

Abstract
The PglZ family of proteins belongs to the alkaline phosphatase superfamily, which consists of metallohydrolases with limited sequence identity but similar metal-coordination architectures in otherwise divergent active sites. Proteins with a well-defined PglZ domain are ubiquitous among prokaryotes as essential components of BREX phage defence systems and two-component systems (TCSs). Whereas other members of the alkaline phosphatase superfamily are well characterized, the activity, structure and biological function of PglZ family proteins remain unclear. We therefore investigated the structure and function of PorX, an orphan response regulator of the Porphyromonas gingivalis TCS containing a putative PglZ effector domain. The crystal structure of PorX revealed a canonical receiver domain, a helical bundle, and an unprecedented PglZ domain, similar to the general organization of the phylogenetically related BREX-PglZ proteins. The PglZ domain of PorX features an active site cleft suitable for large substrates. An extensive search for substrates revealed that PorX is a phosphodiesterase that acts on cyclic and linear oligonucleotides, including signalling molecules such as cyclic oligoadenylates. These results, combined with mutagenesis, biophysical and enzymatic analysis, suggest that PorX coordinates oligonucleotide signalling pathways and indirectly regulates gene expression to control the secretion of virulence factors.
INTRODUCTION
The revolution in genome sequencing has revealed large numbers of protein sequences with entirely unknown biological roles as well as domains with a well-recognized fold or conserved amino acid sequence but no known function. One example is the PglZ family, which belongs to the alkaline phosphatase superfamily (APS) of metalloenzymes. APS proteins hydrolyse P–O, S–O and P–C bonds, and possess a similar metal coordination architecture at the active site despite their limited sequence identity (1). The APS includes well-characterized enzymes such as alkaline phosphatases, phosphodiesterases, phosphotransferases, phosphomutases, sulfamidases and sulfatases (2). However, no activity or biological function has been assigned to members of the PglZ family, even though the Pfam database contains 1744 sequences with this domain derived from 1424 prokaryote species (3). PglZ family proteins are encoded by various polycistronic operons with essential roles in the protection of bacteria against phage infection, including the well-known phage growth limitation (Pgl) and bacteriophage exclusion (BREX) systems (4–9). The PglZ domain is also found in many response regulator (RR) proteins of two-component systems (TCSs), which are essential signal transduction systems that allow bacteria to respond to environmental cues. However, a TCS featuring a PglZ family protein has yet to be characterized in detail.
Multiple TCSs responding to different stimuli are present in all prokaryotes, each featuring a signalling histidine kinase (HK) and corresponding RR protein. A signal detected by the HK sensory input domain leads to the autophosphorylation of a conserved His residue in the cytoplasmic domain, followed by the transfer of the phosphate group to a strictly conserved Asp residue in the receiver domain (RD) of the cognate RR (10). The phosphorylated RR usually activates an effector domain, which in >60% of TCSs is as a direct transcriptional regulator of target genes (10). Effector domains not only control transcription, but also perform many other well-characterized biological functions (11). However, one of the most remarkable exceptions is the PglZ effector domain, which has not been assigned a function. A PglZ effector domain is found in 75 species, including several oral commensals such as Prevotella spp. and periodontal pathogens such as Tannerella forsythia and Porphyromonas gingivalis (http://www.p2cs.org/) (12).
In P. gingivalis, the PorX protein is an orphan RR possessing a canonical RD and a PglZ effector domain. It is thought to regulate the secretion of virulence factors via the type 9 secretion system (T9SS), also known as PorSS or PerioGate (13,14). Most proteinaceous virulence factors are secreted via the T9SS (including the gingipains, which are essential for pathogenicity) and deletion of the porX gene renders the mutant avirulent (15). PorX is thought to interact with PorY, a classical (but orphan) signalling HK in the inner membrane (13,16). Three distinct (but not mutually exclusive) signal transduction pathways affecting the function of the T9SS have been proposed for PorX. First, an unknown stimulus is assumed to trigger the autophosphorylation of PorY, which transfers the phosphate to the conserved Asp residue in the RD of PorX, and the latter induces genes encoding components of the T9SS by binding to the extracytoplasmic function (ECF) sigma factor, SigP (13,17). Second, activated PorX has been shown to bind directly to the sigP and porT promoters, facilitating SigP-dependent transcription (15,18). However, this mechanism is controversial because PorX lacks a canonical DNA-binding motif (16). Notably, PorT is one of the essential components of the T9SS (19). Finally, activated PorX can bind to PorL, the inner membrane component of the T9SS, which enhances protein secretion (16). In all these pathways, the mechanistic involvement of the PglZ domain of PorX remains undetermined, despite the well conserved architecture of the catalytic site typical for APS proteins.
To shed light on the role of the PglZ domain in bacterial TCS pathways and in defence systems against phages, we solved the crystal structure of P. gingivalis PorX, determined the enzymatic activity of the PglZ domain and deciphered its molecular mechanism of action. These data not only reveal the biological activity of the PglZ family, but also define the novel regulatory functions of a RR protein, independent of DNA binding, involving the hydrolysis of key signalling molecules such as cyclic and linear oligoadenylates.
MATERIALS AND METHODS
Bacterial strains and general growth conditions
Porphyromonas gingivalis strain W83 was grown in enriched tryptic soy broth (eTSB) comprising 30 g/l trypticase soy broth, 5 g/l yeast extract, 5 mg/l hemin, 0.5 g/l l-cysteine, 2 mg/l menadione (pH 7.5) or on eTSB blood agar (eTSB medium containing 1.5% (w/v) agar and 4% (w/v) defibrinated sheep blood) at 37°C in an anaerobic chamber (Don Whitley Scientific) with an atmosphere of 90% nitrogen, 5% carbon dioxide and 5% hydrogen. Escherichia coli cloning strains were grown in lysogeny broth (LB) and on 1.5% agar LB plates, supplemented with 100 μg/ml ampicillin. P. gingivalis mutants were grown in the presence of 5 μg/ml erythromycin and/or 1 μg/ml tetracycline.
Expression and purification of wild-type PorX and mutants
The porX gene (KEGG locus tag: PG_0928) was amplified by PCR from genomic DNA extracted from P. gingivalis W83 using primers PorX_pGEX_BamHI_F and PorX_pGEX_SalI_R (Supplementary Table S1). The purified DNA fragment was transferred to the BamHI/SalI sites of pGEX-6P-1 and introduced into E. coli strain BL21 (DE3). The transformants were cultured until the OD600 reached ∼0.6 and the expression construct was induced with 0.2 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG). After incubation for 16 h at 20°C, the cells were collected by centrifugation (5000 × g, 15 min, 20°C), resuspended in PBS and lysed by passing once through a cell disruptor (Constant Biosystems) at 0.75 kW and 23 000 psi, followed by ultracentrifugation (200 000 × g, 45 min, 4°C). The supernatant was loaded onto a Glutathione Sepharose 4 Fast Flow column (Cytiva) equilibrated with PBS, washed with PBS, and the tag-free PorX protein was released by cleaving the GST-tag with PreScission Protease for 24 h at 4°C. The eluted protein was dialysed against 20 mM Bis–Tris (pH 6.5), loaded onto a Resource Q column (Cytiva) and eluted with a linear gradient of NaCl up to 1 M in 20 column volumes (CV). A final SEC step was performed in 5 mM Tris, 50 mM NaCl (pH 7.5) using a HiLoad 16/60 Superdex 200 column (Cytiva). A selenomethionine (SeMet) variant of PorX was similarly obtained, except that minimal medium containing Se-Met (Chem-Impex) was used instead of LB.
BeF3-induced dimerization of PorX
PorX was incubated in a reaction mixture containing 50 mM Tris, 150 mM NaCl, 5 mM BeSO4, 30 mM NaF and 15 mM MgCl2 (pH 8.0) for 1 h at room temperature to obtain the dimer. PorX incubated without with BeF3 is hereinafter described as the monomer. Dimerization was confirmed by SEC using a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with 10 mM Tris, 100 mM NaCl (pH 8.0) at a flow rate of 1 ml/min.
Ion/AcP-induced dimerization of PorX
PorX (100 μg) was incubated in buffer A (50 mM Tris, 150 mM NaCl, pH 8.0) supplemented with (i) 10 mM MgCl2; (ii) 43 μM ZnCl2; (iii) 10 mM MgCl2 + 43 μM ZnCl2; or (iv) 20 mM AcP, 10 mM MgCl2, for 1 h at room temperature. The mixture was loaded on a Superdex 200 Increase 10/300 GL column equilibrated with 20 mM Tris, 50 mM NaCl, pH 7.6 at a flow rate of (1 ml/min).
Reaction of PorX with 32P-AcP and [γ-32P]-ATP
32P-AcP was synthesized as previously described (20). Briefly, the reaction mixture including 2.5 U of E. coli acetate kinase (Merck), 10 μCi of [γ-32P]-ATP (3000 Ci/mmol, Hartmann Analytic) in reaction buffer containing 25 mM Tris, 60 mM KOAc, 10 mM MgCl2 (pH 7.6) was incubated for 20 min at room temperature. 32P-AcP was purified from acetate kinase by filtration through a micro concentrator (10 000 MW cut-off, Amicon).
PorX, PorX variants and BSA (negative control) at a concentration of 1 μM were incubated in 50 mM Tris, 150 mM NaCl, 3 μM ZnCl2 (pH 8.0) containing 1 μCi of 32P-AcP or 1 μCi of [γ-32P]-ATP for 1 h at room temperature. Samples were mixed with SDS loading buffer (without boiling) and subjected to SDS-PAGE in 10% polyacrylamide gels. After electrophoresis, gels were dried and exposed in Personal Molecular Imager FX (BIO-RAD). Densitometry data were generated using Quantity One 1-D Analysis Software (BIO-RAD).
Bis(4-nitrophenyl) phosphate phosphodiesterase activity assay
Phosphorylated PorX (P-PorX) was prepared by incubating PorX with buffer A supplemented with 20 mM AcP and 10 mM MgCl2 for 1 h at 37°C. The AcP and MgCl2 were then removed using Zeba spin desalting columns equilibrated with buffer A. The phosphodiesterase reaction was initiated by adding 50 μl 2 μM P-PorX (diluted in a test buffer, see below) to 50 μl 10 mM bis(4-nitrophenyl)phosphate (bis- pNPP, Merck) and monitoring the release of the reaction product (p-nitrophenol) for 90 min at 37°C by measuring the absorbance at 405 nm on a SpectraMAX microplate reader (Molecular Devices). The assay was performed in triplicate. For activity pH screening, P-PorX was incubated in 150 mM NaCl, 3 μM ZnCl2, 100 mM Tris, 50 mM MES, 50 mM acetic acid (pH 5.0–9.5) or CAPS buffer (pH 9.5–10.5) for 30 min at 37°C. For metal screening, P-PorX was incubated in buffer A containing 0.5 mM ZnCl2, CuCl2, MnCl2, MgCl2 or CaCl2. PorX was used as a control of the reaction.
Nucleotide cleavage assay
PorX was incubated in dimerization buffer (buffer A supplemented with 20 mM AcP, 10 mM MgCl2, 465 μM ZnCl2) for 1 h at 37°C. Zn2+ and AcP were then removed by SEC using a Superdex 200 Increase 10/300 GL column. P-PorX (1 μM) was incubated with 100 μM of the following nucleotides: adenosine 5′-monophosphate (AMP); adenosine 5′-diphosphate (ADP); adenosine 5′-triphosphate (ATP); guanosine 5′-monophosphate (GMP); guanosine 5′-diphosphate (GDP); guanosine 5′-triphosphate (GTP); 3′,5′-cyclic diguanylate (c-di-GMP); 3′,5′-cyclic diadenylate (c-di-AMP); adenosine-guanosine 3′,3′-cyclic monophosphate (3′,3′-cGAMP); adenosine-guanosine 2′,3′-cyclic monophosphate (2′,3′-cGAMP); 3′,5′-cyclic guanylate (3′,5′-cGMP); 3′,5′-cyclic adenylate (3′,5′-cAMP); cyclic triadenosine monophosphate (cA3); cyclic tetraadenosine monophosphate (cA4); cyclic hexaadenosine monophosphate (cA6); 5′-phosphoadenylyl-(3′→5′)-adenosine (pApA); 5′-phosphoadenylyl-(3′→5′)-guanosine (pApG); 5′-phosphoguanylyl-(3′→5′)-guanosine (pGpG); P1-(5′-adenosyl) P3-(5′-adenosyl) triphosphate (AP3A); P1-(5′-adenosyl) P3-(5′-adenosyl) tetraphosphate (AP4A); or P1-(5′-adenosyl) P4-(5′-guanosyl) tetraphosphate (AP4G). The reaction was carried out overnight at room temperature in buffer A supplemented with 10 mM MgCl2 (total volume = 70 μl). After centrifugation (16 000 × g, 10 min, 20°C) to remove precipitates, 50 μl of the sample was loaded onto an Aeris 5 μm PEPTIDE XB-C18 (150 × 4.6 mm) reversed-phase high-performance liquid chromatography (HPLC) column (Phenomenex) at a flow rate of 1.5 ml/min with a linear gradient (0–30% buffer 2 in 12 column volumes) using buffer 1 (10 mM triethylammonium acetate, pH 8.0) and buffer 2 (80% acetonitrile, 10 mM triethylammonium acetate, pH 8.0) in 12 CV.
