

Abstract
Rationale
Despite the increased recognition of TBX4 (T-BOX transcription factor 4)-associated pulmonary arterial hypertension (PAH), genotype–phenotype associations are lacking and may provide important insights.
Objectives
To compile and functionally characterize all TBX4 variants reported to date and undertake a comprehensive genotype-phenotype analysis.
Methods
We assembled a multicenter cohort of 137 patients harboring monoallelic TBX4 variants and assessed the pathogenicity of missense variation (n = 42) using a novel luciferase reporter assay containing T-BOX binding motifs. We sought genotype–phenotype correlations and undertook a comparative analysis with patients with PAH with BMPR2 (Bone Morphogenetic Protein Receptor type 2) causal variants (n = 162) or no identified variants in PAH-associated genes (n = 741) genotyped via the National Institute for Health Research BioResource–Rare Diseases.
Measurements and Main Results
Functional assessment of TBX4 missense variants led to the novel finding of gain-of-function effects associated with older age at diagnosis of lung disease compared with loss-of-function effects (P = 0.038). Variants located in the T-BOX and nuclear localization domains were associated with earlier presentation (P = 0.005) and increased incidence of interstitial lung disease (P = 0.003). Event-free survival (death or transplantation) was shorter in the T-BOX group (P = 0.022), although age had a significant effect in the hazard model (P = 0.0461). Carriers of TBX4 variants were diagnosed at a younger age (P < 0.001) and had worse baseline lung function (FEV1, FVC) (P = 0.009) than the BMPR2 and no identified causal variant groups.
Conclusions
We demonstrated that TBX4 syndrome is not strictly the result of haploinsufficiency but can also be caused by gain of function. The pleiotropic effects of TBX4 in lung disease may be in part explained by the differential effect of pathogenic mutations located in critical protein domains.
Keywords: pulmonary arterial hypertension, TBX4, interstitial lung disease, lung developmental disease, gain-of-function
At a Glance Commentary
Scientific Knowledge on the Subject
TBX4 (T-BOX transcription factor 4) is the second most common gene culprit for development of pulmonary arterial hypertension, especially in pediatric-onset cases. A growing body of literature has expanded its phenotypic spectrum, which now includes developmental lung lesions, ranging from mild to lethal.
What This Study Adds to the Field
Our study identifies genetic determinants of TBX4 disease heterogeneity, inclusive of newly described gain-of-function missense variants associated with later-onset lung disease. It draws important conclusions on genotype–phenotype associations, informative to TBX4 variant interpretation and genetic counseling of affected families.
Pulmonary arterial hypertension (PAH) is a rare progressive vasculopathy characterized by abnormal cell proliferation in the pulmonary arterioles leading to increased pulmonary artery pressure and, ultimately, right ventricular failure (1). It can occur idiopathically or in association with other medical conditions, such as congenital heart disease, or exposure to certain drugs and toxins. Since the landmark discovery of the BMPR2 (Bone Morphogenetic Protein Receptor type 2) gene in 2000 as the main cause of heritable PAH (2), high-throughput sequencing has implicated more than 20 genes with identified causal variants in up to 25% of individuals diagnosed with idiopathic PAH (3–5). Monoallelic pathogenic variants in the TBX4 (T-BOX transcription factor 4) gene are the second most common heritable cause of PAH, often enriched in pediatric cohorts (6–8). TBX4 belongs to the T-BOX family of transcription factors, playing a critical role in early hindlimb development (9–11) and branching of the lungs (12) and regulating the expression of FGF10 (Fibroblast Growth Factor 10) together with TBX5 (13, 14). Transcriptome analysis suggests it continues to be active after organogenesis and plays an important role in the cellular homeostasis of adult lung fibroblasts (15).
TBX4 sequence variants and contiguous gene deletions, as part of the recurrent chromosome 17q23.2 microdeletion, were originally reported in association with small patella syndrome (SPS; MIM# 147891) (16–18). More recently, evidence is emerging that TBX4 single-nucleotide variants and/or deletions are causative not only of PAH but also of a wide spectrum of developmental parenchymal lung disorders (19, 20). Partial or complete loss of a single TBX4 functional allele is sufficient for the production of the phenotype, termed haploinsufficiency. Although the above disorders are dominantly inherited, both variable expressivity and reduced penetrance have been observed (6).
Herein, we assembled a large cohort of patients harboring TBX4 sequence variants to establish genotype–phenotype correlations. We developed in vitro assays to assess the pathogenicity of nontruncating variants. This led to the novel finding of functionally distinguishable missense variation, causing either gain of function (GoF) or loss of function (LoF).
Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2022.02.06.22270467).
Methods
Using the term “TBX4”, our PubMed search identified 22 publications limited to human studies dating from 2004 to 2021 (online supplement). Cases with the recurrent 17q23.2 deletion were excluded from this study’s scope. We collected phenotypic information on 137 heterozygous carriers of TBX4 sequence variants, including 15 novel cases from the National Institute for Health Research BioResource–Rare Diseases (NBR) study (21), the UK National Cohort Study of Idiopathic and Heritable PAH (3, 5), the Spanish registry of PAH (22), the registry of the French PAH network (23, 24), and the DECIPHER database (25). Demographic and phenotypic information at the time of diagnosis was captured from relevant publications and databases (online supplement). Follow-up information was obtained, where available.
Annotation of variants was harmonized to the TBX4 canonical transcript NM_018488.3 of the human reference genome assembly GRCh37/hg19 using the Mutalyzer and Ensembl Variant Effect Predictor web services. We assessed variant pathogenicity according to the American College of Medical Genetics and Genomics (ACMG) guidelines using the VarSome Clinical tool followed by manual curation (26). Nontruncating variants (missense, indels) and variants predicted to affect splicing were selected for functional characterization (online supplement). Truncating variants (frameshift, nonsense) were presumed to cause LoF; p.Tyr127Ter pathogenic variant was used as a positive control in functional experiments.
Among the T-BOX family members, TBX4 has the highest similarity to TBX5 (27). Because no structure for TBX4 has been reported, we analyzed the effect of variants using the crystal structure of the TBX5 complex with DNA (Figure 3) (28). Variants were subgrouped by protein domains, including the highly conserved T-BOX domain (codons 71–251) containing the first nuclear localization segment (NLS1; 91–103), and the predicted NLS2 (338–351) and transactivating region (351–393) (29, 30).
Figure 3.

Structural analysis of TBX4 (T-BOX transcription factor 4) sequence variants. The crystal structure of TBX5 (T-BOX transcription factor 5) bound to DNA, Protein Database code 2X6V, was used for structural analysis. They share 52.6% sequence together with the full-length proteins and 93.9% in the T-BOX domain. (A) Sequence alignment of TBX4 and TBX5 in the T-BOX region (highlighted in gray) containing the DNA-binding motif as well as the nuclear localization segment 1 (NLS1, in cyan) and the nuclear export segment (NES, in yellow) (29, 30). TBX4 missense variants are indicated in bold/blue, with indels highlighted in magenta. Residues visible in the TBX5 structure are shown in light blue letters. (B) Mutations plotted on the TBX5 crystal structure as spheres. Cyan, yellow, and magenta spheres correspond to the NLS1, NES regions, and indels as indicated in A. When annotating loss-of-function variants on the TBX4 sequence, they are highly enriched in the T-BOX, particularly the NLS1 and NES. (C) Some mutations of the noninterface residues, such as TBX4 p.Glu86 and p.Tyr127 (corresponding residues p.Glu73 and p.Tyr114 in TBX5, respectively), make essential interactions to stabilize the secondary structural elements required for T-BOX binding to DNA. Clustal Omega was used for sequence alignment. Figures were generated using PyMOL Molecular Graphics System.
