

Abstract
Circular RNAs are formed in all domains of life and via different mechanisms. There has been an explosion in the number of circRNA papers in recent years; however, as a relative young field, circRNA biology has an urgent need for common experimental standards for isolating, analysing, expressing and depleting circRNAs. Here we propose a set of guidelines for circRNA studies based on the authorś experience. This perspective will specifically address the major class of circular RNAs in Eukarya that are generated by a spliceosome-catalyzed back-splicing event. We hope that the implementation of best practice principles for circRNA research will help move the field forward and allow a better functional understanding of this fascinating group of RNAs.
1. Introduction
Covalently closed circular RNA molecules (circRNAs) were first reported as virion replication intermediates1,2, side products from self-splicing introns3, a product of RNA splicing in Archaea4 or unusual splicing products in eukarya5,6 but for many years, most circRNAs were thought to be rare and reflect errors7. This changed with the emergence of high-throughput sequencing and the discovery that circRNAs can accumulate to levels exceeding mRNAs and show tissue- and development-specific expression8–10. We are now beginning to understand circRNA biogenesis and function but there are many experimental difficulties specifically associated with circRNA research. In the following, we discuss the experimental challenges of studying circRNAs and propose a set of guidelines based on the authors’ experience. The focus is on circRNAs generated by spliceosome-mediated back-splicing since they constitute the largest and most well-studied group to date. Other types of circRNAs, such as intron-derived nuclear circRNAs, may require different experimental approaches. For a more comprehensive view of the circRNA field, we refer to other excellent reviews in the literature11–15. In addition, two recent reviews provide further experimental guidelines for circRNA research16,17.
Most circRNAs are generated co-transcriptionally in a spliceosome-dependent process known as back-splicing (Fig. 1A). During back-splicing, a 5’ splice site attaches to an upstream 3’ splice site on the same pre-mRNA, circularising the intervening exon(s). CircRNAs are typically formed by exons also found in the cognate linear RNA, meaning that the splice junction generated during back-splicing (the Back-Splice Junction or BSJ) is the only sequence that allows unambiguous identification of the circular form. Furthermore, the circRNA and linear RNA are normally