

Abstract
Multimodal single-cell sequencing technologies provide unprecedented information on cellular heterogeneity from multiple layers of genomic readouts. However, joint analysis of two modalities without properly handling the noise often leads to overfitting of one modality by the other and worse clustering results than vanilla single-modality analysis. How to efficiently utilize the extra information from single cell multi-omics to delineate cell states and identify meaningful signal remains as a significant computational challenge. In this work, we propose a deep learning framework, named SAILERX, for efficient, robust, and flexible analysis of multi-modal single-cell data. SAILERX consists of a variational autoencoder with invariant representation learning to correct technical noises from sequencing process, and a multimodal data alignment mechanism to integrate information from different modalities. Instead of performing hard alignment by projecting both modalities to a shared latent space, SAILERX encourages the local structures of two modalities measured by pairwise similarities to be similar. This strategy is more robust against overfitting of noises, which facilitates various downstream analysis such as clustering, imputation, and marker gene detection. Furthermore, the invariant representation learning part enables SAILERX to perform integrative analysis on both multi- and single-modal datasets, making it an applicable and scalable tool for more general scenarios.
INTRODUCTION
Single cell sequencing (sc-seq) offers genome-wide measurements of genetic information from individual cells (1–7). Recent technology advances allow simultaneous profiling of multiple modalities in the same cells (8,9), allowing us to dissect cellular heterogeneity from multiple layers and investigate the transcriptomic and epigenomic interplays at the finest possible resolution.
Several computational methods have been developed to deal with some key factors of data integration, such as correcting batch effect while maintaining biological patterns for scRNA-seq data (scVI, scANVI, Scanorama, Harmony etc.) (10–13), and embedding multi-modal data to the same embedding without corresponding information (14–20). Readers can refer to (15) for a more detailed comparison of data integration methods. However, it is still remaining a challenge to effectively utilize information cross different modalities due to problems such as unbalanced signal-to-noise ratio (SNR), datasets with missing modalities, handling modality-specific noise factors and batch effects. Recently, many computational methods have been developed to analyze multimodal single cell data (21–26). A common strategy used by many methods is to project data from different modalities to a shared latent space. For example, existing methods like scAI, scMM, scMVAE, BABEL and Cobolt (23–27) use either Nonnegative Matrix Factorization (NMF) or Encoder-Decoder types of neural networks to project multiple modalities to a common latent space. Their underlying assumption is that measurements from different modalities are equally informative and share a common distribution, which does not hold under many circumstances. For instance, a typical scATAC-seq experiment usually reports 1000–20 000 mappable fragments per cell over the entire 3.2 billion base pair genome, resulting in noticeably higher dropout rates and coverage variations as compared to the RNA modality from the same cell. As a result, lines of literatures pointed out that direct fusion of modalities with neural networks can introduce severe overfitting across modalities, resulting in poor separation of cell clusters in learned latent representation (28). In observance of this, Sigh et al. proposed Schema framework by learning an affine transformation of similarity matrices through metric learning to find a joint representation of cells which is regularized to be similar to a reference embedding (28). However, the flexibility of the transformation could limit the expressiveness of the joint embedding, and it does not explicitly handle batch effect and other technical noises. In another strategy, Signac (29) used weighted nearest neighbor (WNN) graph to generate a joint embedding based on predictability of data from two modalities of each cell. However, information fusion is done after separate embeddings are generated without considering latent interaction between the two modalities, potentially limiting the overall performance. Besides, most existing methods cannot handle sc-multiome data with missing modalities (due to either possible QC failures in one modality or data integrations from different sequencing protocols) or contain explicit mechanisms to handle technical noises in each modality, which are common in real data analysis (Table 1).
Table 1.
Comparisons on the functionality of benchmarked methods
| Method | Approach | Nonlinear | Scalability | Multiome | Missing modality | Bias correction |
|---|---|---|---|---|---|---|
| Signac | LSI | ✗ | ✗ | ✓ | ✗ | ✗ |
| Schema | QP | ✓ | ✗ | ✓ | ✗ | ✗ |
| SAILER | VAE-Inv | ✓ | ✓ | ✗ | ✗ | ✓ |
| Cobolt | MVAE | ✓ | ✓ | ✓ | ✓ | ✗ |
| SAILERX | VAE-Inv | ✓ | ✓ | ✓ | ✓ | ✓ |
Hereby, to tackle these issues, we propose a deep learning framework, named SAILERX, to improve analysis of multiomics or hybrid of single- and multi-modal single cell sequencing datasets (Table 1). Distinct from existing methods, SAILERX can handle both parallel scRNA-seq and scATAC-seq multiome data, single modal scATAC-seq data, and a hybrid of these two types of data. To address the modality heterogeneity and avoid overfitting, we use the more robust gene expression information as a reference modality, to regularize the learning process of the chromatin accessibility modality. Specifically, scATAC-seq data is modeled with a Variational Autoencoder (VAE) and embeddings of scRNA-seq data are pre-trained and not explicitly modeled at training time. We further impose regularization via minimizing the distance between the pairwise similarity in the embedding space between two modalities (Figure 1), which encourages local structures of cells to be similar to the reference modality while accommodating substantially different technical noises across modalities. The resulting representation of cells implicitly contains information from two modalities and avoids the risk of overfitting. In the meantime, an invariant representation learning objective (30,31) is used in the VAE framework to eliminate observable technical noises and allows integration of multiple datasets through end-to-end training. The modeling choice of SAILERX allows hybrid integration of datasets with scATAC-seq measures and datasets with paired scRNA-seq and scATAC-seq, effectively utilize the information from high quality multimodal data to improve the analysis of single-modal datasets.
Figure 1.

Overall design of SAILERX. (A) SAILERX takes co-assayed single cell RNA-seq and ATAC-seq data as input. scATAC-seq data is modeled with invariant representation learning through VAE, while embedding of scRNA-seq is processed during pre-training and not explicitly modeled in the training process. A regularization is imposed to encourage the local structure of cells in the embedding space to be similar between two modalities through minimizing the distance between pairwise cosine similarity matrices of two modalities. Latent scATAC-seq feature is further used to perform downstream analysis. (B) SAILERX is also capable of integrating single modal scATAC-seq with multimodal datasets through hybrid training, which could further enhance the clustering performance on single modality data.
We benchmark SAILERX with existing state-of-the-art (SOTA) methods for multi/single-modal single cell data analysis on three popular single cell datasets with different sequencing technologies and types of tissues. We show that SAILERX generates representations of cells that provide better clustering and imputation. We also demonstrate how the single modal scATAC-seq dataset could benefit from hybrid training. For biological applications, those improvements significantly benefit the downstream analysis of chromatin accessibility data. SAILERX is implemented in a python package freely to the community.
