

To the Editor —
Ribosome profiling (Ribo-seq) has extended our understanding of the translational ‘vocabulary’ of the human genome, uncovering thousands of open reading frames (ORFs) within long noncoding RNAs (lncRNAs) and presumed untranslated regions (UTRs) of protein-coding genes. However, reference gene annotation projects have been circumspect in their incorporation of these ORFs because of uncertainties about their experimental reproducibility and physiological roles. Yet, it is clear that certain ‘Ribo-seq ORFs’ make stable proteins, others mediate gene regulation, and many have medical implications. Ultimately, the absence of standardized ORF annotation has created a circular problem: while Ribo-seq ORFs remain unrecognized by reference annotation databases, this lack of recognition will thwart studies examining their roles. Here, we outline a community-led effort involving Ensembl/GENCODE, the HUGO Gene Nomenclature Committee (HGNC), UniProtKB, HUPO/HPP and PeptideAtlas to produce a standardized catalog of 7,264 human Ribo-seq ORFs; a path to bring protein-level evidence for Ribo-seq ORFs into reference annotation databases; and a roadmap to facilitate research in the global community.
Ribo-seq1 provides an RNA-sequencing-based readout of mRNA translation by isolating ribosome-bound RNA fragments of ~30 nucleotides in length. Sequencing of these fragments offers genome-wide footprints of ribosome–RNA interactions, detecting translated ORFs with sub-codon resolution2–8. Although Ribo-seq circumnavigates the experimental difficulties of working with protein molecules (for example, using mass spectrometry (MS) analytical tools) and readily finds translations missed by in silico evolutionary methods, it does not demonstrate the actual existence of proteins, and most translations do not show signs of constraint as coding sequences (CDS). A wide range of ‘functional’ scenarios are therefore plausible for Ribo-seq ORFs (Table 1).
Table 1 |.
Approaches to interpreting Ribo-seq ORFs
| Possible cellular interpretation of Ribo-seq ORF translation | Comments |
|---|---|
| A Ribo-seq ORF encodes a ‘missing’ conserved protein | Ribo-seq ORFs may be recognized as canonical—in accordance with existing protein annotations—on the basis that the sequence of the proteins they encode shows clear evidence of being maintained by evolutionary selection over a significant period of evolutionary time. |
| A Ribo-seq ORF encodes a taxonomically restricted protein | Ribo-seq ORFs may encode proteins whose sequences and molecular activities are specific to one species or lineage. Evidence for selection or conservation across distant species or lineages is lacking for these ORFs, either because the protein sequence has diverged beyond recognition from its orthologs, or because the protein evolved recently from previously noncoding material and homologs do not exist in other species or lineages. |
| A Ribo-seq ORF regulates protein or RNA abundance | Ribosome engagement of regulatory ORFs does not result in a protein product under selection but regulates the abundance of a canonical protein or RNA. This paradigm is well established for uORFs and uoORFs, as noted in Table 2, though it is applicable to other transcript scenarios. Regulatory ORFs may compete for ribosomes with their downstream canonical ORFs or produce nascent peptides that stall ribosomes, leading to the controlled ‘dampening’ of protein expression. Alternative modes of action, such as the induction of RNA decay pathways, the processing of small RNA precursors or the adjustment of RNA stability, have also been inferred. |
| A Ribo-seq ORF is the result of random translation | The translation of some Ribo-seq ORFs may simply be ‘noise’. Because translation has a high bioenergetic cost, a protein that results from random translation is likely to be translated at lower levels than a canonical CDS and evolve neutrally; it may also be comparatively unstable and could be rapidly degraded. Nonetheless, it is theoretically possible that certain proteins do exist as stable ‘junk’ proteins, or that random translation events affect the expression of canonical proteins. The detection of random Ribo-seq ORFs is less likely to be reproducible. |
| A Ribo-seq ORF encodes a disease-specific protein | This protein would not be produced under normal physiological homeostasis but could be of major interest for diagnostics and therapeutics. Insights of this sort are especially emerging in cancer biology, where transcription and translation are known to be dysregulated. This leads to the production of ‘aberrant’, possibly rapidly degraded proteins that are commonly antigenic and presented on the cell surface by the HLA system, potentially acting as neoantigens. Furthermore, antigens resulting from disease-specific dysregulated ribosome activity—sometimes called defective ribosomal products (DRiPs)—have also been explored. |
Note: a given ORF may encompass several of these possibilities: for example, a translated ORF could be both regulatory and implicated in disease neoantigen production.
