

Abstract
Background and Objectives
Diagnostic criteria emphasize the use of sensitive and disease-specific tests to distinguish patients with rapidly progressive dementia (RPD) due to Creutzfeldt-Jakob disease (CJD) vs other causes (mimics). These tests are often performed in specialized centers, with results taking days to return. There is a need to leverage clinical features and rapidly reporting tests to distinguish patients with RPD due to CJD from those due to other causes (mimics) early in the symptomatic course.
Methods
In this case-control series, clinical features and the results of diagnostic tests were compared between mimics (n = 11) and patients with definite (pathologically proven, n = 33) or probable CJD (with positive real-time quaking-induced conversion [RT-QuIC], n = 60). Patients were assessed at Mayo Clinic Enterprise or Washington University from January 2014 to February 2021. Mimics were enrolled in prospective studies of RPD; mimics met the diagnostic criteria for probable CJD but did not have CJD.
Results
Mimics were ultimately diagnosed with autoimmune encephalitis (n = 6), neurosarcoidosis, frontotemporal lobar degeneration with motor neuron disease, dural arteriovenous fistula, cerebral amyloid angiopathy with related inflammation, and systemic lupus erythematous with polypharmacy. Age at symptom onset, sex, presenting features, and MRI and EEG findings were similar in CJD cases and mimics. Focal motor abnormalities (49/93, 11/11), CSF leukocytosis (4/92, 5/11), and protein >45 mg/dL (39/92, 10/11) were more common in mimics (p < 0.01). Positive RT-QuIC (77/80, 0/9) and total tau >1149 pg/mL (74/82, 2/10) were more common in CJD cases (all p < 0.01). Protein 14-3-3 was elevated in 64/89 CJD cases and 4/10 mimics (p = 0.067). Neural-specific autoantibodies associated with autoimmune encephalitis were detected within the serum (5/9) and CSF (5/10) of mimics; nonspecific antibodies were detected within the serum of 9/71 CJD cases.
Discussion
Immune-mediated, vascular, granulomatous, and neurodegenerative diseases may mimic CJD at presentation and should be considered in patients with early motor dysfunction and abnormal CSF studies. The detection of atypical features—particularly elevations in CSF leukocytes and protein—should prompt evaluation for mimics and consideration of empiric treatment while waiting for the results of more specific tests.

Sporadic Creutzfeldt-Jakob disease (CJD) is a common cause of rapidly progressive dementia (RPD),1 attributable to the relentless propagation of misfolded prion proteins throughout the brain. Increasing awareness of CJD has increased diagnostic consideration and testing for CJD, which has improved estimates of disease prevalence and the accuracy of diagnostic criteria.2-4 However, increased awareness may also lead to diagnostic closure in select patients with RPD, contributing to misdiagnosis with dire consequences.5 Case series from prion disease referral centers identify other causes of rapid decline in 8.8%–47.1% of patients with suspected CJD.4-7 Although atypical presentations of common neurodegenerative diseases (i.e., Alzheimer disease and related dementias) account for the majority of CJD mimics, autoimmune/inflammatory and other potentially treatable causes of RPD are increasingly recognized.4-6,8 The discovery of treatment-responsive CJD mimics underscores the need to determine the specific causes of RPD early in the symptomatic course. Early recognition of potentially treatment-responsive mimics is especially important to ensure that disease-modifying treatments are provided as early as possible.