MATERIALS AND METHODS
In this section, we provide details on our SAILERX model and datasets for benchmarking, as well as describe methods.
Datasets
In this study, we focus on multimodal single cell sequencing data with paired scRNA-seq and scATAC-seq measurements. For this purpose, three popular public single cell multiomics datasets with different cell types and sequencing technologies are used in this study, namely 10x Genomics PBMC dataset (29), Share-seq dataset (9) and SNARE-seq dataset (8).
PBMC dataset
10X Genomics offers multiple datasets with PBMC cells, we collect PBMC 10k Multiome and PBMC 3k from the 10X genomics website. The PBMC 10k dataset is mainly used for benchmarking cross modality integration performance. For the PBMC 3k dataset, we only use the chromatin accessibility data for hybrid joint analysis with 10k dataset. The gene expression modality of 3k dataset is not used in hybrid training and only used for identifying ground truth labels of cells from the 3k dataset in this case. For integration of two sc-multiome datasets, the gene expression modality is used normally. For these two datasets, cell types are annotated through label transfer using an existing PBMC reference dataset via tools in the Seurat (29) and SeuratDisk package. Specifically, we use a high-quality dataset (29) as the reference dataset to transfer cell type labels to PBMC 3k and PBMC 10k datasets respectively.
For scenario one (cross modality integration), the 10k Multiome data is acquired from 10X genomics website. We first download PBMC 10k expression matrix and chromatin accessibility matrix as well as its fragment file from 10X Genomic Multiome dataset, and we follow the same quality control protocol as Signac (32) to filter out low quality cells. This retains 11 331 cells for further analysis. For scRNA-seq, we then normalize scRNA-seq data using SCTransform function with default parameters. After that, principal component analysis (PCA) is used to extract top 50 PCs for further clustering and joint analysis with scATAC-seq. As for scATAC-seq, since the set of peaks identified using CellRanger often merges nearby peaks, which would potentially cause bias in tasks like motif enrichment analysis, in our study, peak calling is performed on PBMC 10x dataset by using fragment file to generate unique peaks using MACS2 software (33). After that, we follow the same process described in (25) and keep the autosome data and get the final scATAC-seq peak-by-cell matrix. This matrix is further used to process and benchmark with all the other methods. For instance, in Signac, TF-IDF is performed on the scATAC-seq matrix and then SVD is adopted on the TF-IDF output matrix to get the 50-dimension latent embedding, which is further used for clustering and joint analysis with scRNA-seq data.
Regarding to the second scenario (hybrid joint analysis), we use the aforementioned multimodal PBMC 10k data, which consists of scRNA-seq and scATAC-seq data as a reference and perform joint analysis with the chromatin accessibility data from PBMC 3k dataset. We retrieve PBMC 3k scATAC-seq data from 10X Genomics and treat it as a single modality dataset. We reason that 3k dataset with scATAC-seq contains less information than the multiomics dataset, however, since they come from the same types of cells, we could use 10k multiomics dataset as a reference to assist the analysis of 3k scATAC-seq data. We use reduce function from GenomicRanges package (34) to merge common peaks from scATAC-seq 10k and 3k dataset, and the peak by cell matrix is reconstructed separately for the two scATAC-seq data, which is further used to train and evaluate our model, as illustrated in Figure 1B.
Share-seq dataset
For Share-seq dataset, we retrieve Share-seq mouse skin dataset from Ma et al. (9), which contains 34 474 cells of both modalities of scRNA-seq and scATAC-seq data. For scRNA-seq data, we normalize its gene by cell matrix by using SCTransform function with default parameters from Signac package, then PCA is utilized to get top 50 PCs for further analysis. For scATAC-seq data, we keep the preprocessed peak by cell matrix used in Ma et al. The gene by cell and peak by cell matrices are used for evaluation on other methods.
Snare-seq dataset
For Snare-seq dataset, we download adult brain cortex data of two modality matrices from Chen et al. (8). For scRNA-seq data, we follow the same processed steps as previous by normalizing gene by cell matrix using SCTransform function (29) with default parameters. After that we adopt PCA on the normalized matrix and use top 50 PCs as latent embedding for further analysis. As for the scATAC-seq data, after retrieving the processed scATAC-seq matrix from Chen et al, we also follow the same processed procedure as BABEL (25) and filter out low quality cells while keeping the original peaks unchanged. In details, genes that are encoded on sex chromosomes are first removed, and cells expressing fewer than 200 genes, or >2500 genes are also filtered.
Model
Here, we describe details and implementation of our SAILERX model. SAILERX combines information from the gene expression measures to improve the downstream analysis of chromatin accessibility. SAILERX could also perform integrative analysis on multiple datasets with one or multiple modalities.
The model takes the co-assayed single cell multimodal data 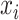 ,
, 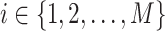 as input. We denote the gene expression data as
as input. We denote the gene expression data as 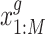 and the peak data as
and the peak data as 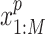 (M indicates the total number of multimodal data samples). Our model could also take single modal scATAC-seq datasets
(M indicates the total number of multimodal data samples). Our model could also take single modal scATAC-seq datasets 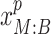 (B indicates total number of sample batches) as input and perform integrative analysis among all
(B indicates total number of sample batches) as input and perform integrative analysis among all 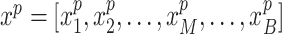 . The overall method follows the invariant representation learning framework based on Variational Autoencoders (VAEs) (30,31). Denoting the confounding variables (read depth and batch indicator) as
. The overall method follows the invariant representation learning framework based on Variational Autoencoders (VAEs) (30,31). Denoting the confounding variables (read depth and batch indicator) as 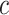 , our method minimizes the VAE objective with an extra mutual information penalty (Equation 1), to encourage the posterior of latent variable
, our method minimizes the VAE objective with an extra mutual information penalty (Equation 1), to encourage the posterior of latent variable 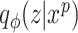 to be independent with confounding variable
to be independent with confounding variable 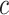 . This objective can be approximated by a variational bound as shown in (30,31).
. This objective can be approximated by a variational bound as shown in (30,31).
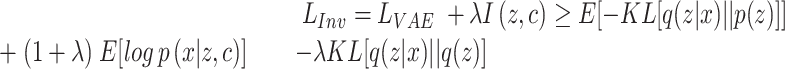 |
(1) |
In order to utilize the gene expression information provided by multimodal single cell samples, we add an extra term to regularize the local data structure in the chromatin accessibility posterior 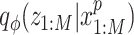 to be close to the local structure measured by gene expression.
to be close to the local structure measured by gene expression.