Several public resources already process and/or display Ribo-seq datasets, including sORFs.org9, GWIPS-viz10 and Trips-Viz11, whereas OpenProt12 and nORFs.org13 incorporate Ribo-seq into whole-translatome catalogs. Meanwhile, McGillivray et al. have produced a catalog of upstream ORFs (uORFs) with predicted biological activity14. Such efforts have made important contributions in Ribo-seq ORF interpretation. Nonetheless, the global scientific community is constrained by the absence of ‘reference’ gene annotation, which supports most large-scale genomics projects and provides the framework for human variant interpretation (Fig. 1a, Supplementary Fig. 1).
Fig. 1 |. Characterization of a consensus set of Ribo-seq ORFs for annotation by GENCODE.
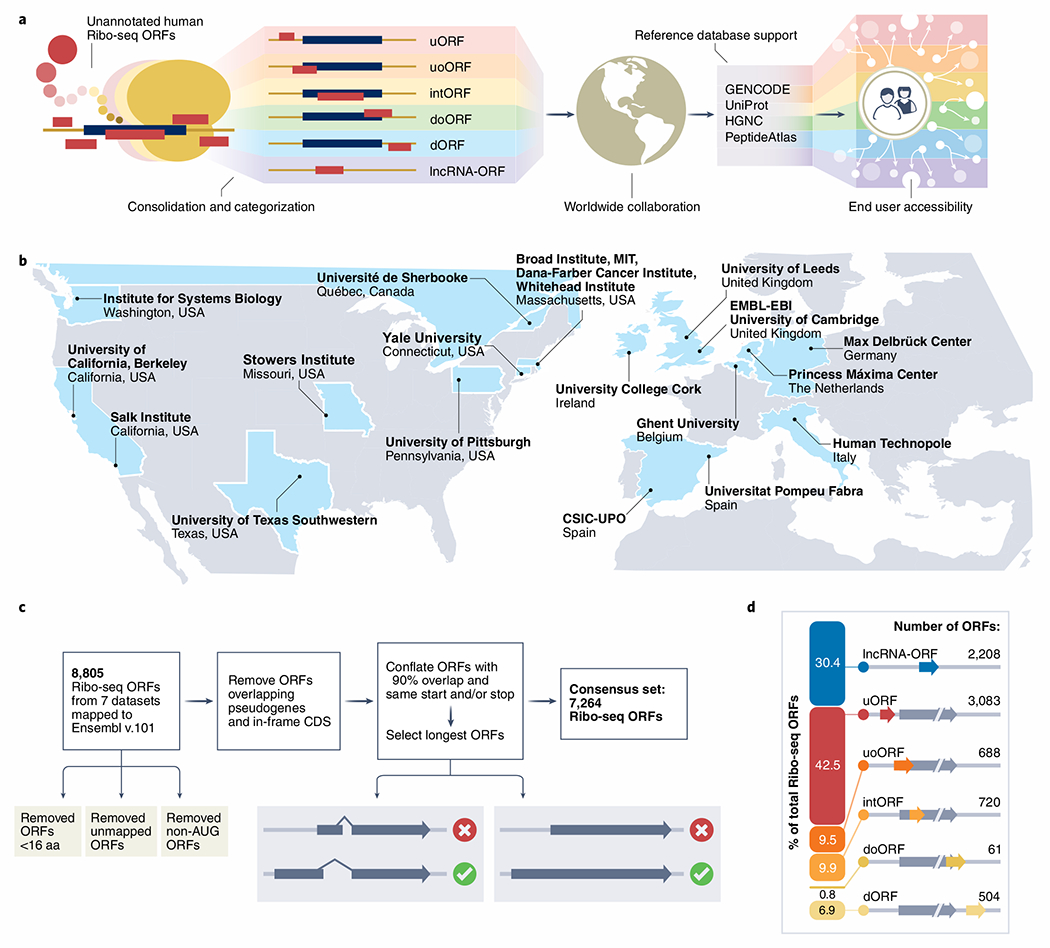
a, Schematic of the main steps and goals for this consortium effort. b, Map showing the participating institutions included in this effort. c, Schematic overview of filtering steps used to create the consensus set of ribosome profiling (Ribo-seq) ORFs. d, Diagrammatic representation of all Ribo-seq ORFs according to ORF type (see Table 2 for more information).
