

Abstract
Rationale
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and often fatal disorder. Two U.S. Food and Drug Administration–approved antifibrotic drugs, nintedanib and pirfenidone, slow the rate of decline in lung function, but responses are variable and side effects are common.
Objectives
Using an in silico data-driven approach, we identified a robust connection between the transcriptomic perturbations in IPF disease and those induced by saracatinib, a selective Src kinase inhibitor originally developed for oncological indications. Based on these observations, we hypothesized that saracatinib would be effective at attenuating pulmonary fibrosis.
Methods
We investigated the antifibrotic efficacy of saracatinib relative to nintedanib and pirfenidone in three preclinical models: 1) in vitro in normal human lung fibroblasts; 2) in vivo in bleomycin and recombinant Ad-TGF-β (adenovirus transforming growth factor-β) murine models of pulmonary fibrosis; and 3) ex vivo in mice and human precision-cut lung slices from these two murine models as well as patients with IPF and healthy donors.
Measurements and Main Results
In each model, the effectiveness of saracatinib in blocking fibrogenic responses was equal or superior to nintedanib and pirfenidone. Transcriptomic analyses of TGF-β–stimulated normal human lung fibroblasts identified specific gene sets associated with fibrosis, including epithelial–mesenchymal transition, TGF-β, and WNT signaling that was uniquely altered by saracatinib. Transcriptomic analysis of whole-lung extracts from the two animal models of pulmonary fibrosis revealed that saracatinib reverted many fibrogenic pathways, including epithelial–mesenchymal transition, immune responses, and extracellular matrix organization. Amelioration of fibrosis and inflammatory cascades in human precision-cut lung slices confirmed the potential therapeutic efficacy of saracatinib in human lung fibrosis.
Conclusions
These studies identify novel Src-dependent fibrogenic pathways and support the study of the therapeutic effectiveness of saracatinib in IPF treatment.
Keywords: Src family kinase, tyrosine kinase, lung fibrosis, preclinical models, idiopathic pulmonary fibrosis
At a Glance Commentary
Scientific Knowledge on the Subject
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fatal disorder. Two antifibrotic drugs were approved by the U.S. Food and Drug Administration in 2014 and, although on average both drugs slow the rate of decline in lung function, responses are variable and do not cure IPF or improve symptoms. Thus, there is an urgent need for development of more effective therapies that safely modify the course of IPF and restore quality of life.
What This Study Adds to the Field
This study identifies novel Src-dependent fibrogenic pathways and supports the study of the therapeutic effectiveness of saracatinib in IPF treatment. Herein, we used an innovative disease-agnostic computational biology-based approach, identified the Src kinase inhibitor saracatinib (AZD0530) as a potential therapeutic agent for IPF, and validated its efficacy in blocking fibrogenic responses in several complementary preclinical models of pulmonary fibrosis. Our data provide strong evidence that saracatinib is equal or superior to the two U.S. Food and Drug Administration–approved drugs, nintedanib and pirfenidone, at inhibiting pulmonary fibrosis in experimental models and support its use in clinical trials in humans with IPF.
Idiopathic pulmonary fibrosis (IPF) is a chronic, relentless, and ultimately fatal disorder characterized by progressive scarring (fibrosis) of the lung parenchyma (1–3). The median survival for patients with IPF is 3 years from diagnosis, with most patients dying from respiratory failure due to disease progression (4).
The mechanisms driving pulmonary fibrosis (PF) are incompletely understood, and both genetic and environmental factors appear to be important in disease pathogenesis (5, 6). A widely accepted hypothesis is that, in a genetically susceptible individual, the lung is repeatedly injured (via an unknown cause), and aberrant repair provokes the release and activation of fibrogenic mediators, myofibroblast accumulation, and deposition of excess extracellular matrix (ECM), resulting in progressive fibrosis (7–9). Cellular responses to profibrotic mediators are usually transduced through transmembrane receptors via intracellular signaling pathways that are controlled in part by SFKs (Src family tyrosine kinases) that include Src, Yes, Fyn, Fgr, Lck, Hck, Blk, Lyn, and Frk (10, 11). SFKs are involved in a range of signaling pathways essential for cellular homeostasis, such as proliferation, differentiation, motility, adhesion, and cytoskeletal organization. Studies in preclinical models of IPF suggest that several Src-dependent processes contribute to IPF pathogenesis, including myofibroblast differentiation and fibrogenic gene expression (12, 13).
Treatment of IPF remains suboptimal. Two antifibrotic drugs, pirfenidone (Esbriet) and nintedanib (OFEV), were approved by the U.S. Food and Drug Administration (FDA) in 2014 for the treatment of IPF (14–16). Clinical trials and real-world experience demonstrate that, although on average both drugs slow the rate of decline in lung function, responses are variable, and these drugs neither cure IPF nor improve the symptoms (17). Thus, there remains an urgent need for the development of more effective therapies that safely modify the course of IPF and restore quality of life.
An evolving understanding of the molecular underpinnings of human diseases has provided opportunities to precisely target disease-specific pathways using bioinformatic methods to mine genomic, molecular, and clinical data (18–20). As part of a data-driven approach to repurpose phase II–ready compounds for new diseases, we identified saracatinib as a potential therapeutic compound for IPF. Saracatinib is a potent and selective Src kinase inhibitor, originally developed for oncological indications (21–23). Building on the initial insights gleaned from transcriptomic connections and the evidence strongly supporting a pivotal role for Src kinase in IPF pathophysiology, we sought to determine the antifibrotic efficacy of saracatinib relative to nintedanib and pirfenidone in preclinical cell culture and animal models of IPF and to elucidate the molecular signatures of pathological fibrogenesis and drug responsiveness.
Some of the results of these studies have been previously reported (24, 25).
Methods
Detailed methods are provided in the online supplement.
In Vitro
Experiments were performed using primary normal human lung fibroblasts (NHLFs) from three different donors (Lonza), and all experiments were performed using cells of passages three to five, with six technical replicates.
In Vivo
PF was induced in C57Bl/6 mice by intrapulmonary delivery of bleomycin or recombinant Ad-TGF-β (adenovirus transforming growth factor-β). Bleomycin (1.5 U/kg) or saline administered by oropharyngeal instillation and Ad-TGF-β1 (VQAd CMV mTGF-β1-Viraquest; 2 × 109 pfu per mouse) or empty vector were administered via the intranasal route. In both animal models, treatment with drugs was started on Day 10, and drugs were administered daily by oral gavage (saracatinib 20 mg/kg; nintedanib 60 mg/kg; pirfenidone 300 mg/kg) until Day 28. Mouse lungs were harvested on Day 28 for fibrosis analysis.
Ex Vivo
Mouse precision-cut lung slices
Mouse precision-cut lung slices (PCLSs) were generated by using mouse lungs from both bleomycin (Day 14) and Ad-TGF-β (Day 21) models (26–28). The PCLSs were cultured and treated with saracatinib (0.6 μM), nintedanib (1 μM), pirfenidone (1 mM), or vehicle control in the first 24 hours after slicing. Lung slices were isolated 5 days after treatment for analysis.
Human PCLS
Cryopreserved sections of human lungs, derived from the patients with IPF or healthy donors, were cultured and used in two independent experiments. The human PCLSs (hPCLSs) harvested from healthy donors were cultured and treated with a medium containing a profibrotic cocktail (FC) (containing TGF-β, PDGF-AB [platelet-derived growth factor-AB], lysophosphatidic acid, TNF-α [tumor necrosis factor-α]) or control cocktail. The hPCLSs were isolated from patients with IPF as well as individual PCLSs treated with FC or control cocktail supplemented with saracatinib (0.6 μM) or vehicle for 5 days and used for histological assessment or gene expression analysis.
Results
The Computational Drug Repurposing Approach Identifies a Connection Between Saracatinib and IPF
We undertook a disease-agnostic, data-driven approach to explore novel connections between diseases and compounds previously tested in clinical studies. A set of 32 compounds was selected based on a combination of factors that included experience in clinical studies and potential for further clinical development beyond the disease for which the compound was originally designed (drug repositioning). The method applied was based on a modified connectivity mapping approach (29, 30), where the transcriptomic signature of each compound was compared computationally with transcriptomic signatures of human diseases. Briefly, differential gene expression signatures for each compound were generated by performing RNA sequencing on two different cell lines (A549 and MCF7) after exposure to two concentrations of each of the 32 compounds (e.g., compound-A A549 high dose, compound-A MCF7 high dose, compound-A A549 low dose, compound-A MCF7 low dose). This analysis was done blinded to the compound identity or chemistry and was generated using the genome-wide pattern of mRNA changes in cell-matched compound versus vehicle-treated samples (Figure 1A). A disease transcriptomic library, consisting of more than 700 unique disease signatures, was built from publicly available gene expression data (19, 31). To identify novel clinical indications for each compound, a connectivity score was calculated by comparing each disease signature to each compound signature. The connectivity score aims to summarize the transcriptomic relationship between each compound and disease, such that a strongly negative score indicates that the compound will induce transcriptomic changes that may revert or “normalize” the disease signature (29). Using this method, significant negative connectivity scores were found between the Src kinase inhibitor (saracatinib) and IPF disease signatures derived from patient lung biopsies (GSE24206) (32) and cultured fibroblasts (GSE44723) (33) (see Table E1 in the online supplement).
Figure 1.
