti F., Saieva L., Li D.K., Kariya S., Monani U.R., Burghes A.H., Pellizzoni L.. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol. Cell. Biol. 2012; 32:126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Simon C.M., Dai Y., Van Alstyne M., Koutsioumpa C., Pagiazitis J.G., Chalif J.I., Wang X., Rabinowitz J.E., Henderson C.E., Pellizzoni L.et al.. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017; 21:3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Simon C.M., Van Alstyne M., Lotti F., Bianchetti E., Tisdale S., Watterson D.M., Mentis G.Z., Pellizzoni L.. Stasimon contributes to the loss of sensory synapses and motor neuron death in a mouse model of spinal muscular atrophy. Cell Rep. 2019; 29:3885–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Van Alstyne M., Simon C.M., Sardi S.P., Shihabuddin L.S., Mentis G.Z., Pellizzoni L.. Dysregulation of mdm2 and mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 2018; 32:1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Son A., Park J.E., Kim V.N.. PARN and TOE1 constitute a 3′ end maturation module for nuclear non-coding RNAs. Cell Rep. 2018; 23:888–898. [DOI] [PubMed] [Google Scholar]
- 96. Lardelli R.M., Lykke-Andersen J.. Competition between maturation and degradation drives human snRNA 3′ end quality control. Genes Dev. 2020; 34:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cheng L., Zhang Y., Zhang Y., Chen T., Xu Y.Z., Rong Y.S.. Loss of the RNA trimethylguanosine cap is compatible with nuclear accumulation of spliceosomal snRNAs but not pre-mRNA splicing or snRNA processing during animal development. PLoS Genet. 2020; 16:e1009098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Madore S.J., Wieben E.D., Kunkel G.R., Pederson T.. Precursors of U4 small nuclear RNA. J. Cell Biol. 1984; 99:1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Madore S.J., Wieben E.D., Pederson T.. Intracellular site of U1 small nuclear RNA processing and ribonucleoprotein assembly. J. Cell Biol. 1984; 98:188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ezzeddine N., Chen J., Waltenspiel B., Burch B., Albrecht T., Zhuo M., Warren W.D., Marzluff W.F., Wagner E.J.. A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3′-end formation. Mol. Cell. Biol. 2011; 31:328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shukla S., Parker R.. Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion. Proc. Natl Acad. Sci. USA. 2014; 111:E3277–E3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Roithova A., Feketova Z., Vanacova S., Stanek D.. DIS3L2 and LSm proteins are involved in the surveillance of sm ring-deficient snRNAs. Nucleic Acids Res. 2020; 48:6184–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Reimao-Pinto M.M., Manzenreither R.A., Burkard T.R., Sledz P., Jinek M., Mechtler K., Ameres S.L.. Molecular basis for cytoplasmic RNA surveillance by uridylation-triggered decay in drosophila. EMBO J. 2016; 35:2417–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ramamurthy L., Ingledue T.C., Pilch D.R., Kay B.K., Marzluff W.F.. Increasing the distance between the snRNA promoter and the 3′ box decreases the efficiency of snRNA 3′-end formation. Nucleic Acids Res. 1996; 24:4525–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Guiro J., Murphy S.. Regulation of expression of human RNA polymerase II-transcribed snRNA genes. Open Biol. 2017; 7:170073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. So B.R., Wan L., Zhang Z., Li P., Babiash E., Duan J., Younis I., Dreyfuss G.. A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat. Struct. Mol. Biol. 2016; 23:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Venters C.C., Oh J.M., Di C., So B.R., Dreyfuss G.. U1 snRNP telescripting: suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harb. Perspect. Biol. 2019; 11:a032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhao D.Y., Gish G., Braunschweig U., Li Y., Ni Z., Schmitges F.W., Zhong G., Liu K., Li W., Moffat J.et al.. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016; 529:48–53. [DOI] [PubMed] [Google Scholar]
- 109. Kannan A., Bhatia K., Branzei D., Gangwani L.. Combined deficiency of senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018; 46:8326–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Reimer K.A., Mimoso C.A., Adelman K., Neugebauer K.M.. Co-transcriptional splicing regulates 3′ end cleavage during mammalian erythropoiesis. Mol. Cell. 2021; 81:998–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rosa-Mercado N.A., Zimmer J.T., Apostolidi M., Rinehart J., Simon M.D., Steitz J.A.. Hyperosmotic stress alters the RNA polymerase II interactome and induces readthrough transcription despite widespread transcriptional repression. Mol. Cell. 2021; 81:502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Alpert T., Straube K., Carrillo