Revised diagnostic criteria emphasize the use of real-time quaking-induced conversion (RT-QuIC) measures of prion protein to identify patients with RPD due to CJD3,9,10 and for good reason. Expanded access to sensitive and specific RT-QuIC assays capable of detecting malformed prions in CSF has greatly improved antemortem recognition of CJD, increasing diagnostic confidence and improving prevalence estimates of CJD.2-4,9,11,12 However, despite the acronym, RT-QuIC cannot be performed quickly. The assay takes days to run and verify.13 In addition, time required to ship the sample to specialty centers contributes to delays in reporting of results. Similar turnaround times are observed with other send-out tests that may aid in the diagnosis of CJD (e.g., CSF total tau) or detection of potential mimics (e.g., disease-specific autoantibodies). These reporting delays present a practical challenge when evaluating rapidly declining patients with suspected CJD. There is a need to leverage clinical features and rapidly reporting tests to distinguish patients with RPD due to CJD from those with other causes (mimics). Recognizing this, we assessed the clinical features and available diagnostic tests that distinguished mimics from patients with probable or definite CJD enrolled in a prospective study of RPD.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
CJD mimics (n = 11) and cases (n = 14) were enrolled from February 14, 2016, to April 30, 2021, within prospective studies of RPD at Washington University in St. Louis (Saint Louis, MO) and Mayo Clinic Florida (Jacksonville, FL). Patients in RPD studies were directly assessed by members of the study team (S.R.D. and G.S.D.) and followed for up to 2 years. Written informed consent was obtained from individuals in these studies or their delegates, and study procedures were approved by the Institutional Review Boards at the respective organizations. Additional CJD cases (n = 79) were identified through an automated search of Mayo Clinic Enterprise records identifying patients diagnosed with probable or definite CJD from January 1, 2014 (time at which specific biomarkers of prion disease were incorporated within clinical practice) to February 28, 2021, at Mayo Clinic in Rochester, MN; Jacksonville, FL; and Scottsdale, AZ. A waiver of consent was granted by the Mayo Clinic Institutional Review Board to incorporate clinical data from these patients. The Mayo Clinic Institutional Review Board approved all study procedures.
Participant Selection and Evaluation
Mimics
Research records from patients enrolled in prospective studies of RPD were independently reviewed by members of the study team (E.L. and G.S.D.). Twenty-five patients met the diagnostic criteria for probable CJD.10 CJD was the suspected diagnosis at the time of presentation in all patients. Subsequent evaluation confirmed alternate (non-CJD) diagnoses in 11 patients, labeled mimics. Two mimics (cases G and K, Table 1) were transferred to hospice care with a diagnosis of probable CJD before seeking reevaluation via our tertiary care center.
Table 1.
Presenting Features, Results of Diagnostic Tests for Probable CJD, Final Diagnoses, and Outcomes in Mimics

Cases
Two hundred fifty-eight patients with suspected CJD were identified through review of research records and medical charts. Records were independently reviewed by 2 authors to identify patients who met the criteria for CJD. One hundred thirty-two patients met the diagnostic criteria for probable CJD, defined as patients with neuropsychiatric disorder with positive CSF RT-QuIC, or RPD with ≥1 of myoclonus, visual/cerebellar signs, pyramidal/extrapyramidal signs, akinetic mutism, and consistent brain MRI.10 To increase diagnostic confidence, CJD cases were restricted to those with definite (neuropathologically proven; n = 33) or probable CJD with positive RT-QuIC (n = 60).
Relevant clinical data were extracted from clinical and research records for cases and mimics, including demographics (age, sex, and race), medical comorbidities, time of onset of the first symptom of CJD, clinical features at presentation, response to treatment (when relevant), and the results of rapidly reporting diagnostic tests (MRI, EEG, and routine CSF tests) and disease-specific biomarkers (CJD CSF biomarkers: protein 14-3-3, total tau, RT-QuIC assays for prions [measures performed via the National Prion Disease Pathology Surveillance Center, Case Western Reserve, Cleveland, OH]; neural-specific autoantibodies with high specificity for CNS autoimmunity in blood and CSF [measures performed via Mayo Clinic Laboratories, Rochester, MN]). Death date and results of genetic or neuropathologic evaluation were recorded where appropriate.
Statistical Analyses
Demographics, clinical features, results of investigations, and outcomes were summarized using descriptive statistics. Investigations were divided into 2 groups based on time from testing to reporting. Rapidly reporting tests included measures performed within in-house laboratories, with results reporting within 24 hours of sampling. Late-reporting tests included measures sent to outside (reference) laboratories, with results generally expected >7 days from testing. Differences between CJD cases and mimics were explored using univariate comparisons (Fisher exact test for categorical variables; nonparametric Mann-Whitney U test for continuous variables). Statistical analyses were conducted using SPSS Statistics (IBM Corp, Version 25.0, Armonk, NY). Statistical significance was established at p < 0.05.