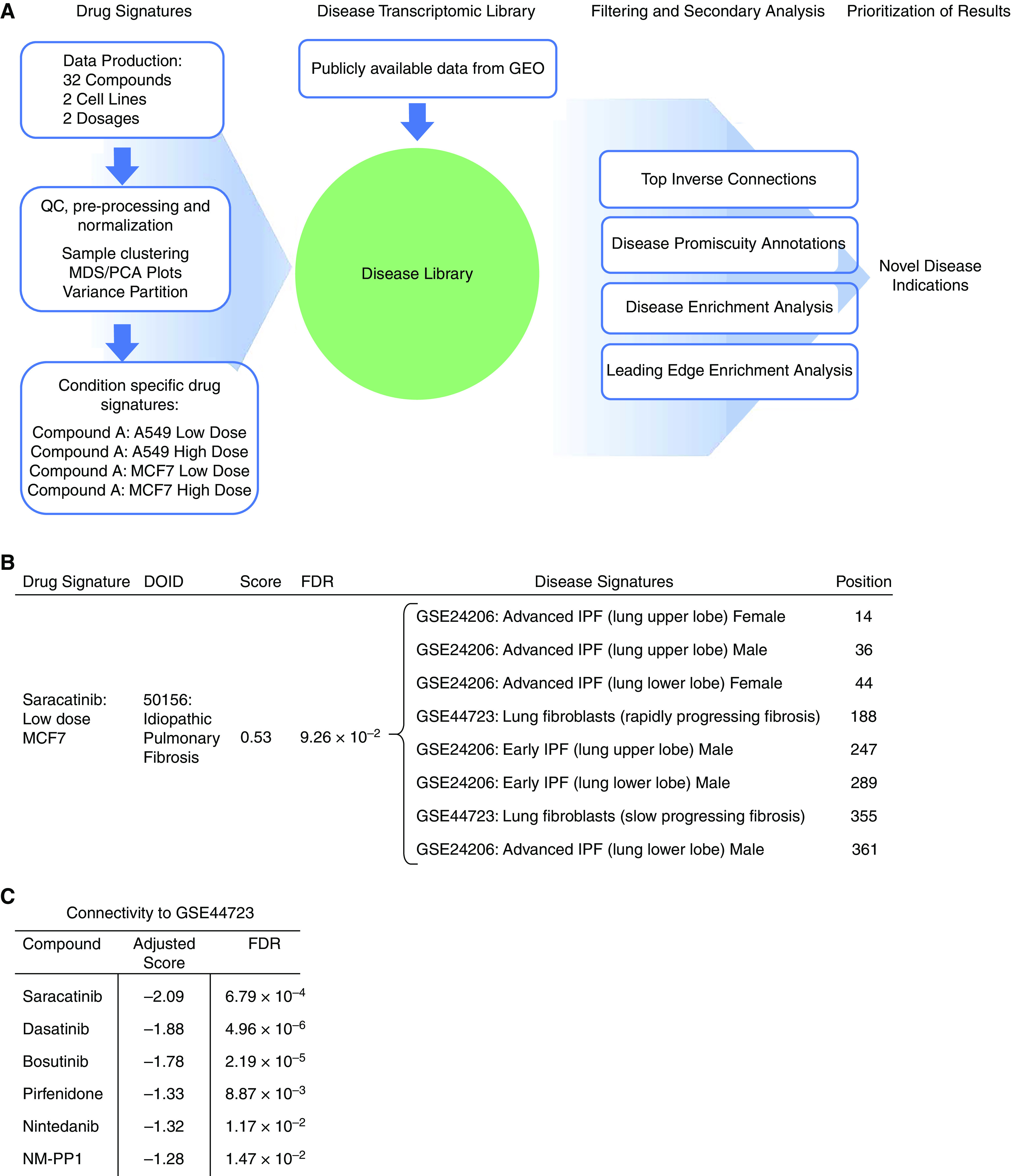
Computational drug repurposing identifies saracatinib as a potential therapeutic for idiopathic pulmonary fibrosis (IPF). (A) Schematic of the in silico approach used to identify novel disease indications for compounds. Drug signatures were obtained for 32 compounds in two different cell lines at two dosages. Each drug signature was compared with a library of disease signatures generated from publicly available data, and a connectivity score was generated for each disease–compound pair. Filtering and secondary analyses were performed to identify novel disease indications for each of the compounds. (B) Disease enrichment analysis results show enrichment of Disease Ontology identifier: 50156/IPF signatures among disease signatures that are transcriptomically connected to saracatinib. (C) Connectivity scores between an IPF disease signature and publicly available drug signatures (saracatinib, dasatinib, bosutinib, pirfenidone, nintedanib, and NM-PP1; obtained from LINCS L1000). FDR = false discovery rate; GEO = Gene Expression Omnibus; MDS/PCA = multi-dimensional scaling/principal components analysis; NM-PP1 = PP1 analog II; QC = quality control.
Accordingly, we performed a disease enrichment analysis, intending to identify high-level disease categories that are transcriptomically connected to saracatinib. This analysis identified that IPF disease signatures were overrepresented in connecting with saracatinib (Figure 1B), indicating that saracatinib is globally relevant to IPF disease.
To probe the biology driving the connection between saracatinib and the IPF signatures, we conducted a leading-edge enrichment analysis to identify overrepresented gene sets that point to pathways by which saracatinib may affect IPF disease. This analysis identified numerous gene sets, including IFNγ response, epithelial–mesenchymal transition (EMT), and TNF-α signaling pathways, all of which have been implicated in the pathogenesis of IPF. In addition, kinase enrichment analysis of these data identified enrichments for RIPK3 (receptor-interacting serine/threonine-protein kinase 3) together with multiple members of the MAP (mitogen-activated protein) kinase family (Table 1).
Table 1.
Leading Edge Enrichment Analysis
| Enrichments: ↑ IPF, ↓ Saracatinib |
Enrichments: ↓ IPF, ↑ Saracatinib |
|||||||
|---|---|---|---|---|---|---|---|---|
| Library | Gene Set | FDR | Genes in Overlap | Fold Change | Gene Set | FDR | Genes in Overlap | Fold Change |
| Hallmark | IFNγ response | 1.79 × 10−12 | ADAR, CD74, HLA-A, HLA-B, HLA-DMA, HLA-G, ICAM1, IFI30, IFIT3, IFITM3, IRF1, LGALS3BP, LY6E, MT2A, MX1, MYD88, NFKBIA, PTPN6, SERPING1 | 9.48 | Myc targets | 3.47 × 10−9 | CCT2, DEK, DHX15, ERH, GLO1, HNRNPA1, IARS, LDHA, NPM1, PSMA6, RPL6, RPLP0, SLC25A3, YWHAQ | 9.28 |
| EMT | 5.94 × 10−11 | CAPG, COL1A1, COL4A2, COL6A2, CYR61, FLNA, FN1, GEM, HTRA1, ID2, IGFBP2, IGFBP4, SERPINE1, SERPINH1, TAGLN, TGFBI, VIM | 8.54 | Allograft rejection | 1.35 × 10−4 | B2M, HIF1A, IFNGR1, MRPL3, NPM1, RPS3A, TIMP1 | 8.59 | |
| TNF-α signaling via NF-κB | 4.03 × 10−10 | CCND1, CD83, CYR61, EGR1, FOSB, GEM, ICAM1, ID2, IER2, IRF1, JUNB, KDM6B, LAMB3, LIF, NFKBIA, SERPINE1, SLC2A3, ZFP36 | 7.88 | Apoptosis | 4.39 × 10−4 | CAV1, GPX3, IFNGR1, PPT1, TGFBR3, TIMP1, TXNIP | 6.91 | |
| Complement | 2.50 × 10−8 | APOC1, C3, COL4A2, CSRP1, CTSD, CTSH, DUSP6, FN1, GNAI2, GNB2, IRF1, PFN1, SERPINE1, SERPING1, TIMP2 | 7.81 | Inflammatory response | 1.49 × 10−2 | ADM, HIF1A, LAMP3, NAMPT, TIMP1 | 6.08 | |
| Coagulation | 3.70 × 10−8 | APOC1, C3, CRIP2, CSRP1, CTSH, DUSP6, FN1, GNB2, HTRA1, SERPINE1, SERPING1, USP11 | 10.07 | Reactive oxygen species | 1.81 × 10−2 | GPX3, PRDX1, PRDX6 | 8.95 | |
| IFNα response | 1.61 × 10−6 | ADAR, CD74, HLA-C, IFI30, IFIT3, IFITM3, IRF1, LGALS3BP, LY6E, MX1 | 9.03 | Oxidative phosphorylation | 1.84 × 10−2 | COX6C, COX7A2, LDHA, NDUFA4, SLC25A3, SLC25A5 | 3.98 | |
| Allograft rejection | 2.79 × 10−5 | CAPG, CD74, FLNA, HLA-A, HLA-DMA, HLA-E, HLA-G, ICAM1, LIF, PTPN6 | 6.67 | Androgen response | 3.27 × 10−2 | B2M, MYL12A, SGK1, SLC38A2 | 5.77 | |
| Apical junction | 4.75 × 10−5 | ACTB, ACTG1, ACTN4, CAP1, EVL, GNAI2, ICAM1, LAMB3, PFN1, TGFBI, VASP, ZYX | 5.63 | EMT | 4.94 × 10−2 | CD59, GJA1, TGFBR3, TIMP1 | 3.70 | |
| KEA | RIPK3 | 3.42 × 10−6 | ACTB, ACTG1, ACTN4, FLNA, GNB2, HSPA8, RPS27, AUB52, UBB, VIM | 8.31 | MAP3K14 | 3.89 × 10−10 | HNRNPA1, HSPA1A, IARS, NPM1, RPL24, RPL4, RPL6, RPL7A, RPL10, RPS11, RPS23, RPS3A, RPS7 | 12.74 |
| MAP3K3 | 5.42 × 10−6 | ACTB, ACTG1, ACTN4, FLNA, GNAI2, GNB2, HSPA8, NFKBIA, RPS27, TUBA1C, UBA52, UBB, VIM | 5.66 | RIPK3 | 3.42 × 10−6 | CCt2, HNRNPA1, HSPA1A, PPP1CB, PPP1CC, PRDX1, SLC25A3, SLC25A5 | 12.17 | |
| MAP3K1 | 1.56 × 10−5 | ACTB, ACTG1, FLNA, HSPA8, LGALS3BP, NFKBIA, RPL35, RPS27, UBA52, UBB, VIM | 6.52 | MAP3K1 | 1.71 × 10−3 | HSPA1A, RPL24, RPL4, RPS11, RPS23, SLC25A5 | 6.50 | |
| MAP3K14 | 4.89 × 10−4 | ACTB, ACTG1, FAU, HSPA8, NFKBIA, RPL35, RPS10, RPS18, RPS20 | 4.82 | MAP3K3 | 7.38 × 10−3 | CCT2, HSPA1A, IARS, PPP1CC, SLC25A5, YWHAQ | 4.79 | |
Definition of abbreviations: EMT = epithelial–mesenchymal transition; IPF = idiopathic pulmonary fibrosis; KEA = kinase enrichment analysis; MAP = mitogen-activated protein; NF-κB = nuclear factor-κB; RIPK3 = receptor-interacting serine/threonine-protein kinase 3; TNF = tumor necrosis factor.
Enrichment analysis was performed on the leading edge of saracatinib drug signature versus IPF disease signature (GSE24206). The drug–disease pair that showed the highest connectivity was used for the analysis, namely “Saracatinib MCF7 Low Dose” versus “Advanced_IPF_explant_upper_lobe obtained from GSE24206”. The fold change reflects the results from a Fisher exact test, which assesses the overlap in gene sets that inversely perturbed between IPF and saracatinib (genes that are both downregulated in IPF and upregulated by saracatinib) and informative gene sets. Gene sets and overlapping genes are listed.