The creation of Ribo-seq annotations within existing reference gene and protein databases presents specific challenges that were not faced by previous cataloging efforts9–13. In particular, it is necessary to consider how these annotations can be integrated into the broad range of user workflows that are already supported by global annotation resources. For such reasons, reference annotation projects are generally conservative when it comes to the incorporation of new data types. Thus, rather than attempt to describe a ‘maximal’ set of potential Ribo-seq translations from the outset, our strategy is to build up a comprehensive resource in stages that is reciprocally improved by input from the scientific community (Fig. 1b).
Here, as ‘Phase I’ of this work, we present a consolidated catalog of Ribo-seq ORFs from seven publications2–8 annotated onto GENCODE version 35 (Fig. 1c; Supplementary Tables 1–9). A detailed description of the Ribo-seq datasets, our analysis methods and ORF characteristics is available in the Supplementary Methods. We removed ORFs smaller than 16 amino acids (aa) and those translated from non-ATG (‘near-cognate’) initiation codons, and merged redundant sense overlapping ORFs, resulting in a collated set of 7,264 unique ORFs (Fig. 1c). We classified these ORFs according to their spatial relationship with existing gene annotations (Fig. 1d), as presented in Table 2. We hope community usage of this catalog will help address the key technical and biological questions necessary to move this work into ‘Phase IT’, where we aim to create a more comprehensive resource as outlined below.
Table 2 |.
Terminology and categories of Ribo-seq ORFs
| Term | Definition | Biological role(s) |
|---|---|---|
| Ribo-seq ORF | Translated sequences identified by the Ribo-seq assay that have not already been annotated by reference annotation projects. Also known as: noncanonical ORFs, alternative ORFs (altORFs), novel ORFs (nORFs) or, when <100 amino acids in size, small ORFs (smORFs), short ORFs (sORFs). Putative encoded proteins in smORFs/sORFs are also known as: microproteins, micropeptides, short ORF-encoded polypeptides (SEPs). | See below. |
| Upstream ORFs (uORFs) | Translated sequences located within the exons of the 5’ untranslated region (UTR) of annotated protein-coding genes. | Regulate translational efficiency of the downstream canonical protein. Cellular-stress-related translation. May produce independently functional proteins. |
| Upstream overlapping ORFs (uoORFs) | Translated sequences beginning in the 5′ UTR of an annotated protein-coding gene and partially overlapping its coding sequence in a different reading frame. | Similar to uORFs. Regulate translation of their overlapping CDS, but with potentially stronger regulatory potential than uORFs. May produce independently functional proteins. |
| Downstream ORFs (dORFs) | Translated sequences located within the 3′ UTR of annotated protein-coding genes | Less commonly detected and generally poorly understood. May act as cis translational regulators. |
| Downstream overlapping ORFs (doORFs) | Translated sequences beginning in the genomic coordinates of an annotated CDS but continuing beyond the annotated CDS and terminating in the 3′ UTR of the annotated protein-coding gene. | Similar to dORFs. |
| Internal out-of-frame ORFs (intORFs) | Translated sequences located on the mRNA of an annotated protein-coding gene and completely encompassed within the canonical CDS, but translated via a different reading frame. Also known as: altCDSs, nested ORFs, dual-coding regions. |
May regulate translation similarly to uORFs in some cases. Detection of intORFs with Ribo-seq is possible but difficult due to the convolution of triplet periodicity signals from two reading frames; it largely depends on the length and translation level of the intORF relative to the overlapping canonical CDS. |
| Long noncoding RNA ORFs (lncRNA-ORFs) | Translated sequences located within transcripts currently annotated as long noncoding RNAs (lncRNAs), including long intervening/intergenic noncoding RNAs (lincRNAs), long noncoding RNAs that host small RNA species (encompassing microRNAs, snoRNAs, etc.), antisense RNAs and others. | May produce independently functional proteins. Typically lack strong sequence conservation. |
For Phase I, we investigated repeated ORF identifications between studies, observing that 3,085 of 7,264 Ribo-seq ORFs were found by more than one publication (Supplementary Fig. 2; Supplementary Tables 2 and 3). However, although such ‘reproducibility’ can demonstrate consistency in Ribo-seq signal, it neither provides insights into biological function nor indicates that the 4,179 non-replicated ORFs are ‘false’. A major goal of Phase II will be to incorporate a greater diversity of human cell types and tissues for improved estimates of ORF reproducibility, expression patterns and potential cell type specificity, along with further evaluation of criteria to quantify the technical confidence in Ribo-seq ORF calls.