Data Availability
Anonymized study data will be shared pending review of a request from qualified individuals by the corresponding author.
Results
Eleven patients enrolled in prospective studies of RPD (11/175, 6.3%) met the clinical criteria for probable CJD (Table 1) but had alternate diagnoses (CJD mimics). CJD mimics included patients with definite autoimmune encephalitis (n = 6), dural arteriovenous fistula, cerebral amyloid angiopathy with related inflammation, frontotemporal dementia with motor neuron disease, neurosarcoidosis, and systemic lupus erythematous with polypharmacy (each, n = 1). Demographic and clinical features were similar between mimics and cases (Table 2), except for early motor signs (e.g., faciobrachial dystonic seizure, myoclonus, dyskinesias, or parkinsonism), which were detected in all mimics, but only 53% of CJD cases (p = 0.002).
Table 2.
Demographic and Clinical Features in CJD Cases and Mimics

Table 3 outlines the results of diagnostic tests, stratified by speed of reporting. Initial MRI findings suggestive of CJD were reported in similar proportions of CJD cases (66/93, 71%) and mimics (6/11, 55%; p = 0.307), including increased T2-FLAIR or diffusion-weighted sequence signal within the caudate/putamen or ≥2 cortical areas (temporal, parietal, or occipital). Neuroimaging findings in selected mimics and cases are shown in Figure 1. Abnormalities on routine EEG were also detected in similar proportions of CJD cases (70/86, 81%) and mimics (9/11, 82%; p > 0.99). By contrast, CSF leukocytosis (mimics: 5/11, 45%; cases: 4/92, 4%; p < 0.001) and elevated protein (mimics: 10/11, 90%; cases: 39/92, 42%; p = 0.003) predominated in mimics. All mimics had elevations in either CSF leukocytes or protein; by contrast, cell count and protein levels were normal in the majority of patients with CJD (50/92, 54%; p < 0.001). Only mimics exhibited both CSF leukocytosis and elevated protein (4/11, 36%; p < 0.001). Hypoglycorrhachia (glucose 31 mg/dL) was noted in 1 mimic—a 65-year-old man with RPD and motor dysfunction, rendering him akinetic mute within 6 months of symptom onset (Case G). This unexpected finding prompted further investigations, which established neurosarcoidosis as the cause of RPD.
Table 3.
Test Results in CJD Cases and Mimics, Organized by Speed of Reporting
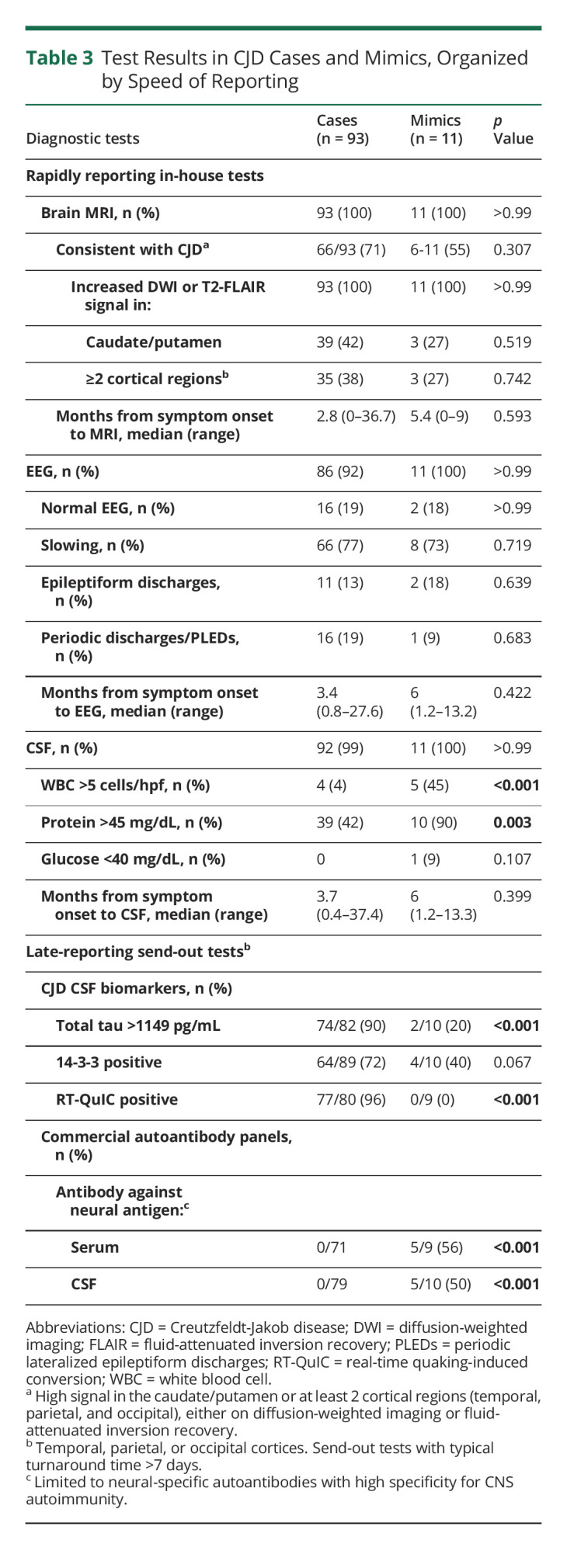
Figure 1. MRI Findings in Selected Creutzfeldt-Jakob Disease (CJD) Cases and Mimics.