To clarify if this connectivity was a feature among other Src inhibitors or unique to saracatinib, we used data from the Connectivity Map (30), a collection of publicly available expression data from cultured human cells treated with small molecules. This data collection contains transcriptomic data in a range of experimental conditions for saracatinib as well as other Src kinase inhibitors. This resource also includes data from the two IPF FDA-approved drugs pirfenidone and nintedanib. We compared each of these drug signatures to the IPF disease signatures in our disease library. One IPF disease signature connected significantly with all six compounds (saracatinib, dasatinib, bosutinib, pirfenidone, nintedanib, and NM-PP1), and the strongest connection was with saracatinib (Figure 1C). In summary, using complementary bioinformatics approaches, we identified a robust transcriptomic connection between saracatinib and IPF, thus providing a strong foundation for the hypothesis that saracatinib might have a potential therapeutic benefit in IPF.
Saracatinib Inhibits TGF-β–induced Phenotypic Changes in Human Lung Fibroblasts
As the compound signature was derived from A549 and MCF7 cells, we next investigated the effects of saracatinib in a more disease-relevant setting. We assessed the effect of saracatinib on TGF-β–induced fibrogenic processes in cultured NHLFs to study signaling pathways relevant to human IPF disease. We confirmed that TGF-β stimulation induces a significant increase in Src phosphorylation of Y416, a response that correlates with activation of Src kinase activity (34) (Figures E1A and E1B). It has been well demonstrated that saracatinib treatment efficiently inhibits TGF-β–induced Src kinase activity in these cells (13). We chose to compare the effect of saracatinib to the two FDA-approved antifibrotic drugs, nintedanib and pirfenidone, in this in vitro system. We selected the optimum dose for all three compounds based on the established clinically relevant doses (35, 36) and our initial screening experiments (Figures E2A and E2B). We observed that saracatinib significantly inhibited TGF-β–induced expression of many profibrotic genes, including smooth muscle alpha (α)-2 actin (Acta2), Collagen type I alpha 1 chain (Col1a1), and serpin1 or plasminogen activator inhibitor 1 (PAI-1), to a similar or greater extent than that observed for nintedanib or pirfenidone (Figures 2A–2C). To validate this finding, we repeated the experiment in primary human lung fibroblasts isolated from three different donors and confirmed the consistency of these findings across donors (Figures E3A and E3B). We next compared the effects of the three drugs on TGF-β–induced Smad3 phosphorylation in NHLFs, as a readout of canonical TGF-β fibrogenic signaling. Saracatinib, but not nintedanib or pirfenidone, significantly inhibited TGF-β–induced Smad3 phosphorylation (Figure 2D). We extended our investigation by comparing the effects of saracatinib to the other two drugs on TGF-β–induced morphological changes in human fibroblasts. Confocal immunofluorescence imaging of these cells for α-SMA (α smooth muscle actin) and F-actin (filamentous actin) demonstrated a strong inhibitory effect of saracatinib on TGF-β–induced alterations in cell shape and stress fiber formation characteristic of myofibroblast transformation (fold change > 2; P value ⩽ 0.001) (Figures 2E–2G). Nintedanib exhibited a similar, but less potent, inhibitory response compared with saracatinib on these phenotypic changes. Concordant with our earlier findings, pirfenidone did not show any effects on TGF-β–induced structural changes in human lung fibroblasts.
Figure 2.
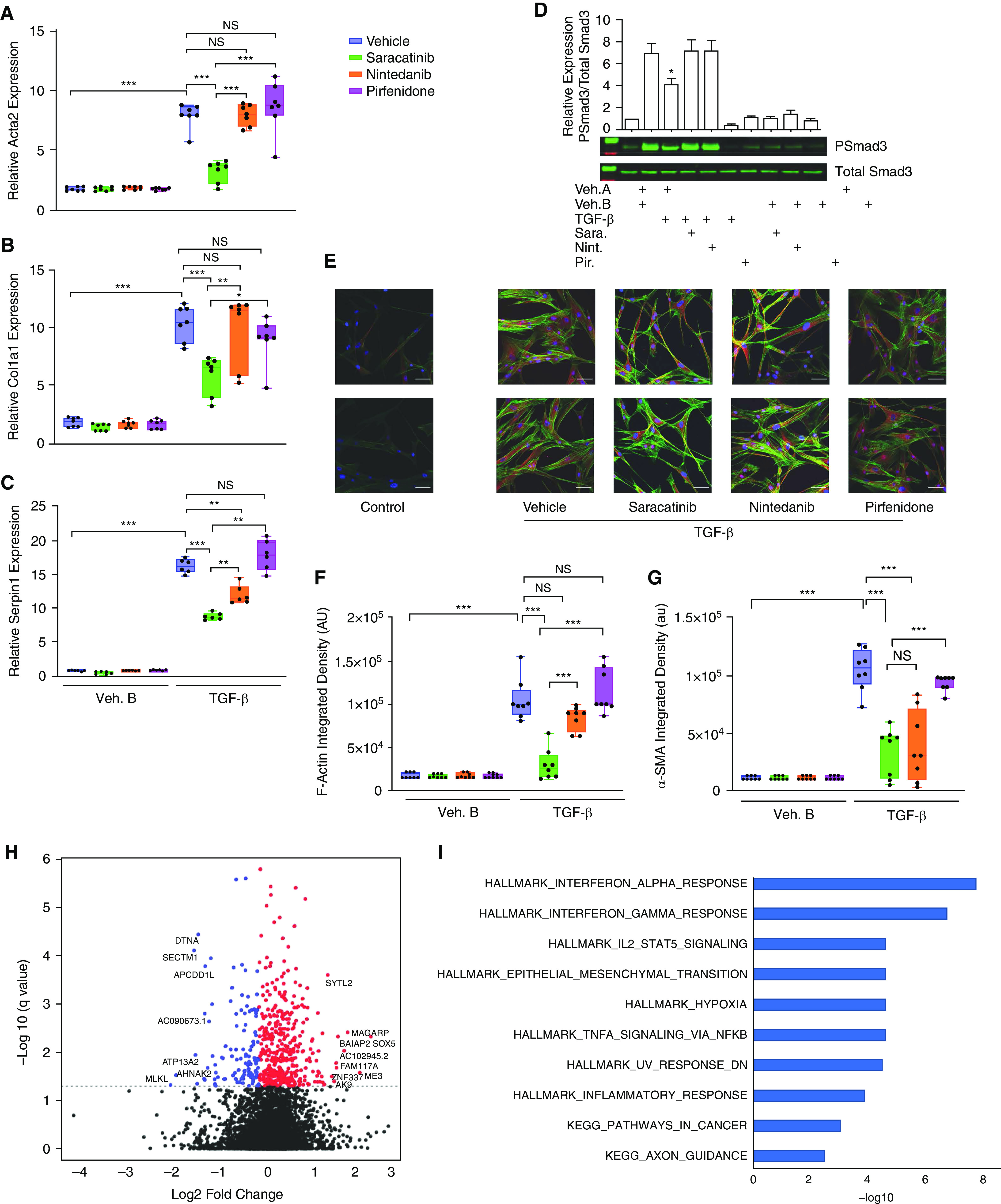
Saracatinib inhibits TGF-β (transforming growth factor-β)-induced phenotypic changes in human lung fibroblasts (NHLFs). Cells were serum-starved overnight and then incubated with inhibitors (saracatinib 0.3 μM, nintedanib 1 μM, pirfenidone 20 μg/ml, or vehicle [DMSO]) for 60 minutes followed by stimulation with human recombinant TGF-β (2 ng/ml) or vehicle control for the indicated times. (A–C) Quantitative real-time PCR (qRT-PCR) analysis for (A) smooth muscle alpha (α)-2 actin (Acta2), (B) collagen type I alpha 1 chain (Col1a1), and (C) serpin1 or plasminogen activator inhibitor 1(PAI-1) in the indicated treatment groups of NHLF (mean + SEM); *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6). (D) Representative western blots showing saracatinib inhibition of TGF-β–induced phosphorylation of Smad3 in human lung fibroblasts; data presented as mean + SEM; n = 6; *P < 0.05. (E) Representative images of α-SMA (α smooth muscle actin) staining (red) together with F-Actin (filamentous actin; green) and DAPI (blue) show fluorescent staining in human fibroblasts after TGF-β stimulation in the indicated treatment groups using confocal microscopy. (F and G) Quantification of α-SMA and F-Actin staining shown as integrated density; ***P < 0.001. (H) Volcano plot showing genes that are differentially expressed in cells treated with TGF-β and saracatinib compared with TGF-β alone (false discovery rate [FDR] < 0.05), negative fold change (blue), and positive fold change (red). (I) Functional enrichment of significantly differentially expressed genes (FDR < 0.05) in response to saracatinib (only the top 10 gene sets are shown). All gene sets shown are significant at FDR < 0.05 and are from Hallmark (H) or Kegg (K). AU or au = average intensity; NS = nonsignificant.
To extend these observations and investigate the broader effects of saracatinib in this disease-relevant in vitro model, we generated transcriptomic drug signatures by performing bulk RNA sequencing on NHLF cells treated with saracatinib, nintedanib, pirfenidone, or vehicle control in the presence or absence of TGF-β stimulation. We identified the transcriptomic effects that were unique to saracatinib and could be used to differentiate the effects of saracatinib from nintedanib and pirfenidone (Figure E4). We discovered that saracatinib altered the expression of more than 500 individual genes (adjusted P value < 0.05) in TGF-β–treated cells (Figure 2H). Gene set enrichment analysis (GSEA) revealed that saracatinib uniquely induced alterations in numerous pathways identified from the Hallmark and KEGG databases, with IFNα, IFNγ, EMT, and inflammatory responses among the top pathways (Figure 2I and Table E2).
In summary, saracatinib inhibits TGF-β–induced fibrogenic responses in this in vitro system more effectively than nintedanib and pirfenidone.