We use pairwise cosine similarity to describe the local data structure, where the cosine similarity is computed as
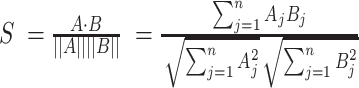 |
(2) |
For each sample batch 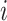 ,
, 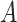 and
and 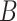 are two single cell data vectors from
are two single cell data vectors from 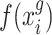 (where
(where 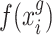 is a transformation of raw gene expression data) and
is a transformation of raw gene expression data) and 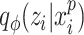 for
for 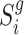 and
and 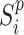 respectively. In general,
respectively. In general, 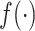 can be any embeddings of gene expression data preferred by user (e.g. a VAE or top PCs from PCA) since it is not parameterized by our neural network model here and only serving as a reference. For the convenience of comparing with existing methods, in our study, we mainly use the PCA results generated by Signac/Seurat (29) as the reference embedding. Some other scRNA-seq embedding methods (scVI (10), scANVI (13), Scanorama (11)) are also tested.
can be any embeddings of gene expression data preferred by user (e.g. a VAE or top PCs from PCA) since it is not parameterized by our neural network model here and only serving as a reference. For the convenience of comparing with existing methods, in our study, we mainly use the PCA results generated by Signac/Seurat (29) as the reference embedding. Some other scRNA-seq embedding methods (scVI (10), scANVI (13), Scanorama (11)) are also tested.
During the training, we minimize a distance-based objective 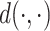 between the local pairwise cosine similarity matrix for each sample batch
between the local pairwise cosine similarity matrix for each sample batch 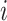 calculated by gene expression data
calculated by gene expression data 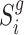 and the pairwise similarity matrix calculated by latent distribution of peak data modeled by invariant VAE
and the pairwise similarity matrix calculated by latent distribution of peak data modeled by invariant VAE 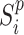 , where both
, where both 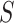 ’s are b by b symmetric matrices with batchsize b for each minibatch during training.
’s are b by b symmetric matrices with batchsize b for each minibatch during training.
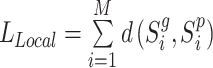 |
(3) |
By choosing a proper differentiable distance metric 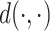 , we can fuse this term into the end-to-end training of our deep generative model. The overall loss function would be the sum of the canonical VAE objective, a mutual information penalty, and the local similarity regularization. Here, we multiply a weight vector
, we can fuse this term into the end-to-end training of our deep generative model. The overall loss function would be the sum of the canonical VAE objective, a mutual information penalty, and the local similarity regularization. Here, we multiply a weight vector 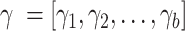 with length b equals to the number of cells in current mini batch. This weight
with length b equals to the number of cells in current mini batch. This weight 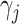 is calculated based on the ratio between read depth from gene expression modality and read depth from chromatin accessibility modality for each cell j. This weight vector is then subject to log transformation and min-max normalization to ensure stability
is calculated based on the ratio between read depth from gene expression modality and read depth from chromatin accessibility modality for each cell j. This weight vector is then subject to log transformation and min-max normalization to ensure stability 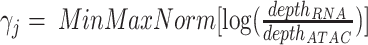 . After scaling it with a constant scalar, we have our final weight vector
. After scaling it with a constant scalar, we have our final weight vector  . The relationship between the scaling factor and the final
. The relationship between the scaling factor and the final 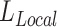 is shown in Supplementary Figure S1A. We note that after certain point, further increase of this scaling factor will no longer reduce the final
is shown in Supplementary Figure S1A. We note that after certain point, further increase of this scaling factor will no longer reduce the final 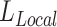 . We recommend using this point as the choice for the scaling factor, as further increase of this weight does not transfer more information from the reference modality. Meanwhile, it may compromise the invariant representation learning objective, which could lead to problems in confounding factor removal or imputations. Also, from Supplementary Figure S1B, we can see clustering metrics of SAILERX are robust in a relatively large range of weight values. In terms of choice of
. We recommend using this point as the choice for the scaling factor, as further increase of this weight does not transfer more information from the reference modality. Meanwhile, it may compromise the invariant representation learning objective, which could lead to problems in confounding factor removal or imputations. Also, from Supplementary Figure S1B, we can see clustering metrics of SAILERX are robust in a relatively large range of weight values. In terms of choice of 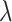 and dimension of latent variable, similar as in (31), the framework is robust against the choice of
and dimension of latent variable, similar as in (31), the framework is robust against the choice of 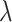 and dimension of latent variable.
and dimension of latent variable.
The final loss of SAILERX is a summation of the invariant representation learning objective from Equation (1) and the local alignment loss from Equation (3) weighted by 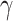 .
.
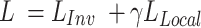 |
(4) |
In our implementations, we chose the Euclidean distance for 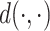 since it is differentiable and easy to calculate.
since it is differentiable and easy to calculate.
For the architecture of neural networks, we adapt the encoders and decoders structures from BABEL (25), where each chromatin is independently modeled by a two-layer dense encoder network, and outputs from each encoder network are concatenated with each other before being input to the final linear layer which yields the latent variable. The decoder is symmetric to the encoder network, taking the latent and confounding variables as input and reconstructing the data. The assumption here is that interactions between genes and regulating factors are mainly within each chromatin. This type of modeling is efficient in memory consumption since it significantly reduces the total number of parameters. For fair comparison, the original SAILER encoder and decoder networks are also updated to the same structure.
Hybrid training
One characteristic of SAILERX is that it allows integration of datasets with missing modalities (when 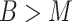 ). In this scenario, for datasets with both modalities measured (
). In this scenario, for datasets with both modalities measured (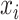 ,
, 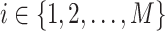 ), the loss function follows the form of equation (4), where a reference embedding is available for calculating
), the loss function follows the form of equation (4), where a reference embedding is available for calculating 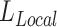 ; for datasets with only one modality (
; for datasets with only one modality (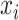 ,
, 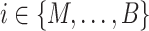 ), we no longer calculate or backpropagate the gradient for
), we no longer calculate or backpropagate the gradient for 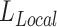 , since no reference embeddings are available for these datasets. For these scATAC-seq datasets, we still perform batch effect correction through the invariant representation learning objective (Equation (1)), where the batch effect is represented as the confounding variable
, since no reference embeddings are available for these datasets. For these scATAC-seq datasets, we still perform batch effect correction through the invariant representation learning objective (Equation (1)), where the batch effect is represented as the confounding variable 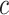 , along with the read depth for each single cell.
, along with the read depth for each single cell.