We sought genotype–phenotype associations in carriers of likely pathogenic/pathogenic TBX4 variants (excluded functionally assessed variants classed as benign/likely benign or of uncertain significance) and undertook a comparative analysis with genotyped patients with PAH recruited to the NBR study, termed in this paper as BMRP2 (n = 162) and no identified causal variant (n = 741) groups (Table 1). The latter included patients with idiopathic PAH and likely pathogenic/pathogenic BMRP2 variants or no identified likely pathogenic/pathogenic variants in PAH-associated genes (BMPR2, TBX4, EIF2AK4, SMAD1/4/9, CAV1, KCNK3, ENG, ALK1, GDF2, AQP1, ATP13A3, SOX17) (3). We additionally obtained computed tomography chest images from a subset of patients from the BMPR2 (n = 34) and no identified causal variant (n = 143) groups and compared these to 11 TBX4 cases from the NBR study. Details of the radiological substudy can be found in the online supplement. We assessed genotype–phenotype differences in the range of phenotypic features expected to differentiate the three selected genotypes. Because this analysis was preplanned and the results were consistent, we did not perform correction for multiple comparisons.
Table 1.
Demographic and Clinical Characteristics at Diagnosis of the Patient Population Included in the Genotype–Phenotype Dataset
| BMPR2 (n = 162) | No Causal Variant Identified (n = 741) | TBX4 (n = 98) | P Value | Available (Total n) | |
|---|---|---|---|---|---|
| Primary diagnosis | 1,001 | ||||
| 1.1 Idiopathic PAH, including drug and toxin induced | 107 (66.0) | 741 (100) | 58 (59.2) | ||
| 1.2 Heritable PAH | 53 (32.7) | — | 17 (17.3) | ||
| 1.4.1 PAH associated with connective tissue disease | 1 (0.62) | — | 2 (2.04) | ||
| 1.4.4 PAH associated with congenital heart disease | 1 (0.62) | — | 11 (11.2) | ||
| 1.6 PAH with overt features of venous/capillary (PVOD/PCH) involvement | — | — | 1 (1.02) | ||
| 3.5 Developmental lung disorders | — | — | 9 (9.18) | ||
| Sex: female | 107 (66.0) | 530 (71.5) | 62 (63.9) | 0.156 | 1,000 |
| Smoking history: past/current, adults only | 53 (39.3) | 293 (53.3) | 10 (47.6) | 0.039 | 706 |
| Exposure to drug or toxins: yes | 6 (3.70) | 45 (6.07) | 7 (19.4) | 0.007 | 939 |
| Ethnicity | 966 | ||||
| European | 137 (84.6) | 630 (85.0) | 53 (84.1) | ||
| Finnish European | — | 1 (0.13) | — | ||
| African | 2 (1.23) | 20 (2.70) | 5 (7.94) | ||
| East Asian | 2 (1.23) | 6 (0.81) | 1 (1.59) | ||
| South Asian | 6 (3.70) | 48 (6.48) | 1 (1.59) | ||
| Other | 15 (9.26) | 36 (4.86) | 3 (4.76) | ||
| Age at diagnosis of lung disease, yr | 39 (31–51) | 51 (38–66) | 14 (2–48) | <0.001 | 997 |
| Age at transplantation or death, yr | 52 (43–61) | 67 (53–75) | 64 (1–71) | <0.001 | 302 |
| WHO functional class | 0.277 | 918 | |||
| I | 2 (1.24) | 15 (2.11) | 2 (4.44) | ||
| II | 32 (19.9) | 144 (20.2) | 10 (22.2) | ||
| III | 96 (59.6) | 466 (65.4) | 27 (60.0) | ||
| IV | 31 (19.3) | 87 (12.2) | 6 (13.3) | ||
| Exercise test | |||||
| Distance, m | 350 (276–420) | 330 (210–410) | 371 (308–422) | 0.028 | 810 |
| Pretest saturation | 96.0 (94.0–98.0) | 96.0 (93.0–97.0) | 97.5 (95.5–98.0) | 0.006 | 738 |
| Post-test saturation | 94.0 (89.0–96.8) | 91.0 (85.0–95.0) | 95.5 (86.5–97.0) | 0.001 | 683 |
| Lung function, % predicted | |||||
| FEV1 | 91.0 (79.0–100) | 85.0 (72.3–96.0) | 82.0 (70.0–98.0) | 0.001 | 719 |
| FVC | 99.7 (17.2) | 93.2 (19.4) | 88.0 (17.7) | <0.001 | 703 |
| Kco | 83.4 (74.2–96.5) | 68.0 (49.0–83.0) | 72.0 (59.8–89.8) | <0.001 | 516 |
| TLC | 96.0 (89.0–106) | 94.0 (85.0–104) | 104 (98.0–110) | 0.044 | 511 |
| Hemodynamics | |||||
| mPAP, mm Hg | 57.0 (52.0–66.8) | 52.0 (42.0–61.0) | 60.5 (48.2–82.2) | <0.001 | 916 |
| mPAWP, mm Hg | 10.0 (7.00–12.0) | 9.00 (7.00–12.0) | 9.00 (7.00–11.0) | 0.841 | 822 |
| PVR, WU | 14.5 (10.8–20.4) | 10.3 (7.06–13.9) | 12.8 (8.25–16.2) | <0.001 | 767 |
| CO, L/min | 3.30 (2.69–3.94) | 4.04 (3.25–5.10) | 3.65 (3.09–4.61) | <0.001 | 852 |
| CI, L/min/m2 | 1.90 (1.51–2.23) | 2.30 (1.80–2.80) | 2.63 (1.98–3.20) | <0.001 | 503 |
| Vasoresponders | 1 (1.28) | 51 (17.7) | 6 (10.2) | 0.001 | 425 |
| Increased BNP (>50 pg/ml) or NT-proBNP (>300 pg/ml) | 34 (97.1) | 140 (79.1) | 5 (71.4) | 0.011 | 219 |
Definition of abbreviations: BMPR2 = Bone Morphogenetic Protein Receptor type 2; BNP = brain natriuretic peptide; CI = cardiac index; CO = cardiac output; mPAP = mean pulmonary artery pressure; mPAWP = mean pulmonary artery wedge pressure; NT-pro-BNP = N-terminal pro-brain natriuretic peptide; PCH = pulmonary capillary hemangiomatosis; PVOD = pulmonary venoocclusive disease; PVR = pulmonary vascular resistance; TBX4 = T-BOX transcription factor 4; WHO = World Health Organization; WU = Wood units = mm Hg/L/min.