Saracatinib Inhibits PF in Preclinical Animal Models
Next, we compared the effects of saracatinib, nintedanib, and pirfenidone at clinically relevant doses (37–41) in two preclinical models of PF in mice (42, 43). In the first model, fibrosis was induced using a single dose of bleomycin (1.5 U/kg) administered into the lung by oropharyngeal aspiration, and mice received either saracatinib, nintedanib, pirfenidone, or vehicle control once daily via oral gavages on Days 10–27 after bleomycin administration. On Day 28, the mouse lungs were harvested and the antifibrotic effects of all three drugs were assessed. Mice receiving vehicle control exhibited prominent weight loss as expected after the administration of bleomycin, whereas all drug-treated mice, except the mice treated with pirfenidone, recovered their weight loss by the end of the experiment (Figures E5A and E5B). The bleomycin-induced increase in lung collagen content was significantly attenuated in mice receiving saracatinib, nintedanib, or pirfenidone (P value ⩽ 0.001) (Figure 3A). Consistent with the biochemical analysis, saracatinib significantly attenuated the bleomycin-induced lung expression of fibrogenic genes, including Acta2, Col1a1, and Col3a1 (Figures 3B, 3C, and E6A) (P value ⩽ 0.001 for Acta 2 and Col3a1; P value ⩽ 0.01 for Col1a1), whereas neither nintedanib nor pirfenidone treatments resulted in any significant changes in the expression of these fibrogenic genes. Histopathological evaluation of these lungs using Masson’s Trichrome staining demonstrated a greater reduction in bleomycin-induced PF by saracatinib and nintedanib (P value ⩽ 0.001) than pirfenidone (P value ⩽ 0.01) (Figures 3D and 3F). Micro–computed tomography analysis supported the biochemical and histopathological observations; aerated lung volume measurements showed saracatinib and nintedanib significantly attenuated the bleomycin-induced radiographic alterations in the lung parenchyma (P value ⩽ 0.001) and to a greater extent than pirfenidone (P value ⩽ 0.01) (Figures 3E, 3G, and E7). Finally, lung function measurements and physiological evaluation of these mouse lungs identified that saracatinib also significantly attenuated bleomycin-induced alterations in lung physiology (static compliance and elastance) as measured using the flexiVent system (fold change = 2; P value ⩽ 0.001) compared with nintedanib or pirfenidone (P value ⩽ 0.01) (Figure 3H).
Figure 3.
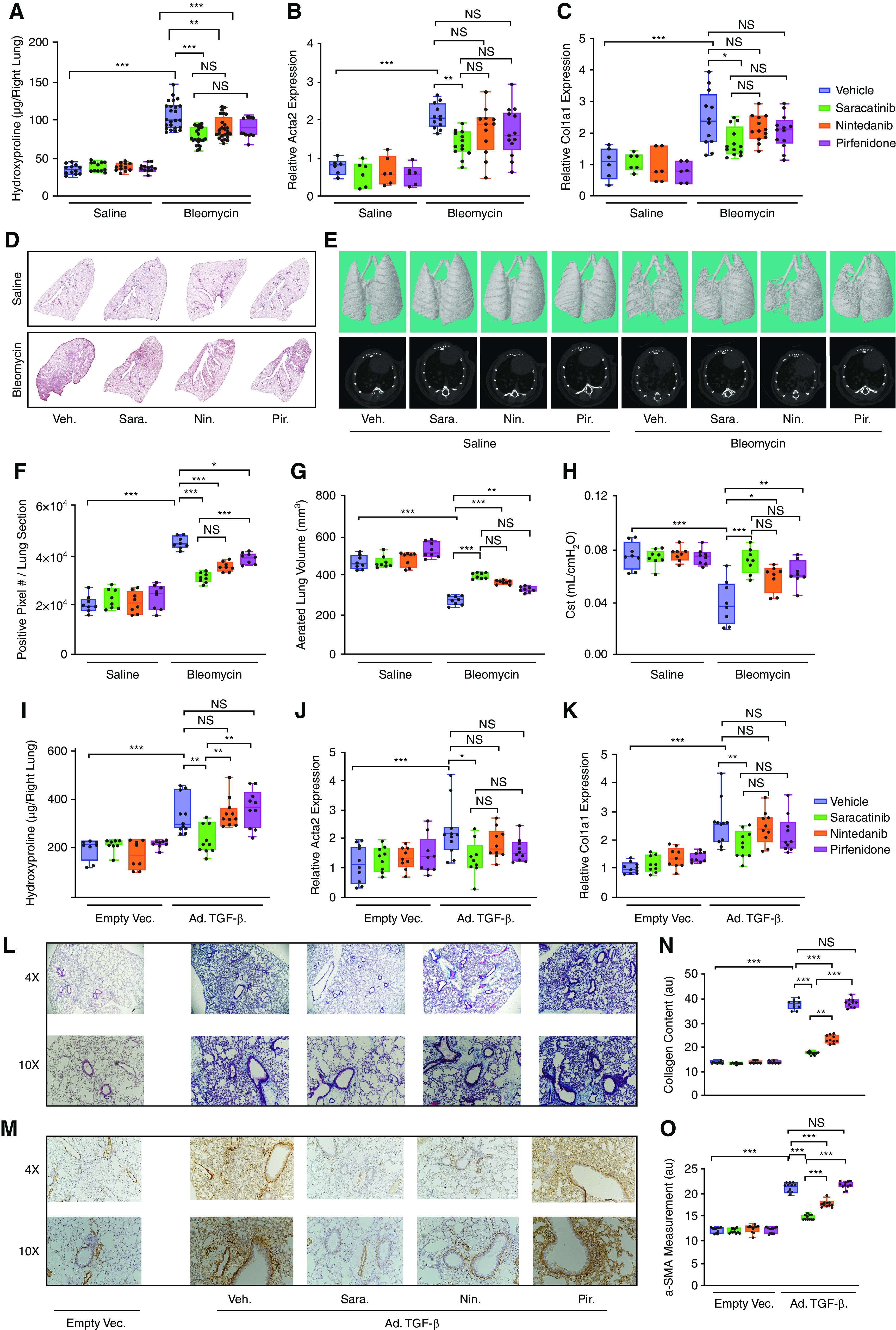
Saracatinib inhibits pulmonary fibrosis in bleomycin and adenovirus TGF-β (transforming growth factor-β) mouse models. (A–H) Evaluation of bleomycin-induced lung fibrosis. (A) Quantitative analysis of hydroxyproline in lung homogenates from indicated groups of mice. Lung collagen content increased significantly in bleomycin-treated mice receiving vehicle control (fold change = 2.9); **P < 0.01 and ***P < 0.001. (B and C) Quantitative real-time PCR analysis on mouse lungs for (B) smooth muscle alpha (α)-2 actin (Acta2) and (C) collagen type I alpha 1 chain (Col1a1) in the indicated treatment groups. (D and F) Representative images and quantitative measurements of Masson’s Trichrome staining of lung sections in the indicated groups of mice. (E and G) Representative images (dorsal view of three-dimensional reconstructions and axial view) and quantifications of micro–computed tomography on mouse lung tissues in the indicated groups. Gross abnormality resulting from bleomycin-induced lung fibrosis is alleviated after treatment. Aerated lung volume measurements showed a significant decrease in bleomycin-treated mouse lungs (P value ⩽ 0.001; fold change > 2), whereas saracatinib and nintedanib significantly attenuated the bleomycin-induced radiographic alterations in the lung parenchyma (P value ⩽ 0.001). (H) Lung compliance measurements of the lungs in the indicated groups are shown as static compliance (Cst). (I–O) Evaluation of Ad-TGF-β (adenovirus transforming growth factor-β)-induced lung fibrosis. (I) Quantitative analysis of hydroxyproline in lung homogenates from indicated groups of mice. The hydroxyproline assay revealed a significant increase in lung collagen content for mice receiving Ad-TGF-β (fold change = 1.8; P value ⩽ 0.001), which is decreased significantly by saracatinib. (J and K) Quantitative real-time PCR analysis on mouse lungs for (J) Acta2 and (K) Col1a1 in the indicated treatment groups. (L and N) Representative images and quantification of Masson’s Trichrome staining of lung sections in the indicated groups of mice. (M and O) Representative images and quantification of α-SMA (α smooth muscle actin) staining of lung sections in the indicated groups of mice. All data are presented as mean + SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 6 in saline and n ⩾ 12 in bleomycin and Ad-TGF-β–treated groups). NS = nonsignificant.
A second model of PF was established using the expression of recombinant murine TGF-β using adenoviral-mediated gene delivery (Ad-TGF-β) administered to the lung intranasally. As in the bleomycin model, all three drugs were given to mice once daily via oral gavages from Days 10 to 27 after TGF-β administration. Mice receiving vehicle control exhibited prominent weight loss after the administration of Ad-TGF-β, and, notably, only the mice treated with saracatinib recovered their weight loss by the end of the experiment (Figures E5C and E5D). In this model, only saracatinib demonstrated antifibrotic effects as assessed by a reduction in lung collagen content (P value ⩽ 0.001) (Figure 3I) and expression of Acta2, Col1a1, and Col3a1 mRNA (P value ⩽ 0.001) (Figures 3J, 3K, and E6B). By contrast, neither nintedanib nor pirfenidone attenuated fibrosis in this model. Histopathological assessment of the lungs using Masson’s Trichrome and α-SMA staining confirmed these observations (Figures 3L–3O and E8A and E8B).
Given the fact that TGF-β signaling cascades are consistently activated in fibrotic tissues, regardless of the etiology of the initial injury, and that the Smad3 pathway is a key intermediary in this cascade (44, 45), we evaluated the effect of saracatinib on Smad3 phosphorylation in the lung tissues after bleomycin and Ad-TGF-β treatments. We identified significant inhibition of this pathway by saracatinib in both preclinical animal models of PF (Figures E9A–E9D).
In summary, in two complementary mouse models of PF, saracatinib attenuated lung fibrosis more effectively than pirfenidone or nintedanib at clinically relevant doses.
Saracatinib Inhibits PF in Mouse PCLSs
We next sought to confirm these observations in an ex vivo model using PCLSs to compare the antifibrotic effects of saracatinib with nintedanib and pirfenidone (26). PCLS is a useful method to test the therapeutic effects of different compounds in a system with relatively preserved lung architecture containing various lung-resident cell types and ECM (46). In this study, mice were treated with either bleomycin or Ad-TGF-β and killed at the peak of the fibrotic response. Lung slices were prepared and treated ex vivo with each of the three drugs or vehicle for 5 days (Figures 4A–4L). These lung slices remain viable for at least 5 days (120 h), (Figure E10). Analysis of these lung slices revealed a reduction in Col1a1 and Acta2 mRNA expression by saracatinib in PCLSs harvested from mice treated with either bleomycin or Ad-TGF-β (P values ⩽ 0.001), whereas the fibrosis remained sustained in the vehicle-treated groups (Figures 4A, 4B, 4G, and 4H). Live serial imaging of the lung slices using second harmonic generation microscopy demonstrated that saracatinib significantly attenuated collagen accumulation in both models (P values ⩽ 0.001) (Figures 4C, 4D, 4J, and 4I). It was not possible to assess the effects of nintedanib on collagen content by second harmonic generation microscopy in this model because of a strong autofluorescence signal generated by nintedanib. Treatment with pirfenidone attenuated lung collagen content (P values ⩽ 0.001) only in the TGF-β model. Histological evaluation of the lung slices using Masson’s Trichrome staining revealed that saracatinib strongly attenuated lung fibrosis in both models (P values ⩽ 0.001) (Figures 4E, 4F, 4K, and 4L).
Figure 4.