UHPLC-MS analysis
Samples were analysed by ultrahigh-performance liquid chromatography/mass spectrometry (UHPLC-MS) on an Acquity I-class UPLC system and Synapt G2-Si Q-Tof mass spectrometer with an electrospray ion source (Waters) follow. Samples were loaded onto an ACQUITY UPLC 1.7 μm (2.1 × 100 mm) BEH Amide column (Waters) maintained at 45°C, followed by isocratic elution in a mobile phase of 10 mM NH4HCO3 (pH 9.0) in water and acetonitrile (1:2 v/v). The total analysis time was 6 min at a flow rate of 0.16 ml/min. The auto-sampler temperature was set to 8°C, and 2 μl of each sample was injected. A full MS scan in electrospray negative ionization mode (ESI–) was applied (capillary voltage = 2.5 kV, sampling cone = 40, source offset = 30, source temperature = 120°C, desolvation temperature = 450°C, desolvation gas flow = 800 l/h, cone gas flow = 40 l/h). Data were acquired in resolution mode using MassLynx v4.1. Leucine enkephalin (50 pg/μl) was used as a lock spray interface at a flow rate of 10 μl/min (m/z = 554.2615).
Site-directed mutagenesis of the porX gene
Mutations were introduced into the PorX-pGEX-6P-1 vector using primers with overhangs containing the required modifications (Supplementry Tables S1 and S2). The DNA was ligated using the NEBuilder HiFi DNA Assembly Cloning Kit (21). The resulting plasmids were analysed by DNA sequencing.
Crystallization, X-ray data collection and crystal structure determination
Crystallization screens were carried out at 20°C in a sitting drop setup, at the Barcelona Science Park Crystallization Platform. We mixed 300–400 nl of protein solution (PorX-SeMet in all cases) containing 5 mM Tris pH 7.5, 150 mM NaCl (PorX-BeF3 crystal) or 50 mM NaCl (PorX-HR and T272A-pGpG crystals) with the same volume of precipitant solution, using nano-dispensing robots and 96-well plates (MRC96T-PS). BeF3 reagents (at final concentrations of 15 mM MgCl2, 30 mM NaF, 5 mM BeSO4) were added directly either to the protein solution (PorX-BeF3 crystal) or to the precipitant solution (PorX-HR and T272A-pGpG crystals). Crystals were cryo-protected by adding 27.3% glycerol to the reservoir solution and dispensing a volume to the crystallization drop so that the final concentration was an estimated 18%. Crystals were harvested using cryo-loops (Molecular Dimensions) and were flash frozen in liquid nitrogen. PorX-BeF3 (at 2.3 mg/ml) crystallized in 100 mM glycine pH 8.0, 100 mM sodium formate and a polyethylene glycol (PEG) mixture (1.5% each of 20K, 10K, 8K and 6K). PorX-HR crystals appeared in 100 mM glycine pH 8.0–8.5, 100 mM sodium formate, 3–4% PEG 8K at a protein concentration of 12.8 mg/ml with 0.1 mM ZnCl2 added to the precipitant and 0.5 mM GMP to the cryo-protectant solution. Crystals grew as a cluster which was broken with CrystalProbes (Hampton Research) into single crystals after adding cryo-protectant. A second crystal from the PorX-HR crystal cluster was soaked for 20 min in solution containing 0.5 mM MnCl2. T272A-pGpG (12 mg/ml) crystallized like crystal PorX-HR but with 0.5 mM of ZnCl2 and pGpG. X-ray data were collected at the ALBA synchrotron (Cerdanyola del Vallès, Spain; T272A-pGpG) and at the European Synchrotron Radiation Facility, beamline ID29 (ESRF, Grenoble, France; PorX-BeF3 and PorX-HR) at 100 K with Pilatus 6M and Pilatus 6M-F detectors, respectively. PorX-BeF3 and T272A-pGpG crystals were collected at the Se-absorption edge (0.97893 and 0.97923 Å, respectively), whereas PorX-HR data were collected at the Zn2+ absorption edge (1.28202 Å). PorX-BeF3 was collected at a single position, PorX-HR at five positions with three kappa orientations, and T272A-pGpG at two positions with different kappa orientations. The Mn-soaked fragment from the PorX-HR crystal cluster was diffracted at the Mn (1.89608 Å) and Zn absorption edges to confirm metal identity.
Data were processed with XDS (22). Correction factors were applied, and multiple datasets were merged with XSCALE (22). PorX-BeF3 was further scaled with AIMLESS (23). PorX-HR and T272A-pGpG datasets were scaled, merged and truncated, and the resolution cut-off based on CC1/2 >50 was applied using ap_SCALE (Staraniso suite) (24). Structures were solved using the MR-SAD PHASER-EP pipeline (25) as implemented in the CCP4i suite (23). The model used for MR-SAD was built on a previous SAD solution using SHELX C/D/E (26) and several merged datasets. Models were refined and maps calculated with PHENIX.refine (27) and BUSTER.refine (28) by keeping unmerged Friedel pairs, alternating with manual model building and real space refinement using COOT (29). Figures and RMSD values were generated with UCSF Chimera (30) and COOT (29). The overall structures of PorX-HR and PorX-BeF3 are very similar, yet they provide complementary structural information. The LH subunit of PorX-BeF3 was fully traced (chain B and D) at loops Lα3b-α3c (amino acids 298–307) while these could not be traced in PorX-HR molB and molD. The high resolution of the PorX-HR data allowed us to unambiguously identify BeF3 at the PglZ active site, place metals and coordinating waters with confidence, and the strong anomalous signal from zinc permitted metal identification. Further, the PorX-HR structure shows two clear conformations of loop LβIc-β2 of the capping domain (aa 431–436) in molecules A and C, while in PorX-BeF3 the same loop could only be tentatively traced due to weak density. The dataset collected at the Mn absorption edge did not give any anomalous signal, so an Mg was placed at the RD active site.
Small angle X-ray scattering (SAXS)
The PorX monomer was concentrated using Amicon Ultra 0.5-ml centrifugal filters to concentration between 15.5 and 17 mg/ml and then diluted with appropriate buffers. Buffer A was used for PorX-monomer samples and BeF3-buffer (buffer A plus 5 mM BeSO4, 30 mM NaF and 15 mM MgCl2) for PorX + BeF3 dimer samples. The final concentrations of the PorX monomer were 0.5, 2.0 and 10 mg/ml. The final concentrations of the PorX + BeF3 dimer were 0.5, 1.0 and 2.0 mg/ml. Samples PorX + Zn2+ and PorX + BeF3 + Zn2+ were diluted to 0.5 mg/ml with buffer A or BeF3-buffer supplemented with 43 μM ZnCl2 (to avoid high zinc concentrations while still saturating all zinc binding sites) and concentrated to 2–3 mg/ml before redilution to 0.5, 1.0 and 2.0 mg/ml.
Samples were centrifuged before SAXS data collection, and collected in batch mode at the BM29 BIOSAXS beamline (ESRF Grenoble, France). Data were processed and analysed with BioXTAS RAW (31), which included data reduction, Guinier analysis, molecular weight (MW) estimation and indirect Fourier transform (IFT) analysis with GNOM (32) from the ATSAS 3.0 package (33). The MW was calculated based on the Porod volume (Vp) (34), or the volume of correlation (Vc) (35). For monomeric PorX, datasets at 0.5, 2 and 10 mg/ml were superimposed and corresponding regions with the best signal/noise ratio were selected and merged into a new curve. PorX + Zn2+ samples at 0.5 and 2 mg/ml showed different behaviour at the Guinier region and were processed separately, whereas the data at 1 mg/ml were discarded due to polydispersity. PorX + BeF3 data were best at 2 mg/ml, whereas PorX + BeF3 + Zn2+ data were best at 1 mg/ml. The datasets were processed and analysed, whereas low-q regions showing intramolecular interactions were discarded. Ab initio electron density was reconstructed with DENSS (36) using the IFT calculated with GNOM. For the electron density maps of PorX and BeF3 + PorX, 100 ab initio maps were calculated and averaged in slow mode with default parameters and refined against the P(r) function with an oversampling ratio of 5 to optimize map quality. The PorX monomer was modelled manually with CHIMERA v1.16 (30) against the DENSS electron density (37), and the model was then refined and validated with SREFLEX (38) and CRYSOL (39) with 1000 points of the computed SAXS curve, 33 maximum order of harmonics (as suggested by CRYSOL) and constant background subtraction.
Multi-angle laser light scattering (MALLS)
MALLS data were collected at the Barcelona Science Park Protein Purification Facility. A DAWN-HELEOS-II detector (Wyatt Technology) equipped with a 664.3-nm laser was coupled to a Superdex 200 Increase 10/300 GL column. We separated 50 μl of each sample at room temperature. The system was first equilibrated with the specific buffer and calibrated with bovine serum albumin (BSA). The samples included SeMet-PorX, protein variants D58A and D361A/H365A, and the corresponding phosphorylated forms. The running buffer contained buffer A plus 10 mM MgCl2 (or in some cases 20 mM AcP + 10 mM MgCl2 and/or 43 mM ZnCl2). Proteins were concentrated to between 4.6 and 6.9 mg/ml, as measured on a GE NanoVUE spectrophotometer (at 280 nm) and diluted with concentrator filtrate to 2.7 mg/ml for the Zn2+-containing samples, or 2.6 mg/ml for the other samples. Salts were then added from stock solutions diluting protein concentrations to 2.0 mg/ml. Phosphorylated samples were pre-incubated with 20 mM AcP for 1 h at 37°C before the corresponding run, whereas the Zn2+-containing samples were pre-incubated with 0.7 mM Zn2+. During incubation with Zn2+, partial protein precipitation was observed for the wild-type and D58A variant. Before injection, all samples were passed through 0.22-μm cut-off filters and kept at 4°C in the MALLS device autosampler. For each buffer, injections were done every 1.5 CV at a flow rate of 0.5 ml/min. Phosphorylated samples were injected first to minimize dephosphorylation.
PglZ comparison with BREX ortholog proteins
Protein sequences taken from the NCBI database included PorX (GenBank: BAG33538.1), BREX_PglZ_1_B (NF033450), BREX_PglZ_2 (NF033446), BREX_PglZ_3 (NF033449), BREX_PglZ_4 (NF033445), BREX_PglZ_5 (NF033444) and BREX_PglZ_6 (NF033443). For BREX-PglZs, we used the consensus sequences of all members of the corresponding protein family within the database. The 3D models were predicted using ColabFold, which is a combination of AlphaFold2 (40) and MMseqs2 (41). First, proteins were predicted as monomers, whose structures were then subdivided into smaller subdomains such as globular N-terminus, helical bundle, PglZ and for BREX-2, -4 and -6, an additional C-terminal domain. Predictions were limited by the number of 1000 residues. Therefore, in some cases, the prediction was calculated for fragments that overlapped at their ends, so that the structures of the fragments could be connected for further analysis. Sequences were aligned using the secondary structure predictions from the AlphaFold models, as well as predictions from JPRED 4.0 (41) in combination with subsequence alignments by T-coffee, MAFFT, MUSCLE and CLUSTAL Ω (42) followed by manual comparisons and readjustments in JALVIEW (43). JALVIEW was also used to calculate and export phylogenetic trees by average distance using Blosum62, and the trees were visualized using Dendroscope (44). The identity matrix was calculated using Clustal 2.1 (45) in test mode (without realignment). Structures/models were compared, aligned and visualized with UCSF CHIMERA (30).