Data are presented as n (%) or median (interquartile range). Heterozygous carriers of TBX4 variants shown to be benign by our functional studies were excluded.
Statistical analysis was performed using R (www.r-project.org). The R package “survival” was used to compare event-free survival between different groups. Survival was estimated by the Kaplan-Meier method from the time of diagnosis to death or transplantation. To avoid immortal time bias, this was limited to a 10-year interval. Sex and age at diagnosis of lung disease were included as covariates in the semiparametric Cox proportional hazard models.
Results
Study Population
We identified 137 heterozygous carriers of TBX4 variants, the majority of whom were sporadic cases (n = 127, 93%). Out of four identified families, eight related individuals with available detailed phenotypic information were included in our analyses. Twenty-one cases had a primary phenotype of SPS. In the remaining 116 individuals presenting with lung disease, subsequent assessment for SPS was lacking in most, with only 29 cases (25%) reported having associated skeletal features. PAH was the predominant primary lung phenotype (Table 1). Median age at diagnosis of lung disease was 14 years (interquartile range [IQR], 2–47 yr). Fifty-three individuals (45%) presented in adulthood, 41 (36%) in childhood, and 22 (19%) in the perinatal period. In the overall patient cohort, there was an equal female-to-male ratio, with an observed female predominance (62%) in the lung disease group.
Spectrum and Functional Assessment of TBX4 Variants
A total of 108 distinct TBX4 variants were retrieved from the literature and aforementioned databases (Figure 1). Of these, 43 were missense, 39 frameshift, 15 nonsense, 6 indels including an additional deletion of whole exon 5, and 3 variants predicted to affect splicing (online supplement). A single case with a variant in the TBX4 promoter region was excluded from our genotype–phenotype analyses (23).
Figure 1.
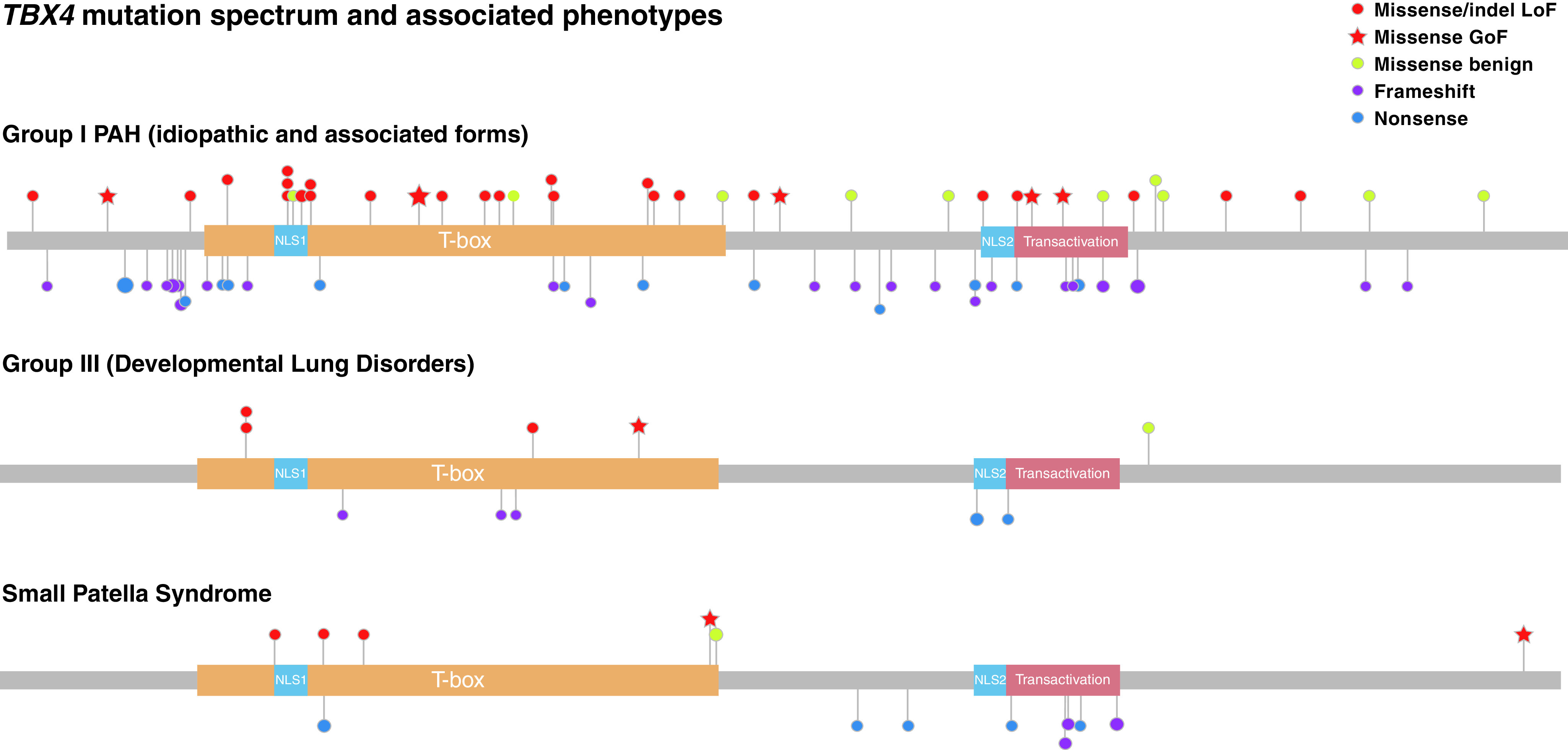
Lolliplot depicting TBX4 (T-BOX transcription factor 4) mutation spectrum. Recurrently mutated positions are represented by a proportionally sized lollipop. Critical protein domains are highlighted, inclusive of the DNA binding T-BOX, nuclear localization segments (NLS1 and NLS2), and transactivation domains. Variants are grouped by primary associated phenotype and color-coded, taking into account the functional assessment of missense and inframe insertion/deletions (indels). GoF = gain-of-function; LoF = loss-of-function; PAH = pulmonary arterial hypertension.
We assessed the pathogenicity of all indels and 42/43 missense variants using a luciferase reporter assay (Figure 2). Variant c.1021G>C was assessed by a minigene assay instead as it was predicted to affect the same donor splice site as in c.1021 + 1G>A, which resulted in a double exon skipping. All indels and 23 missense variants caused LoF, with another 11 shown to be benign. Eight missense variants resulted in GoF with mean relative luciferase units of approximately twice the amount of the wild-type construct. We confirmed that GoF was not an artifact by checking TBX4 protein expression amount of several constructs with different outcomes in the luciferase assay by quantitative PCR and western blot (see Figure E1 in the online supplement). Independent of their functional activity, all assessed variants translocated to the nucleus, replicating the translocation of wild-type and green fluorescent protein–tagged TBX4 (Figures E2 and E3).
Figure 2.