co-expressed, which complicates the analysis. CircRNA biogenesis is not fully understood but base-pairing between inverted repeats (e.g. Alu-elements) in the flanking introns and/or association of specific RNA-binding proteins (RBPs) that bring the relevant splice sites into proximity have been shown to drive back-splicing (Fig. 1A, reviewed in11,13,14). Unlike transcripts arising from incomplete splicing, BSJ-containing circRNAs are usually exported to the cytoplasm. The absence of free ends renders circRNAs resistant to exonucleases18,19 and allows some circRNAs to accumulate to levels that may exceed the linear form9,18,20.
Figure 1: Overview of circRNA biogenesis and function.
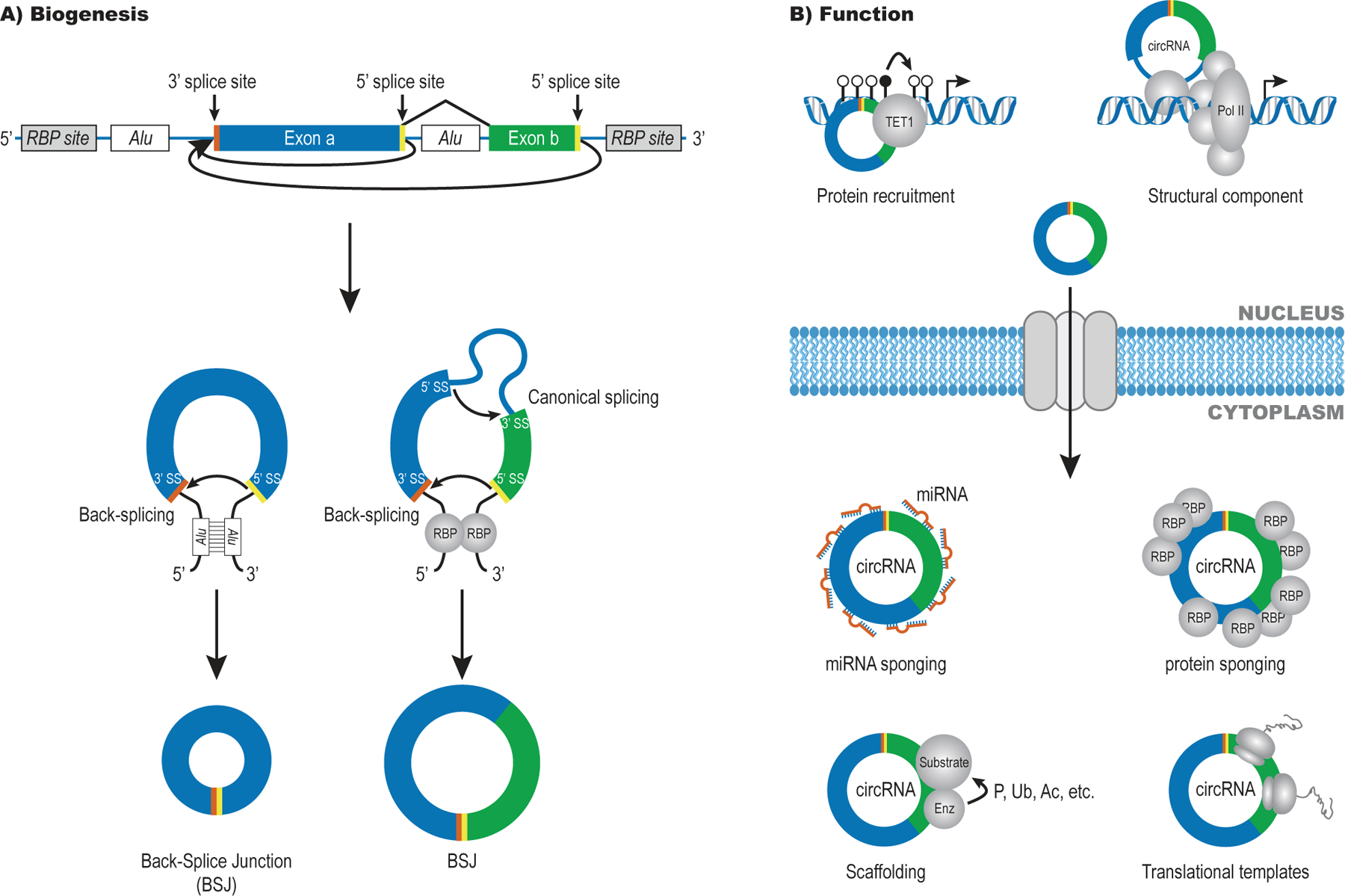
A) Spliceosome-dependent circRNAs are formed by back-splicing of one or more exons that can also undergo linear splicing. The splice sites used to form the circRNA-specific Back-Splice Junction (BSJ) are labelled in yellow (5’SS) and orange (3’SS).
B) Nuclear circRNAs have been linked to transcriptional regulation via protein recruitment or as structural components. Cytoplasmic circRNAs are thought to act in a number of ways including as sponges for miRNAs and/or proteins, as scaffolds for complex assembly that may facilitate post-translational modification (e.g., phosphorylation (P), ubiquitylation (Ub) and acetylation (Ac)) and as templates for translation.
Functional studies on circRNAs are challenging due to the difficulty in depleting or generating the circular form without affecting the linear counterpart. One of the most well-described circRNAs is CDR1as/ciRS-7, a circRNA originating from a locus that produces only negligible amounts of linear RNA20,21. CDR1as is highly expressed in the brain, bound by miR-7 and miR-671 and forms part of a regulatory network of non-coding RNAs8,9. Deletion of the CDR1as locus in mice led to deregulation of miR-7 and miR-671 and resulted in impaired sensorimotor gating22. In addition to miRNA sponging effects, circRNAs can serve as scaffolds for protein binding, act as templates for translation and regulate transcription (Fig. 1B, reviewed in 11,13).
There are currently no approved HUGO symbols for circRNAs but a recent guideline review recommends to use circ[gene symbol]-n, where n is a five-digit number listing the order of discovery for circRNAs originating from the same host gene23. Several circRNA databases are available online24–26 and we strongly encourage everyone to use them both for newly identified and known circRNAs. Furthermore, circRNA names should be supplemented with specific coordinates and reference genome used.
