

To the Editor:
Lower respiratory tract infections (LRTI), including community-acquired pneumonia (CAP), are major contributors to morbidity and mortality worldwide, especially in children. Major causes of CAP include Streptococcus pneumoniae, Haemophilus influenzae, and potentially Moraxella catarrhalis (1). Viruses are also major drivers of CAP (2). Determining the microbial cause of nonbacteraemic pneumonia is difficult in children and relies on nasopharyngeal samples, but bacteria associated with CAP are also frequent colonizers of healthy children (3, 4). Most clinical studies using nasopharyngeal samples are on the basis of culturing or the use of DNA-based methods such as sequencing the 16S ribosomal RNA (rRNA) gene (5). Little is known about which bacteria and/or viruses are actively transcribed during health and LRTI.
Here, we developed a novel metatranscriptomic pipeline with ultrasensitive sequencing of nasopharyngeal aspirates to map the expression of the nasopharyngeal microbiota and associated host responses in 20 healthy children and 20 children with CAP (https://doi.org/10.6084/m9.figshare.20452026.v2) (6). For each child with CAP, a matched control subject on age and calendar time was selected. No control subjects, and only one case, received antibiotics before sampling. DNA and RNA were isolated simultaneously from the aspirates, and deep sequencing was performed using Illumina Novaseq 6000S1, two lanes with 2 × 150 bp read lengths with an estimated output of around 3 billion reads. The metatranscriptomic sequencing yielded 15–60 million reads per sample of which 50–75% were human, 25–40% bacterial, and less than 1% viral reads. Because of sensitivity limitations, we could not capture DNA viruses. The K-mer–based taxonomic classification method Kraken2 combined with Bracken estimation was used for the classification of bacterial and viral species.
This research letter has an online data supplement, which is accessible online at: https://doi.org/10.6084/m9.figshare.20452026.v2
Bacterial and Viral Transcriptomic Analyses Associated with Health and Pneumonia and Microbial Properties Could Be Distinguished
Between 1,114 and 3,084 species found in the cases and healthy control subjects were corrected for the negative control (57–462 species). However, only one or two species dominated in a sample, comprising more than 80% abundance (https://doi.org/10.6084/m9.figshare.20452026.v2). Permanova was calculated on the microbial composition using the Bray-Curtis method and fourth-root transformed abundances with adonis2 function from the vegan R package, using 10,000 permutations. This revealed significance for CAP cases versus healthy children (P = 0.0272). The most commonly transcribed bacteria in the healthy children were Dolosigranulum pigrum (17/20), M. catarrhalis (12/20), S. pneumoniae (8/20), and H. influenzae (4/20) (Figure 1A). In the CAP cases, the most frequently found bacteria were M. catarrhalis (13/20), and H. influenzae (13/20), followed by S. pneumoniae (11/20) (Figure 1A). A total of 19/20 of the cases and 17/20 of the healthy control subjects harbored one or more of S. pneumoniae, H. influenzae, and/or M. catarrhalis. When only these abundant species were included, permanova analysis yielded P = 0.00619 for CAP cases versus control subjects, and the data suggest that H. influenzae proliferate more in the nasopharynx in CAP (P = 0.002) and that the reverse is found for D. pigrum (P = 0.042). D. pigrum has been associated with healthy microbiota and a negative correlation with S. pneumoniae and Staphylococcus aureus (7, 8).
Figure 1.