PorX surface conservation
Representation of PorX surface conservation was done with USCF CHIMERA (30). Its 5 closest homologs within the PDB were structurally superposed at the APS catalytic core, sequences aligned and colored in a gradient by conservation. Zinc atom radii were enlarged to 2.5 Å to visualize buried Zn2 at the surface. Coloring thresholds used for identical residues were 100%, similar residues 75%, and not conserved 50% or below.
RESULTS
PorX dimerizes in the presence of acetyl phosphate
In a canonical TCS, the activation of a RR protein leads to phosphorylation within the RD, which often results in the formation of RR dimers, but this has not yet been demonstrated for RR proteins containing a PglZ domain (10). RD phosphorylation in vitro can be achieved by incubating the protein with low-molecular-weight phospho-donors such as acetyl phosphate (AcP) in the presence of Mg2+ (46). Using this approach, PorX was indeed phosphorylated with radiolabelled AcP (Figure 1A). Size-exclusion chromatography (SEC) confirmed that phosphorylation increased the Stokes radius, which usually indicates a conformational change or the formation of multimers (Figure 1B). We determined the absolute molecular weight (MW) of the protein by multi-angle laser light scattering (MALLS) (Figure 1C, D; Supplementary Table S3). The peak of non-phosphorylated PorX (theoretical MW = 61.1 kDa) corresponded to a monomer (MW = 70.2 kDa) whereas the phosphorylated sample corresponded to a dimer (MW = 118.2 kDa). Mg2+ alone did not induce multimerization (Figure 1C) (10). Due to lability of the acyl phosphate linkage, we incubated PorX with BeF3 to mimic phosphorylation since, in the crystallization drop, this compound makes a reversible ionic interaction with the RR active site in a stable equilibrium, enabling crystallization (47). Subsequent attempts to crystallize the PorX monomer did not yield any crystals after extensive trials, whereas PorX-BeF3 generated crystals that diffracted to a resolution of 1.9 Å (Supplementary Table S4).
Figure 1.

PorX phosphorylation. (A) Phosphorylation of wild type PorX (wt) with 32P-acetyl phosphate (32P-AcP). Bovine serum albumin (BSA) was used as a negative control. (B) Size exclusion chromatography (SEC) elution chromatogram of wild-type PorX (blue curve = non-phosphorylated, orange curve = phosphorylated with AcP) in the presence of Mg2+. (C, D) SEC-MALLS analysis to determine the absolute MW of wild-type PorX (wt) incubated with (C) MgCl2 alone (Mg2+), required for phosphorylation, and (D) AcP (wt-P) and MgCl2 (Mg2+). The mass fraction of the corresponding peak is shown in parentheses.
The PorX-BeF3 crystal structure is an intertwined dimer
The PorX-BeF3 crystal structure shows two dimers (AB and CD chains, respectively; A and C are structurally equivalent, and so are B and D) in the crystallographic asymmetric unit. Relevant conformational differences were found between monomers in a dimer (RMSD between molecules A/B = 1.32 Å, and between molecules C/D = 1.30 Å; differences between monomers are described below) but not between dimers (RMSD between molecules A/C = 0.469 Å, and between molecules B/D = 0.381 Å). We therefore chose dimer AB and molecule A (molA) as the representative structures (Figure 2A). PorX bears an N-terminal RD (Pro7–Ile128), followed by a three-helix bundle domain (HBD) comprising helices H1, H2 and H3 (Ile129–Arg211), and a C-terminal PglZ domain (Pro212–Lys518). Dimerization involves two interfaces, between the RDs and between the PglZ domains, and a binding energy of –39.9 kcal/mol according to QtPISA (23). The monomers intertwine, the RD of one subunit sitting on the HBD of the other (Figure 2A, C).
Figure 2.

Crystal structure of the PorX dimer. (A) The domains of one PorX monomer (molA) are shown in different colours whereas the entire second monomer (molB) is depicted in grey (except at the contacting LH region). At the N-terminus, the receiver domain (RD, in green) interacts via its α4-β5-α5 surface with the RD of molB, both RDs exposing the active sites at the top of the dimer (BeF3 and Mg2+ are shown as ball and stick models). The following three-helical bundle domain (HBD, in orange) interacts with both the RD of molB and the PglZ domain (in blue) of its own molecule. The PglZ domain contacts the PglZ domain of molB via two helices (HH, in pink) that are folded into a loop and helix in molB (LH, in yellow). The PglZ catalytic and cap subdomains are represented in light and dark blue, respectively. The N- and C-terminal ends in both subunits are indicated by N and C, respectively. (B) Close-up view of the RD active site. The residues that coordinate BeF3 (beryllium and fluorine atoms in pistachio and magenta, respectively) and Mg2+ (light green sphere) are shown as sticks and are labelled. Oxygen and nitrogen atoms are represented in red and blue, respectively. Polar contacts are represented as dashed lines. N59, N60, O60 and N87 correspond to the main-chain atoms of the corresponding residues. Water molecules are represented as light blue spheres. (C) Interface between the two RDs. In one monomer, the RD is represented in green and the HBD in orange, the other monomer is depicted in grey. The RD is followed by the HBD, whose α-helices are indicated (H1, H2 and H3). The arrow shows the kink in H1. (D) RD dimer from the top. The interface helices α4 and α5 and β-strand 5 (β5) are indicated. Tyr105 and Thr86 (in blue and green in molA and molB, respectively), BeF3 and Mg2+ are depicted. (E) Sharp bend between RD helix α5 and H1 from the HBD. The hydrophobic core that stabilizes the inter-subunit interaction involves the burial of molB Ile98 (helix α4) into the hydrophobic cavity built by molA Leu117, Lys120 and Lys121 methylene side chains, Gln124 from helix α5, and Gln125, Ile128 and Ile129 from helix H1. (D) and (E) use the same colour code as in (C).
The RDs of the PorX dimer adopt an activated conformation
Within the PorX dimer, both RDs display the canonical (α/β)5 doubly-wound fold, which consists of a central parallel five-stranded β-sheet (strands β1–β5) surrounded by α-helices (helices α1–α5). Helices α1 and α5 are found on one face of the β-sheet, whereas helices α2, α3 and α4 are found on the other. Each strand is connected to an α-helix by a loop (10) (Figure 2; Supplementary Figure S1). At the C-terminal end of the β-sheet, the connecting loops form a cavity that contains the highly conserved active site, which includes Asp14 and Asp15 (end of strand β1), Asp58 (β3 tip), Thr86 (β4 tip) and Lys108 (β5 tip) (Figure 2A, B; Supplementary Figure S1). These residues coordinate BeF3 and Mg2+ via interactions typically found in all activated RDs (47,48). The highly conserved Asp58 is found at the conserved phosphorylation position in RDs, and its terminal carboxyl oxygen Oδ1 contacts the beryllium of BeF3. In addition, Asp58 Oδ2 together with one BeF3 fluorine atom, Asp15 Oδ1 and a water molecule, form the equatorial coordination plane of Mg2+, which occupies the canonical position of this metal in RDs (see methods), and is necessary for phosphorylation (10) (Figure 2B, Supplementary Figure S1). The Mg2+ octahedral coordination sphere is fulfilled with Asn60 C=O (O60 in Figure 2B) and a second water molecule, which is contacted by Asp14 Oδ1. BeF3 is also stabilized by Thr86 Oγ1 and Nζ Lys108, and main-chain amides from Glu59, Asn60 and Lys87 (atoms N59, N60 and N87, respectively; Figure 2B).
Thr86 not only interacts with BeF3 but also forms the highly conserved TY pair (10,49) with Tyr105 in the centre of β5, at the RD dimer interface (Figure 2D). Crystal structures of activated RR dimers have extensively shown that, following phosphorylation, the side chain of the conserved Thr residue in the TY pair re-orients towards the active site phosphate, thereby creating a space that is occupied by the conserved Tyr residue (which is otherwise oriented to the solvent) (10). Because the Tyr residue is in the centre of β5, its reorientation causes the whole α4–β5–α5 surface to be remodelled. In the OmpR subfamily of RR proteins, this remodelling enables the formation of RD dimers (10,49). The PorX structure shows that RDs of molA and molB dimerize precisely via this surface (Figure 2A, C, D), with Tyr105 oriented towards Thr86, the latter providing the Oγ1 atom that contacts BeF3 (Figure 2B). This is consistent with the active conformation of the RDs.
The intermediate HBD joins the RD and PglZ domain
Beyond RD helix α5, the polypeptide chain bends by 80° (Figure 2A, C) and folds as helix H1 (Gln125–Met148) of the HBD. This bend creates a hydrophobic concavity occupied by molB helix α4 (Figure 2E). H1 also contacts molB RD helix α3 through an extensive network of water molecules. It kinks (black arrow in Figure 2C) to pack against the central hydrophobic core of the HBD, which also contains the buried H2 (Phe154–Gln173). H3 (Arg176–Ala206) contacts laterally the hydrophobic core while making extensive contacts with the PglZ domain (Figure 2A). Accordingly, whereas H1 contacts the other subunit's RD, the remaining HBD forms a block that sits on the PglZ domain of its own monomer.
The two PglZ active sites within the dimer adopt different conformations
The PglZ domain displays a canonical AP-core (α/β)6 fold as found in the APS (50), which consists of a β-sheet flanked by α-helices (Figure 3A). In PorX, a β-rich capping subdomain (51), hereafter termed cap subdomain, is inserted between β5 and α6, so the overall topology of the PglZ domain is α1–β1–α2–β2–α3(a–c)–β3–α4–β4–α5(a–d)–β5–cap–α6–β6 (Figure 3A; Supplementary Figure S1). The two PglZ domains from molA and molB are highly similar, except precisely at the conserved phosphodiesterase active site core residues (Figure 3A inset), in proximity to the dimerization interface, where there are striking structural differences. Specifically, the Phe359–Arg367 residues of molA fold as helix α5a (Figure 3B, in magenta) whereas the same residues in molB fold in a well-defined extended loop (Figure 3C, in yellow). The region Arg367–Ser370, which also belongs to helix α5a in molA, forms a new N-terminus in the downstream helix α5b in molB, which is described as helix α5b′. Therefore, molA features two consecutive helices (α5a–α5b, conformation HH), whereas molB features a loop and a helix (loop + α5b′, conformation LH). Furthermore, in helix α5a of molA, Asp361 Oδ1 and His365 Nϵ2 coordinate a Zn2+ ion (Zn1) (Figure 3B, Suppl. Figure S1), which is absent in molB due to the L loop conformation that relocates both side chains and places Met363 near to Zn1 instead (compare Figure 3B and C). In molA, Zn1 interacts with BeF3, which in turn coordinates a second Zn2+ (Zn2). Notably, BeF3 and Zn2 are also present in molB despite the absence of Zn1 (Figure 3B, C, Supplementary Figure S1). Both Zn1 and Zn2 were confirmed by anomalous diffraction at the Zn absorption edge (see methods and Suppl. Fig S1). The protein atoms that coordinate BeF3 in molA include Thr272 Oγ1 and the corresponding backbone amide (atom N272 in loop β3-α3a, also in molB; Figure 3B, C). In both molA and molB, Zn2 is also coordinated by Thr272 Oγ1, as well as Asp415 Oδ2 and His416 Nϵ2 (in the loop connecting to the cap subdomain at one side of the active site), and Asp239 Oδ1 (at the end of PglZ β1) (Figure 3B, C). Thr272 occupies the conserved catalytic Thr/Ser position of phosphodiesterases in the APS (1), whereas the two Zn2+ ions and BeF3 in molA occupy the same positions as the metals and phosphate found in many APS structures (1,50,52) (Supplementary Figure S2). Notably, Zn2+ ions were not added during any production nor crystallization step of PorX-BeF3, indicating that the metal was spontaneously acquired from E. coli. In the unprecedented PglZ dimer, the second Zn therefore reconstitutes the canonical bi-metal site of APS phosphodiesterases in molA, but in molB the site is disrupted due to the LH conformation.
Figure 3.