Evaluations
For all methods, we project the input data to a lower-dimensional space (dimension of embedding is 50 by default, unless specified by other methods) that delineates the latent cell states. For Seurat, we use the scTransform function to normalize the raw counts and use the normalized data as input for PCA; for Signac, we use its multimodal integration analysis, which uses the same normalized gene expression data and additional TF-IDF transformed peak data as input; for SAILER we use the peak data as input; and for Cobolt and Schema, we follow their tutorials and use data from both modalities as input. To generate a lower-dimensional embedding for benchmarking, for Seurat, we use the top 50 PCs after PCA; for Signac, we use the results of Weighted Nearest Neighbor (WNN) analysis as a joint embedding of gene expression and chromatin accessibility modalities; for SAILERX, we extract the mean of the posterior latent distributions as the cell representation; for Cobolt, we use the latent variable z with dimension 50 calculated from its multimodal variational autoencoder; and for Schema, we use the 50-dimensional latent feature retrieved by using its fit_transform function. Other compared reference embeddings are generated with scVI (35), scANVI (13) and Scanaroma (11) using scIB package (36). We set the default dimension as 50 for compared methods, including Seurat, Schema, Cobolt, SAILER, and Signac in our analysis in order to fairly compare all these works. As for the rest methods, we keep the default latent dimension settings in the scIB package for scVI, scANVI and Scanaroma (30, 30 and 100 respectively). 2D visualizations are acquired by running uniform manifold approximation and projection (UMAP) (37) on the latent embeddings.
One major task of these dimensional-reduction methods is to project the input genomics data to a lower dimensional embedding that is informative on cell type identification through clustering. To evaluate how the clustering generated from these embeddings are compared to the ground truth cell labels, we use quantitative metrics of Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and Silhouette Score to assess the performance of different methods. ARI and NMI evaluate how well computational clusters overlap with ground truth labels, and the Silhouette coefficient evaluates the separation of the cell clusters. These metrics are common metrics used for benchmarking single cell clustering methods (38,39). Specifically, to generate cluster assignment for each cell, we construct k-nearest neighbor (KNN) graphs from the lower-dimensional embeddings of different methods respectively, and then apply the Louvain algorithm (40) to assign individual cells to different clusters. Each method generates its own set of clusters, and these clusters are then used to calculate quantitative metrics of ARI, NMI, and Silhouette Score for benchmarking. The calculations of metrics are carried out by functions from scikit-learn (41) library. For analysis in Figure 2 and 3, for fair comparisons, all methods are producing the same number of clusters. To determine the effect of clustering parameters and cluster numbers, we provide a wide range of resolutions and KNN numbers to the Louvain algorithm to determine the final clustering assignments. During the process, we record the number of clusters identified based on each combination of parameters (resolution and KNN number) for each experiment, as well as the metric scores for that clustering assignment. The effect of clustering parameters and cluster numbers are summarized in Supplementary Figure S4.
Figure 2.

Results on PBMC 10k Multiome dataset. Cells colored by ground truth label. (A) UMAP visualizations of embeddings on PBMC 10k Multiome dataset generated by different methods. Red circles show separation of sub clusters of B cells under Seurat (scRNA-seq only), SAILER (scATAC-seq only) and SAILERX (multimodal). (B) Quantitative metrics of ARI, NMI, and Silhouette Score on clustering generated by different methods. Error bars are generated by repeating experiments with 90% randomly subsampling. (C) Quantitative metrics of ARI Score on Reference Embeddings on gene expression modality and Integrated Embeddings generated by SAILERX.
Figure 3.

Results on SNARE-seq dataset. (A) UMAP visualizations of embeddings generated by different methods on SNARE-seq dataset. Cells are colored by ground truth labels. (B) Quantitative metrics of ARI, NMI, and Silhouette Score on clustering generated by different methods. Error bars are generated by repeating experiments with 90% subsampling.
Imputation
We generate the imputation data via a reconstruction conditioned on the invariant representation and fixed confounding factors. Specifically, we first push the raw data through the encoder network, and obtain the mean parameters for latent distributions. Unlike the training process, where we calculate the depth of the raw data and load the one-hot embedding according to the real batch information, here we fix the depth and batch indicator for reconstruction. As a result, we use only the invariant component 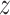 to reconstruct the chromatin landscape during the imputation process, while keeping the other confounding factors at a fixed level.
to reconstruct the chromatin landscape during the imputation process, while keeping the other confounding factors at a fixed level.
When evaluating imputation results, we first generate imputed data with each method. Then use randomized PCA to project the imputed data to a lower dimension. We then use UMAP to visualize the landscape of imputed data in 2D. For benchmarking against MAGIC, we use both graphs generated by scRNA-seq modality and scATAC-seq modality for fair comparison. The RNA graph is based on the Seurat embedding and ATAC graph is based on MAGIC’s own pipeline. For benchmarking with scOpen, we follow the manual on its GitHub site to generate a dense imputation matrix. The imputed matrices are then subject to randomized PCA and visualized with UMAP. Quantitative scores (ARI, NMI, Silhouette Score) are calculated based on clustering results generated from the top PCs.
Marker gene expression analysis
To further evaluate the quality of cell clusters, we visualize the expression of marker genes in clusters labelled as CD4 naïve cells and B naïve cells from the PBMC dataset and L4 cells and Pvalb cells from the SNARE-seq dataset. To associate the cell clusters to biological cell types, the cell cluster labels are called based on a majority vote of the ground truth labels of the cells contained in each cluster. The four types of cells are chosen for this analysis because they are similar to other cell types and are challenging to cluster them. The CD4 cluster sits very close to CD8 naïve and other CD4 subtype clusters in the embedding space. The L4 cluster sits close to the L2/3 and L6 IT cell clusters. In particular, gene expression information alone cannot well separate subtypes of B cells.
The cell-type specific marker genes used for the visualization are called by the FindMarker function in Seurat (42). These genes are identified as marker genes because they show significant differential RNA expression in the cells labeled with the corresponding cell types versus other cells. Cell-type labels are based on ground-truth labels. The top 10 chosen marker genes associated with each cell type are shown in Supplementary Table S1.
For each cell type, we use boxplots to visualize the mean normalized expression of marker genes (Supplementary Figure S3) of the cells from the cluster labeled with the corresponding cell type. The gene expression values are normalized by scTransform, and the mean values are shown in Supplementary Table S2. Pairwise t-tests between SALIERX and other methods indicate whether the marker genes from SALIERX show significantly higher expression than those from other methods. t-test P-values are indicated by ns (P-value > 0.05, i.e. not significant), * (P-value < 0.05), ** (P-value < 0.01) and *** (P-value < 0.001).