Furthermore, Phase I excluded many translations by restricting the consensus set to ATG-initiated ‘cognate’ translations of at least 16 aa in length. Although these tiny ORFs may provoke skepticism in the absence of additional evidence—the smallest annotated human protein is 24 aa—there may be no lower size limit for a functional ORF15. For example, the tarsal-less (tal) gene produces a polycistronic transcript translated into proteins as short as 11 aa in several insect species16. Furthermore, the inclusion of ORFs initiated with near-cognate start codons can be complicated by ambiguous predictions of initiation site positions17. Ribo-seq following treatment with lactimidomycin or homoharringtonine, which inhibit translation elongation and result in accumulation of sequencing reads at the putative start sites, can help to identify near-cognate start sites17,18. Such datasets will be leveraged by our future Phase II efforts. For our current annotation resource, we have separately aggregated the Ribo-seq ORFs with near-cognate start codons or translations shorter than 16 codons (Supplementary Fig. 3a–c and Supplementary Tables 4 and 5), rather than including them in the Phase I catalog.
A core aim of Phase II will be to identify which Ribo-seq ORFs participate in cell physiology and how they do so. One aspect is distinguishing between cellular function mediated by a stable protein and functionality imparted at the level of translation itself. We here use ‘protein’ as an umbrella term for protein, peptide and polypeptide, although we recognize that the terms polypeptide, micropeptide or microprotein are commonly used for small protein molecules (Table 2). Because of the challenges of protein sequencing, evolutionary analysis has played a major historical role in ORF annotation, which is based on the assumption that the evolution of translated sequences is driven by selection at the protein level. Within our Phase I dataset, 75 Phase I replicated Ribo-seq ORFs (2.4%) present evidence of potential protein-level constraint as measured by PhyloCSF19 (Supplementary Fig. 3d–f); among these, ten have now been classified as protein coding by GENCODE (Supplementary Table 6).
Nonetheless, the evolutionary profile of many Phase I Ribo-seq ORFs remains hard to interpret. In part, this is because distinguishing ORF selection at the protein and DNA levels can be especially difficult for very small regions, and Ribo-seq ORFs are typically much smaller than those of known annotated proteins (Supplementary Fig. 3g–j). A second drawback is that evolutionary analysis cannot infer the protein-coding or regulatory potential of evolutionarily ‘young’ de novo Ribo-seq ORFs20. Reference annotation projects remain skeptical about the existence of proteins that are not deeply conserved, despite the fact that some young proteins clearly do participate in cellular physiology20,21. Furthermore, there is a substantial knowledge gap in regard to the mode and tempo of regulatory ORF evolution. Here, genetic variation within human populations may provide insights. For example, Whiffin et al.22 recently used the gnomAD human variation dataset to identify 3,191 genes in which uORF-perturbing variants are likely to be deleterious, thereby inferring the physiological importance of these translations. Meanwhile, Neville et al.23 used the same dataset to find aggregate evidence of selective pressure against deleterious variants in their nORFs.org catalog13, which is especially pronounced for STOP-gain variants in uORFs. In prostate cancer, a recent analysis of 5′ UTR variants found regulatory roles for several uORFs23.