Diffusion-weighted imaging (DWI) and acute diffusion coefficient (ADC) sequences are shown in radiologic convention for patients with autoimmune encephalitis (A, Case A), neurosarcoidosis (C, Case G) and dural arteriovenous fistula (E, Case J), and CJD (B, D, and F). White arrowheads highlight areas of suspected cortical diffusion restriction (cortical ribboning). Black arrowheads indicate suspected diffusion restriction within the deep nuclei. Qualitative comparisons emphasize the subtle nature of findings in mimics vs cases.
Late-reporting CSF tests demonstrated excellent sensitivity and specificity. RT-QuIC assays were positive in 77/80 CJD cases (96%) and negative in mimics (0/9; p < 0.001). Median total tau was substantially higher in CJD cases (median = 4,000 pg/mL, range 78.0–23586 pg/mL) than mimics (median = 402.5 pg/mL, range 100.0–1528.0 pg/mL; p < 0.001). In addition, more CJD cases (74/82, 90%) than mimics (2/10, 20%; p < 0.001) exhibited total tau levels >1149 pg/mL—a threshold associated with CJD.4 Disease-associated autoantibodies were detected in the CSF of 5 mimics, establishing the diagnoses of definite NMDA receptor (NMDAR) encephalitis (n = 2), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) encephalitis (n = 1), leucine-rich glioma-inactivated 1 (LGI1) antibody–associated encephalitis (n = 1), and type 1 antineuronal nuclear antibody (ANNA-1)-associated encephalitis (n = 1). Matching autoantibodies were detected in the serum of 4/5 patients. Glycine receptor autoantibodies were detected in the serum but not CSF of a sixth patient supporting the diagnosis of progressive encephalopathy with rigidity and myoclonus. Antibodies in CJD cases were detected in the serum only and were limited to low levels of antibodies against GAD-65 (n = 5; median 0.06 nmol/L, range 0.05–0.13; expected ≤0.02 nmol/L), voltage-gated calcium channels (n = 2; 0.08 and 0.16 nmol/L; expected ≤0.02 nmol/L), acetylcholine receptors (n = 1; 3.19 nmol/L; expected ≤0.02 nmol/L), and striated muscle (n = 1; titer 1:120; expected <1:120). Neural-specific autoantibodies with high specificity for CNS autoimmunity were not identified in CJD cases.
Empiric immunomodulatory therapies (most commonly high-dose methylprednisone) were provided to 22/93 CJD cases (24%) and 10/11 mimics (90%). Clinical improvement was exclusive to mimics, with 8/10 (80%) patients improving within 2 weeks of treatment. No treatment-related serious adverse effects were noted in CJD cases or mimics.