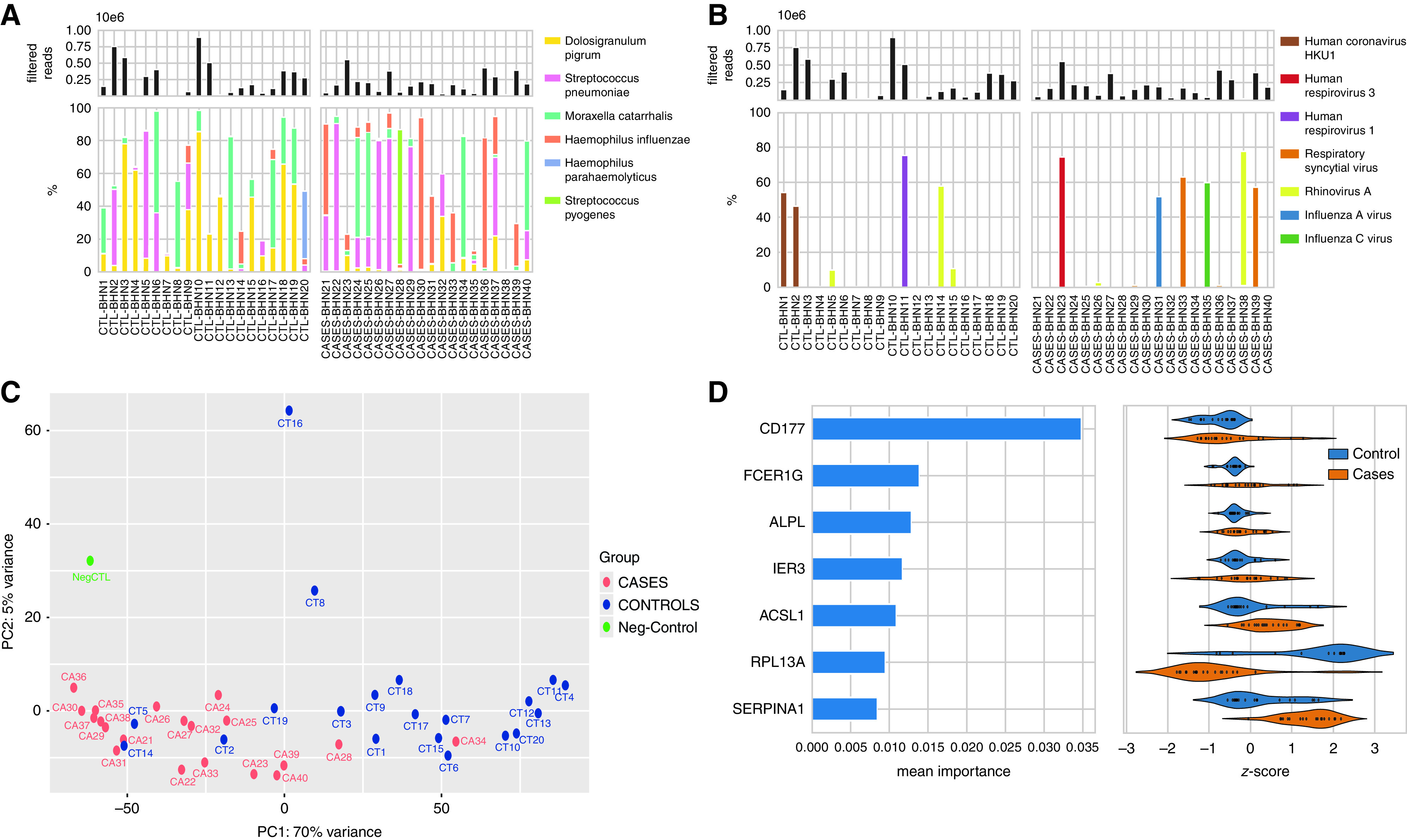
(A) The top panel shows the total filtered non-rRNA (non-ribosomal RNA) reads (bacterial, viral, and human non-rRNA reads) across the samples. The bottom panel shows the percentage of reads that belong to the most abundant bacterial taxa identified. (B) The top panel shows the total filtered non-rRNA reads, and the bottom panel shows the percentage of reads that belong to the most abundant viral species. (C) The principal component analysis (PCA) was performed on human reads using variance stabilizing normalization from DESeq2, R package using the plotPCA function that includes the top 500 genes selected by highest row variance. The normalization employed considers the sequencing depth and the RNA composition and uses the median of ratios method. Blue color circles indicate the control subjects, and red color circles show the cases. Names have been shortened to read CTRL-BHN1 = CT1 and Case-BHN21 = CA21, etc. (D) Random forest feature importance averaged over 20 model runs (left) for the main features in the best-performing classification model, and their corresponding transcription degrees expressed as z-scores in cases and control subjects. Z-scores were calculated by subtracting the mean and dividing by the standard deviation for each sample, resulting in samples having a mean of 0 and a variance of 1. PC1 = principal component 1; PC2 = principal component 2.
We identified transcription of bacterial virulence properties such as the capsular serotype. For H. influenzae, none of the capsular regions were detected. For S. pneumoniae, we performed in silico serotyping by capturing the unique regions of specific serotypes. The results were in agreement with Quellung serotyping (Table 1).
Table 1.