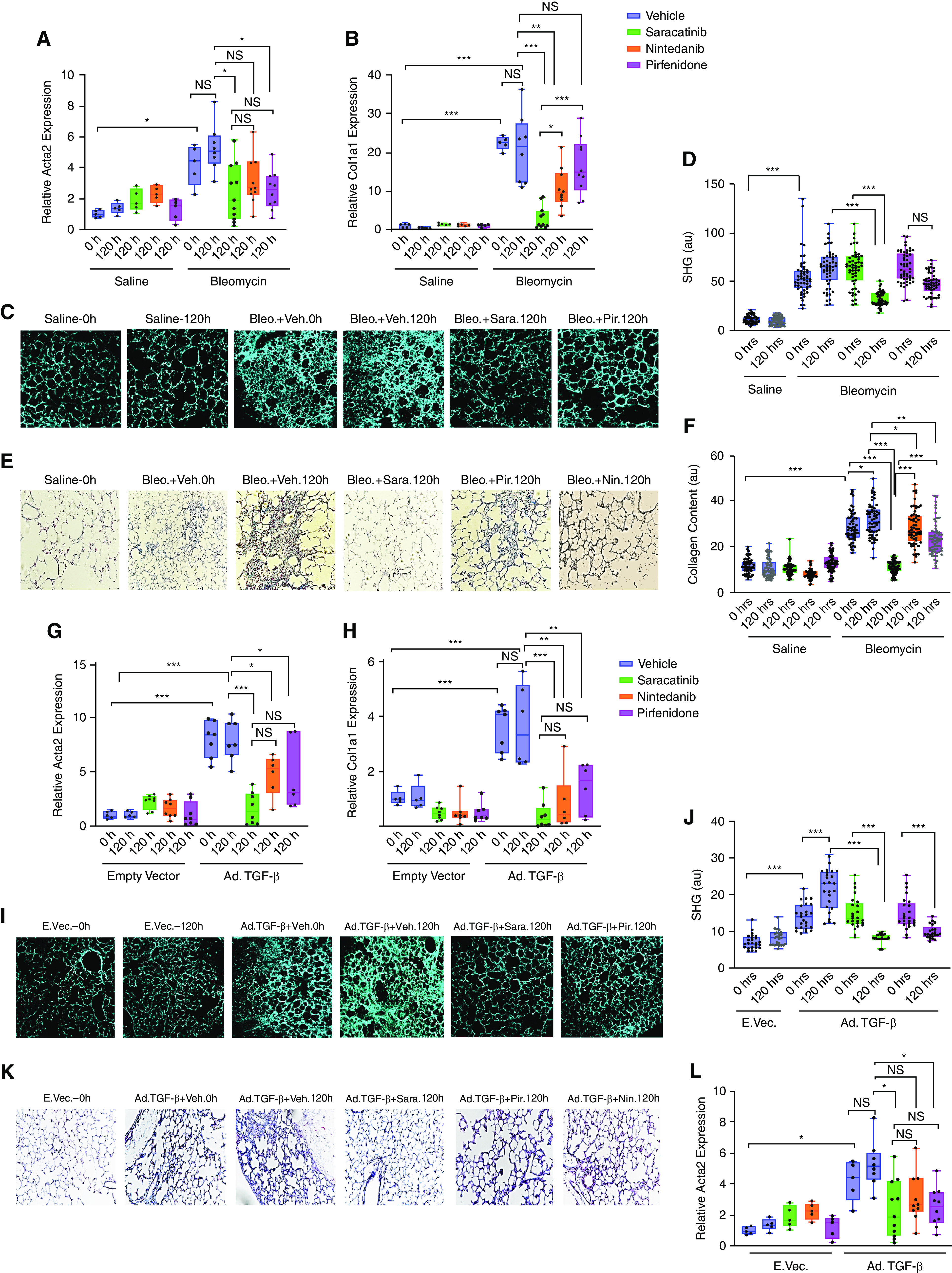
Saracatinib inhibits pulmonary fibrosis in ex vivo murine precision-cut lung slices (PCLSs) in bleomycin and Ad-TGF-β (adenovirus transforming growth factor-β) models. Mouse PCLSs were generated from both bleomycin (Day 14) and Ad-TGF-β (Day 21) models. Treatment with saracatinib (0.6 μM), nintedanib (1 μM), pirfenidone (1 mM), or vehicle control was administered in the first 24 hours after slicing (time 0 h). All lung slices were isolated 5 days after treatment for analysis (time 120 h). (A–F) Effect of saracatinib, nintedanib, and pirfenidone in PCLSs isolated from the bleomycin mouse model of pulmonary fibrosis. (A and B) Quantitative real-time PCR analysis on mice PCLSs in bleomycin model for smooth muscle alpha (α)-2 actin (Acta2) and collagen type I alpha 1 chain (Col1a1) in the indicated treatment groups. (C and D) Representative live images using second harmonic generation microscopy (SHG) and quantification assessments of PCLS samples at times 0 and 5 days (120 h) after indicated treatments. (E and F) Representative images and quantification assessments of Masson’s Trichrome staining of PCLS slides at times 0 and 5 days (120 h) after indicated treatments. (G–L) Effect of saracatinib, nintedanib, and pirfenidone in PCLSs isolated from Ad-TGF-β–treated mouse models of pulmonary fibrosis. (G and H) Quantitative real-time PCR analysis of mouse PCLSs in the Ad-TGF-β model for Acta2 and Col1a1 in the indicated treatment groups (I and J). Representative live images using SHG and quantification assessments of PCLS samples at times 0 and 5 days (120 h) after indicated treatment. (K and L) Representative images and quantification assessments of Masson’s Trichrome staining of PCLS samples at times 0 and 5 days (120 h) after indicated treatment. All data are presented as (mean + SEM); *P < 0.05, **P < 0.01, and ***P < 0.001 (n ⩾ 6 in all groups). au = average intensity; NS = nonsignificant.
In summary, using murine PCLS, we demonstrated that saracatinib is more effective than either nintedanib or pirfenidone in attenuation of PF in an ex vivo model and confirmed our findings in the in vivo murine models.
A Comparison of Transcriptional Changes in Mouse Models Identifies Core Genes Relevant for IPF and Points to a Mechanism of Action of Saracatinib
We undertook bioinformatic analysis to elucidate the molecular mechanisms underlying the antifibrotic effects of saracatinib shown in both murine models of PF. RNA sequencing was performed on lungs isolated from mice treated with either bleomycin or Ad-TGF-β in the presence or absence of saracatinib treatment. Consistent with the favorable clinical safety profile of saracatinib, there were relatively few gene expression changes in mice receiving saracatinib alone compared with the control group. Importantly, saracatinib reversed the expression of many of the genes that were altered by bleomycin (Figures 5A and 5B). GSEA demonstrated that saracatinib reversed alterations in numerous gene sets induced by bleomycin (including Myc targets, E2F targets, EMT, and G2M checkpoint gene sets) (Figure 5C). Analysis of the pulmonary transcriptional changes observed in the Ad-TGF-β model revealed that TGF-β induced changes in approximately 4,500 genes; however, in contrast to the observed changes in the bleomycin model, saracatinib treatment had a much smaller effect with fewer significant changes in expression. Further evaluation using GSEA, taking expression levels of all genes into account, demonstrated that saracatinib attenuated many pathways altered by TGF-β expression, including IFNγ, IL6, JAK-STAT (janus kinase-signal transducer and activator of transcription) signaling, and IFNα responses (Figure 5D).
Figure 5.
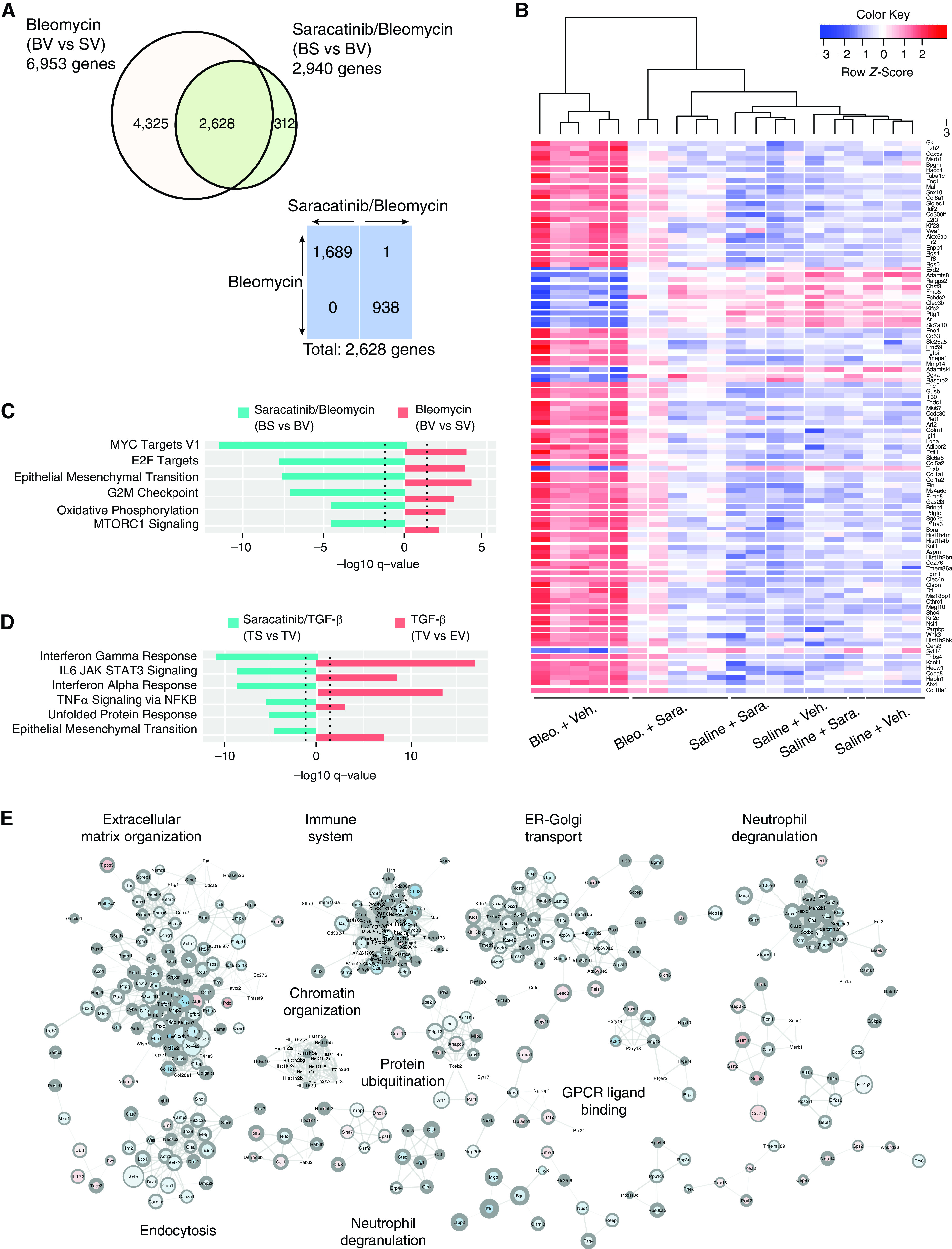
Saracatinib treatment results in the reversal of transcriptional changes observed in idiopathic pulmonary fibrosis (IPF) mouse models. (A) Comparison of number and direction of significantly differentially expressed (DE) genes (false discovery rate [FDR] < 0.05) in bleomycin and bleomycin/saracatinib treatment groups. Bleomycin administration induced significant differential expression of almost 7,000 genes (adjusted P value < 0.05) in the bleomycin treatment group when compared with the control group. Bleomycin-treated mice that also received saracatinib had significant changes in 2,940 genes compared with mice treated with bleomycin alone. A total of 2,628 differentially expressed genes are common between treatment groups. Of these, 1,689 are upregulated by bleomycin and downregulated by saracatinib, and 938 are downregulated by bleomycin and upregulated by saracatinib. (B) Heatmap of top 100 differentially expressed genes (BS vs. BV). (C) Top Hallmark gene set enrichment analysis (GSEA) in bleomycin murine experiments (adjusted P value < 0.05) ranked by saracatinib effect. Blue bars show BS versus BV and red bars show BV versus SV. The positive or negative signs indicate log fibrotic cocktail (FC) directions. (D) Top Hallmark GSEA in Ad-TGF-β (adenovirus transforming growth factor-β) experiments ranked by saracatinib effect (adjusted P value < 0.05). Blue bars show TS versus TV and red bars show TV versus EV. The positive or negative signs indicate logFC directions. (E) Cytoscape network analysis of common differentially expressed genes shared by both the bleomycin and TGF-β mouse models that are reversed by saracatinib. Node size: expression level; node color: logFC (red is up, blue is down); node border width: negative log adjusted P value. Bleomycin experimental conditions: SV = saline + vehicle; BV = bleomycin + vehicle; BS = bleomycin + saracatinib; BV versus SV = bleomycin effect; BS versus BV = saracatinib on bleomycin. Ad-TGF-β experimental conditions: EV = control + vehicle; TV = TGF-β + vehicle; TS = TGF-β + saracatinib; TV versus EV = TGF-β effect; TS versus TV = saracatinib on TGF-β. ER = endoplasmic reticulum; GPCR = G protein coupled receptors.