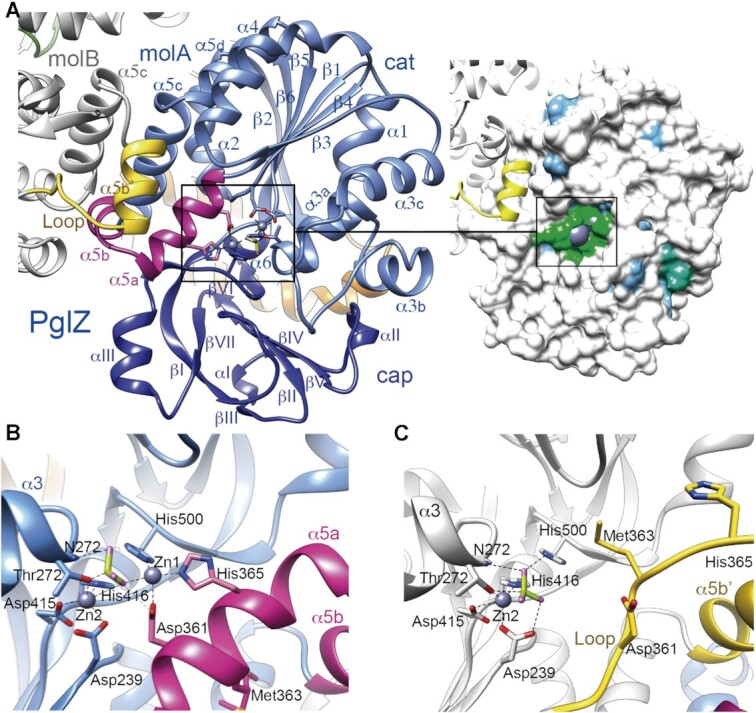
Crystal structure of the PglZ domain. (A) Left panel, Overall structure of the PglZ domain. Monomers A (molA, in blue) and B (molB, in grey) are shown. The catalytic (cat) and cap subdomains are shown in light and dark blue, respectively. Secondary structure elements are labelled. MolA helices α5a and α5b are depicted in magenta, whereas the same region in molB is folded into a loop and helix α5b′, shown in yellow. The two Zn2+ ions, BeF3 and coordinating side chains at the active site are depicted as ball and stick models. Note that without α5a–α5b, the root mean square deviation (RMSD) between molecules drops from 1.66 Å to 1.07 Å for Cα atoms 212–518. Right panel, the Connolly surface with the conservation map depicted on the PglZ domain, which shows low conservation to its five closest structural homologs (all of them are phosphodiesterases), except for the metal-binding residues. Identical amino acids are coloured in green, similar (75%) in blue and non-conserved (<50% in white). See also Supplementary Figure 2. (B) PglZ domain with helices α5a and α5b (HH conformation) in the PorX molA active site. (C) PglZ domain with the loop and helix α5b′ (LH conformation) in PorX molB. Panels (B) and (C) use the same colour code as in (A).
The contacts between PglZ domains not only involve helices α5a and α5b, but also the following helix α5c, which makes an antiparallel interaction with α5c from the other monomer. Remarkably, none of the other available APS structures dimerise by this surface, and despite they possess helix α5a (in which Asp361 and His365 are fully conserved for phosphoesterases; the active site amino acids are highly conserved among APS, see Figure 3A, Supplementary Fig S2) they do not show helices α5b and α5c (Supplementary Fig S2). In these other structures, a short turn connects α5a with a long helix that eventually coincides with the C-terminus of PorX α5d, suggesting a more rigid region. Thus, among APS members, α5b and α5c are PorX-specific insertions that configure the dimer interface.
The PorX RD and PglZ domains show extensive crosstalk
To gain more insight into the functional relationship between dimerization, phosphorylation, the binding of Mg2+ in the RD, and the binding of Zn2+ in the PglZ domain, we prepared a series of structure-guided protein variants mutated at the active site or dimerization interfaces, and analysed their ability to be phosphorylated by 32P-AcP and [γ-32P]-ATP (Figure 4A), as well as their behaviour as un- and phosphorylated proteins in SEC (Figure 4B–G; please see Figure 1B for SEC of the wt protein for comparison). Notably, RDs are generally phosphorylated by 32P-AcP (46), and the PglZ domain from APS phosphoesterases can be transiently phosphorylated at the conserved Thr/Ser residue (Thr272 in PorX) during the catalytic turnover of [γ-32P]-ATP (51,53,54). Consistent with this, we found that wild-type PorX bound efficiently to the radioactive moiety from both 32P-AcP and [γ-32P]-ATP (Figure 4A). Mutation of the RD phosphorylation site Asp58 (D58A) abolished phosphorylation by 32P-AcP and prevented dimerization (Figure 4A, B). Surprisingly, it also prevented phosphorylation by [γ-32P]-ATP (Figure 4A), suggesting that the RD regulates the PglZ domain. Accordingly, the mutation of residues involved in Zn-binding or hydrolysis (D361A/H365A and T272A, respectively) caused the anticipated complete loss of phosphorylation by [γ-32P]-ATP but also attenuated phosphorylation by 32P-AcP as well as dimerization, suggesting the PglZ domain can attenuate the effect of the RD (Figure 4A, C, D). Disruption of the interface between RDs (M94K/D104A/I129A) or PglZ domains (S385E/S389E, S385E/S389E/D361A/H365A) completely abolished dimerization but, interestingly, also blocked phosphorylation by 32P-AcP or [γ-32P]-ATP (Figure 4A, E–G). These results suggest that the activating signal at the catalytic site (of either the RD or PglZ domain) was transmitted to the corresponding dimerization surface. The converse was also true: intact interfaces were required for the active site to function. The two domains were therefore interdependent, with mutations causing functional impairment in one domain also disrupting the activity of the other.
Figure 4.
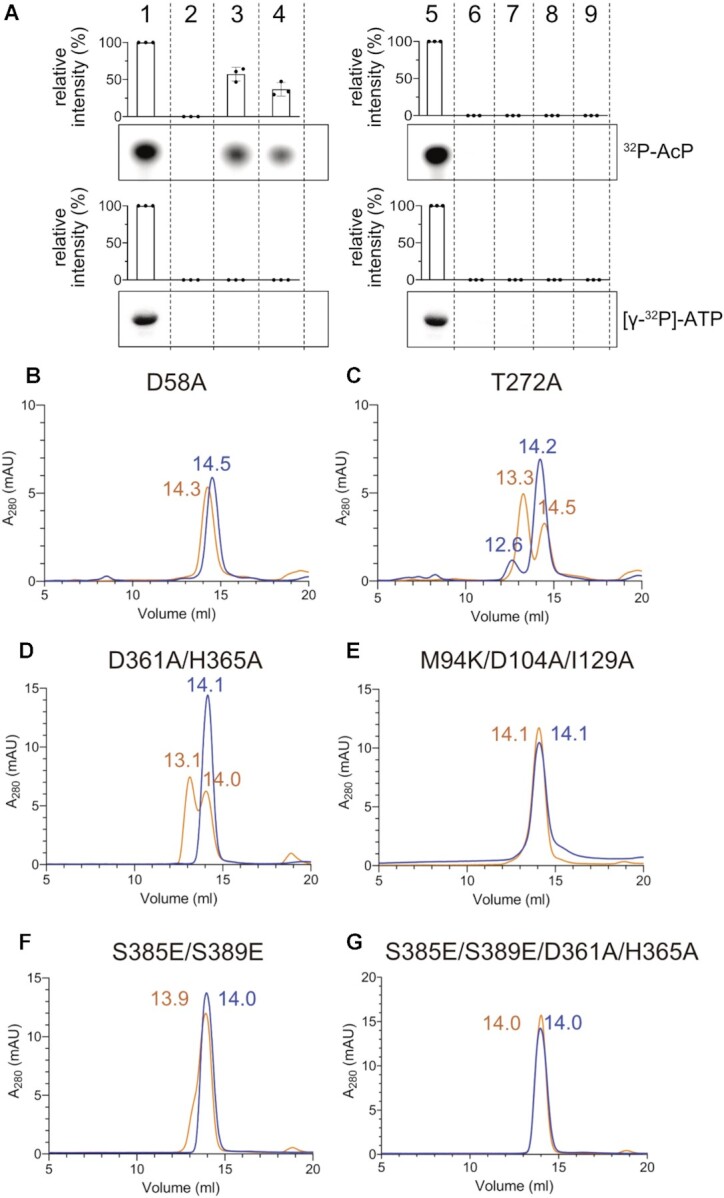
Phosphorylation of PorX mutants analysed by size-exclusion chromatography (SEC). The profiles represent PorX structure-based mutants at the RD or PglZ domain active sites or at domain interfaces following incubation with AcP (orange curves) and corresponding non-phosphorylated controls (blue curves). The SEC corresponding to the wild-type protein is shown in Figure 1B. (A) Representative radiograms showing phosphorylation of wild-type and mutant PorX with 32P-acetyl phosphate (32P-AcP, upper panel) or [γ-32P]-ATP (lower panel). Bovine serum albumin (BSA) was used as a negative control. Intensities were quantified by densitometry. The percentage values are intensities relative to the wt, which is set at 100%. Graphs show the mean of three independent replicates, error bars represent standard deviation (SD). Dots show the individual replicates. 1, wt; 2, D58A; 3, T272A; 4, D361A/H365A; 5, wt; 6, M94K/D104A/I129A; 7, S385E/S389E; 8, S385E/S389E/D361A/H365A; 9, BSA. (B) D58A mutated at the RD phosphorylation site. (C) T272A mutated at the PglZ domain active site. (D) D361A/H365A with mutated Zn-coordinating amino acids at the PglZ active site. (E) M94K/D104A/I129A mutated at the RD dimer interface. (F) S385E/S389E mutated at the PglZ domain dimer interface. (G) D361A and H365A with mutated PglZ Zn-coordinating ions were added to the substitutions in (F). Elution volumes are indicated above each peak.
To analyse the effect of Zn2+ in more detail, a three-fold molar excess of Zn2+ was added to the SEC buffer, which surprisingly induced the dimerization of wild-type PorX, RD mutants (D58A, M94K/D104A/I129A) and PglZ mutants (T272A), but neither D361A/H365A nor S385E/S389E were able to dimerize (Supplementary Figure S3). These results suggest that, by binding to Asp361 and His365, Zn1 facilitates PglZ dimerization by inducing the HH fold to create the appropriate interface contacts abolished by bulky residues in the S385E/S389E mutants. These effects were not observed in the presence of Mg2+ alone (Supplementary Figure S4). To confirm the nature of the SEC elution peaks, we used MALLS to compare the absolute MW of wild-type PorX and that of the D58A and D361A/H365A mutants (affecting the metal-binding sites) in the presence of Mg2+ and Zn2+, with or without phosphorylation. The results confirmed that the peak shifts induced by adding Zn2+ were due to dimerization (Supplementary Figure S5; Supplementary Table S3).
The PorX PglZ domain shows phosphodiesterase but not monophosphatase activity
RR proteins can be assigned to functional families by looking for similar architectures (55). We therefore screened for structural homology using DALI (56), and found that the top 12 closest structural homologs (Z-scores > 14.9) were phosphodiester hydrolases, phosphonoacetate hydrolases or alkaline phosphatases, and contained two zinc metal centres each (Supplementary Table S5). The highest-ranked hits (PhnA, XAC and Ecto-Nucleotide Pyrophosphatases/Phosphodiesterases, ENPPs 1 to 7) showed the closest structural similarity near the Zn2+-binding sites whereas the surrounding cavities were more diverse. Indeed, ENPPs hydrolyse mononucleotides, dinucleotides and (cyclic) polynucleotides (including Ap3A and Ap4A) and feature large active site clefts with several pockets similar to PorX, suggesting that PorX may accept dinucleotides or longer (cyclic) polynucleotides (Supplementary Figure S6). We therefore tested whether non-activated and AcP-activated PorX (P-PorX) were able to hydrolyse the phosphodiesterase substrate analogue bis(p-nitrophenyl) phosphate (bis- pNPP) in the presence of different divalent cations (Zn2+, Cu2+, Mn2+, Mg2+ and Ca2+) or without. Only Zn2+ stimulated the activity of PorX (Figure 5A, B), and this was dependent on the pH (Figure 5C, D). The optimal pH differed between PorX (pH 8–8.5) and P-PorX (pH 8.5–9), confirming that RD phosphorylation and dimerization alters the activity of PglZ bound to Zn2+ (Figure 5C, D). In contrast, there was no activity against the monophosphatase substrate p-nitrophenyl phosphate (pNPP) under any conditions (data not shown).
Figure 5.