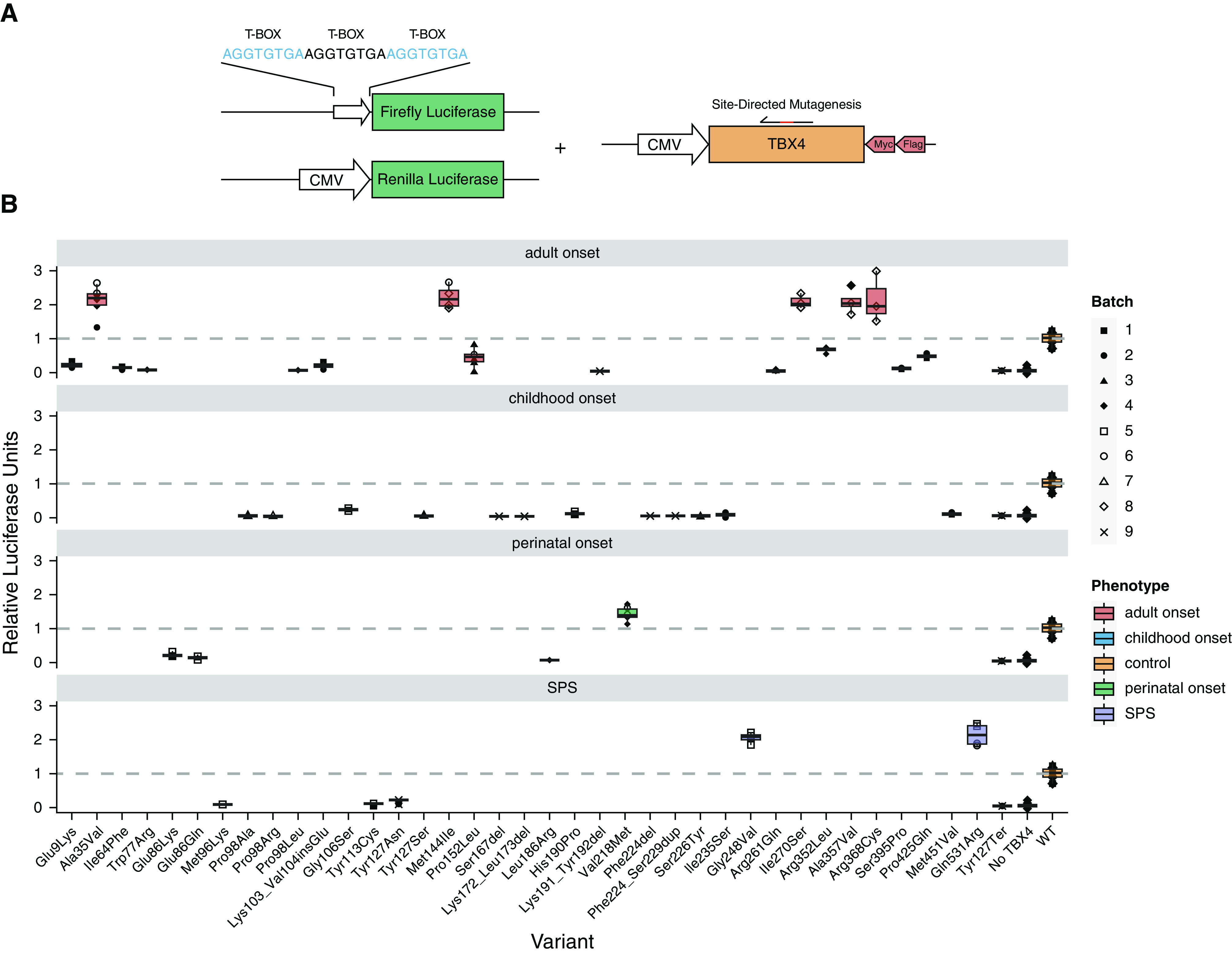
Functionally assessed TBX4 (T-BOX transcription factor 4) variants by luciferase assay inducing gain or loss of function. (A) Schematic representation of the in vitro dual-luciferase reporter assay. We cotransfected three different plasmids: Firefly luciferase with x3 T-BOX motifs as the promoter, Renilla luciferase, and TBX4 overexpression plasmid (wild type/mutated). (B) Variants inducing gain or loss of function grouped by primary phenotype; lung disease (perinatal-, childhood-, and adult-onset) or small patella syndrome (SPS). The y-axis represents the relative luciferase units; the dashed line marks the level of the wild type. Data are shown as box plots representing median ± quartiles. Dots represent biological replicates with corresponding batches in different shapes. CMV = Cytomegalovirus promoter; Flag = Flag tag; Myc = partial cMyc tag.
As per ACMG guidelines, our luciferase functional data altered the classification of the majority of respective variants, with a total of 33/48 (67%) initially classified as variants of uncertain significance and an equivalent number (32/48) of likely pathogenic/pathogenic variants after application of the PS3 criterion (in vitro functional studies supportive of a damaging effect) where appropriate (Figure E4). We evaluated the performance of in silico tool predictions for likely pathogenic/pathogenic TBX4 missense variants, with SIFT (Sorting Intolerant From Tolerant) generating the highest overall percentage of correct calls (Figure E5). Overall, a total of 4/8 GoF and 22/23 LoF missense variants were classed as likely pathogenic/pathogenic and included in the genotype–phenotype analyses (online supplement).
Finally, we assessed the functional impact of all previously reported TBX4 variants predicted to affect splicing with recurrent variants c.702 + 1G>A and c.1021 + 1G>A inducing exon skipping events (Figure E6). Key findings of the structural variant analysis are summarized in Figure 3.
Genotype–Phenotype Associations
Patients with lung disease and variants within the DNA binding T-BOX domain presented at a younger age (median [IQR], 7.5 [1–18.5] yr) than carriers of variants outside this domain (18 [3–51.5] yr; P = 0.028). This remained true for sequence variants located in the T-BOX domain containing either NLS1 or NLS2 at the C terminus (P = 0.005; Figure 4). Individuals with LoF variants (missense, indels) were also diagnosed at a younger age than GoF variant carriers (14 [5–31] yr vs. 57 [42.5–59.5] yr; P = 0.038; Figure 4).
Figure 4.

Age at diagnosis of lung disease by TBX4 (T-BOX transcription factor 4) genotype. (A) Variants in the T-BOX and second nuclear localization segment (NLS2) versus other domains. (B) Likely pathogenic/pathogenic gain-of-function (GoF) versus loss-of-function (LoF) missense variants.
Baseline clinical features and hemodynamic parameters did not differ significantly between protein domain groups, with the exception of interstitial lung disease, which was more frequently reported in carriers of TBX4 variants located in the T-BOX domain alone (75% vs. 21%; P = 0.003) or in combination with NLS2 variants (P = 0.001). Similarly, no significant differences were observed between LoF and GoF variation, and this was consistent when protein-truncating variants were added to the LoF group.
The observed primary phenotypes varied significantly between protein domains, with a greater frequency of developmental lung disorders (including acinar dysplasia and congenital alveolar dysplasia) in the T-BOX group (15.4% vs. 3.8%; overall P = 0.046); when combined with variants in the NLS2 domain, this remained statistically significant (overall P = 0.003). Lung histology was available in 17 previously published TBX4 cases (see online supplement for details). A greater frequency of a confirmed secondary phenotype of SPS was observed in carriers of variants located outside the T-BOX and NLS2 domains (29.7% vs. 11.4%; P = 0.038) and was more prevalent in protein-truncating versus missense variation (27.8% vs. 7.1%; P = 0.044). Variant exon location did not appear to have any phenotypic impact (data not shown).