Although Ribo-seq ORFs may have regulatory roles irrespective of an encoded protein, the first step in confirming a protein-level physiological role for such an ORF is to demonstrate the existence of the protein in the cell. MS is a widely accepted approach to catalog the proteome, and its utility will be an important area of investigation for Phase II. At present, 609 of 7,264 Ribo-seq ORFs have been reported to have support in published MS datasets (Supplementary Table 10). However, different groups use distinct methodologies and parameters for MS, and for Phase I these findings are simply reported in Supplementary Tables 2 and 3 without further investigation. Reference annotation projects have historically favored high-stringency MS approaches, and the Human Proteome Organization (HUPO)/Human Proteome Project (HPP)—which aims to produce a full annotation of the human proteome—has published guidelines to standardize the nature of MS evidence required to annotate a human protein24. As one facet of our development of an MS workflow, these Ribo-seq ORFs have been added to the PeptideAtlas analytical pipeline, which is used by HUPO. In Phase II, our projects will jointly examine the question of how best to use MS data to define which Ribo-seq ORFs produce proteins. For reference annotation, we see two aspects to this: first, how to set standards for accepting and reporting potential MS support for a prospective Ribo-seq ORF protein; and second, how to define the point at which the body of evidence supports protein-coding annotation.
These aspects are illustrated by a preliminary analysis, which took advantage of the fact that 333 of our Ribo-seq ORFs are present in sequences previously queried by the PeptideAtlas workflow (Supplementary Methods). We find single-mapping peptide-spectrum matches (PSMs) for 13 Ribo-seq ORFs (Supplementary Table 11); all but one are supported by a single PSM each, whereas most of the peptides identified are not fully tryptic (two examples are presented in Supplementary Fig. 4). The majority of observed PSMs derive from human leukocyte antigen (HLA) peptidome datasets, which is consistent with prior proteomic analyses demonstrating enrichment for peptides mapping to Ribo-seq ORFs in immunopeptidome data25–27. We emphasize that this preliminary analysis was not a full remapping of MS data and involved only a fraction of the Ribo-seq ORFs; a larger, focused effort will be forthcoming.
There are multiple causes contributing to the fact that Ribo-seq ORFs and certain classes of canonical proteins are infrequently detected in MS data, which are summarized elsewhere28. One consideration for HUPO is that an MS-based ‘canonical’ protein assignment requires multiple PSMs, ideally based on non-overlapping tryptic peptides. Although we recognize the value of these guidelines, very small proteins may be ‘less discoverable’ by MS, especially due to a paucity of identifiable tryptic fragments28. Notably, nearly 1,500 protein-coding genes annotated by GENCODE, UniProt and HGNC do not presently have MS support recognized by HUPO24. Moving forward, we are committed to examining all potential protein-coding Ribo-seq ORF cases with full manual gene annotation processes, and we plan to expand this workflow to include manual analysis of the peptide spectra by PeptideAtlas.
Although the value of MS in identifying translated proteins is indisputable, we believe a broader ‘gold standard’ for evidence should employ additional methodologies, such as epitope tagging combined with western blot imaging or endogenous antibody work; HUPO already incorporates such data in collaboration with the Human Protein Atlas24. Consideration also needs to be given to emerging proteomics technologies, such as targeted proteomics workflows and immunopeptidomics, and progress is being made in medium-throughput functional screening assays. For example, recent large-scale studies have translated hundreds of Ribo-seq ORFs in mammalian cells through exogenous expression, finding that nearly 50% may stably produce proteins, despite little evidence of evolutionary constraint2,6,27.