Analysis of PorX phosphodiesterase activity. (A) Screening the phosphodiesterase activity of non-phosphorylated PorX in the presence of divalent cations and using bis- pNPP as a substrate, measuring the newly generated pNPP. The percentage values are activities relative to the reaction including Zn2+, which is set at 100%. (B) As in (A) but for phosphorylated PorX (P-PorX). (C) Analysis of PorX activity in the presence of Zn2+ using buffers with different pH values. Tris, MES and acetic acid activity points (from pH 4.0–9.5 are shown as dots, and CAPS (pH 9–10.5) as squares. (D) As in (C), with P-PorX. (E) Phosphodiesterase kinetics of PorX and PorX mutants at increasing concentrations of bis-pNPP, in the presence of Zn2+. (F) Same as in (E) but previously incubated with AcP and analysed by size-exclusion chromatography. The T272A, D361A/H365A, and S385E/S389E/D361A/H365A mutants showed no activity.
We next tested the cleavage kinetics of bis- pNPP for wild-type PorX and mutants affecting the active site and domain interfaces, in the presence of Zn2+. Apparently, the Zn-induced dimerization can induce enzymatic activity even in the absence of AcP. Therefore, the observed effect of AcP was only moderate and only slightly increased the reaction turnover rate (kcat) of wild-type PorX (Figure 5E, F; Supplementary Table S6). In contrast, AcP did not stimulate the activity of the D58A or M94K/D104A/I129A RD interface mutants (Figure 5E, F; Supplementary Table S6), confirming that dimeric PglZ activity depends on the RD. Mutating the PglZ phosphate binding site (T272A), PglZ interface (S385E/S389E) or the residues that coordinate Zn1 (D361A/H365A, which border the PglZ dimer interface) decreased the activity dramatically, in some cases to undetectable values (Figure 5E,F; Suppl. Table S6). AcP only slightly stimulated the PglZ interface mutants S385E/S389E, and no activity was detected for the other mutants. We concluded that PorX is a Zn2+-dependent phosphodiesterase by virtue of the PglZ domain.
The crystal structure of PorX in a complex with pGpG
We initially considered the cyclic dinucleotide c-di-GMP as a potential natural substrate for PorX because this is a major signalling molecule that controls motility, biofilm formation and virulence (57). P. gingivalis produces diguanylate cyclase PGN_1932 (58) that catalyses the synthesis of c-di-GMP, but it does not possess EAL or HD-GYP homologues, which degrade this cyclic dinucleotide (59,60). Both c-di-GMP and its derivative pGpG would fit in the wide active site of PorX, similar to mouse ENPP1 and rat ENPP3, which were crystallized with pApG and Ap4A, respectively (61,62) (Supplementary Figure S6). Therefore, we attempted to crystallize PorX and inactive variant T272A as complexes with c-di-GMP, pGpG and GMP, to catch any of the three steps of c-di-GMP cleavage, as well as GTP, GTPγS analogue and ATP, by co-crystallization or crystal soaking. Only pGpG appeared in the active site of the T272A mutant crystal structure, T272A-pGpG (Figure 6A, B; Supplementary Table S4). pGpG binds all four molecules (A and B chains in one dimer, C and D in the other, all of which with HH conformation) and the pGpG 5′ phosphate is coordinated by Zn1 and Zn2, similar to BeF3 in the unliganded structure (Figure 6A; Supplementary Figure S6A, S7). The presence of two Zn2+ ions in all molecules may reflect the high Zn2+ concentration (0.5 mM). However, PorX under the same conditions (0.5 mM Zn2+) but in the presence of c-di-GMP instead of pGpG crystallized as dimers with LH/HH conformation, without a bound nucleotide. Therefore, pGpG stabilized dimers with di-Zn and HH/HH conformation.
Figure 6.
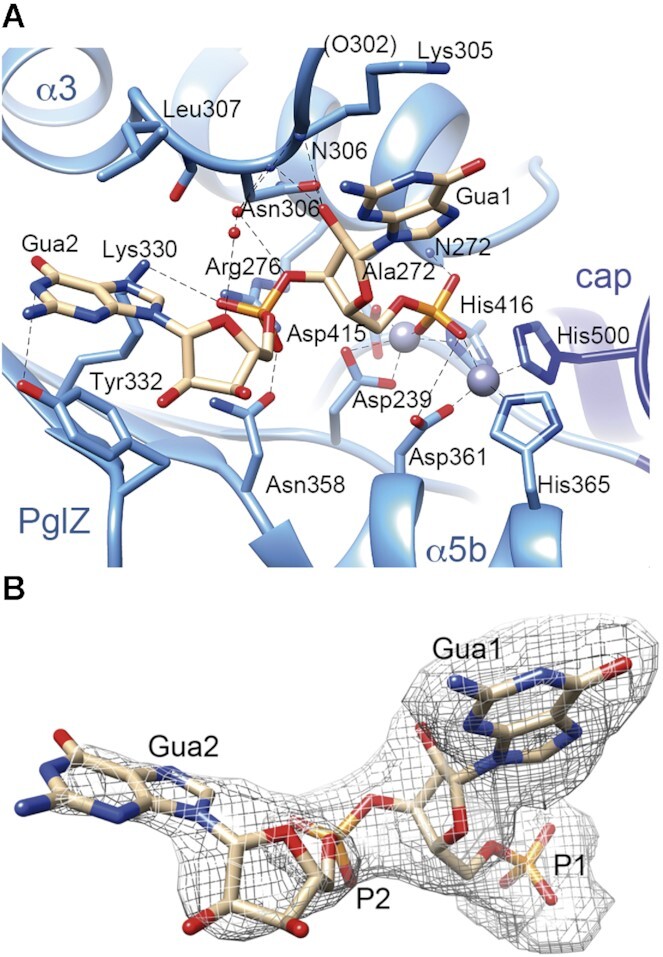
Analysis of nucleotide binding. (A) The pGpG molecule is bound at the PglZ domain active site of the T272A mutant, with interacting residues represented as sticks, water molecules as red spheres, and nitrogen and oxygen atoms coloured in blue and red, respectively. Phosphate is depicted in orange, and Zn2+ in grey. Electrostatic interactions are shown as dashed lines. Ala272 corresponds to the site of the T272A mutation. ‘cap’ refers to cap subdomain. (B) Structure of the active site-bound pGpG molecule surrounded by the 2Fo – Fc electron density map at 0.8 σ. Guanines 1 and 2 are indicated, together with the corresponding phosphates.
From the Zn binding site, pGpG emerges occupying part of the wide catalytic centre, which features several additional pockets (Supplementary Figure S6A). The 5’ guanine (Gua1) is located between pockets A and B, aligned with a loop between helices α3b and α3c (Lα3b-α3c, residues 297–307; Figure 6A). Notably, two guanine-specific atoms, Gua1 N2 and O6, are contacted by the protein. Gua1 N2 is contacted by main chain C=O Glu302 in molecules B, C and D (O302 in brackets in molA; Figure 6A), whereas Lys305, also at Lα3b-α3c, stacks its extended methylene chain against the guanine rings. In addition, Lys305 Nζ in molC contacts Gua1 O6, a contact that could easily be established also in molecules A, B and D with a slight reorientation of the side chain tip. In all molecules, the amide from Asn306 backbone (atom N306 in Figure 6A), at the end of loop Lα3b–α3c, contacts O2′ from the Gua1 sugar, further stabilizing the ligand in the active site. Note that in molB and molD from the ligand-free PorX structures with the LH conformation, loop Lα3b–α3c is highly flexible and difficult to trace (in molA and molC, the loop is stabilized by contacts with the second dimer) whereas in the T272A-pGpG structure, molB and molD show this loop stabilized by adding further contacts to the ligand. Gua2 extends along pocket A towards the protein surface (Figure 6A), but the electron density is well defined only at its phosphate (Figure 6B). Next to it, Asn358 and Arg276 contact each other, close to where a third metal ion (Mg2+) is found in the active sites of alkaline phosphatases (Figure 6A). This contact creates a platform, which is occupied by an electronegative phosphate oxygen from Gua2. Beyond the phosphate, the density around Gua2 purine rings becomes fainter close to the solvent. Gua2 is sandwiched between Leu307 and poorly-defined Tyr332, making stacking interactions with the latter that rotates the Tyr side chain by 60° compared to PorX-BeF3 structure (Supplementary Figure S7). Apart from the local changes, the main differences between PorX with and without bound pGpG were mostly found in the flexible loop of the cap subdomain (Ser432–Thr435; Suppl. Figure S1, S7) and are probably caused by crystal packing. The pGpG molecule does not fill all pockets of the active site cavity, as also shown in the structures of ENPP1-pApG and ENPP3-Ap4A (61,62) (Supplementary Figure S6A–C), suggesting the site accepts larger substrates or allows the reorientation of the ligand during catalysis.
P-PorX cleaves cyclic oligoadenylates and linear oligonucleotides
The structure of PorX in a complex with pGpG suggested that the physiological substrate could be a signalling nucleotide, so we screened the activity of PorX against a library of nucleotides (AMP, ADP, ATP, GMP, GDP, GTP, c-di-GMP, c-di-AMP, 2′,3′-cGAMP, 3′,3′-cGAMP, c-AMP, c-GMP, cA3, cA4, cA6, pApA, pApG, pGpG, AP3A, AP4A and AP4G) by high-performance liquid chromatography (HPLC). PorX showed moderate reactivity in the presence of the cyclic adenylates (cOAs) cA3, cA4 and cA6 (Figure 7A–C). The main product of these reactions was a linear oligoadenylate, resulting from a single cleavage (pApApA for cA3, pApApApA for cA4 and pApApApApApA for cA6), together with intermediate hydrolysis products en route to the final product AMP (Figure 7A–C; Supplementary Table S7). We used pApA as a positive control (Figure 7D) and tested guanine-containing linear dinucleotides such as pApG (Figure 7E) and pGpG (Figure 7F), which were cleaved to single nucleotides with similar efficiency, indicating that PorX also accepts guanine. PorX showed high selectivity for cyclic oligoadenylates or linear oligonucleotides because it did not cleave other nucleotides from the library (Supplementary Figure S8).
Figure 7.

HPLC analysis of nucleotides cleavage by PorX. Nucleotides: (A) cA3, (B) cA4, (C) cA6, (D) pApA, (E) pApG, (F) pGpG, were incubated with phosphorylated P-PorX (orange curves) or in reaction buffer without PorX (blue curves). New peaks following cleavage are indicated with labelled arrows.
The monomer in solution reveals the reorientation of helix H1, the cap subdomain and a highly flexible HH/LH region
We were unable to crystallize the PorX monomer, so we analysed the structure of PorX and PorX-BeF3 in solution by small-angle X-ray scattering (SAXS) (Figure 8, Suppl. Figure S9). The oligomeric state of PorX (monomer) did not change at protein concentrations up to 10 mg/ml according to the MW based on the Porod volume (Vp). In contrast, PorX-BeF3 was already a dimer at concentrations of 1 mg/ml (not shown) and 2 mg/ml (Supplementary Table S8). The dimerization of PorX was therefore concentration-independent but required activation. The monomers and dimers were both compact, non-flexible molecules (see Kratky plot in Supplementary Figure S9B). In the absence of BeF3, the addition of Zn2+ had different effects depending on the protein concentration, inducing the formation of dimers at 2 mg/ml but not at 0.5 mg/ml. The overall volume of all samples recorded as the radius of gyration (Rg, based on P(r) in Supplementary Table S8) was 29.6 Å for the monomer and 33.4 Å for the dimer. The Rg of the PorX-Zn2+ dimer was 39.0 Å and the largest maximum dimension (Dmax) was 118 Å (Supplementary Figure S9B; Supplementary Table S8). In contrast, the remaining monomers and dimers had similar Dmax values of 103 and 105 Å, respectively. These similar Rg and Dmax values are consistent with monomers of elongated shape that dimerize along the largest dimension, as we see in the X-ray structure. In contrast, Zn2+ ions stimulated a non-native dimerization of the non-phosphorylated monomers.
Figure 8.

Analysis of the PorX monomer in solution. (A) Experimental scattering-intensity curve (blue dots) represented on a logarithmic scale as a function of the momentum transfer. The curve of the fitted model (panel (B), in red) describes the complete q-range with a χ2 of 1.15. Below, the normalized residuals show the quality of fit along the q-range. (B) PorX monomer model fitted into the ab initio electron density map (loops excluded). (C) Superposition of the monomer in the crystallographic dimer onto the monomer in solution. The arrows show the main direction of the displacement for the RD (in green), helix 1 from the helical bundle domain (in yellow), the cap domain (in blue) and the dimer interface region (in red). Compared to (B), the superposition was slightly rotated (black arrow on the horizontal axis) for better visualization of the differences.