Comparison of Traditional Pneumococcal Serotyping with Serotyping Performed Using Metatranscriptomics
| Sample | Culture Pnc | Metatranscriptomics Pnc | Culture Serotype | Meta Serotype |
|---|---|---|---|---|
| CTL-BHN1 | + | (+) | 11A | ND* |
| CTL-BHN2 | + | + | 19A | 19A |
| CTL-BHN3 | — | (+) | — | ND* |
| CTL-BHN4 | — | + | — | ND* |
| CTL-BHN5 | + | + | 15B | 15B/15C |
| CTL-BHN6 | + | + | 21 | 21 |
| CTL-BHN7 | — | (+) | — | ND* |
| CTL-BHN8 | — | — | — | — |
| CTL-BHN9 | + | + | 35F | ND* |
| CTL-BHN10 | — | (+) | — | ND* |
| CTL-BHN11 | — | — | — | — |
| CTL-BHN12 | — | — | — | — |
| CTL-BHN13 | — | — | — | — |
| CTL-BHN14 | + | + | 35F | ND* |
| CTL-BHN15 | — | — | — | — |
| CTL-BHN16 | — | + | — | ND* |
| CTL-BHN17 | — | — | — | — |
| CTL-BHN18 | — | (+) | — | ND* |
| CTL-BHN19 | — | — | — | — |
| CTL-BHN20 | — | + | — | ND* |
| CASES-BHN21 | + | + | 35F | 35F |
| CASES-BHN22 | + | + | 21 | 21 |
| CASES-BHN23 | — | (+) | — | ND* |
| CASES-BHN24 | + | + | 35B | ND* |
| CASES-BHN25 | — | + | — | ND† |
| CASES-BHN26 | + | + | 15A | 15A/15F |
| CASES-BHN27 | + | + | 3 | 3 |
| CASES-BHN28 | — | (+) | — | ND* |
| CASES-BHN29 | + | + | 22F | 22F |
| CASES-BHN30 | — | — | — | — |
| CASES-BHN31 | — | — | — | — |
| CASES-BHN32 | + | + | 35F | ND* |
| CASES-BHN33 | — | — | — | — |
| CASES-BHN34 | + | (+) | 15B | ND* |
| CASES-BHN35 | + | + | 23B | ND* |
| CASES-BHN36 | — | (+) | — | ND* |
| CASES-BHN37 | + | + | 35F | 35F |
| CASES-BHN38 | — | — | — | — |
| CASES-BHN39 | — | — | — | — |
| CASES-BHN40 | + | + | 15B | 15B/15C |
Definition of abbreviation: Pnc = pneumococci.
(+) indicates low abundance reads.
Capsular region not determined.
Only cpsA was captured, and the serotype could not be determined.
We also identified RNA viruses/particles down to the species (Figure 1B). Among the control subjects, six children harbored viruses, including three children with rhinovirus (BHN5, BHN14, and BHN15), two with human coronavirus HKU1 (BHN1 and BHN2), and one with human respirovirus 1 (BHN11). Six cases showed RNA viruses, of which four are known causes of LRTI: human respirovirus 3 (BHN23), influenza A (BHN31), and respiratory syncytial virus (RSV) (BHN33 and BHN39). We obtained complete genome sequences for two HKU1 viruses and found that they showed divergent spike mutational profiles and belonged to different sublineages.
The transcriptomic analyses were validated using 16S rRNA (12 out of 40 samples) and culturing for bacteria and real-time PCR for viruses (2), wherein both revealed highly similar taxonomic profiles (https://doi.org/10.6084/m9.figshare.20452026.v2).
Host Transcriptional Profiles Were Specific for CAP Cases, and Three Potential Biomarkers Were Identified
A majority of the RNA sequences corresponded to human-associated RNA. The global expression profile of the human transcriptome was analyzed with DESeq2 and principal component analysis and revealed significant distinct clustering patterns for cases and control subjects for most children, irrespective of microbial species (Figure 1C). Permanova was calculated with the adonis2 function from the vegan R package, using 10,000 permutations, and revealed significant differences between the two groups (P = 9.999 × 10−5). The two outliers among the cases (BHN28 and BHN34) that clustered with control subjects had S. pyogenes and M. catarrhalis or D. pigrum as dominating bacterial species, and none contained pathogenic RNA viruses (Figure 1). Of control subjects that clustered with cases, BHN2 and BHN5 had high reads of S. pneumoniae, and BHN14 had high reads of H. influenzae (Figure 1).
Differential gene expression was studied using P-adjusted values (Wald test) with Benjamini-Hochberg false discovery rate < 0.05 with a twofold difference annotated using gene ontology. A total of 3,635 transcripts were downregulated, whereas 232 were upregulated in the cases as compared with control subjects. To find predictors for CAP, we used random forest analysis to classify samples into “case” and “control” groups using microbial species abundance and human transcriptome profiles. We found that the highest mean classification accuracy was achieved using both species abundances and transcriptome datasets. Specifically, species abundances inferred from non-rRNA microbial reads using Bracken and with contaminant removal using Recentrifuge resulted in the highest mean accuracy of 0.93. Using the result of the best performing classifier, we identified three host transcripts, CD177, FCPER1G, and ALPL, among the top 10 most important features in the majority of model runs and potential predictors to distinguish between CAP and healthy (Figure 1D). The strongest predictor was CD177, a specific marker for neutrophil activation that was recently shown to be a hallmark for severe coronavirus disease (COVID-19) and death (9). FCER1G is a high-affinity IgE receptor involved in integrin-mediated neutrophil activation. A transcriptomic study of airway neutrophils from infants with severe respiratory syncytial virus (RSV) showed high activation of FCER1G (10, 11).