As previously reported (31), patients with TBX4-associated lung disease presented at a younger age (median [IQR], 14 [2–48] yr) than patients in the BMPR2 (39 [31–51] yr) and no identified causal variant (51 [38–66] yr; P < 0.001) groups. They performed better at the 6-minute-walk test, with no significant differences in functional class at presentation (Table 1). They also had worse baseline lung function (FEV1, FVC), despite presentation at a younger age (Table 1). The frequency of airway and/or acinar abnormalities was significantly greater in the TBX4 (87.5%) versus the BMPR2 (32.4%) and no identified causal variant groups (33.6%; P = 0.009) (Table 2).
Table 2.
Radiological Features of Computed Tomography of the Chest in Pulmonary Arterial Hypertension Cases Analyzed as Part of the Imaging Substudy
| BMPR2 (n = 34) | No Causal Variant Identified (n = 143) | TBX4 (n = 8) | P Value | Available (Total n) | |
|---|---|---|---|---|---|
| Vascular features | |||||
| Axial MPA diameter, mm | 34.2 (30.9–37.4) | 34.6 (31.4–38.3) | 34.0 (30.7–38.5) | 0.674 | 185 |
| PA to Ao ratio | 1.23 (1.12–1.40) | 1.14 (0.99–1.29) | 1.18 (0.94–1.32) | 0.083 | 185 |
| Neovascularity | 7 (23.3) | 10 (7.69) | — | 0.048 | 167 |
| Lung and mediastinal features | |||||
| Ground-glass opacities | 18 (52.9) | 62 (43.4) | — | 0.016 | 185 |
| Centrilobular pattern | 0.088 | 185 | |||
| None | 17 (50.0) | 90 (62.9) | 8 (100) | ||
| Subtle | 4 (11.8) | 21 (14.7) | — | ||
| Present | 13 (38.2) | 32 (22.4) | — | ||
| Nonspecific mosaic pattern | 0.441 | 180 | |||
| None | 31 (91.2) | 130 (90.9) | 3 (100) | ||
| Subtle | — | 7 (4.90) | — | ||
| Present | 3 (8.82) | 6 (4.20) | — | ||
| Fibrosis | 1.000 | 185 | |||
| None | 34 (100) | 137 (95.8) | 8 (100) | ||
| Present | — | 6 (4.2) | — | ||
| Pleural effusion | |||||
| None | 33 (97.1) | 132 (92.3) | 8 (100) | 1.000 | 185 |
| Subtle | — | 3 (2.10) | — | ||
| Present | 1 (2.94) | 8 (5.59) | — | ||
| Interlobular septal thickening | 0.157 | 179 | |||
| None | 27 (87.1) | 122 (87.1) | 5 (62.5) | ||
| Subtle | 3 (9.68) | 9 (6.43) | 1 (12.5) | ||
| Present | 1 (3.23) | 9 (6.43) | 2 (25.0) | ||
| Mediastinal lymphadenopathy | 0.721 | 185 | |||
| None | 29 (85.3) | 122 (85.3) | 6 (75.0) | ||
| Present | 5 (14.7) | 21 (14.7) | 2 (25.0) | ||
| Airway/acinar features | 11 (32.4) | 48 (33.6) | 7 (87.5) | 0.009 | 185 |
| Bronchial wall thickening | 0.051 | 179 | |||
| None | 26 (83.9) | 126 (90.0) | 6 (75.0) | ||
| Subtle | 3 (9.68) | 11 (7.86) | — | ||
| Present | 2 (6.45) | 3 (2.14) | 2 (25.0) | ||
| Emphysema | 0.026 | 185 | |||
| None | 32 (94.1) | 120 (83.9) | 5 (62.5) | ||
| Subtle | — | 13 (9.09) | — | ||
| Present | 2 (5.88) | 10 (6.99) | 3 (37.5) | ||
| Air trapping | 0.040 | 185 | |||
| None | 28 (82.4) | 121 (84.6) | 4 (50.0) | ||
| Subtle | 4 (11.8) | 7 (4.90) | 1 (12.5) | ||
| Present | 2 (5.88) | 15 (10.5) | 3 (37.5) | ||
| Suspected PVOD | 1 (3.03) | 8 (5.59) | 1 (12.5) | 0.500 | 184 |
Definition of abbreviations: Ao = aortic; MPA = main pulmonary artery; PA = pulmonary artery; PVOD = pulmonary venoocclusive disease; TBX4 = T-BOX transcription factor 4.
Data are presented as n (%) or median (interquartile range).
Impact of Genotype on Survival
Clinical outcomes were available for 89/115 TBX4 cases presenting with lung disease, with a median follow-up of 8 years (IQR, 3–10 yr). Event-free survival was shorter in the T-BOX domain variant group (P = 0.022 for log-rank test), although age at diagnosis also had a significant effect (P = 0.0461 for Cox proportional hazard model; Figure 5). There were no observed differences in outcomes between GoF and LoF variants or missense versus protein-truncating variants. Overall, event-free survival was longer in the TBX4 group compared with BMPR2 and no identified causal variants groups (P = 0.0025). Pairwise comparisons showed no significant differences between TBX4 and BMPR2 variant carriers (P = 0.69). Compared with individuals with no causal sequence variants, both patients with TBX4 (P = 0.035) and BMPR2 variants (P = 0.016) had higher event-free survival rates. However, genotype did not have a significant effect on survival after correction for age and sex; both male sex (P = 0.0002) and older age at the time of diagnosis (P < 2 × 10−16) were associated with shorter survival in the Cox proportional hazard model.
Figure 5.

Time to death or lung transplantation (years) by TBX4 (T-BOX transcription factor 4) protein domain genotype. Event-free survival was shorter in the T-BOX domain variant group, although age had a significant effect in the hazard model. CI = confidence interval; *Variables with P < 0.05.
Discussion
Distinct Mutational Mechanisms Disrupt TBX4 Function
A wide range of TBX4 mutations can result in human disease. Intragenic sequence variants occur throughout the gene and are mostly private. The functional impact of TBX4 variants was not previously investigated, and a haploinsufficient effect was assumed. Our assessment of TBX4 missense variants reported to date led to the discovery of two distinct functional classes of variants, GoF or LoF. Although this constitutes a novel finding, GoF variants have been reported in other members of the T-BOX gene family in association with a similar phenotypic spectrum caused by LoF. This includes carriers of missense variants in the TBX1 gene with velocardiofacial syndrome, a single TBX5 variant in a family with Holt-Oram syndrome, and a TBX20 variant in a family with atrial septal defects and valvular disease (32–34). All of the above were located in the highly conserved T-BOX domain, with the exception of a single TBX1 variant in exon 8 of 9 (c.928G>A). Structural analyses were suggestive of increased protein stability; our respective analysis was indicative of the detrimental effects of LoF variants, with no simple explanation for GoF variation (Figure 3).