We then tested different models to find the one that fitted best the SAXS data obtained from the PorX monomer. We calculated theoretical scattering curves from molA and molB from the crystal structure, but they fitted the experimental data poorly, indicating significant structural deviations. In parallel, an ab initio 3D electron density map (not biased by any model) was calculated from the SAXS data (Supplementary Table S9). We tried to fit crystal monomers against such ab initio map but the HBD H1, PglZ cap subdomain and LH/HH segments of the Zn-binding site did not fit inside the density (Supplementary Figure S9). After relocating these regions by applying iterative calculations guided by automated domain border definition, we generated a final model that fitted precisely to both the ab initio map and experimental SAXS curve (Figure 8A, B). This model features a rotated RD that connects to the HBD via displaced helix H1 (Figure 8C, see Supplementary Figure S9 for details). The cap subdomain faces away from the PglZ dimerization surface and apparently covers the active site (Figure 8C). The SAXS map showed no electron density in the HH or LH region, which suggests high flexibility in the monomer (Supplementary Figure S9). Note that the cap subdomain contacts the HH/LH region in the crystal structure and probably stabilizes it, so the displacement of the cap subdomain in the soluble monomer likely affected the HH/LH region. In contrast, the crystallographic dimer model fitted reasonably the PorX-BeF3 data (Supplementary Figure S9E, F). Therefore, our SAXS results suggest that, during dimerization, RD and H1 are reoriented to contact the second monomer appropriately, whereas the cap subdomain near the PglZ domain is repositioned in close proximity to the HH or LH region at the dimer interface and increases its stability.
Comparison of the PglZ domain of PorX with BREX orthologues
Multiple sequence alignments and structural predictions were carried out using the AlphaFold algorithm to compare PorX with BREX PglZ homologues (40). The predicted 3D structure of the BREX proteins (Supplementary Figure S10) features three major domains: an N-terminal α/β domain followed by an all α-helix domain and an apparently larger C-terminal PglZ-domain, in some cases followed by an extra globular domain. The predicted fold of the PglZ domains in the BREX proteins was similar to the crystal structure of the PglZ domain in PorX (Suppl. Figure S10) despite the low sequence identity of 19–25% (Figure 9A, B). Notably, the BREX-3 PglZ domain is the most similar to PorX, which is consistent with the level of protein sequence conservation (Figure 9C, D). It shares the overall fold and secondary structures of PorX (Figure 9D). The structural similarity between PorX and BREX PglZ domains is especially important with regard to the core residues of this fold, namely L268, P269, T270 (Ser in BREX-1/5), T414 (Ser in BREX-2/3/4), G417, R438, F481 (Tyr in BREX-2), G501, G502, E507 and P511. This conservation extends to the metal-coordinating residues D239, T272, D361, H365 (Asp in BREX-2 and Glu in BREX-5/6), D415, H416 and H500 (Figure 9A). However, among the residues that bind the nucleotide in our pGpG structure, only Arg276 (Lys in BREX-1/4/5 and Met in BREX6) and Asn358 (Asp in BREX-1/3/4, Glu in Brex-6) are conserved, and these interact with Gua2 phosphate group, not coordinated by Zn2+ ions.
Figure 9.

Comparison of the primary and tertiary structures of the PglZ domains in PorX and the BREX1–6 PglZ proteins. (A) Multiple sequence alignment of the PorX PglZ domain with all PglZ domains of BREX-1 to BREX-6. (B) Percent identity matrix and (C) the phylogenetic tree from the primary sequence alignment. The BREX PglZ proteins are a highly divergent group with sequence identities within the PglZ domain of 19–30%, but the conserved fold predicted by AlphaFold 2 modelling nevertheless closely resembles the PglZ domain of PorX (Supplementary Figure S10). The BREX-3 PglZ shows the highest similarity to the PorX PglZ domain, with an identity of 25%. (D) Superposition of PorX-PglZ and BREX-3 PglZ. Regions with major differences are marked in colour in the structural superposition and as coloured rectangles in the alignment. The shared PglZ folds catalytic subdomain are coloured in blue and the cap subdomains are coloured in light blue.
We also launched the prediction of dimers for each BREX protein, and BREX-1 to 3 and BREX-6 yielded symmetric dimers. Strikingly, these models showed a dimerization interface similar to that of PorX, described above, with slightly different interaction modes (Supplementary Figure S10). For BREX-4 and 5, dimers were not predicted at this surface, yet a dimerization interface cannot be excluded. For BREX-1 and 6, the interface shows helices similar to PorX α5a, α5b and α5c, hereafter B-α5a, B-α5b, B-α5c (Supplementary Figure S10). B-α5b interacts with B-α5c* (* refers to the other monomer), yet B-α5c and B-α5d form a continuous helix. In addition, in BREX-1, B-α5a interacts with B-α5a*. In BREX-2, α5b equivalent is missing. A long loop (B-Lα5a-α5c) connects B-α5a to B-α5c and interacts with B-α5a*, B-α5c* and B-Lα5a-α5c*. In BREX-3, a short loop connects B-α5a to B-α5c and interacts with α5a* and α5c*, and both α5c make a strong antiparallel interaction as in PorX.
DISCUSSION
The PglZ domain is often found in the RR proteins of TCSs (11)—including PorX, which is required for the activation of the T9SS (18). In an apparently unrelated context, proteins containing a PglZ domain are necessary for the BREX anti-phage defence system (7,9). Here, we determined the crystal structure of PorX, its structure–function relationships, and the enzymatic activity of the PglZ domain. Biochemical experiments and mutagenesis revealed that phosphorylation at RD Asp58 regulates the dimerization of PorX via the RD and PglZ domain, as well as PglZ enzymatic activation. The structure of the dimer indicated that Zn2+ is present at both PglZ active sites, which adopt different conformations in both PorX-BeF3 and PorX-HR structures, which confirmed the findings. Specifically, two consecutive helices α5a and α5b at one edge of the active site feature the ‘HH conformation’ in one monomer, and these helices are remodelled as a loop and a longer helix α5b’ in the ‘LH conformation’ of the other monomer. Whereas the LH conformation shows a Zn bound at the active site, only HH features the canonical bi-metal (Zn2+) site typical for APS phosphodiesterases (1,50,52). This changed to a double-HH conformation following the binding of pGpG, suggesting that one HH conformation is sterically necessary for PorX dimerization, while ligand binding may induce the HH conformation in the other monomer.
In PorX, conserved Asp361 and His365 belong to helix α5a and bind the second Zn in the HH conformation. However, helix α5a is also present in all available structures of bimetallo- (two Zn ions) APS, which do not dimerize by this surface, therefore the transition from loop to helix α5a is required but seems not sufficient for PglZ dimerization in PorX. Notably, following helix α5a there are helices α5b and α5c that also contribute to the dimer interface. Interestingly, our SAXS data shows that precisely helices α5a, α5b and α5c are disordered in the monomer, suggesting rearrangement during dimerization, which might be facilitated by bi-metal Zn binding that stabilizes α5a in at least one subunit. Surprisingly, whereas in APS helices α5b and α5c are exclusive to PorX, a similar helical arrangement is found in the BREX dimers predicted by AlphaFold, which dimerize precisely by this region. This is an intriguing feature that might be related to a dimerization-dependent activity at the nearby active sites.
We found evidence for extensive crosstalk between the PorX domains. Phosphodiesterase activity was abolished by mutating the PglZ catalytic amino acid Thr272 (homologous to Ser/Thr in the active site of APS proteins) but also by disrupting the PglZ dimerization interface, which borders the active sites and probably influences their stability. The same phenomenon was observed for Asp58 and the RD interface. However, the effect propagated beyond the mutated domain. For example, mutation T272A at the PglZ active site impaired PorX dimerization induced by phosphorylation at Asp58. Conversely, mutation D58A in the RD prevented dimerization and phosphorylation at PglZ Thr272. Excess Zn2+ compensated for the disassembly of the D58A dimer and promoted phosphodiesterase activity. However, due to the limited intercellular concentration of free Zn2+ (63), this phenomenon probably does not contribute to the biological function of PorX in vivo. A previous crystallization study with PorX from Flavobacterium johnsoniae also showed crystals containing dimers of non-phosphorylated protein and without addition of Zn (64). Despite the corresponding structure not being available, this indicates that under their crystallization conditions dimers also might be formed spontaneously.
Structural homology searches revealed that the PorX catalytic center is similar to that of enzymes that cleave mononucleotides, dinucleotides and (cyclic) polynucleotides such as cyclic and linear oligoadenylates (Suppl. Table S5). The similarity was high for mouse ENPP1 and rat ENPP3, which have been crystalized with the dinucleotides pApG and Ap4A, respectively (61,62). Like the PglZ domain of PorX, ENPP1 and ENPP3 also feature broad active site clefts with connected pockets. The broad active site of PorX could also accommodate cyclic oligoadenylate (cOA) bases and, in all cases facilitate the flipping of adenosines between pockets, so that all phosphodiester bonds reach the catalytic site, as proposed for substrate catalysis in ENPP1 (61). By screening a library of nucleotides, we showed that dimeric PorX cleaves cOAs and linear oligoadenylates to NMPs. Considering these results, we can hypothesize that the T272A-pGpG structure is representative of a reaction state intermediate with incomplete substrate degradation due to the T272A mutation. All polynucleotides were linked 3’-5’. Therefore, the scissile bond was between the O3’ from one adenine and the P from the phosphate of the following adenine. PorX did not cleave mononucleotides, cyclic dinucleotides or other nucleotides. The cleavage mechanism of PorX must differ from that of ring nucleases, which do not require metals, and oligoribonucleases, which are Mn2+-dependent 3′→5′ exonucleases (65,66).
Cyclic oligoadenylates are generated by type III CRISPR-Cas systems and cyclic oligonucleotide-based anti-phage signalling systems (CBASS) (67–70). In type III CRISPR-Cas, when the effector complex detects foreign RNA, the Cas10 subunit converts ATP into adenylates cA2 to cA6 (68,69), which bind to the CARF domain of RNAse Csm6/Csx1 to induce the non-specific degradation of invading RNAs (68,69). On the other hand, each CBASS employs a cGAS/DncV-like nucleotidyltransferase (CD-NTase), which synthesizes a nucleotide second messenger (including cyclic trinucleotides) in response to viral infection (67,70) Subsequently, the nucleotide messenger activates an associated Cap effector protein which kills the host cell and, as a consequence, blocks phage propagation (71). All sequenced P. gingivalis strains encode at least one CRISPR-Cas system, but only 36% possess a type III CRISPR-Cas that synthesizes cOAs. Moreover, P. gingivalis strains do not encode CD-NTase nor any known effector of CBASS. Therefore, unless there are other, yet unknown cOA synthesis pathways, it seems unlikely these molecules play a critical role in PorX-regulated signalling. Given that the BREX system and recently discovered associated genes including brxU (72) and brxR (73,74) are also absent in P. gingivalis, it is puzzling that bacteriophages have not yet been shown to infect P. gingivalis, which would indicate the presence of a resistance system (75). Because cOAs regulate defence systems, it is tempting to speculate that they activate an unknown anti-phage mechanism involving PorX, and this should be investigated in more detail.
In addition to cOAs, PorX also cleaves linear oligoadenylates (Figure 7A–C) and ribodinucleotides (Figure 7D–F) to mononucleotides. Accordingly, PorX may facilitate RNA turnover, the cycle of transcription and RNA degradation used by cells when they adjust to environmental changes, and this would match the role of PorX as a RR protein. In E. coli, mRNA decay is initiated by the endonucleolytic activity of RNase E, followed by 3′→5′ exonucleolytic degradation catalysed by other enzymes (76). The exonucleases cannot complete the degradation cycle because they are unable to cleave dinucleotides through to pentanucleotides (nanoRNAs), which are digested by oligoribonucleases such as Orn in E. coli (77), NrnA and NrnB in Bacillus subtilis (78), and NrnC in Bartonella spp. (79) NanoRNAs are not only the intermediate products of RNA decay, they are also produced during abortive transcriptional initiation (80) and the single-step cleavage of cyclic dinucleotide second messengers (65,66). NanoRNAs play important roles in cellular physiology including the priming of transcriptional initiation (81), changing promoter-specific gene expression (81), and changing auto-aggregation and biofilm phenotypes by inhibiting enzymes that break down c-di-GMP (66). P. gingivalis W83 encodes NrnA homologue PG_1091, but this does not exclude the possibility that the oligoribonuclease activity of PorX is physiologically relevant, because many bacteria produce multiple oligoribonucleases with redundant functions (78). It is tempting to speculate that an additional route of PorX-dependent gene regulation involves the cleavage of nanoRNAs.