In addition to their evaluation as proteins or regulatory units, the reference annotation of Ribo-seq ORFs necessitates the creation of integrated workflows to interpret overlapping variants, and notwithstanding great community interest in this field, standardized approaches are not yet available. We emphasize that variant interpretation pipelines designed to classify CDS mutations may be unsuitable for Ribo-seq ORFs (Table 1), and that a minority of overlapping variants fall within sequences displaying amino-acid-level constraint. Neville et al.13 found that their nORFs.org catalog contains 48 Human Gene Mutation Database or ClinVar variants that are already considered pathogenic or likely to be pathogenic, even though they do not disrupt annotated CDSs. Although these variants may affect noncanonical ORFs, it will be important to define their mechanisms of action through experimental studies, as alternative explanations for pathogenicity, such as the creation of cryptic splice sites, are supported in certain cases. After exclusion of variants in Ribo-seq ORFs that overlap annotated CDSs, a total of 1,142 single-nucleotide variants present in the ClinVar database29 were located within our aggregated set of Phase I Ribo-Seq ORFs (Supplementary Methods). Fewer than 2% of these variants have been classified as pathogenic or likely to be pathogenic, but this is likely to be an underestimate because the absence of pathogenesis is commonly inferred from the absence of overlap with known coding features, and because ClinVar variant coverage is heavily skewed toward annotated CDSs.
Furthermore, there is major interest in the application of Ribo-seq to study human disease. In particular, it is being widely used to explore the dynamics of translation in cancer cells with aberrant proteins as diagnostic markers or targets for immunotherapy25,26,30. At present, reference annotation projects do not attempt to distinguish aberrant translation events from those that contribute to ‘normal’ physiology. It will be important to deduce the fraction of Ribo-seq ORFs that encode proteins that exist in normal cellular conditions. Conversely, we envisage the value of classifying potentially aberrant translations within Phase II through a distinct annotation framework.
Our intention is for the Ribo-seq Phase I catalog to be seen as a pragmatic interim solution to a long-term problem. We believe that reference annotation databases can advance both scientific and clinical research through the propagation and standardization of Ribo-seq ORF datasets, even—and perhaps especially—while the phenotypic impact of these features remains uncertain. As biological knowledge improves, this will support the development of more accurate annotations and variant interpretations, with the potential to yield substantial insights across all aspects of human biology. In this spirit, we hope the results of Phase I of this project will be useful and beneficial to the community and invite interested labs to join our future Phase II efforts.
Endorsement
HUPO/HPP Executive Committee members affirming support for this work are Rudolf Aebersold, Cecilia Lindskog Bergström, Yu-Ju Chen, Fernando Corrales, Lydie Lane, Siqi Liu, Edward Nice, Gilbert Omenn, Christopher Overall, Young-Ki Paik, Charles Pineau, Michael Roehrl and Susan Weintraub. This work is further endorsed by Piero Carninci from Human Technopole and RIKEN.
Supplementary Material
Acknowledgements
A.F., J.M.M., F.C. and P.F. are supported by the Wellcome Trust (grant number 108749/Z/15/Z), the National Human Genome Research Institute (NHGRI) of the US National Institutes of Health (NIH) under award number 2U41HG007234 and the European Molecular Biology Laboratory (EMBL). For the purpose of open access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Ensembl is a registered trademark of EMBL. M.G. and Y.T.Y. are supported by NHGRI, NIH, under award number 2U41HG007234. I.J. and M.K. are supported by National Human Genome Research Institute of the NIH under award numbers 2U41HG007234 and R01 HG004037. UniProt is supported by the NHGRI, NIH, under award number (U24HG007822), EMBL core funds and the Swiss Federal Government through the State Secretariat for Education, Research and Innovation (SERI). J.R.P. is supported by the Harvard K-12 in Central Nervous System tumors (5K12 CA 90354-18), the Alex’s Lemonade Stand Foundation Young Investigator Award (no. 21-23983) and the Musella Foundation for Brain Tumor Research. T.F.M. is supported by the NIH under award number F32GM123685. M.M.A. acknowledges funding from the Spanish Government grant PGC2018-094091-B-I00 (MCI/AEI/FEDER, EU) and AGAUR grant 2017SGR01020. J.C. is supported by the NIH Pathway to Independence Award (R00 GM134154) and the Cancer Prevent and Research Institute of Texas (RR200095). J.S.W. is supported by the Howard Hughes Medical Institute. A.A.B. is supported by the Stowers Institute for Medical Research and the NIH (R01 GM136849). A.