At the time of data collection (April 2021), 91/93 (98%) CJD cases had died; median survival was 6.0 months (range 1.3–56.9 months). Diagnoses of CJD were confirmed in all autopsied cases (n = 33). Two mimics died 11.1 and 19.8 months following symptom onset. Autopsy confirmed autoimmune encephalitis in 1 patient. The other patient died of complications of motor neuron disease; the patient's family declined autopsy.
Discussion
Readily assessable clinical features and widely available rapidly reporting tests may distinguish patients with RPD due to CJD from mimics at the time of presentation. Stereotyped motor abnormalities on neurologic examination and abnormalities on routine CSF testing (pleocytosis and elevated protein) were more common in CJD mimics in this series. Accordingly, detection of these findings should raise the prospect of a mimic, prompting consideration of empiric treatment while awaiting the results of disease-specific tests (e.g., RT-QuIC assays for prions and disease-specific autoantibodies). High-dose methylprednisolone represents a reasonable first-line empiric immunotherapy, acknowledging the high prevalence of steroid-responsive conditions in this series and others5,7 and the low risk of serious adverse effects observed in CJD cases treated with steroids.
MRI did not reliably discriminate CJD cases and mimics in this series. On the contrary, the detection of stereotyped T2-FLAIR hyperintensities and changes on diffusion-weighted imaging sequences increased diagnostic suspicion of CJD in 6/11 (55%) mimics. Although cortical ribboning and signal changes in the deep nuclei often signify CJD,14-16 these findings are also reported in patients with autoimmune encephalitis,17,18 seizures,19 venous/arterial congestion,20 selected infections,21,22 chronic medication use,23 cerebral lymphoma,6 and other disorders associated with rapid neuronal dysfunction or death.24 Clinicians should consider this differential diagnosis when interpreting MRIs in patients with subtle abnormalities, recognizing that the high pretest probability may contribute to overcalling of features suggestive of CJD in patients with RPD (Figure 1). Our results suggest that it is even more important to consider alternate diagnoses when subtle imaging findings are present in patients with early clinical features and abnormalities on routine CSF studies. When doubt persists, repeat neuroimaging is recommended, expecting that neuroimaging changes will become more apparent in patients with CJD12 and may resolve in mimics. Similar to MRI, EEG findings were not useful in discriminating CJD cases and mimics. Despite their emphasis in CJD diagnostic criteria,3,10 periodic sharp wave complexes were rarely observed in CJD cases or mimics at presentation, calling into question their diagnostic utility early in the symptomatic course.12
CJD-specific biomarkers performed as expected. RT-QuIC was negative in all mimics and positive in the overwhelming majority of cases, consistent with reported test performance.3,4,12,13 Total tau levels, while nonspecific, may discriminate between CJD cases and mimics, with 90% of cases having levels >1149 pg/mL. Notably, suprathreshold total tau was also observed in mimics with ANNA-1–associated autoimmune encephalitis and frontotemporal dementia with motor neuron disease, emphasizing the nonspecific nature of this biomarker. Protein 14-3-3 was neither sensitive nor specific for CJD, calling into question the diagnostic utility of this measure in the modern era.12
The detection of autoantibodies against neural antigens established diagnoses of definite autoimmune encephalitis in 6 mimics. Several points can be drawn from these findings. First, and most importantly, autoimmune encephalitis should be considered in all patients with disease-specific antibodies targeting cell surface neural antigens—especially antibodies against LGI1, NMDAR, and AMPAR.25,26 Second, when practical, antibodies should be measured in CSF and serum, recognizing the variable sensitivity and specificity of measures in biofluids (e.g., disease-associated NMDAR antibodies are more likely to be detected in the CSF,27 whereas LGI1 antibodies are more likely to be detected in the serum28). Third, antibody results must be interpreted in light of the clinical syndrome and prevalence in the general population,29,30 acknowledging the uncertain relevance of low levels of selected antibodies that may be detected in CJD cases.31-33
Mimics in this study were selected from prospectively accrued cohorts with RPD, applying strict criteria to identify individuals who fulfilled the diagnostic criteria for probable CJD. This approach differs from prior studies, which focused on patients referred to prion disease reference centers for evaluation of suspected CJD and included patients referred for postmortem analyses.4-7 These differences may explain the predominance of treatment-responsive CJD mimics in this series. We acknowledge that the selection criteria applied in this study may affect generalizability, recognizing that other centers may prioritize other tests when evaluating patients with suspected or possible CJD. However, by focusing on patients who met the stringent clinical criteria for probable CJD, our study results may improve diagnostic accuracy and early management in the subpopulation of patients who are at the greatest risk of misdiagnosis of CJD. Multicenter studies including greater numbers of CJD mimics are needed to increase confidence in these results.