Out of 31 pathogenic missense TBX4 variants reported in our study, 8 resulted in GoF, including the recurrent variant c.432G>T (22). These were located across the gene, with three in the T-BOX region and two in the transactivation domain (Figure 1). Two variants (c.743G>T and c.1592A>G) were originally reported in 2004 in two Dutch families with classic SPS and constituted one of the first reports implicating TBX4 in this phenotype (16). The remaining variants were described in association with adult-onset lung disease diagnosed as late as 81 years, with the exception of a neonate presenting with interstitial lung disease (Table 3). Notably, six of eight GoF variants were present in control populations (gnomAD database) including variant c.104C>T, with the highest overall population frequency in the cumulative TBX4 variant dataset. As a result, many of these were classified as likely benign or of uncertain clinical significance and cannot be considered as responsible for the underlying phenotype at present (Table 3).
Table 3.
Clinical Features of Individuals Heterozygous for TBX4 Gain-of-Function Missense Variants
| Variant | Demographics | Primary Lung Phenotype; Age at Diagnosis | Details of Lung Disease and Comorbidities | Skeletal Features | Follow-Up | Pedigree Information | ACMG Classification; Population Frequency (gnomAD Exomes) |
|---|---|---|---|---|---|---|---|
| c.104C>T; p.Ala35Val | Dutch PAH patient cohort (40) | PAH; adult onset | N/A | N/A | Deceased (unavailable details) | N/A | Likely benign; 0.009 |
| c.432G>T; p.Met144Ile | Female, White Hispanic (22) | IPAH; 28 yr | Clinical diagnosis of possible PVOD due to radiological findings and reduced diffusion capacity. Histopathological findings (explanted lung tissue): typical PAH features, no evidence of PVOD or ILD. Unknown smoking status. | Absent | Clinical deterioration with bilateral lung transplantation 9 yr after diagnosis | Maternally inherited variant, PAH ruled out at 67 yr | Pathogenic; 3.98 × 10−6 |
| c.432G>T; p.Met144Ile | Male, White Hispanic (22) | PVOD; 62 yr | Radiological findings typical of PVOD. Signs of heart failure, severe respiratory insufficiency. Not eligible for lung transplant because of advanced age and comorbidities. Unknown smoking status. | Absent | Deceased (26 mo after diagnosis) | No known family history of PAH/SPS. Deceased parents before PAH diagnosis. Genetic counseling provided to the rest of the family (declined genetic testing) | Pathogenic; 3.98 × 10−6 |
| c.652G>A; p.Val218Met | Female, White European (24) | chILD; 5 mo | Lung biopsy at 7 yr: diffuse alveolar simplification, moderate thickened PA muscular wall, and PA fibrointimal proliferation. PFO, microcephaly, hearing loss. | Short stature, no SPS features | 10 yr: chILD, moderate pulmonary hypertension | Parents not tested | VUS; 1.23 × 10−4 |
| c.743G>T; p.Gly248Val | Dutch family (16, 40) | — | Family members screened for PAH by echocardiography with none fulfilling diagnostic criteria (unavailable details). | Classical SPS phenotype, including patellar and pelvic anomalies | N/A | Three-generation pedigree (family A) (16) | Pathogenic; not reported |
| c.809T>G; p.Ile270Ser | Female, White European (37) | IPAH; 57 yr | Functional class III at diagnosis. Asthma, HTN, obesity, sleep apnea. Never smoked. | N/A | Alive (1-yr follow-up) | N/A | Likely pathogenic; 4.00 × 10−6 |
| c.1070C>T; p.Ala357Val | Female, White British (3) | IPAH; 81 yr | Functional class III at diagnosis. Radiological findings included minor bronchial wall thickening, no evidence of PVOD except mediastinal nodes, no interstitial lung disease. Hypothyroidism, IHD, T2DM, HTN, breast cancer. Never smoked. | N/A | Died in hospice 7 mo after diagnosis (progressive heart failure) | N/A | VUS; 1.99 × 10−5 |
| c.1102C>T; p.Arg368Cys | Female, African Caribbean (3) | IPAH; 39 yr | Functional class III at diagnosis. Radiological findings are typical of PAH, no lung disease, PFO, hypothyroidism, chronic subdural hematoma. Unknown smoking status. | N/A | Deceased (50 yr) | N/A | VUS; 5.58 × 10−5 |
| c.1592A>G; p.Gln531Arg | Dutch family (16, 40) | — | Family members screened for PAH by echocardiography with none fulfilling diagnostic criteria (unavailable details) | Classical SPS phenotype, including patellar and pelvic anomalies | N/A | Three-generation pedigree (family C) (16) | Likely pathogenic; not reported |
Definition of abbreviations: ACMG = American College of Medical Genetics and Genomics; chILD = childhood-onset interstitial lung disease; HTN = systemic hypertension; IHD = ischemic heart disease; ILD = interstitial lung disease; IPAH = idiopathic pulmonary arterial hypertension; N/A = not available; PA = pulmonary artery; PAH = pulmonary arterial hypertension; PFO = patent foramen ovale; PVOD = pulmonary venoocclusive disease; SPS = small patella syndrome; T2DM = type 2 diabetes mellitus; TBX4 = T-BOX transcription factor 4; VUS = variant of uncertain clinical significance.
It remains to be seen whether these GoF variants are purely hypermorphic, increasing the protein’s function, or neomorphic, causing ectopic expression or acquisition of a new function. This may in turn influence their exerted phenotypic effects. From a mechanistic perspective, our knowledge of the consequences of TBX4 overexpression during embryogenesis and/or after organogenesis is lacking. Transcription can be affected not only by decreased but also by excessive amounts of transcription factors (35). Comparison of likely pathogenic/pathogenic GoF and LoF missense variation showed a later-onset lung disease in the former group (Figure 4), which may suggest a milder influence on the phenotype, with additional genetic modifiers likely at play (20). A number of functionally assessed TBX4 missense variants appeared to be hypomorphic, resulting in reduced amounts of transcriptional activity but not complete LoF (Figures 2 and E7); the vast majority of null variants were located in the T-BOX domain, whereas hypomorphic variants spread across the gene. However, the above differences in transcriptional activity alone were not sufficient to explain the diverse phenotypic spectrum of TBX4 disease (no significant genotype–phenotype correlations).
Modifiers of TBX4 Disease Spectrum
Our study captures the variable expressivity of TBX4 pathogenic variation, with additionally observed phenotypic differences in recurring variants (online supplement). Aside from hypertensive pulmonary vascular disease, distal lung development can be disrupted to a variable degree, raising the issue of TBX4 disease classification under World Health Organization group 3, pulmonary hypertension associated with hypoxia and lung disease, as opposed to group 1, idiopathic/heritable PAH. Although the underlying mechanisms are not yet fully elucidated, the impact of genotype was discernible with a greater frequency of developmental lung disorders and interstitial lung disease in individuals harboring pathogenic variants located in the T-BOX domain and nuclear localization segments NLS1 and NLS2. In contrast, SPS was more frequently observed as a secondary phenotype when variants occurred outside the above TBX4 regions.