Motif analysis
We perform motif analysis on several key motifs to demonstrate a case of discovering cell-type specific motif enrichment between different cell types. The putative cell types are determined by same procedures in previous section through clustering and majority vote. We first compute a per-cell motif activity score by running chromVAR (43). It converts the peak by cell matrix to a motif by cell matrix, allowing us to get the motif activity score per cell, which provides an alternative method for identifying differentially active motifs between diverse cell types. In order to discover differential motif activities, we also utilize z-score, calculated by chromVAR, and FindMarkers function, provided by Signac (32), to get the average z-score differences between different cell types. Then these motifs are sorted according to their P-values. We set the parameter mean.fxn = ‘rowMeans’ and fc.name = ‘avg_diff’ in the FindMarkers function following Signac tutorial to compute the average difference in z-score in terms of fold-change calculation between the groups.
After that we apply MotifPlot to plot the four of the top 6 motifs that represents the most differential expressed motifs between the two cell types. Finally, we also get the clustering result with regards to specific cell types that we use to compare differential motifs. We use Louvain algorithm to assign a specific cluster number to each cell cluster, and then collect all the cells that belong to the same cluster number, which overlaps with the most cells of that specific ground truth cell type. We refer the z-score of those cells of that motif calculated from chromVAR and draw barplot to show the z-score distribution on that plot.
RESULTS
Joint analysis of single cell multi-omics data with paired measurements often suffers from imbalanced SNR from different modalities (28). In our study, we mainly focus on paired measurements of scRNA-seq data and scATAC-seq data. In practice, data from the scATAC-seq modality is often more affected by read depth variations and limited coverage rate, which would greatly impact the joint embedding when fusing data from two modalities together. In order to address the aforementioned issues, we design a framework SAILERX by using the structural similarity for the integration of the two-modality data and achieve satisfactory result. Here, we benchmark SAILERX with other methods that are able to cluster single/multi-modal single cell data. We also demonstrate that SAILERX could be used to align datasets with missing modality and improve analysis by applying joint analysis with a high-quality multimodal dataset. We include Table 1 to better illustrate the differences between our methods and others. After that, we further demonstrate the benefits of our method on downstream analysis such as motif discovery. Details are described in the following subsections.
SAILERX generates better clustering by fusing information from two modalities
We first benchmark our framework on PBMC 10k dataset, which consists of paired transcription and chromatin accessibility sequenced on 11 331 cells of human PBMC. This dataset is generated by 10X genomics. Some mature and differentiated blood cells from PBMC dataset have clear separation of cell types such as B cells and T cells. However, within those cell types, some sub-cell types such as monocytes are still ongoing differentiation process, resulting in continuously distributed cell clusters which often pose challenges to clustering algorithms.
During training, the regularization term in SAILERX encourages the local structure of the posterior distribution on the scATAC-seq data to be close to its scRNA-seq correspondence. The embedding from scATAC-seq is generated by the encoder network of a VAE, and the embedding for scRNA-seq modality for this dataset is generated by one of the scRNA-seq embedding methods. In this study, we mainly use PCA from Seurat as the scRNA-seq reference embedding, but other methods are also demonstrated in this dataset (Figure 2C). During training, we also assign a weight for each cell on this regularization term based on the read depth of two modalities (Materials and Methods). Cells with poor quality on scATAC-seq measurements will have higher weights. With this flexible weighting mechanism, cells with poor scATAC-seq measurements could get more information from its scRNA-seq correspondence, and cells with better data quality from scATAC-seq side could preserve their informative parts. After training, we retrieve the posterior mean of latent variable as our final embedding and cluster those cells accordingly. We benchmark our methods with three state-of-the-art (SOTA) methods that could handle multiomics data integration, i.e. Signac, Schema and Cobolt, as well as SOTA methods that only work on single modality data (i.e. Seurat, scVI, scANVI, Scanorama on scRNA-seq, and SAILER on scATAC-seq).
2D visualizations of the embeddings generated by different methods are shown in Figure 2A, with cells colored by ground truth cell type labels. The ground truth cell type labels are inferred through Seurat-style mapping strategy from (29). We validate these ground truth cell type labels by visualizing some enriched expressions of known cell type-specific marker genes (Figure S2), such as pDC cells (with known marker genes CLEC4C and NRP1) (44–47), and Treg cells (with known marker gene FOXP3 and RTKN2) (48,49). From the results, we can see that the ground truth cell types here correspond well with the well-known cell-type markers, so we consider these labels as ‘ground truth’ labels for the following analyses.
To quantitatively assess these clustering methods, we use ARI, NMI, and Silhouette metrics to evaluate the clustering results. ARI and NMI evaluate how well the computational clusters derived from lower-dimensional embeddings overlap with ground truth cell labels; and the Silhouette coefficient measures the separation of the cell clusters in the embedding space. Higher scores indicate better matchings and separations. The metric scores are shown in Figure 2B, C and Supplementary Figure S4, with SAILERX achieving the highest scores in ARI, NMI, and Silhouette coefficient. From the scores, we can see Seurat achieves a great performance on overall clustering results. In the figure, we can see it forms tight and separable clusters for most cell types. Some other multimodal integration methods do not perform as well as Seurat when adding extra information from chromatin accessibility, showing that adding extra information without properly handling the noise could harm the overall clustering result. However, when we compare SAILERX with Seurat, we can see the embedding generated by SAILERX keeps the robust separation of cell clusters inherited from its reference gene expression modality, while preserves the useful signals appearing in the chromatin accessibility modality. This could also be demonstrated by the separation of sub clusters of B cells colored in red and blue (Figure 2A, red circles), and the higher marker gene expressions for cells identified as B naïve cells (Supplementary Figure S3A and Table S2). This shows that through proper integration of information from both modalities, SAILERX could discover new (sub)types of cells previously unidentifiable with gene expression modality only. Also, from the results, we can see that our integration benefits the delineation of continuously distributed cell types, e.g. CD4 cells. CD4 cells are previously reported to be more identifiable using chromatin accessibility information (50). This can be demonstrated when we try to identify subtypes of CD4 cell. Compared with other methods, CD4 naïve cells identified by SAILERX have higher marker gene expressions (Supplementary Table S2). This shows our cross-modality integration can also benefit the cell type identifications for ambiguous subtypes.
For robustness evaluation, we further test if our method could consistently improve upon different reference embeddings. Here, we use three other scRNA-seq embedding methods (scVI, scANVI, and Scanorama) to generate reference embeddings and then use these embeddings to help train SAILERX models. As shown in the Figure 2C and Supplementary Figure S5, the joint embeddings combine information from two modalities and constantly outperform their reference embeddings. This shows effectiveness and robustness of SAILERX’s information fusing strategy.