Next, we compared the transcriptional changes observed in these two animal models and identified a set of genes common to both models (consisting of 1,233 upregulated genes and 1,256 downregulated genes). A subset of these genes is reversed by saracatinib (225 upregulated, 560 downregulated). To interrogate this subset further, a protein–protein interaction network analysis identified modules including ECM organization, immune system processes, endoplasmic reticulum (ER)–Golgi transport, and neutrophil degranulation. Within this ECM cluster are numerous known markers of fibrosis, including members of the collagen family (Col3a1, Col4a1, Col4a2, Col5a2, Col12a1, Col16a1), Fn1 (fibronectin), Lama1 (laminin, Tnc (tenascin), Fbn1 (fibrillin), TGF-βr1 (TGF-β receptor1), and the ECM receptor Cd44 (Figures 5E and E11).
Saracatinib Ameliorates PF in hPCLSs
We sought to assess how saracatinib affects collagen production and profibrotic gene expression in human lungs. To this end, we used ex vivo culture of hPCLSs prepared from human IPF lungs as well as lungs from healthy donors. Briefly, hPCLSs isolated from patients with IPF were cultured and treated with saracatinib or vehicle for 5 days. In an independent experiment, the hPCLS harvested from healthy donors were also treated with saracatinib or vehicle in the medium containing an FC (TGF-β, PDGF-α, TNF-α, and lysophosphatidic acid) or the control cocktail as previously described (27) (Figure 6A). Histological evaluation of these slices revealed a significant reduction in collagen accumulations by saracatinib in both experiments as indicated by Masson’s Trichrome (Figures 6B–6D) and α-SMA (Figures 6E–6G) staining.
Figure 6.
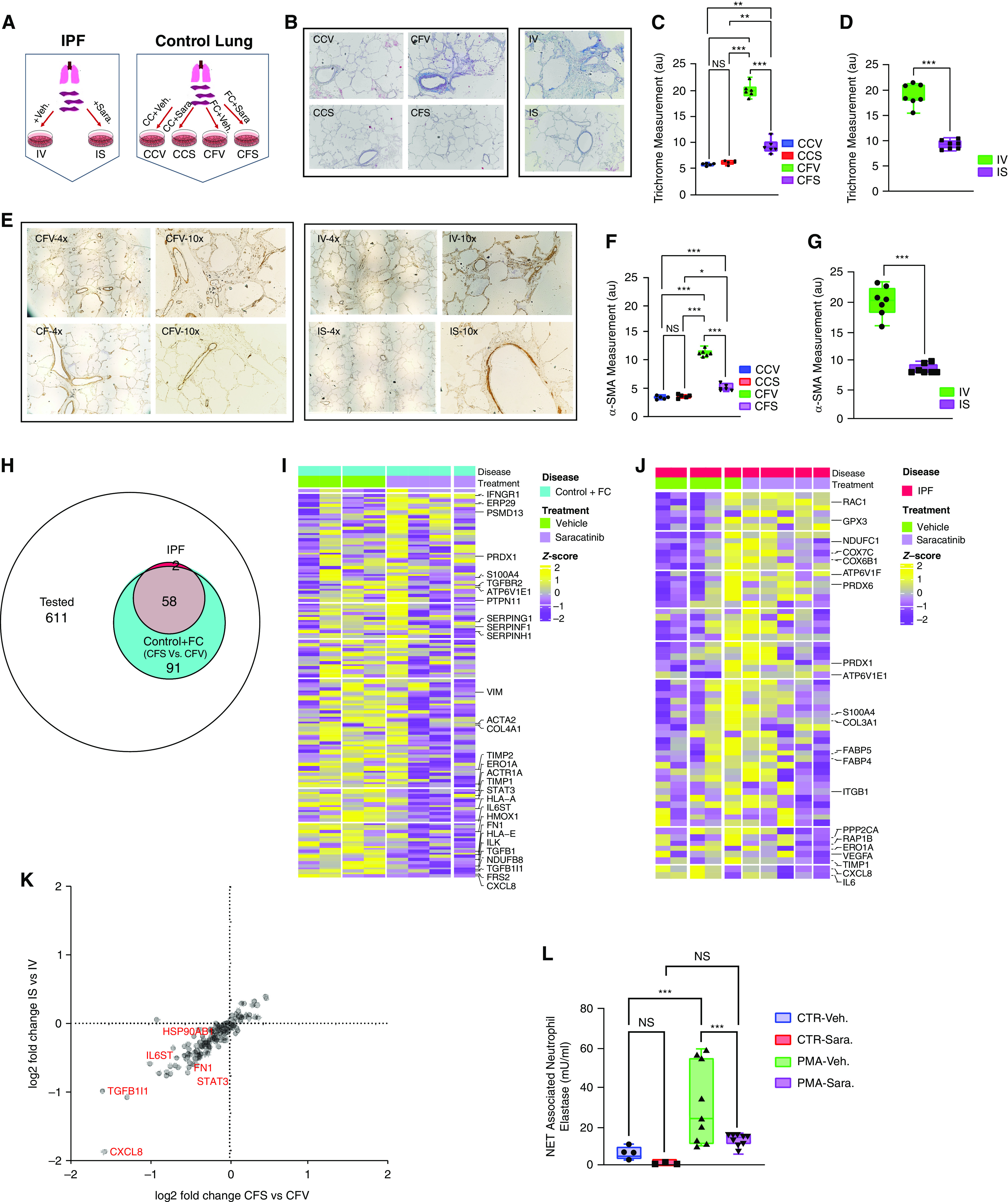
Saracatinib attenuates pulmonary fibrosis at the level of gene expression and collagen protein accumulation in ex vivo human precision-cut lung slice (hPCLS) models. (A) Schematic view of the experimental design. hPCLSs isolated from patients with idiopathic pulmonary fibrosis (IPF) were cultured and treated with saracatinib (0.6 μM) or vehicle for 5 days; IS (IPF + saracatinib) and IV (IPF + vehicle). In an independent experiment, the hPCLSs harvested from healthy donors were also cultured and treated with fibrotic cocktail (FC), (containing 5 μg TGF-β [transforming growth factor-β], 50 μg PDGF-AB [platelet-derived growth factor-AB], 10 ng TNF-α [tumor necrosis factor-α], and 10 mg lysophosphatidic acid), or control cocktail (CC), with saracatinib or vehicle for 5 days; CCV (control lung + CC + vehicle), CCS (control lung + CC + saracatinib), CFV (control lung + FC + vehicle), and CFS (control lung + FC + saracatinib). (B–D) Representative images and quantitation measurements of Masson’s Trichrome staining on the harvested slides from all groups at the end of the time points. (E–G) Representative images and quantitation measurements of α-SMA (α-smooth muscle actin) staining on the harvested slides from all groups at the end of the time points. (H) Venn diagram of the number of genes differentially expressed after saracatinib treatment in PCLSs from control hPCLSs with FC (CFV vs. CFS) (number of genes = 149) or IPF (IV vs. IS) (number of genes = 60) among all measured genes (number of genes = 761). (I) Heatmap of 149 differentially expressed genes (DEGs) in control hPCLSs with FC (CFV vs. CFS). Genes are ordered from highest to lowest fold change; z-scores are calculated across samples. (J) Heatmap of 60 DEGs in IPF PCLSs (IV vs. IS). Genes are ordered from highest to lowest fold change; z-scores are calculated across samples. (K) Volcano plot of the combined DEG results of both data sets; x-axis = log2 fold change of DEGs between CFS versus CFV; y-axis = log2 fold change of DEGs between IS versus IV. (L) Neutrophil extracellular trap (NET)-associated neutrophil elastase (mU/ml) measured from the phorbol myristate acetate (PMA)-induced neutrophil extracellular traps after 6 hours of incubation with saracatinib or vehicle. All data are presented as mean + SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 (n ⩾ 6 in all groups). CTR = control; NS = nonsignificant.