Conclusions
This novel ultrasensitive expression platform can be used for studies of the microbiota and host signatures in respiratory infections and pave the way for the identification of new biomarkers and pathways that can be targeted for treatment.
Acknowledgments
Acknowledgment
The authors thank Centre for Translational Microbiome Research (CTMR) at Karolinska Institutet for helping with the 16S rRNA sequencing; Staffan Normark for scientific discussions; and the Swedish National Infrastructure for Computing (SNIC)/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure.
Footnotes
Supported by the Knut and Alice Wallenberg Foundation; Region Stockholm (grants ALF and HMT); the Swedish Research Council; Torsten Söderberg Foundation; and the National Bioinformatics and Genomics Infrastructure Stockholm funded by Science for Life Laboratory.
Author Contributions: P. Nannapaneni and B.H.-N. designed the study. S.P.-N. and P. Nannapaneni performed experiments. S.R., P. Naucler, Å.Ö., and B.H.-N. were involved in collecting samples. J.S. and P. Nannapaneni performed statistical and data analyses. All authors analyzed the data. P. Nannapaneni and B.H.-N. wrote the manuscript. All authors contributed to the writing of the manuscript.
Originally Published in Press as DOI: 10.1164/rccm.202203-0463LE on August 10, 2022
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1. Pneumonia Etiology Research for Child Health (PERCH) Study Group. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: the PERCH multi-country case-control study. Lancet . 2019;394:757–779. doi: 10.1016/S0140-6736(19)30721-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rhedin S, Lindstrand A, Hjelmgren A, Ryd-Rinder M, Öhrmalm L, Tolfvenstam T, et al. Respiratory viruses associated with community-acquired pneumonia in children: matched case-control study. Thorax . 2015;70:847–853. doi: 10.1136/thoraxjnl-2015-206933. [DOI] [PubMed] [Google Scholar]
- 3. García-Rodríguez JA, Fresnadillo Martínez MJ. Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J Antimicrob Chemother . 2002;50:59–73. doi: 10.1093/jac/dkf506. [DOI] [PubMed] [Google Scholar]
- 4. Henriqus Normark B, Christensson B, Sandgren A, Noreen B, Sylvan S, Burman LG, et al. Clonal analysis of Streptococcus pneumoniae nonsusceptible to penicillin at day-care centers with index cases, in a region with low incidence of resistance: emergence of an invasive type 35B clone among carriers. Microb Drug Resist . 2003;9:337–344. doi: 10.1089/107662903322762761. [DOI] [PubMed] [Google Scholar]
- 5. Pendleton KM, Erb-Downward JR, Bao Y, Branton WR, Falkowski NR, Newton DW, et al. Rapid pathogen identification in bacterial pneumonia using real-time metagenomics. Am J Respir Crit Care Med . 2017;196:1610–1612. doi: 10.1164/rccm.201703-0537LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindstrand A, Galanis I, Darenberg J, Morfeldt E, Naucler P, Blennow M, et al. Unaltered pneumococcal carriage prevalence due to expansion of non-vaccine types of low invasive potential 8 years after vaccine introduction in Stockholm, Sweden. Vaccine . 2016;34:4565–4571. doi: 10.1016/j.vaccine.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 7. Brugger SD, Eslami SM, Pettigrew MM, Escapa IF, Henke MT, Kong Y, et al. Dolosigranulum pigrum cooperation and competition in human nasal microbiota. MSphere . 2020;5:e00852-20. doi: 10.1128/mSphere.00852-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Steenhuijsen Piters WA, Heinonen S, Hasrat R, Bunsow E, Smith B, Suarez-Arrabal MC, et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am J Respir Crit Care Med . 2016;194:1104–1115. doi: 10.1164/rccm.201602-0220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lévy Y, Wiedemann A, Hejblum BP, Durand M, Lefebvre C, Surénaud M, et al. French COVID cohort study group CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience . 2021;24:102711. doi: 10.1016/j.isci.2021.102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Besteman SB, Callaghan A, Langedijk AC, Hennus MP, Meyaard L, Mokry M, et al. Transcriptome of airway neutrophils reveals an interferon response in life-threatening respiratory syncytial virus infection. Clin Immunol . 2020;220:108593. doi: 10.1016/j.clim.2020.108593. [DOI] [PubMed] [Google Scholar]
- 11. Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol . 2011;127:153–160, 160 e151–159. doi: 10.1016/j.jaci.2010.10.024. [DOI] [PubMed] [Google Scholar]