Similar analyses are performed on the SNARE-seq dataset (8) with a different sequencing technology. SNARE-seq data are from mouse brain tissue. A great majority of cells in this dataset are found in a quiescent state, and thus is more stable compared with PBMC cells. Compared with PBMC 10K from 10X genomics, the SNARE-seq data tends to have much shallower read depth in chromatin accessibility reads, which makes this chromatin accessibility data here sparser than the scATAC-seq data in the previous analysis. From the results (Figure 3), we can see some integration methods severely suffer from this when projecting data from two modalities into one shared latent space. In this scenario, embedding generated by SAILERX forms tighter clusters (Figure 3A) and achieves the best performance in terms of quantitative results (Figure 3B). The separation of cell types is also demonstrated by marker gene expressions of cells identified by different methods (Supplementary Figure S3B and Table S2), where SAILERX shows higher results compared with other methods.
We also perform clustering analyses on a more recent Share-seq dataset (9) on mouse skin tissues. The results are shown in Supplementary Figure S6, where SAILERX achieves better results in terms of quantitative scores. Among all different types of tissues and sequencing technologies, the integration strategy used by SAILERX robustly outperforms other methods, showing the effectiveness of our framework.
SAILERX improves analysis of single modal scATAC-seq dataset by aligning it to multimodal datasets
Besides fusing information from two modalities within one dataset, SAILERX is also capable of performing multi-sample data alignment even for datasets with missing modalities. This is achieved by the invariant representation learning objective of our framework. By assigning a batch indicator variable as a confounding factor, the model automatically corrects for the batch effect during training. When integrating datasets with missing modalities, we ignore the regularization term for those cells with only one type of measurements. For this case, we use PBMC 10k Multiome dataset with paired scRNA-seq and scATAC-seq measurements, together with a single-modal PBMC 3k dataset with scATAC-seq only as described in Materials and Methods. Two datasets are jointly trained as described above. We then obtain the latent representation and perform clustering on cells from PBMC 3k dataset using Louvain community detection. The results are shown in Figure 4, and ground truth cell types are identified by marker genes as in Hao et al. (29). Here we evaluate the clustering metric, and compare it with Cobolt (27), which is also capable of integrating multimodal data with missing modality, and Signac, which only performs integration with scATAC-seq modalities. The Cobolt method adopts a multimodal VAE with shared latent space. As shown in Figure 4B–C, SAILERX achieves the best clustering metrics, showing that the flexible fusing mechanism works better on the noisy single cell multiomics data compared with Cobolt, and the single modal data with lower data quality could benefits a lot from this type of multi-sample alignment.
Figure 4.

Hybrid training result on PBMC 3k dataset. (A) Datasets used for training. (B) UMAP visualizations of PBMC 3k dataset. (C) Metrics on clustering for PBMC 3k dataset.
In addition to batch alignment between one multi-modal and one single modal dataset, SAILERX could also align data from multiple multimodal datasets. We demonstrate this with complete PBMC 3k and 10k datasets. As shown in Supplementary Figure S7, SAILERX could align data from different batches when there exists a clear batch effect while preserving a high quality of clustering results. And in Supplementary Figure S8, SAILERX is trained in a situation with cell type heterogeneity: we mimic this by dropping one unique cell type from each batch. When these data are processed together for batch alignment, we find that the unique cell clusters are preserved. This shows that SAILERX can preserve biological signals when performing batch effect corrections.
Cross modality integration facilitates downstream analysis of chromatin accessibility data
In previous sections we have demonstrated that SAILERX is able to generate better embeddings under different scenarios. Here, we explore how this advantage could benefit downstream analysis of chromatin accessibility data. Here, we perform motif enrichment and motif activity analysis on the SNARE-seq data mentioned above, which suffers more from the sparsity and dropouts on the chromatin accessibility signals.
We first perform differential testing using the chromVAR (43) deviation z-score as described in Methods. Here we use Pvalb and Sst cells (colored in red and purple in Figure 3A) to calculate the differential motifs between these two cell types. Then we plot the top 6 motifs that are mostly enriched between the two cell types by p-value calculated by FindMarkers function from Seurat. As shown in Figure 5. Mef-family motifs are greatly enriched in Pvalb-specific peaks in scATAC-seq data, with four out of six Mef-family motifs enriched in those Pvalb-specific regions. These findings are consistent with previous reports (42,51). Moreover, the Mef2c motif is also reported to be involved in the development of Pvalb interneurons (52), and also shown enriched as one of the differential motifs (Figure 5, Supplementary Figure S9). To quantify the performance of these enriched motifs, we select those groups of cells from clustering results of each method, which most likely represent Pvalb cells, and then we calculate the value of z-score within those cells (details in Methods). We compare the results generated by five other methods that are able to integrate multimodal scRNA-seq and scATAC-seq data or work only on scATAC-seq modality. As shown in Figure 5, our method achieves the highest value of motif deviation z-score among all the methods with the differential significance of pairwise t-test p-values all less than 0.05, showing that SAILERX is more likely to discover novel motifs based on this clustering. In addition, we compare L4 and L5 PT cells and compute the enriched motifs between those cells. Previous reports claim that POU3F2 protein associates with bipolar disorder and is involved in the neocortex development in mice (53). From the top 6 enriched motifs we could find, there are several POU family related motifs enriched in the cells including POU3F2. Therefore, we explore the motif enrichment results on L5 PT cells using POU1F1 and POU3F2 motif deviation z-score calculated by chromVAR. Results are shown in Figure 5B. We find that SAILERX still achieves the highest motif deviation z-score, further demonstrates the effectiveness of our method on facilitating downstream analysis of chromatin accessibility data.
Figure 5.

Motif enrichment scores. Motif deviation z-scores on cells identified as (A) Pvalb and (B) L5 PT by different methods from SNARE-seq dataset and the imputed dataset (imputation done by SAILERX). For each cell type, four enriched motifs are selected. Pairwise t-tests are performed between SAILERX and all other methods. Three-stars refers to differential significance between two methods (P-value < 0.05).
SAILERX recovers the cell type landscape in chromatin accessibility space through imputation
The high throughput of sc-seq measurements provides expressions and chromatin accessibility information at the finest resolution. However, due to the limitations of read depth and coverage, sc-seq data suffers from severe sparsity due to random dropouts during the sequencing stage. Imputation is often applied during data analysis to recover the missing values. Here we test how our methods denoise the raw scATAC-seq data after integrating information from the scRNA-seq modality. We benchmark against MAGIC (54), which utilizes data diffusion to perform data imputation, and scOpen which is a matrix factorization based method.