We also aimed to assess the effects of saracatinib on gene expression alterations in human lung fibrosis by screening the expression of 761 fibrosis-related genes in these lung slices. Analysis of transcriptional genes using the nCounter Analysis System by NanoString revealed that the FC induced the expression of the numerous fibrotic genes in the control lungs (Figure E12). The comparison between control lungs treated with FC plus saracatinib versus control lungs treated with FC plus vehicle revealed that saracatinib treatment altered the expression of 149 of these genes (Figure 6H). Considering that explanted lungs from patients with IPF are generally in a very advanced and heterogenic fibrotic state, it was of interest that treatment with saracatinib altered the expression of 60 genes in the IPF slices; 58 of these were in common with the FC plus saracatinib group, and two of these genes (VEGF-A [vascular endothelial growth factor-A] and IL-6) were uniquely altered in the IPF lung. Further analysis of this transcriptomic data revealed that saracatinib altered many of the profibrotic genes, including ACTA2, COL4A1, COL3A1, FN, TIMP metallopeptidase inhibitors (TIMPs), TGFβ-R1, and TGFβ1 in both datasets (Figures 6I and 6J). In accordance with the transcriptomic data from the mouse experiments, the expression of numerous genes related to the immune system and inflammatory cascades were also altered in these human lung slices (Tables E3 and E4). Combining these two independent gene expression datasets revealed that the patterns of gene alterations by saracatinib in both human fibrotic lung models were very similar, with CXCL8 and TGF-β1L1 as the most dominant downregulated gene in both experiments (Figure 6K).
CXCL8 has been introduced as one of the most critical genes in neutrophil activation (47, 48). Given that the murine transcriptomic data highlighted a possible regulatory role for saracatinib in neutrophil degranulation and activation in PF (Figure 5E) and the importance of neutrophil extracellular traps (NETs) in innate immune response in the pathogenesis of IPF (49, 50), we sought to test the role of saracatinib on the activation of neutrophils isolated from human blood of healthy control subjects. As is shown in Figure 6L, saracatinib dramatically suppressed phorbol myristate acetate–induced NET formation compared with control, as indicated by the measurements of NET-associated neutrophil elastase.
In summary, the analysis of two human ex vivo lung models of fibrosis demonstrated that saracatinib has potent antifibrotic effects in human lung tissue as assessed at the level of gene expression as well as at the level of lung collagen protein content.
Discussion
In this study, we used an innovative disease-agnostic computational biology-based approach and identified the Src kinase inhibitor saracatinib (AZD0530) (23) as a potential therapeutic agent for IPF and delineated multiple fibrogenic pathways targeted by this agent at the molecular level. Furthermore, we validated the efficacy of saracatinib in blocking fibrogenic responses in several complementary preclinical models of PF. Our data provide strong evidence that saracatinib is equal or superior to the two FDA-approved drugs, nintedanib and pirfenidone, at inhibiting PF in experimental models. Analysis of the transcriptional changes induced by saracatinib in the in vitro and in vivo models of PF demonstrated that saracatinib was able to reverse the expression of diverse fibrogenic genes and pathways, such as myofibroblast differentiation, EMT, ECM organization, immune system processes, endoplasmic reticulum–Golgi transport, and neutrophil degranulation. Importantly, many of these differentially expressed gene sets and pathways were selectively modified by saracatinib compared with nintedanib and pirfenidone. We also demonstrated a profound change in the fibrotic gene expression and collagen accumulation in several independent ex vivo models, including bleomycin and Ad-TGF-β mouse PCLSs, hPCLSs treated with FC, and PCLSs obtained from patients with IPF. Taken together, our in vitro, in vivo, and ex vivo data support the validity of our in silico–derived hypothesis that saracatinib is to be considered a candidate for drug repurposing in IPF.
At the inception of this study, we used genome-wide transcriptional profiling of human IPF lungs cross-referenced with distinct drug transcriptomic signatures to identify Src kinase–dependent signaling pathways as critical checkpoints that control profibrotic responses in the lung. Importantly, we established that saracatinib was a potent and effective therapeutic agent that significantly attenuated experimental PF in several preclinical models with equal or greater efficacy than the two FDA-approved drugs nintedanib and pirfenidone. This approach, anchored in transcriptomic profiles in human lung cells and tissues, led to the identification of novel pathways that are relevant to human IPF.
Our study provides additional information and is complementary to proteomics-based analyses of human tissues that have yielded therapeutic targets for PF (51–53). Repurposing drugs to treat conditions other than the one for which they were developed and/or approved, can shorten timelines, decrease costs, and increase success rates. Using a computational biology approach in combination with Connectivity Map can help in identifying new drugs, predicting drug candidates, and discovering connections among small molecules sharing a mechanism of action. Based on the connectivity scores of saracatinib to IPF relative to nintedanib and pirfenidone using L1000 data, we identified that saracatinib had greater connectivity to IPF than the other two FDA-approved drugs. This approach has been widely used as a resource in cancer drug discovery (29, 54), and our study extends this methodology to IPF treatment.
Several studies have shown that SFKs control key signaling pathways relevant to PF pathogenesis (55), including TGF-β–mediated myofibroblast differentiation and fibrogenic responses in lung fibroblasts (13). SFKs are also required for TGF-β–driven EMT and are necessary for the recruitment and activation of immune cells in pulmonary inflammation (55, 56). We have reported that mice genetically deficient in PTPα (protein tyrosine phosphatase α), a known activator of SFKs, are protected from experimental PF (57, 58). We also reported that SHP2 (SH2 domain–containing tyrosine phosphatase 2) is an antifibrotic regulator in PF (59, 60). These studies underscore the importance of reversible tyrosine phosphorylation reactions, controlled by specific tyrosine kinases and phosphatases, in the pathogenesis of PF and provide a mechanistic foundation for the use of saracatinib in the treatment of this disease. Our studies using normal human lung fibroblasts and animal models of PF and PCLS provide strong evidence that saracatinib inhibits fibrogenic responses more potently than either nintedanib or pirfenidone. Our study revealed that saracatinib perturbed ‘EMT’, ‘MYC targets’, and ‘IL6_JAK_STAT signaling’ gene sets and that saracatinib uniquely induced perturbations in ‘E2F target’, ‘G2M checkpoints’, and ‘oxidative phosphorylation’ and IFN gene sets in the in vivo models. This analysis provides important insights into the mechanisms by which SFKs modulate fibrogenic signaling pathways.
Although our study presents comprehensive evidence for the potential antifibrotic effects of saracatinib, questions of interest concerning the effects of saracatinib on fibrogenic pathways remain to be addressed. In the current study, we focused on TGF-β–dependent pathways because of the well-accepted importance of this growth factor in organ fibrosis (61, 62). In addition to TGF-β, other growth factors, including PDGF (63, 64), IGF-1 (insulin-like growth factor-1) (65), and FGFs (fibroblast growth factors) (66), are known to trigger profibrotic cellular responses through signaling pathways that are controlled by SFKs. Similarly, kinase enrichment analysis of differentially expressed gene sets altered by saracatinib identified enrichment for RIPK3 and the MAP kinases. Importantly, RIPK3 has been implicated in the pathogenesis of renal fibrosis (67). SFKs are known to regulate MAP kinase–dependent signaling pathways relevant to fibrogenesis (68, 69). Whether RIPK3 is regulated by SFK is unknown and the subject of current investigations. In future studies, we aim to investigate the effects of saracatinib on PDGF, IGF-1, FGF, and RIPK3, as well other MAP kinase–dependent signaling pathways to better dissect their relative contributions.
In conclusion, based on a computational drug repurposing strategy, we have identified saracatinib as a potential therapeutic for IPF (40). To our knowledge, this is the first study that has used a computational approach to link a compound to IPF in silico and then validated the efficacy of the drug in in vitro, in vivo, and ex vivo models culminating in a human clinical trial in the treatment of IPF (70).
Footnotes
The original connectivity mapping studies were supported by funding from AstraZeneca (J.T.D.). The in vitro and preclinical animal model studies and bioinformatics analysis were supported by National Institutes of Health National Center for Advancing Translational Sciences grants UG3TR002445 (G.P.D., N.K., and J.T.D.) and R01HL132950 (G.P.D.) and National Institutes of Health grants R01HL127349, U01HL145567, U01HL122626, and U54HG008540 (N.K.).
Author Contributions: G.P.D., N.K., and J.T.D. conceptualized, acquired funding, and supervised the study. L.P.C. provided saracatinib and necessary guidance for this project. All in vitro studies were performed by F.A., M.N., C.M., K.C., H.M.R., K.W.K., N.B., and Y.A. All in vivo studies were performed by F.A., D.G.F., K.B., K.-A.R., S.D., J.C.S., Q.L., T.B., and N.O. All murine ex vivo studies were performed by F.A., M.C., and K.-A.R. The human precision-cut lung slices for ex vivo studies were provided by I.R., and all experiments and analysis were performed by F.A., A.J., and T.A. The neutrophil isolations and assays were performed by F.A. and L.S. under C.S.D.C.’s protocol. RNA sequencing on in vitro and in vivo models was performed by F.A., D.G.F., and G.D. Transcriptomic data were processed, curated, and visualized by C.B., X.W., B.R., and L.N.L. The original RNA sequencing to identify the compound signatures was performed by B.M., R.H., and A.B. Connectivity mapping and disease enrichment analysis were performed by C.B. and B.R. The manuscript was drafted by F.A., C.B., G.P.D., and N.K. and was reviewed and edited by all other authors.