Despite the younger age at presentation in TBX4 compared with BMPR2 variant carriers, baseline lung function was worse, with evidence of possible disrupted alveolarization on computed tomography imaging suggestive of underlying developmental lung lesions. It can be postulated that this partly accounts for the variable age of onset of TBX4-associated pulmonary hypertension with less pulmonary reserve and increased susceptibility to external insults acting as environmental modifiers of penetrance. There are insufficient data at present to suggest a specific PAH treatment approach, distinct from other etiologies, for TBX4 disease. However, at the point of diagnosis, it is important to evaluate the potential impact of any parenchymal abnormalities versus pure vascular disease to adapt treatment accordingly.
Gene-Specific Variant Classification and Prognostic Value
Inconsistent variant interpretation can not only lead to misdiagnosis of individual patients but also have significant consequences for at-risk relatives through inappropriate predictive testing. Gene-specific knowledge overcomes some of these pitfalls, especially when semiautomated impact analysis tools are used for variant classification. In silico predictions did not reliably reflect the true effects of TBX4 missense variants on gene activity (Figure E4). Structural analysis was also not capable of discerning TBX4 GoF variants. In light of these limitations, an integrated pipeline incorporating molecular testing and functional assessment of novel TBX4 missense variants by standardized assays would be of high diagnostic value.
Our results indicate that both protein-truncating and missense variants contribute to TBX4 disease, with the majority of the latter (26/42) classified as likely pathogenic/pathogenic after functional assessment. In light of this, we removed the ACMG BP1 criterion (missense variant in a gene for which primarily truncating variants are known to cause disease) from TBX4 variant annotation. In addition, the PM1 criterion (variant located in critical or well-established functional domain) could be expanded to include not only variants located in the T-BOX domain but also the predicted nuclear localization segments (NLS1 and NLS2), as our study was suggestive of an equivalent effect on produced phenotypes, including a higher rate of developmental (early-onset) lung disorders.
Estimating the true penetrance of TBX4 disease remains a high priority and impacts variant interpretation as well as genetic counseling of respective families. A characteristic example exhibiting inter- and intrafamilial variability is that of splice variant c.1021 + 1G>A reported by three independent studies (online supplement); index cases had adult-onset lung disease with positive family history, including first-degree relatives with severe PAH resulting in death in infancy or childhood or absent patella only (22, 36, 37). Leaky splicing variants (reducing but not completely abolishing the production of normal transcripts) can result in reduced penetrance, although this phenomenon would still not explain the variable phenotypes observed in association with other types of recurrent variants (online supplement). We applied the ACMG BS1 criterion (allele frequency is greater than expected for the disorder) using a maximum tolerated population frequency of 5.00 × 10−8 arising from a generous estimate of 50% for lung disease penetrance (38); out of 17/42 missense TBX4 variants present in gnomAD, several GoF and LoF variants remained of uncertain clinical significance (online supplement). Where there is enough evidence to support (likely) pathogenicity for missense variation in critical protein domains, important prognostic information can be inferred based on our genotype–phenotype analysis.
Limitations
We were limited by the multicenter nature of this study including retrospective data collection with variable follow-up duration. This methodology introduced selection bias, because the vast majority of cases were previously published, a proportion of which consisted of severe cases with lung histopathology from postmortem examination or explants at the time of lung transplantation. Published cases may not be truly representative of the natural history of TBX4 disease at large, which is yet to be elucidated via recall-by-genotype studies. We were unable to account for missing phenotypic information (presence or absence of SPS features in individuals with a primary lung phenotype), although estimation of TBX4 disease penetrance was not the focus of this work. The main limitation of our functional assay is that we used overexpression plasmids, forcing the expression of variants whose effect could be lowered by post-transcriptional regulation. Our reporter system was designed using the standard T-BOX motif and FGF10 promoter sequences. A recent report of TBX4 chromatin immunoprecipitation in human fetal lung fibroblasts identified several other potential genome-wide target sites whose effects we have not tested (39). Therefore, a variant shown to have a modest or benign effect by our luciferase assay may still have significant damaging effects on interactions with other crucial binding sites in biologically relevant tissues.
Conclusions
We combined functional and phenotypic characterization of all reported TBX4 sequence variants to date to determine the hallmarks of TBX4-mediated lung disease. We used in vitro analyses to assess the pathogenicity of missense, indel, and splice variants, resulting in either LoF or GoF effects with phenotypic and prognostic implications when also taking into account variant location in functional domains. Our knowledge of TBX4 genotype–phenotype associations can only be furthered by active collaborations between molecular scientists and clinicians, requiring both an in-depth understanding of the biological aspects of the disease and a systematic approach to phenotyping.
Acknowledgments
Acknowledgments
The authors thank Sebastián Comesaña, Verónica Outeiriño and Inés Pazos of the Centro de Apoio Científico-Técnico á Investigación (CACTI) for their help in sequencing and imaging. They also thank Carlos Solarat, Berta Estévez and Cristina González Abalde for their colony picking skills, Martin R. Wilkins for his contribution to cohort data, and A.C. Houweling for counseling patients from the Dutch cohort (Amsterdam).
Footnotes
Supported by NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Hospital Charity, Medical Research Council grant MR/K020919/1, Dinosaur Trust, Fundación Contra la Hipertensión Pulmonar, NIHR Cambridge Biomedical Research Centre, Dutch Heart Foundation grant CVON2017-4 DOLPHIN-GENESIS, British Heart Foundation grant FS/18/13/3328, Instituto de Salud Carlos III grant PI18/01233, and Consellería de Cultura, Educación e Ordenación Universitaria, Xunta de Galicia grant ED431G/02. M.L.D. was supported by a Xunta de Galicia predoctoral fellowship (ED481A-2018/304). A.S. is funded by a Wellcome Trust fellowship (205188/Z/16/Z).