Here, imputed data is generated by SAILERX, MAGIC and scOpen respectively. For MAGIC, since one key factor for imputation quality is the neighborhood graph, we provide graphs generated by scRNA-seq and scATAC-seq to MAGIC (details in Methods), and show the visualizations of imputation results in Figure 6. As we can see, compared with MAGIC and scOpen, imputed data generated by SAILERX better preserves the cell type landscape, where cells of different types are forming distinct clusters. Since SAILERX can control the read depth at imputation stage, imputed data is free of these technical artifacts. Compared with other imputation strategies, imputation done by deep generative models better preserves the cell clusters and keeps distinct features of cells. To further validate the imputation result, we use imputed SNARE-seq data generated by SAILERX and redo the motif enrichment analysis on Pvalb and L5 PT cells (previous section). Motif deviation z-scores are visualized with violin plots as shown in Figure 5 (see SAILERX_imp column). From the results, we can see that data imputed by SAILERX shows significantly higher enrichment score, which indicates that some missing peaks are imputed for certain cell types.
Figure 6.

Results of imputations on PBMC 10k. (A) UMAP visualizations of imputed 10x Genomics PBMC chromatin accessibility data generated by SAILERX, scOpen, and MAGIC (MAGIC imputations are done with graphs generated by scRNA-seq and scATAC-seq respectively). Cells are colored by ground truth labels. (B) Quantitative metrics on the cell landscape.
DISCUSSION
Multimodal single cell data provides a more comprehensive way of measuring cell manifold. However, it is computationally challenging to leverage these multiomics data to better depict the biological view of cell-cell specificity still poses challenges for researchers due to imbalanced SNR cross modalities. Some modalities in nature have lower coverage rate, thus suffers more from noises like dropout. Current methods often fuse these multimodal data by projecting them to a same latent space(27,28,32). These approaches assume measurements from two modalities have the same distribution, and both modalities are equally informative on cell state information. In reality, these assumptions barely hold because chromatin accessibility changes usually prior to the changes of gene expression states (9); and scATAC-seq measurements tend to suffer more from sparsity but could potentially provide more detailed information on cell states. In the meantime, since there exist technical noises during sequencing process, which could bias the observed state of a cell toward different directions, projecting the observed data from different modalities to a same point could be problematic. Experiments have shown that projecting two modalities to a shared latent space could result in overfitting of noises and lead to worse delineation of cell state landscape, especially when using powerful models like neural networks (28).
To tackle these issues, in SAILERX, we use a more stable way by representing the more robust gene expression modality as a reference embedding, and guide the inference of a VAE modeling chromatin accessibility data. Instead of regularizing the latent variable for different modalities to be the same, we encourage the pairwise distances between cells to be similar across different modalities, in the meantime, use invariant representation learning to remove technical noises that are observable at training time. This flexible information fusing framework encourages the local structure of data to be similar and weights cells differently to better retrieve information from heterogeneous modalities. According to our results, this type of information fusion is able to preserve the informative parts from both modalities and constantly achieves better embeddings and downstream analysis. The final clustering results implicitly contain information from two modalities and can constantly improve upon any single modalities. SAILERX could also be used on dataset with missing reference modality. This allows SAILERX to be used under more scenarios (when datapoints from reference modality is missing during QC or analyzing a dataset with different sequencing protocols), using multimodal single cell data as a reference to facilitate the analysis of scATAC-seq data which usually suffers from low signal-to-noise ratio. With the help of SAILERX, researchers could rescue those low-quality single modality data through hybrid data integration and discover more informative features underneath those noises.
DATA AVAILABILITY
All data, code, and materials used in the analyses is available at https://github.com/uci-cbcl/SAILERX.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S Xu, A Hwang, and members of Xie lab for helpful discussions.
Author contributions: Conceived and supervised the study: XX; Contributed to model design: YC, LF and XX; Conducted model implementation: YC and LF; Contributed to experimental design: QN, JZ and XX; Contributed to experimental analysis: All; Wrote the paper: YC, LF and XX with input from other authors.
Contributor Information
Yingxin Cao, Department of Computer Science, University of California, Irvine, CA 92697, USA; Center for Complex Biological Systems, University of California, Irvine, CA 92697, USA; NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine, CA 92697, USA.
Laiyi Fu, Department of Computer Science, University of California, Irvine, CA 92697, USA; Systems Engineering Institute, School of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an, Shannxi 710049, China.
Jie Wu, Department of Biological Chemistry, University of California, Irvine, CA 92697, USA.
Qinke Peng, Systems Engineering Institute, School of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an, Shannxi 710049, China.
Qing Nie, Department of Mathematics, University of California, Irvine, CA 92697, USA; Center for Complex Biological Systems, University of California, Irvine, CA 92697, USA; NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine, CA 92697, USA.
Jing Zhang, Department of Computer Science, University of California, Irvine, CA 92697, USA.
Xiaohui Xie, Department of Computer Science, University of California, Irvine, CA 92697, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [IIS-1715017, DMS-1763272]; National Institutes of Health [U54-CA217378, R01HG012572]; National Institute of Mental Health [MH123896]; Simons Foundation [594598]. Funding for open access charge: National Science Foundation [IIS-1715017, DMS-1763272]; National Institutes of Health [grant number U54-CA217378].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A.et al.. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009; 6:377–382. [DOI] [PubMed] [Google Scholar]
- 2. Cao J., O’Day D.R., Pliner H.A., Kingsley P.D., Deng M., Daza R.M., Zager M.A., Aldinger K.A., Blecher-Gonen R., Zhang F.et al.. A human cell atlas of fetal gene expression. Science. 2020; 370:eaba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Domcke S., Hill A.J., Daza R.M., Cao J., O’Day D.R., Pliner H.A., Aldinger K.A., Pokholok D., Zhang F., Milbank J.H.et al.. A human cell atlas of fetal chromatin accessibility. Science. 2020; 370:eaba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cusanovich D.A., Hill A.J., Aghamirzaie D., Daza R.M., Pliner H.A., Berletch J.B., Filippova G.N., Huang X., Christiansen L., DeWitt W.S.et al.. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018; 174:1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P.. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013; 502:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karemaker I.D., Vermeulen M.. Single-Cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol. 2018; 36:952–965. [DOI] [PubMed] [Google Scholar]