Oesterreich F., Herzel L., Neugebauer K.M.. Widespread transcriptional readthrough caused by nab2 depletion leads to chimeric transcripts with retained introns. Cell Rep. 2020; 33:108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ottesen E.W., Luo D., Seo J., Singh N.N., Singh R.N.. Human survival motor neuron genes generate a vast repertoire of circular RNAs. Nucleic Acids Res. 2019; 47:2884–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pagliarini V., Jolly A., Bielli P., Di Rosa V., De la Grange P., Sette C.. Sam68 binds Alu-rich introns in SMN and promotes pre-mRNA circularization. Nucleic Acids Res. 2020; 48:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Arnold M., Bressin A., Jasnovidova O., Meierhofer D., Mayer A.. 2021) A BRD4-mediated elongation control point primes transcribing RNA polymerase II for 3′-processing and termination. Mol. Cell. 81:3589–3603. [DOI] [PubMed] [Google Scholar]
- 116. Drexler H.L., Choquet K., Merens H.E., Tang P.S., Simpson J.T., Churchman L.S.. Revealing nascent RNA processing dynamics with nano-COP. Nat. Protoc. 2021; 16:1343–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Osman E.Y., Van Alstyne M., Yen P.F., Lotti F., Feng Z., Ling K.K., Ko C.P., Pellizzoni L., Lorson C.L.. Minor snRNA gene delivery improves the loss of proprioceptive synapses on SMA motor neurons. JCI Insight. 2020; 5,:e130574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kannan A., Jiang X., He L., Ahmad S., Gangwani L.. ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain. 2020; 143:69–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K.et al.. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017; 377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 120. Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De Vivo D.C., Yamashita M., Rigo F., Hung G., Schneider E.et al.. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016; 388:3017–3026. [DOI] [PubMed] [Google Scholar]
- 121. Darras B.T., Masson R., Mazurkiewicz-Beldzinska M., Rose K., Xiong H., Zanoteli E., Baranello G., Bruno C., Vlodavets D., Wang Y.et al.. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N. Engl. J. Med. 2021; 385:427–435. [DOI] [PubMed] [Google Scholar]
- 122. Baranello G., Darras B.T., Day J.W., Deconinck N., Klein A., Masson R., Mercuri E., Rose K., El-Khairi M., Gerber M.et al.. Risdiplam in type 1 spinal muscular atrophy. N. Engl. J. Med. 2021; 384:915–923. [DOI] [PubMed] [Google Scholar]
- 123. Chaytow H., Faller K.M.E., Huang Y.T., Gillingwater T.H.. Spinal muscular atrophy: from approved therapies to future therapeutic targets for personalized medicine. Cell Rep. Med. 2021; 2:100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Blatnik A.J. 3rd, McGovern V.L., Burghes A.H.M.. What genetics has told us and how it can inform future experiments for spinal muscular atrophy, a perspective. Int. J. Mol. Sci. 2021; 22:8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hosseinibarkooie S., Schneider S., Wirth B.. Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Expert Rev. Proteomics. 2017; 14:581–592. [DOI] [PubMed] [Google Scholar]
- 126. Sumner C.J., Crawford T.O.. Two breakthrough gene-targeted treatments for spinal muscular atrophy: challenges remain. J. Clin. Invest. 2018; 128:3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Talbot K., Tizzano E.F.. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017; 24:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Costa-Roger M., Blasco-Perez L., Cusco I., Tizzano E.F.. The importance of digging into the genetics of SMN genes in the therapeutic scenario of spinal muscular atrophy. Int. J. Mol. Sci. 2021; 22:9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wirth B. Spinal muscular atrophy: in the challenge lies a solution. Trends Neurosci. 2021; 44:306–322. [DOI] [PubMed] [Google Scholar]
- 130. Janzen E., Mendoza-Ferreira N., Hosseinibarkooie S., Schneider S., Hupperich K., Tschanz T., Grysko V., Riessland M., Hammerschmidt M., Rigo F.et al.. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain. 2018; 141:2343–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Edgar R., Domrachev M., Lash A.E.. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom Python scripts used for the analysis of 3′ RACE data are available at https://github.com/cmroake/NAR_TGS1.
The custom R scripts used for the analysis of transcriptome data produced via Illumina sequencing are available at https://github.com/roozbehdn/TGS1_SMN.
The RNAend-Seq data discussed in this publication have been deposited with links to BioProject accession number PRJNA628085 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
The Illumina and Nanopore RNA-Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (131) and are accessible through GEO Series accession numbers GSE189413 and GSE189138, respectively.