Immune-mediated, vascular, granulomatous, and neurodegenerative diseases may mimic CJD at presentation and should be considered in patients with early motor dysfunction and abnormal CSF studies. The detection of atypical features in a patient with suspected CJD—particularly elevations in CSF leukocytes and protein—should prompt evaluation for possible mimics and consideration of empiric treatment while waiting for the results of disease-specific tests.
Appendix. Authors

Study Funding
This work was funded by NIH/NIA: K23AG064029 (G.S. Day).
Disclosure
E.B. Lazar, A.L. Porter, C.C. Prusinski, and S.R. Dunham report no conflicts. A.S. Lopez-Chiriboga has served on advisory boards for Genentech and Horizon Therapeutics. M.B. Hammami reports no conflicts. D. Dubey has received research support from Grifols Pharmaceuticals. He has consulted for UCB, Immunovant, and Astellas Pharmaceuticals. All compensation for consulting activities is paid directly to Mayo Clinic. Dr Dubey has patents pending for Kelch-like protein 11 (KLHL11) IgG and Leucine Zipper 4 (LUZP4) IgG and as markers of neurological autoimmunity. G.S. Day serves as a consultant for Parabon NanoLabs Inc, as a Topic Editor (Dementia) for DynaMed (EBSCO), and as the Clinical Director of the Anti-NMDA Receptor Encephalitis Foundation (Inc, Canada; uncompensated). He owns stock in ANI Pharmaceuticals. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
TAKE-HOME POINTS
→ Immune-mediated, vascular, granulomatous, and neurodegenerative diseases may mimic CJD at presentation.
→ Disease-specific tests for CJD and other causes of rapidly progressive dementia may take days to result; widely accessible clinical features and diagnostic tests are needed to improve early recognition of mimics.
→ Early motor dysfunction and abnormalities on routine CSF studies (leukocytosis and elevated protein) were more common in mimics than CJD cases.
→ Early recognition of discriminating features may facilitate earlier treatment of CJD mimics.
References
- 1.Day GS, Tang-Wai DF. When dementia progresses quickly: a practical approach to the diagnosis and management of rapidly progressive dementia. Neurodegenerative Dis Manag. 2014;4(1):41-56. [DOI] [PubMed] [Google Scholar]
- 2.Hermann P, Appleby B, Brandel JP, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021;20(3):235-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hermann P, Laux M, Glatzel M, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. 2018;91(4), e331-e338. [DOI] [PubMed] [Google Scholar]
- 4.Rhoads DD, Wrona A, Foutz A, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. 2020;95(8), e1017-e1026. [DOI] [PubMed] [Google Scholar]
- 5.Maat P, de Beukelaar JW, Jansen C, et al. Pathologically confirmed autoimmune encephalitis in suspected Creutzfeldt-Jakob disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(6), e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudge P, Hyare H, Green A, Collinge J, Mead S. Imaging and CSF analyses effectively distinguish CJD from its mimics. J Neurol Neurosurg Psychiatry. 2018;89(5):461-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011;70(3):437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Day GS. Rapidly progressive dementia. CONTINUUM: Lifelong Learn Neurol. 2022;28(3):901-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson N, Hermann P, Ladogana A, et al. Validation of revised international Creutzfeldt-Jakob disease surveillance network diagnostic criteria for sporadic Creutzfeldt-Jakob disease. JAMA Netw Open. 2022;5(1), e2146319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Center for Disease Control and Prevention. CDC's diagnostic criteria for Creutzfeldt-Jakob Disease (CJD), 2018 [online]. Accessed August 11, 2022. cdc.gov/prions/cjd/diagnostic-criteria.html.