Data and materials availability: All data, code, and materials used in the analysis are available to any researcher for purposes of reproducing or extending the analysis. All raw count expression data from in vitro and in vivo studies were deposited to the Gene Expression Omnibus (GEO), and the accession numbers are GSE178518 and GSE17845, respectively.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202010-3832OC on August 23, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet . 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 2. Martinez FJ, Chisholm A, Collard HR, Flaherty KR, Myers J, Raghu G, et al. The diagnosis of idiopathic pulmonary fibrosis: current and future approaches. Lancet Respir Med . 2017;5:61–71. doi: 10.1016/S2213-2600(16)30325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med . 2018;379:797–798. doi: 10.1056/NEJMc1807508. [DOI] [PubMed] [Google Scholar]
- 4. Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med . 2014;2:566–572. doi: 10.1016/S2213-2600(14)70101-8. [DOI] [PubMed] [Google Scholar]
- 5. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med . 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med . 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 7. King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet . 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 8. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med . 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thannickal VJ, Henke CA, Horowitz JC, Noble PW, Roman J, Sime PJ, et al. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol . 2014;184:1643–1651. doi: 10.1016/j.ajpath.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amanchy R, Zhong J, Hong R, Kim JH, Gucek M, Cole RN, et al. Identification of c-Src tyrosine kinase substrates in platelet-derived growth factor receptor signaling. Mol Oncol . 2009;3:439–450. doi: 10.1016/j.molonc.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Twamley-Stein GM, Pepperkok R, Ansorge W, Courtneidge SA. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc Natl Acad Sci USA . 1993;90:7696–7700. doi: 10.1073/pnas.90.16.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tatosyan AG, Mizenina OA. Kinases of the Src family: structure and functions. Biochemistry (Mosc) . 2000;65:49–58. [PubMed] [Google Scholar]
- 13. Hu M, Che P, Han X, Cai GQ, Liu G, Antony V, et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis. J Pharmacol Exp Ther . 2014;351:87–95. doi: 10.1124/jpet.114.216044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hughes G, Toellner H, Morris H, Leonard C, Chaudhuri N. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med . 2016;5:78. doi: 10.3390/jcm5090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med . 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 16. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med . 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 17. Corte T, Bonella F, Crestani B, Demedts MG, Richeldi L, Coeck C, et al. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir Res . 2015;16:116. doi: 10.1186/s12931-015-0276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med . 2011;3:96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med . 2011;3:96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brum AM, van de Peppel J, van der Leije CS, Schreuders-Koedam M, Eijken M, van der Eerden BC, et al. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc Natl Acad Sci USA . 2015;112:12711–12716. doi: 10.1073/pnas.1501597112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene . 2008;27:6365–6375. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol . 2009;3:248–261. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baselga J, Cervantes A, Martinelli E, Chirivella I, Hoekman K, Hurwitz HI, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin Cancer Res . 2010;16:4876–4883. doi: 10.1158/1078-0432.CCR-10-0748. [DOI] [PubMed] [Google Scholar]
- 24. Ahangari F, Becker C, Foster D, Chioccioli M, Nelson M, Beke K, et al. Saracatinib is a potential novel therapeutic for pulmonary fibrosis [abstract] Am J Respir Crit Care Med . 2020;201 [Google Scholar]
- 25.Ahangari F, Becker C, Foster DG, Chioccioli M, Nelson M, Beke K, et al. Saracatinib, a selective Src kinase inhibitor, blocks fibrotic responses in in vitro, in vivo, and ex vivo models of pulmonary fibrosis [preprint] 2022. https://www.biorxiv.org/content/10.1101/2022.01.04.474955v1 [DOI] [PMC free article] [PubMed]
- 26. Lehmann M, Buhl L, Alsafadi HN, Klee S, Hermann S, Mutze K, et al. Differential effects of nintedanib and pirfenidone on lung alveolar epithelial cell function in ex vivo murine and human lung tissue cultures of pulmonary fibrosis. Respir Res . 2018;19:175. doi: 10.1186/s12931-018-0876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alsafadi HN, Staab-Weijnitz CA, Lehmann M, Lindner M, Peschel B, Königshoff M, et al. An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am J Physiol Lung Cell Mol Physiol . 2017;312:L896–L902. doi: 10.1152/ajplung.00084.2017. [DOI] [PubMed] [Google Scholar]
- 28. Uhl FE, Vierkotten S, Wagner DE, Burgstaller G, Costa R, Koch I, et al. Preclinical validation and imaging of Wnt-induced repair in human 3D lung tissue cultures. Eur Respir J . 2015;46:1150–1166. doi: 10.1183/09031936.00183214. [DOI] [PubMed] [Google Scholar]
- 29. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science . 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 30. Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell . 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Z, Monteiro CD, Jagodnik KM, Fernandez NF, Gundersen GW, Rouillard AD, et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat Commun . 2016;7:12846. doi: 10.1038/ncomms12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meltzer EB, Barry WT, D’Amico TA, Davis RD, Lin SS, Onaitis MW, et al. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: proof-of-principle. BMC Med Genomics . 2011;4:70. doi: 10.1186/1755-8794-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng R, Sridhar S, Tyagi G, Phillips JE, Garrido R, Harris P, et al. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS One . 2013;8:e59348. doi: 10.1371/journal.pone.0059348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiong J, Wu JS, Mao SS, Yu XN, Huang XX. Effect of saracatinib on pulmonary metastases from hepatocellular carcinoma. Oncol Rep . 2016;36:1483–1490. doi: 10.3892/or.2016.4968. [DOI] [PubMed] [Google Scholar]
- 35. Jin J, Togo S, Kadoya K, Tulafu M, Namba Y, Iwai M, et al. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-β1. Respir Res . 2019;20:119. doi: 10.1186/s12931-019-1093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang J, Beyer C, Palumbo-Zerr K, Zhang Y, Ramming A, Distler A, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis . 2016;75:883–890. doi: 10.1136/annrheumdis-2014-207109. [DOI] [PubMed] [Google Scholar]
- 37. Wollin L, Togbe D, Ryffel B. Effects of nintedanib in an animal model of liver fibrosis. BioMed Res Int . 2020;2020:3867198. doi: 10.1155/2020/3867198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rothschild SI. Clinical potential of nintedanib for the second-line treatment of advanced non-small-cell lung cancer: current evidence. Lung Cancer (Auckl) . 2014;5:51–57. doi: 10.2147/LCTT.S49490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res . 2014;15:157. doi: 10.1186/s12931-014-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev . 2011;20:85–97. doi: 10.1183/09059180.00001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu YY, Zhao XK, Yu L, Qi F, Zhai B, Gao CQ, et al. Interaction of Src and alpha-V integrin regulates fibroblast migration and modulates lung fibrosis in a preclinical model of lung fibrosis. Sci Rep . 2017;7:46357. doi: 10.1038/srep46357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2008;294:L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 43. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest . 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J Exp Med . 2020;217:e20190103. doi: 10.1084/jem.20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol . 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cedilak M, Banjanac M, Belamarić D, Paravić Radičević A, Faraho I, Ilić K, et al. Precision-cut lung slices from bleomycin treated animals as a model for testing potential therapies for idiopathic pulmonary fibrosis. Pulm Pharmacol Ther . 2019;55:75–83. doi: 10.1016/j.pupt.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 47. Teijeira A, Garasa S, Ochoa MDC, Cirella A, Olivera I, Glez-Vaz J, et al. Differential interleukin-8 thresholds for chemotaxis and netosis in human neutrophils. Eur J Immunol . 2021;51:2274–2280. doi: 10.1002/eji.202049029. [DOI] [PubMed] [Google Scholar]
- 48. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol . 2020;17:433–450. doi: 10.1038/s41423-020-0412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suzuki M, Ikari J, Anazawa R, Tanaka N, Katsumata Y, Shimada A, et al. PAD4 deficiency improves bleomycin-induced neutrophil extracellular traps and fibrosis in mouse lung. Am J Respir Cell Mol Biol . 2020;63:806–818. doi: 10.1165/rcmb.2019-0433OC. [DOI] [PubMed] [Google Scholar]
- 50. Dimmeler S, Zeiher AM. Netting insights into fibrosis. N Engl J Med . 2017;376:1475–1477. doi: 10.1056/NEJMcibr1616598. [DOI] [PubMed] [Google Scholar]
- 51. Yu G, Ibarra GH, Kaminski N. Fibrosis: lessons from OMICS analyses of the human lung. Matrix Biol . 2018;68-69:422–434. doi: 10.1016/j.matbio.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Palfreyman MG. Human tissue in target identification and drug discovery. Drug Discov Today . 2002;7:407–409. doi: 10.1016/s1359-6446(02)02226-2. [DOI] [PubMed] [Google Scholar]
- 53. Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos Ethics Humanit Med . 2009;4:2. doi: 10.1186/1747-5341-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Musa A, Ghoraie LS, Zhang SD, Glazko G, Yli-Harja O, Dehmer M, et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform . 2017;18:903. doi: 10.1093/bib/bbx023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H, Zhao C, Tian Y, Lu J, Zhang G, Liang S, et al. Src family kinases and pulmonary fibrosis: a review. Biomed Pharmacother . 2020;127:110183. doi: 10.1016/j.biopha.2020.110183. [DOI] [PubMed] [Google Scholar]
- 56. Ulsamer A, Wei Y, Kim KK, Tan K, Wheeler S, Xi Y, et al. Axin pathway activity regulates in vivo pY654-β-catenin accumulation and pulmonary fibrosis. J Biol Chem . 2012;287:5164–5172. doi: 10.1074/jbc.M111.322123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aschner Y, Khalifah AP, Briones N, Yamashita C, Dolgonos L, Young SK, et al. Protein tyrosine phosphatase α mediates profibrotic signaling in lung fibroblasts through TGF-β responsiveness. Am J Pathol . 2014;184:1489–1502. doi: 10.1016/j.ajpath.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aschner Y, Nelson M, Brenner M, Roybal H, Beke K, Meador C, et al. Protein tyrosine phosphatase-α amplifies transforming growth factor-β-dependent profibrotic signaling in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol . 2020;319:L294–L311. doi: 10.1152/ajplung.00235.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene . 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 60. Tzouvelekis A, Yu G, Lino Cardenas CL, Herazo-Maya JD, Wang R, Woolard T, et al. SH2 domain-containing phosphatase-2 is a novel antifibrotic regulator in pulmonary fibrosis. Am J Respir Crit Care Med . 2017;195:500–514. doi: 10.1164/rccm.201602-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aschner Y, Downey GP. Transforming growth factor-β: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol . 2016;54:647–655. doi: 10.1165/rcmb.2015-0391TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci USA . 2000;97:1778–1783. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med . 2005;201:925–935. doi: 10.1084/jem.20041393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Veracini L, Franco M, Boureux A, Simon V, Roche S, Benistant C. Two functionally distinct pools of Src kinases for PDGF receptor signalling. Biochem Soc Trans . 2005;33:1313–1315. doi: 10.1042/BST0331313. [DOI] [PubMed] [Google Scholar]
- 65. Xiao H, Huang X, Wang S, Liu Z, Dong R, Song D, et al. Metformin ameliorates bleomycin-induced pulmonary fibrosis in mice by suppressing IGF-1. Am J Transl Res . 2020;12:940–949. [PMC free article] [PubMed] [Google Scholar]
- 66.Guzy RD, Li L, Smith C, Dorry SJ, Koo HY, Chen L, et al. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J Biol Chem. 2017;292:10364–10378. doi: 10.1074/jbc.M117.791764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, et al. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight . 2018;3:e94979. doi: 10.1172/jci.insight.94979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol . 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 69. Pechkovsky DV, Scaffidi AK, Hackett TL, Ballard J, Shaheen F, Thompson PJ, et al. Transforming growth factor beta1 induces alphavbeta3 integrin expression in human lung fibroblasts via a beta3 integrin-, c-Src-, and p38 MAPK-dependent pathway. J Biol Chem . 2008;283:12898–12908. doi: 10.1074/jbc.M708226200. [DOI] [PubMed] [Google Scholar]
- 70.ClinicalTrials.gov. Saracatinib in the Treatment of Idiopathic Pulmonary Fibrosis (STOP-IPF) https://clinicaltrials.gov/ct2/show/NCT04598919