In P. gingivalis, PorX has been associated with PorY (13,16), an orphan signalling HK with a periplasmic sensor domain. However, there is little phenotypic overlap between the ΔporX and ΔporY mutants (18,82), making it unlikely that a PorY/PorX TCS regulates the secretion of virulence factors. Even so, it is possible that PorX and PorY operate as a TCS that regulates T9SS activity in response to environmental cues in other Bacteroidetes. Notably, although P. gingivalis produces only five or six HKs (depending on the strain) including the two orphan proteins PorY and GppX (12), we cannot exclude the possibility that another HK is involved. Alternatively, PorX phosphorylation may depend on an as yet unknown Asp-kinase or AcP. Interestingly, several orphan RR proteins are activated by AcP in vivo (83). The concentration of AcP in P. gingivalis is not known, but in some bacteria such as E. coli it can reach at least 3 mM which suffices for the efficient autophosphorylation of RR proteins (84).
To the best of our knowledge, PorX is unique among RR proteins by combining enzymatic and protein-binding capabilities, both of which depend on PorX phosphorylation and dimerization. PorX regulates the expression of T9SS genes by interacting with the ECF sigma factor SigP (13). In E. coli, a similar case occurs between the RR RssB and the stress-inducible sigma factor RpoS, which regulates the expression of ∼500 genes (85). The C-terminal effector domain of RssB features a PPM/PP2C family phosphatase fold, which it is enzymatically inactive, but binds to RpoS and triggers its degradation. Like the C-terminus of RssB, the C-terminus of PorX may be involved in the interaction with SigP. Specifically, parts of the PglZ cap subdomain or with the PorX-PglZ dimerization interface, which is disordered in the monomer. Despite many similarities, the RssB and PorX RR proteins might have opposite effects on their cognate sigma factors, with RssB marking RpoS for proteolysis but PorX stabilizing SigP (13). Moreover, the phosphorylation of RssB strengthens its interaction with RpoS but the impact of PorX phosphorylation on its interaction with SigP is unknown. PorX also interacts with PorL, which forms an inner membrane complex with PorM to provide energy for the T9SS (16,86). This interaction is reminiscent of CheY binding to the FliM and FliN switch complex proteins of the flagellar C-ring, thus reversing the rotational direction of the filament (87). Indeed, the T9SS was shown to promote the rotary movement of the SprB filamentous adhesin on the cell surface of Flavobacterium johnsoniae (88). Moreover, the binding of CheY to FliN is mediated by a hydrophobic patch on the surface of FliN and a similar patch in PorL is necessary for the interaction with PorX (16).
Although the protein–protein interactions of PorX are well documented, its DNA-binding activity is more controversial. This has been investigated in two independent studies with contradictory results (16,18). The original study reported that full-length PorX does not bind the promoter regions of T9SS-related genes, including porT, sov and porP (13,16), whereas more recent experiments identified two PorX-binding sites in the porT promoter region (18). The structural data obtained herein confirm that PorX lacks canonical DNA-binding motifs, thus a potential mechanism of gene expression regulation by interacting directly with promoters remains unclear. So far, this function has been clearly proven for SigP, which binds directly to the promoter region of porT (13).
Our analysis of sequenced bacterial genomes revealed that PorX is limited to the Fibrobacteres–Chlorobi–Bacteroidetes (FCB) group with most examples found in the phylum Bacteroidetes. This agrees with the phylogenetic distribution of 19 components of the T9SS, which is characterized by monophyletic clades representing all Bacteroidetes classes, and PorX is always present even in species possessing only the core subset of T9SS components (89). This argues that PorX is indispensable for the regulation of T9SS activity. In contrast, although the BREX system is widely distributed across bacterial phyla, it is only sporadically found in the phylum Bacteroidetes, with the exception of the genus Prevotella. Furthermore, whereas PorX and its orthologues are composed of 500–530 amino acids, BREX proteins featuring a PglZ domain are composed of at least 600. Despite the low level of identity between PorX and BREX-PglZ domains (19–25%), the latter possess a remarkably conserved fold with a high degree of similarity to the PglZ domain of PorX. The BREX-PglZ proteins are therefore phylogenetically related to PorX, and it is fascinating that although PorX and its closest homologue BREX-3 apparently evolved from a common ancestor, the BREX-3 system is not found in Bacteroidetes (7) whereas PorX is present in all classes within this phylum (89).
PorX and similar RR proteins in other bacteria are apparently important components of complex signalling pathways that involve protein–protein interactions and the cleavage of signalling nucleotides to modulate the activity of the T9SS. Our structural and functional characterization of PorX provides insight into the biological activity of orphan RR proteins from TCSs, which is based on the specific hydrolysis of cOAs and linear oligoadenylates by the PlgZ domain. This regulatory function might be independent from the interaction of PorX with SigP and PorL. It is tempting to speculate that protein–protein interactions directly affect T9SS activity, whereas the enzymatic activity of the PglZ domain is functionally related to the BREX PglZ proteins, as revealed by their sequence conservation and close but non-overlapping phylogenetic relationship. In the future, we will focus on AcP as a hypothetical regulator of PorX activity, the potential crosstalk between CRISPR-Cas and PorX, and the role of cOAs and/or linear oligoadenylates in the physiology of P. gingivalis and the function of PglZ-domain proteins in the BREX-3 system.
DATA AVAILABILITY
The data supporting the findings of this study are available from the corresponding authors upon reasonable request. Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 7PV7 (PorX-BeF3), 7PVA (PorX-HR) and 7PVK (PorX-pGpG).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank G. Tamulaitis and I. Mogila for reagents and preliminary mass spectrometry experiments. We also thank T. Kantyka and E. Bielecka for help in designing HPLC experiments, M. Bochtler for critical reading of the manuscript and comments, and A.M. Stock for BeF3 reaction protocol. We thank the personnel at synchrotrons ESRF (Grenoble, France), ALBA (Cerdanyola del Vallès, Spain) and from the Automated Crystallography Platform (IBMB-CSIC) for their highly valuable support.
Contributor Information
Claus Schmitz, Department of Structural Biology, Molecular Biology Institute of Barcelona, CSIC, Barcelona Science Park, Barcelona E-08028, Spain.
Mariusz Madej, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland.
Zuzanna Nowakowska, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland.
Anna Cuppari, Department of Structural Biology, Molecular Biology Institute of Barcelona, CSIC, Barcelona Science Park, Barcelona E-08028, Spain.
Anna Jacula, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland.
Miroslaw Ksiazek, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland.
Katarzyna Mikruta, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland.
Jerzy Wisniewski, Department of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wroclaw University of Science and Technology, Wroclaw PL-50-370, Poland.
Natalia Pudelko-Malik, Department of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wroclaw University of Science and Technology, Wroclaw PL-50-370, Poland.
Anshu Saran, Department of Anatomy and Cell Biology, McGill University, Montréal, Quebec H3A 0C7, Canada.
Natalie Zeytuni, Department of Anatomy and Cell Biology, McGill University, Montréal, Quebec H3A 0C7, Canada.
Piotr Mlynarz, Department of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wroclaw University of Science and Technology, Wroclaw PL-50-370, Poland.
Richard J Lamont, Department of Oral Immunology and Infectious Diseases, School of Dentistry, University of Louisville, Louisville, KY 40202, USA.
Isabel Usón, Department of Structural Biology, Molecular Biology Institute of Barcelona, CSIC, Barcelona Science Park, Barcelona E-08028, Spain; ICREA Institució Catalana de Recerca i Estudis Avançats, Barcelona E-08010, Spain.
Virginijus Siksnys, Institute of Biotechnology, Vilnius University, Vilnius 10257, Lithuania.
Jan Potempa, Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków PL-30-387, Poland; Department of Oral Immunology and Infectious Diseases, School of Dentistry, University of Louisville, Louisville, KY 40202, USA.
Maria Solà, Department of Structural Biology, Molecular Biology Institute of Barcelona, CSIC, Barcelona Science Park, Barcelona E-08028, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Centre, Poland [UMO-2015/19/N/NZ1/00322 to M.M., UMO-2016/23/N/NZ1/01513 to Z.N., UMO-2018/31/B/NZ1/03968 to J.P., UMO-2019/35/B/NZ1/03118 to M.K.]; Spanish Ministry of Science, Innovation and Universities [MCIN/AEI/ 10.13039/501100011033 ERDF ‘A way to make Europe’, BFU2015-70645-R, RTI2018-101015-B-100, PID2021-129038NB-I00, MDM-2014–0435 to M.S., BES-2013–063407 to C.S., PGC2018-101370-B-I00 and PID2021-128751NB-I00 (MICINN/AEI/FEDER/UE) to I.U.]; Generalitat de Catalunya [2014-SGR-99, 2017-SGR-1192 to M.S.]; European Social Fund [09.3.3-LMT-K-712-01-0126 to V.S.]; National Institute of Dental and Craniofacial Research (NIDCR) [DE012505, DE011111 to R.J.L.]; Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology. Funding for open access charge: National Science Centre, Poland [UMO-2018/31/B/NZ1/03968].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zalatan J.G., Fenn T.D., Herschlag D. Comparative enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J. Mol. Biol. 2008; 384:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galperin M.Y., Jedrzejas M.J.. Conserved core structure and active site residues in alkaline phosphatase superfamily enzymes. Proteins. 2001; 45:318–324. [DOI] [PubMed] [Google Scholar]
- 3. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chinenova T.A., Mkrtumian N.M., Lomovskaia N.D. [Genetic characteristics of a new phage resistance trait in streptomyces coelicolor A3(2)]. Genetika. 1982; 18:1945–1952. [PubMed] [Google Scholar]
- 5. Sumby P., Smith M.C.. Genetics of the phage growth limitation (Pgl) system of streptomyces coelicolor A3(2). Mol. Microbiol. 2002; 44:489–500. [DOI] [PubMed] [Google Scholar]
- 6. Makarova K.S., Wolf Y.I., Snir S., Koonin E.V.. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011; 193:6039–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldfarb T., Sberro H., Weinstock E., Cohen O., Doron S., Charpak-Amikam Y., Afik S., Ofir G., Sorek R.. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015; 34:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoskisson P.A., Sumby P., Smith M.C.M.. The phage growth limitation system in streptomyces coelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology. 2015; 477:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordeeva J., Morozova N., Sierro N., Isaev A., Sinkunas T., Tsvetkova K., Matlashov M., Truncaite L., Morgan R.D., Ivanov N.V.et al.. BREX system of escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 2019; 47:253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao R., Bouillet S., Stock A.M.. Structural basis of response regulator function. Annu. Rev. Microbiol. 2019; 73:175–197. [DOI] [PubMed] [Google Scholar]
- 11. Galperin M.Y. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 2006; 188:4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mattos-Graner R.O., Duncan M.J.. Two-component signal transduction systems in oral bacteria. J. Oral Microbiol. 2017; 9:1400858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kadowaki T., Yukitake H., Naito M., Sato K., Kikuchi Y., Kondo Y., Shoji M., Nakayama K.. A two-component system regulates gene expression of the type IX secretion component proteins via an ECF sigma factor. Sci. Rep. 2016; 6:23288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lasica A.M., Ksiazek M., Madej M., Potempa J.. The type IX secretion system (T9SS): highlights and recent insights into its structure and function. Front. Cell Infect. Microbiol. 2017; 7:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang C., Yang D., Hua T., Hua Z., Kong W., Shi Y.. A PorX/PorY and sigma(P) feedforward regulatory loop controls gene expression essential for porphyromonas gingivalis virulence. Msphere. 2021; 6:e0042821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vincent M.S., Durand E., Cascales E.. The PorX response regulator of the porphyromonas gingivalis PorXY two-component system does not directly regulate the type IX secretion genes but binds the PorL subunit. Front. Cell Infect. Microbiol. 2016; 6:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yukitake H., Shoji M., Sato K., Handa Y., Naito M., Imada K., Nakayama K.. PorA, a conserved C-terminal domain-containing protein, impacts the PorXY-SigP signaling of the type IX secretion system. Sci. Rep. 2020; 10:21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang D., Jiang C., Ning B., Kong W., Shi Y.. The PorX/PorY system is a virulence factor of porphyromonas gingivalis and mediates the activation of the type IX secretion system. J. Biol. Chem. 2021; 296:100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato K., Sakai E., Veith P.D., Shoji M., Kikuchi Y., Yukitake H., Ohara N., Naito M., Okamoto K., Reynolds E.C.et al.. Identification of a new membrane-associated protein that influences transport/maturation of gingipains and adhesins of porphyromonas gingivalis. J. Biol. Chem. 2005; 280:8668–8677. [DOI] [PubMed] [Google Scholar]