Author Contributions: All authors collected data and provided constructive criticism of the study manuscript. M.P., M.L.D., E.M.S., W.L., A.J.S., P.D.U., N.W.M., S.G., and D.V. designed and interpreted the study. M.P., M.L.D., and E.M.S. analyzed the data.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202203-0485OC on July 19, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Simonneau G, Montani D, Celermajer D, Denton C, Gatzoulis M, Krowka M, et al. Hemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J . 2019;53:1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet . 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun . 2018;9:1416. doi: 10.1038/s41467-018-03672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swietlik EM, Greene D, Zhu N, Megy K, Cogliano M, Rajaram S, et al. Bayesian inference associates rare KDR variants with specific phenotypes in pulmonary arterial hypertension. Circ Genom Precis Med . 2020;14:e003155. doi: 10.1161/CIRCGEN.120.003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu N, Swietlik EM, Welch CL, Pauciulo MW, Hagen JJ, Zhou X, et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med . 2021;13:80. doi: 10.1186/s13073-021-00891-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swietlik EM, Prapa M, Martin JM, Pandya D, Auckland K, Morrell NW, et al. ‘There and back again’—forward genetics and reverse phenotyping in pulmonary arterial hypertension. Genes (Basel) . 2020;11:1408. doi: 10.3390/genes11121408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Welch CL, Chung WK. Genetics and genomics of pediatric pulmonary arterial hypertension. Genes (Basel) . 2020;11:1213. doi: 10.3390/genes11101213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol . 2019;17:85–95. doi: 10.1038/s41569-019-0242-x. [DOI] [PubMed] [Google Scholar]
- 9. Naiche LA, Harrelson Z, Kelly RG, Papaioannou VE. T-box genes in vertebrate development. Annu Rev Genet . 2005;39:219–239. doi: 10.1146/annurev.genet.39.073003.105925. [DOI] [PubMed] [Google Scholar]
- 10. Chapman DL, Garvey N, Hancock S, Alexiou M, Agulnik SI, Gibson-Brown JJ, et al. Expression of the T-box family genes, Tbx1–Tbx5, during early mouse development. Dev Dyn . 1996;206:379–390. doi: 10.1002/(SICI)1097-0177(199608)206:4<379::AID-AJA4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11. Gibson-Brown JJ. Agulnik SI, Silver LM, Papaioannou VE. Expression of T-box genes Tbx2–Tbx5 during chick organogenesis. Mech Dev . 1998;74:165–169. doi: 10.1016/s0925-4773(98)00056-2. [DOI] [PubMed] [Google Scholar]
- 12. Naiche LA, Arora R, Kania A, Lewandoski M, Papaioannou VE. Identity and fate of Tbx4-expressing cells reveal developmental cell fate decisions in the allantois, limb, and external genitalia. Dev Dyn . 2011;240:2290–2300. doi: 10.1002/dvdy.22731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell . 2009;17:290–298. doi: 10.1016/j.devcel.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arora R, Metzger RJ, Papaioannou VE. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet . 2012;8:e1002866. doi: 10.1371/journal.pgen.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Horie M, Miyashita N, Mikami Y, Noguchi S, Yamauchi Y, Suzukawa M, et al. TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am J Physiol Lung Cell Mol Physiol . 2018;314:L177–L191. doi: 10.1152/ajplung.00193.2017. [DOI] [PubMed] [Google Scholar]
- 16. Bongers EMHF, Duijf PHG, van Beersum SEM, Schoots J, van Kampen A, Burckhardt A, et al. Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet . 2004;74:1239–1248. doi: 10.1086/421331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, Thrush DL, et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet . 2010;86:454–461. doi: 10.1016/j.ajhg.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levin ML, Shaffer LG, Lewis RA, Gresik MV, Lupski JR. Unique de novo interstitial deletion of chromosome 17, del(17) (q23.2q24.3) in a female newborn with multiple congenital anomalies. Am J Med Genet . 1995;55:30–32. doi: 10.1002/ajmg.1320550110. [DOI] [PubMed] [Google Scholar]
- 19. German K, Deutsch GH, Freed AS, Dipple KM, Chabra S, Bennett JT. Identification of a deletion containing TBX4 in a neonate with acinar dysplasia by rapid exome sequencing. Am J Med Genet A . 2019;179:842–845. doi: 10.1002/ajmg.a.61096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karolak JA, Vincent M, Deutsch G, Gambin T, Cogné B, Pichon O, et al. Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet . 2019;104:213–228. doi: 10.1016/j.ajhg.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature . 2020;583:96–102. doi: 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hernandez-Gonzalez I, Tenorio J, Palomino-Doza J, Meñaca AM, Ruiz RM, Lago-Docampo M, et al. Clinical heterogeneity of pulmonary arterial hypertension associated with variants in TBX4. PLoS One . 2020;15:e0232216. doi: 10.1371/journal.pone.0232216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thoré P, Girerd B, Jaïs X, Savale L, Ghigna M-R, Eyries M, et al. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur Respir J . 2020;55:1902340. doi: 10.1183/13993003.02340-2019. [DOI] [PubMed] [Google Scholar]
- 24. Galambos C, Mullen MP, Shieh JT, Schwerk N, Kielt MJ, Ullmann N, et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur Respir J . 2019;54:1801965. doi: 10.1183/13993003.01965-2018. [DOI] [PubMed] [Google Scholar]
- 25. Bragin E, Chatzimichali EA, Wright CF, Hurles ME, Firth HV, Bevan AP, et al. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res . 2014;42:D993–D1000. doi: 10.1093/nar/gkt937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med . 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Papaioannou VE. The T-box gene family: emerging roles in development, stem cells and cancer. Development . 2014;141:3819–3833. doi: 10.1242/dev.104471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stirnimann CU, Ptchelkine D, Grimm C, Müller CW. Structural basis of TBX5-DNA recognition: the T-box domain in its DNA-bound and -unbound form. J Mol Biol . 2010;400:71–81. doi: 10.1016/j.jmb.2010.04.052. [DOI] [PubMed] [Google Scholar]
- 29. Kulisz A, Simon H-G. An evolutionarily conserved nuclear export signal facilitates cytoplasmic localization of the Tbx5 transcription factor. Mol Cell Biol . 2008;28:1553–1564. doi: 10.1128/MCB.00935-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murakami M, Nakagawa M, Olson EN, Nakagawa O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc Natl Acad Sci USA . 2005;102:18034–18039. doi: 10.1073/pnas.0509109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med . 2018;11:e001887. doi: 10.1161/CIRCGEN.117.001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zweier C, Sticht H, Aydin-Yaylagül I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet . 2007;80:510–517. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Postma AV, Van De Meerakker JBA, Mathijssen IB, Barnett P, Christoffels VM, Ilgun A, et al. A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ Res . 2008;102:1433–1442. doi: 10.1161/CIRCRESAHA.107.168294. [DOI] [PubMed] [Google Scholar]
- 34. Posch MG, Gramlich M, Sunde M, Schmitt KR, Lee SHY, Richter S, et al. A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J Med Genet . 2010;47:230–235. doi: 10.1136/jmg.2009.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Veitia RA, Birchler JA. Dominance and gene dosage balance in health and disease: why levels matter! J Pathol . 2010;220:174–185. doi: 10.1002/path.2623. [DOI] [PubMed] [Google Scholar]
- 36. Shrivastava S, Kruisselbrink TM, Mohananey A, Thomas BC, Kushwaha SS, Pereira NL. Rare TBX4 variant causing pulmonary arterial hypertension with small patella syndrome in an adult man. JACC Case Rep . 2021;3:1447–1452. doi: 10.1016/j.jaccas.2021.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu N, Pauciulo MW, Welch CL, Lutz KA, Coleman AW, Gonzaga-Jauregui C, et al. PAH Biobank Enrolling Centers’ Investigators Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med . 2019;11:69. doi: 10.1186/s13073-019-0685-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med . 2017;19:1151–1158. doi: 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karolak JA, Gambin T, Szafranski P, Stankiewicz P. Potential interactions between the TBX4-FGF10 and SHH-FOXF1 signaling during human lung development revealed using ChIP-seq. Respir Res . 2021;22:26. doi: 10.1186/s12931-021-01617-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, Leter EM, Douwes MJ, Dijk AV, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet . 2013;50:500–506. doi: 10.1136/jmedgenet-2012-101152. [DOI] [PMC free article] [PubMed] [Google Scholar]