- 11.Foutz A, Appleby BS, Hamlin C, et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol. 2017;81(1):79-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shir D, Lazar EB, Graff-Radford J, et al. Analysis of clinical features, diagnostic tests, and biomarkers in patients with suspected Creutzfeldt-Jakob disease, 2014-2021. JAMA Netw Open. 2022;5(8):e2225098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green AJ. RT-QuIC: a new test for sporadic CJD. Pract Neurol. 2019;19(1):49-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bizzi A, Pascuzzo R, Blevins J, et al. Evaluation of a new criterion for detecting prion disease with diffusion magnetic resonance imaging. JAMA Neurol. 2020;77(9):1141-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geschwind MD, Potter CA, Sattavat M, et al. Correlating DWI MRI with pathologic and other features of Jakob-Creutzfeldt disease. Alzheimer Dis Associated Disord. 2009;23(1):82-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol. 2008;65(10):1341-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Chen L, Yang J, Wang L, Shang H, Chen X. Anti-N-methyl-D-Aspartate receptor encephalitis mimicking sporadic Creutzfeldt-Jakob disease. Front Neurol. 2020;11:593680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budhram A, Britton JW, Liebo GB, et al. Use of diffusion-weighted imaging to distinguish seizure-related change from limbic encephalitis. J Neurol. 2020;267(11):3337-3342. [DOI] [PubMed] [Google Scholar]
- 20.Holekamp TF, Mollman ME, Murphy RKJ, et al. Dural arteriovenous fistula-induced thalamic dementia: report of 4 cases. J Neurosurg. 2016;124(6):1752-1765. [DOI] [PubMed] [Google Scholar]
- 21.Day GS, Tai P, Moharir M, Tang-Wai DF. Teaching NeuroImages: recurrent SSPE presenting as Anton syndrome with cortical ribboning. Neurology. 2015;85(19), e141-e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renard D, Nerrant E, Lechiche C. DWI and FLAIR imaging in herpes simplex encephalitis: a comparative and topographical analysis. J Neurol. 2015;262(9):2101-2105. [DOI] [PubMed] [Google Scholar]
- 23.Harsha KJ, Joshy EV, Aravinda RV, Poornima R. Chronic pregabalin abuse with subacute encephalopathy mimicking autoimmune encephalitis. Neurol India. 2021;69(6):1785-1788. [DOI] [PubMed] [Google Scholar]
- 24.Rosenbloom MH, Atri A. The evaluation of rapidly progressive dementia. The Neurologist. 2011;17(2):67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grau-Rivera O, Sanchez-Valle R, Saiz A, et al. Determination of neuronal antibodies in suspected and definite Creutzfeldt-Jakob disease. JAMA Neurol. 2014;71(1):74-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks J, Yarbrough ML, Bucelli RC, Day GS. Testing for N-methyl-d-aspartate receptor autoantibodies in clinical practice. Can J Neurol Sci. 2020;47(1):69-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadoth A, Pittock SJ, Dubey D, et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann Neurol. 2017;82(1):79-92. [DOI] [PubMed] [Google Scholar]
- 29.Kelkar P. Reader response: unintended consequences of Mayo paraneoplastic evaluations. Neurology. 2019;93(13):602. [DOI] [PubMed] [Google Scholar]
- 30.Brier MR, Bucelli RC, Day GS. An elderly man with dementia. Clin Chem. 2020;66(3):415-420. [DOI] [PubMed] [Google Scholar]
- 31.Valencia-Sanchez C, Pittock SJ, Mead-Harvey C, et al. Brain dysfunction and thyroid antibodies: autoimmune diagnosis and misdiagnosis. Brain Commun. 2021;3(2), fcaa233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebright MJ, Li SH, Reynolds E, et al. Unintended consequences of Mayo paraneoplastic evaluations. Neurology. 2018;91(22), e2057-e2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michael S, Waters P, Irani SR. Stop testing for autoantibodies to the VGKC-complex: only request LGI1 and CASPR2. Pract Neurol. 2020;20(5):377-384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized study data will be shared pending review of a request from qualified individuals by the corresponding author.