- 20. Quon K.C., Marczynski G.T., Shapiro L.. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell. 1996; 84:83–93. [DOI] [PubMed] [Google Scholar]
- 21. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W Xds. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A.et al.. Overview of the CCP4 suite and current developments. Acta. Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tickle I.J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G.. 2018; Cambridge, United Kingdom: Global Phasing Ltd. [Google Scholar]
- 25. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uson I., Sheldrick G.M.. An introduction to experimental phasing of macromolecules illustrated by SHELX; new autotracing features. Acta. Crystallogr. D. Struct. Biol. 2018; 74:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta. Crystallogr. D Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O.S., Vonrhein C.et al.. 2017; Cambridge, United Kingdom: Global Phasing Ltd. [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of coot. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 31. Hopkins J.B., Gillilan R.E., Skou S.. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017; 50:1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992; 25:495–503. [Google Scholar]
- 33. Manalastas-Cantos K., Konarev P.V., Hajizadeh N.R., Kikhney A.G., Petoukhov M.V., Molodenskiy D.S., Panjkovich A., Mertens H.D.T., Gruzinov A., Borges C.et al.. ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021; 54:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piiadov V., Ares de Araujo E., Oliveira Neto M., Craievich A.F., Polikarpov I.. SAXSMoW 2.0: online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein Sci. 2019; 28:454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rambo R.P., Tainer J.A.. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013; 496:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grant T.D. Ab initio electron density determination directly from solution scattering data. Nat. Methods. 2018; 15:191–193. [DOI] [PubMed] [Google Scholar]
- 37. Goddard T.D., Huang C.C., Ferrin T.E.. Visualizing density maps with UCSF chimera. J. Struct. Biol. 2007; 157:281–287. [DOI] [PubMed] [Google Scholar]
- 38. Panjkovich A., Svergun D.I.. Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys. 2016; 18:5707–5719. [DOI] [PubMed] [Google Scholar]
- 39. Svergun D.I., Barberato C., Koch M.H.J.. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 40. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drozdetskiy A., Cole C., Procter J., Barton G.J.. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015; 43:W389–W394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D.et al.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J.. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huson D.H., Scornavacca C.. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012; 61:1061–1067. [DOI] [PubMed] [Google Scholar]
- 45. Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R.et al.. Clustal w and clustal x version 2.0. Bioinformatics. 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 46. Lukat G.S., McCleary W.R., Stock A.M., Stock J.B.. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan D., Cho H.S., Hastings C.A., Igo M.M., Lee S.Y., Pelton J.G., Stewart V., Wemmer D.E., Kustu S.. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:14789–14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wemmer D.E., Kern D. Beryllofluoride binding mimics phosphorylation of aspartate in response regulators. J. Bacteriol. 2005; 187:8229–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu X., Volz K., Matsumura P.. The chez-binding surface of CheY overlaps the CheA- and FliM-binding surfaces. J. Biol. Chem. 1997; 272:23758–23764. [DOI] [PubMed] [Google Scholar]
- 50. Kim E.E., Wyckoff H.W.. Reaction mechanism of alkaline phosphatase based on crystal structures. Two-metal ion catalysis. J. Mol. Biol. 1991; 218:449–464. [DOI] [PubMed] [Google Scholar]
- 51. Kim A., Benning M.M., OkLee S., Quinn J., Martin B.M., Holden H.M., Dunaway-Mariano D. Divergence of chemical function in the alkaline phosphatase superfamily: structure and mechanism of the P-C bond cleaving enzyme phosphonoacetate hydrolase. Biochemistry. 2011; 50:3481–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sunden F., AlSadhan I., Lyubimov A.Y., Ressl S., Wiersma-Koch H., Borland J., Brown C.L. Jr, Johnson T.A., Singh Z., Herschlag D. Mechanistic and evolutionary insights from comparative enzymology of phosphomonoesterases and phosphodiesterases across the alkaline phosphatase superfamily. J. Am. Chem. Soc. 2016; 138:14273–14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Clair T., Lee H.Y., Liotta L.A., Stracke M.L.. Autotaxin is an exoenzyme possessing 5'-nucleotide phosphodiesterase/ATP pyrophosphatase and ATPase activities. J. Biol. Chem. 1997; 272:996–1001. [DOI] [PubMed] [Google Scholar]
- 54. Oda Y., Kuo M.D., Huang S.S., Huang J.S.. The major acidic fibroblast growth factor (aFGF)-stimulated phosphoprotein from bovine liver plasma membranes has aFGF-stimulated kinase, autoadenylylation, and alkaline nucleotide phosphodiesterase activities. J. Biol. Chem. 1993; 268:27318–27326. [PubMed] [Google Scholar]
- 55. Galperin M.Y. Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 2010; 13:150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Holm L. DALI and the persistence of protein shape. Protein Sci. 2020; 29:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Valentini M., Filloux A.. Biofilms and cyclic di-GMP (c-di-GMP) signaling: lessons from pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 2016; 291:12547–12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chaudhuri S., Pratap S., Paromov V., Li Z., Mantri C.-K., Xie H.. Identification of a diguanylate cyclase and its role in porphyromonas gingivalis virulence. AM JSoc Microbiol. 2014; 82:2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Opoku-Temeng C., Zhou J., Zheng Y., Su J., Sintim H.O.. Cyclic dinucleotide (c-di-GMP, c-di-AMP, and cGAMP) signalings have come of age to be inhibited by small molecules. Chem. Commun. (Camb.). 2016; 52:9327–9342. [DOI] [PubMed] [Google Scholar]
- 60. Valentini M., Filloux A.. Multiple roles of c-di-GMP signaling in bacterial pathogenesis. Annu. Rev. Microbiol. 2019; 73:387–406. [DOI] [PubMed] [Google Scholar]
- 61. Kato K., Nishimasu H., Oikawa D., Hirano S., Hirano H., Kasuya G., Ishitani R., Tokunaga F., Nureki O.. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 2018; 9:4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dohler C., Zebisch M., Strater N.. Crystal structure and substrate binding mode of ectonucleotide phosphodiesterase/pyrophosphatase-3 (NPP3). Sci. Rep. 2018; 8:10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Outten C.E., O’Halloran T.V.. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001; 292:2488–2492. [DOI] [PubMed] [Google Scholar]
- 64. Saran A., Weerasinghe N., Thibodeaux C.J., Zeytuni N.. Purification, crystallization and crystallographic analysis of the PorX response regulator associated with the type IX secretion system. Acta Crystallogr. F Struct. Biol. Commun. 2022; 78:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cohen D., Mechold U., Nevenzal H., Yarmiyhu Y., Randall T.E., Bay D.C., Rich J.D., Parsek M.R., Kaever V., Harrison J.J.et al.. Oligoribonuclease is a central feature of cyclic diguanylate signaling in pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:11359–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Orr M.W., Donaldson G.P., Severin G.B., Wang J., Sintim H.O., Waters C.M., Lee V.T.. Oligoribonuclease is the primary degradative enzyme for pGpG in pseudomonas aeruginosa that is required for cyclic-di-GMP turnover. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5048–E5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cohen D., Melamed S., Millman A., Shulman G., Oppenheimer-Shaanan Y., Kacen A., Doron S., Amitai G., Sorek R.. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature. 2019; 574:691–695. [DOI] [PubMed] [Google Scholar]
- 68. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 69. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 70. Whiteley A.T., Eaglesham J.B., de Oliveira Mann C.C., Morehouse B.R., Lowey B., Nieminen E.A., Danilchanka O., King D.S., Lee A.S.Y., Mekalanos J.J.et al.. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature. 2019; 567:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lowey B., Whiteley A.T., Keszei A.F.A., Morehouse B.R., Mathews I.T., Antine S.P., Cabrera V.J., Kashin D., Niemann P., Jain M.et al.. CBASS immunity uses CARF-Related effectors to sense 3'-5'- and 2'-5'-Linked cyclic oligonucleotide signals and protect bacteria from phage infection. Cell. 2020; 182:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Picton D.M., Luyten Y.A., Morgan R.D., Nelson A., Smith D.L., Dryden D.T.F., Hinton J.C.D., Blower T.R.. The phage defence island of a multidrug resistant plasmid uses both BREX and type IV restriction for complementary protection from viruses. Nucleic Acids Res. 2021; 49:11257–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Luyten Y.A., Hausman D.E., Young J.C., Doyle L.A., Higashi K.M., Ubilla-Rodriguez N.C., Lambert A.R., Arroyo C.S., Forsberg K.J., Morgan R.D.et al.. Identification and characterization of the WYL BrxR protein and its gene as separable regulatory elements of a BREX phage restriction system. Nucleic Acids Res. 2022; 50:5171–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Picton D.M., Harling-Lee J.D., Duffner S.J., Went S.C., Morgan R.D., Hinton J.C.D., Blower T.R.. A widespread family of WYL-domain transcriptional regulators co-localizes with diverse phage defence systems and islands. Nucleic Acids Res. 2022; 50:5191–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sandmeier H., Bar K., Meyer J.. Search for bacteriophages of black-pigmented gram-negative anaerobes from dental plaque. FEMS Immunol. Med. Microbiol. 1993; 6:193–194. [DOI] [PubMed] [Google Scholar]
- 76. Condon C. Maturation and degradation of RNA in bacteria. Curr. Opin. Microbiol. 2007; 10:271–278. [DOI] [PubMed] [Google Scholar]
- 77. Niyogi S.K., Datta A.K.. A novel oligoribonuclease of escherichia coli. I. Isolation and properties. J. Biol. Chem. 1975; 250:7307–7312. [PubMed] [Google Scholar]
- 78. Fang M., Zeisberg W.M., Condon C., Ogryzko V., Danchin A., Mechold U.. Degradation of nanoRNA is performed by multiple redundant RNases in bacillus subtilis. Nucleic Acids Res. 2009; 37:5114–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu M.F., Cescau S., Mechold U., Wang J., Cohen D., Danchin A., Boulouis H.J., Biville F.. Identification of a novel nanoRNase in bartonella. Microbiology (Reading). 2012; 158:886–895. [DOI] [PubMed] [Google Scholar]
- 80. Goldman S.R., Ebright R.H., Nickels B.E.. Direct detection of abortive RNA transcripts in vivo. Science. 2009; 324:927–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Goldman S.R., Sharp J.S., Vvedenskaya I.O., Livny J., Dove S.L., Nickels B.E.. NanoRNAs prime transcription initiation in vivo. Mol. Cell. 2011; 42:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sato K., Naito M., Yukitake H., Hirakawa H., Shoji M., McBride M.J., Rhodes R.G., Nakayama K.. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wolfe A.J. Physiologically relevant small phosphodonors link metabolism to signal transduction. Curr. Opin. Microbiol. 2010; 13:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Klein A.H., Shulla A., Reimann S.A., Keating D.H., Wolfe A.J.. The intracellular concentration of acetyl phosphate in escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 2007; 189:5574–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Battesti A., Majdalani N., Gottesman S.. The rpos-mediated general stress response in escherichia coli. Annu. Rev. Microbiol. 2011; 65:189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hennell James R., Deme J.C., Kjr A., Alcock F., Silale A., Lauber F., Johnson S., Berks B.C., Lea S.M. Structure and mechanism of the proton-driven motor that powers type 9 secretion and gliding motility. Nat. Microbiol. 2021; 6:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Paul K., Harmon J.G., Blair D.F.. Mutational analysis of the flagellar rotor protein flin: identification of surfaces important for flagellar assembly and switching. J. Bacteriol. 2006; 188:5240–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shrivastava A., Lele P.P., Berg H.C.. A rotary motor drives flavobacterium gliding. Curr. Biol. 2015; 25:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Emrizal R., Nor Muhammad N.A.. Phylogenetic comparison between type IX secretion system (T9SS) protein components suggests evidence of horizontal gene transfer. PeerJ. 2020; 8:e9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from the corresponding authors upon reasonable request. Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 7PV7 (PorX-BeF3), 7PVA (PorX-HR) and 7PVK (PorX-pGpG).