-R.C. is supported by funds provided by the Searle Scholars program, the Sloan Research Fellowship in Computational and Evolutionary Molecular Biology and the National Institute of General Medical Sciences, NIH, award number DP2GM137422. P.V.B. wishes to acknowledge the support from the Investigator in Science Award (grant number 210692/Z/18/Z) by SFI-HRB-Wellcome Trust Biomedical Research Partnership and from Russian Science Foundation (grant number 20-14-00121). N.H. is the recipient of a European Research Council advanced grant under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. AdG788970). N.H. is supported by a grant from the Leducq Foundation (11 CVD-01). The work of the HGNC is funded by the Wellcome Trust (208349/Z/17/Z) and the NHGRI, NIH (under award number U24HG003345). X.R. and M.A.B. are funded by the Canadian Institutes of Health Research, grant PJT-175322. X.R. is funded as a Canada Research Chair in functional proteomics and discovery of novel proteins. N.T.I. is supported by the NIH under award number R01 GM130996. R.L.M. and E.W.D. are supported by NIH grants R01GM087221, R24GM127667, U19AG023122, 1S10OD026936-01 and US National Science Foundation grant DBI-1933311. J.L.A. is supported by the Medical Research Council (MR/N000471/1). M.A.B. is supported by a Junior 1 fellowship from the Fonds de Recherche du Quebéc-Santé.
Footnotes
Competing interests
P.V.B. is a co-founder of RiboMaps Ltd., which provides Ribo-seq analysis as a commercial service, including identification of translated ORFs. A.R.C. is a member of the scientific advisory board for Flagship Labs 69, Inc. P.F. is a member of the scientific advisory boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01369-0.
References
- 1.Ingolia NT, Ghaemmaghami S, Newman JRS & Weissman JS Science 324,218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Heesch S et al. Cell 178, 242–260.e29 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Ji Z, Song R, Regev A, & Struhl K eLife 4, e08890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cahviello L et al. Nat. Methods 13, 165–170 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Martinez TF et al. Nat. Chem. Biol 16, 458–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J et al. Science 367,1140–1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaertner B et al. eLife 9, e58659 (2020).32744504 [Google Scholar]
- 8.Raj A et al. eLife 5, e13328 (2016).27232982 [Google Scholar]
- 9.Olexiouk V, Van Criekinge W & Menschaert G Nucleic Acids Res. 46, D497–D502 (2018). D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michel AM, Kiniry SJ, O’Connor PBF, Mullan JP &Barancv PV Nucleic Acids Res. 46, D823–D830 (2018). D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiniry SJ, O’Connor PBF, Michel AM & Baranov PV Nucleic Acids Res. 47, D847–D852 (2019). D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunet MA et al. Nucleic Acids Res. 47, D403–D410 (2019). D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neville MDC et al. Genome Res. 31, 327–336 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGillrvray P et al. Nucleic Acids Res. 46, 3326–3338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vattem KM & Wek RC Proc. Natl Acad. Sci. USA 101, 11269–11274 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galindo MI, Pueyo JI, Fouix S, Bishop SA & Couso JP PLoS Biol. 5, e106 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S et al. Proc. Natl Acad. Sci. USA 109, E2424–E2432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingolia NT, Lareau LF & Weissman JS Cell 147, 789–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin MF, Jungreis I & Kellis M Bioinformatics 27, i275–i282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy A Nature 574, 314–316 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Ruiz-Orera J & Albà MM Trends Genet. 35,186–198 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Whiffin N et al. Nat. Commun 11, 1 (2020).31911652 [Google Scholar]
- 23.Lim Y et al. Nat. Commun 12,4217 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adhikari S et al. Nat. Commun 11, 5301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouspenskaia T et al. Nat. Biotechnol 40, 209–217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laumont CM et al. Sci. Transl. Med 10, eaau5516 (2018).30518613 [Google Scholar]
- 27.Prensner JR et al. Nat. Biotechnol 39, 697–704 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omenn GS, Lane L, Lundberg EK, Overall CM & Deutsch EW J. Proteome Res 16,4281–4287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landrum MJ et al. Nucleic Acids Res. 48, D835–D844 (2020). D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chong C et al. Nat. Commun 11,1293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.