- 7. Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P.. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017; 14:865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen S., Lake B.B., Zhang K.. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019; 37:1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma S., Zhang B., LaFave L.M., Earl A.S., Chiang Z., Hu Y., Ding J., Brack A., Kartha V.K., Tay T.et al.. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020; 183:1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N.. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018; 15:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hie B., Bryson B., Berger B.. Efficient integration of heterogeneous single-cell transcriptomes using scanorama. Nat. Biotechnol. 2019; 37:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P., Raychaudhuri S.. Fast, sensitive and accurate integration of single-cell data with harmony. Nature Methods. 2019; 16:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu C., Lopez R., Mehlman E., Regier J., Jordan M.I., Yosef N.. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol. Syst. Biol. 2021; 17:e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu J., Huang Y., Singh R., Vert J.-P., Noble W.S.. Jointly embedding multiple single-cell omics measurements. Algorithms Bioinform. 2019; 143:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Argelaguet R., Cuomo A.S.E., Stegle O., Marioni J.C.. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 2021; 39:1202–1215. [DOI] [PubMed] [Google Scholar]
- 16. Kriebel A.R., Welch J.D.. UINMF performs mosaic integration of single-cell multi-omic datasets using nonnegative matrix factorization. Nat. Commun. 2022; 13:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Welch J.D., Hartemink A.J., Prins J.F.. MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 2017; 18:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stark S.G., Ficek J., Locatello F., Bonilla X., Chevrier S., Singer F., Aebersold R., Al-Quaddoomi F.S., Albinus J., Tumor Profiler Consortium et al.. SCIM: universal single-cell matching with unpaired feature sets. Bioinformatics. 2020; 36:i919–i927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duren Z., Chen X., Zamanighomi M., Zeng W., Satpathy A.T., Chang H.Y., Wang Y., Wong W.H.. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:7723–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao K., Bai X., Hong Y., Wan L.. Unsupervised topological alignment for single-cell multi-omics integration. Bioinformatics. 2020; 36:i48–i56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gayoso A., Steier Z., Lopez R., Regier J., Nazor K.L., Streets A., Yosef N.. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods. 2021; 18:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Welch J.D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E.Z.. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019; 177:1873–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin S., Zhang L., Nie Q.. scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biology. 2020; 21:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Minoura K., Abe K., Nam H., Nishikawa H., Shimamura T.. scMM: Mixture-of-experts multimodal deep generative model for single-cell multiomics data analysis. 2021; bioRxiv doi:02 March 2021, preprint: not peer reviewed 10.1101/2021.02.18.431907. [DOI] [PMC free article] [PubMed]
- 25. Wu K.E., Yost K.E., Chang H.Y., Zou J.. BABEL enables cross-modality translation between multiomic profiles at single-cell resolution. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2023070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuo C., Chen L.. Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data. Brief. Bioinformatics. 2021; 22:bbaa287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gong B., Zhou Y., Purdom E.. Cobolt: joint analysis of multimodal single-cell sequencing data. Genome Biol. 2021; 22:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh R., Hie B.L., Narayan A., Berger B.. Schema: metric learning enables interpretable synthesis of heterogeneous single-cell modalities. Genome Biol. 2021; 22:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M.et al.. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moyer D., Gao S., Brekelmans R., Galstyan A., Ver Steeg G.. Invariant representations without adversarial training. Adv. Neural Inform. Process. Syst. 2018; 31:9084–9093. [Google Scholar]
- 31. Cao Y., Fu L., Wu J., Peng Q., Nie Q., Zhang J., Xie X.. SAILER: scalable and accurate invariant representation learning for single-cell ATAC-seq processing and integration. Bioinformatics. 2021; 37:i317–i326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R.. Single-cell chromatin state analysis with signac. Nat. Methods. 2021; 18:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biology. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N.. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018; 15:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luecken M.D., Büttner M., Chaichoompu K., Danese A., Interlandi M., Müller M.F., Strobl D.C., Zappia L., Dugas M., Colomé-Tatché M.et al.. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods. 2022; 19:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McInnes L., Healy J., Melville J.. Umap: uniform manifold approximation and projection for dimension reduction. JOSS. 2018; 3:861. [Google Scholar]
- 38. Yu L., Cao Y., Yang J.Y.H., Yang P.. Benchmarking clustering algorithms on estimating the number of cell types from single-cell RNA-sequencing data. Genome Biol. 2022; 23:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiong L., Xu K., Tian K., Shao Y., Tang L., Gao G., Zhang M., Jiang T., Zhang Q.C.. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat Commun. 2019; 10:4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Traag V.A., Waltman L., Van Eck N.J.. From louvain to leiden: guaranteeing well-connected communities. Scientific Reports. 2019; 9:5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.et al.. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 42. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. III, Hao Y., Stoeckius M., Smibert P., Satija R.. Comprehensive integration of single-cell data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J.. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017; 14:975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collin M., Bigley V.. Human dendritic cell subsets: an update. Immunology. 2018; 154:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlitzer A., Zhang W., Song M., Ma X.. Recent advances in understanding dendritic cell development, classification, and phenotype. F1000Research. 2018; 7:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mair F., Prlic M.. OMIP-044: 28-color immunophenotyping of the human dendritic cell compartment. Cytometry Part A. 2018; 93:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rhodes J.W., Tong O., Harman A.N., Turville S.G.. Human dendritic cell subsets, ontogeny, and impact on HIV infection. Front.Immunol. 2019; 10:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sakaguchi S., Miyara M., Costantino C.M., Hafler D.A.. FOXP3+ regulatory t cells in the human immune system. Nat. Rev. Immunol. 2010; 10:490–500. [DOI] [PubMed] [Google Scholar]
- 49. Bhairavabhotla R., Kim Y.C., Glass D.D., Escobar T.M., Patel M.C., Zahr R., Nguyen C.K., Kilaru G.K., Muljo S.A., Shevach E.M.. Transcriptome profiling of human foxp3+ regulatory t cells. Hum. Immunol. 2016; 77:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogers D., Sood A., Wang H., van Beek J.J.P., Rademaker T.J., Artusa P., Schneider C., Shen C., Wong D.C., Bhagrath A.et al.. Pre-existing chromatin accessibility and gene expression differences among naive CD4+ t cells influence effector potential. Cell Rep. 2021; 37:110064. [DOI] [PubMed] [Google Scholar]
- 51. Fang R., Preissl S., Li Y., Hou X., Lucero J., Wang X., Motamedi A., Shiau A.K., Zhou X., Xie F.et al.. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021; 12:1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mayer C., Hafemeister C., Bandler R.C., Machold R., Brito R.B., Jaglin X., Allaway K., Butler A., Fishell G., Satija R.. Developmental diversification of cortical inhibitory interneurons. Nature. 2018; 555:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen C., Meng Q., Xia Y., Ding C., Wang L., Dai R., Cheng L., Gunaratne P., Gibbs R.A., Min S.et al.. The transcription factor POU3F2 regulates a gene coexpression network in brain tissue from patients with psychiatric disorders. Sci. Transl. Med. 2018; 10:eaat8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Dijk D., Nainys J., Sharma R., Kaithail P., Carr A.J., Moon K.R., Mazutis L., Wolf G., Krishnaswamy S., Pe’er D.. MAGIC: a diffusion-based imputation method reveals gene-gene interactions in single-cell RNA-sequencing data. 2017; bioRxiv doi:25 February 2017, preprint: not peer reviewed 10.1101/111591. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data, code, and materials used in the analyses is available at https://github.com/uci-cbcl/SAILERX.