2. Purification and profiling of circRNAs
CircRNAs exist in a wide range of cell types, tissues and biofluids and can be isolated via standard RNA preparation protocols. The high stability of circRNAs may increase their yield relative to co-expressed linear RNAs when working with partially degraded samples or post-mitotic cells. On the other hand, many circRNAs are expressed at low levels and require more input material for robust detection. This seems to be particularly relevant for rapidly proliferating cells where circRNAs have less time to accumulate to detectable levels27.
We recommend that circRNA profiling always starts from a total RNA pool (Table 1). The covalently closed circRNAs lack poly(A)-tails and are therefore depleted in poly(A)-enriched samples, traditionally used for mRNA sequencing. Implementing circRNA-enrichment steps can increase read coverage for low-abundance circRNAs but removal of linear RNAs makes it difficult to assess if circRNA expression changes are independent of their linear host gene. For experiments that involve high throughput RNA-sequencing (RNA-seq), depletion of rRNA is preferred as it increases the proportion of reads that map to circRNAs.
Table 1.
Recommended best practices in circRNA research
| Recommendation | Rationale |
|---|---|
| Purification and profiling of circRNAs | |
| Start with a pool of total RNA. | CircRNAs lack poly(A)-tails and will mostly be absent in poly(A)-enriched samples traditionally used for mRNA sequencing. |
| Assess the quantity, quality and purity of the samples, e.g., using Bioanalyzer or NanoDrop. | CircRNA profiling methods have different requirements for the quantity, quality and purity of RNA8,9,29,30,32,34. |
| Consider enriching for circRNA content using: | Many circRNAs are expressed at low levels and may be difficult to detect in the presence of an excess of linear RNAs. It may therefore be beneficial to enrich the circRNA content of samples before analysis, although this precludes estimates of circ-to-linear ratio. |
Optimize RNase R treatment conditions using:
|
RNase R treatment will preferentially degrade linear RNAs while leaving most circRNAs intact. However, depending on the conditions circRNAs may also be degraded (long incubation times) and linear RNAs may escape degradation (short incubation times)18,30,33. |
| Estimate the proportions of different cell types present when extracting RNA from complex tissues (e.g., using marker genes). | When performing circRNA expression profiling in bulk tissue it can be helpful to know what cell types contribute to the signal. This is particularly important for conclusions about circRNA co-expression with other RNAs91. |
| Consider sequencing depth and read lengths when performing RNA-seq experiments. | CircRNAs are quantified based on reads spanning the BSJs as opposed to linear transcripts that can be quantified based on reads that align throughout8–10. Since many circRNAs are lowly expressed, relatively deep sequencing data is needed for reliable quantification12,40. |
| Use more than one bioinformatic algorithm for circRNA quantification in RNA-seq data, e.g., find_circ, CIRI2, CIRCexplorer2, circRNA_finder. | Bioinformatic algorithms for circRNA quantification are prone to false positive results. Using the overlay between more than one algorithm can significantly reduce the number of false positive circRNAs40. |
| CircRNA validation and quantification | |
| Validate new or poorly characterized circRNAs using: | Enzymatic procedures used during library preparation for RNA-seq experiments may produce false positive BSJs, e.g., by template switching44. Therefore, additional validation of the circular structure is recommended. |
Validate the specificity of RT-qPCR primers, e.g., using:
|
Divergent primers used to detect circRNAs may also detect cDNA concatemers and alternative circRNA isoforms. |
| Confer with circRNA databases (e.g., circ_Base, circRNADB and CircInteractome), and list genomic coordinates for new circRNAs. | Avoid reporting known circRNAs as new ones23. |
| Detecting circRNAs by microscopy | |
| Treat fixed cells with protease prior to probe hybridization. | The BSJ needs to be accessible for base-pairing during circRNA-ISH. Association with RNA-binding proteins (RBPs) or packaging in RNA granules may prevent probe binding and cause an absence of signal49. |
| Include positive and negative controls. | Weak signal or partial probe complementarity to abundant, non-related RNAs may skew the analysis. Imaging using cells where the circRNA has been depleted can support probe specificity. |
| CircRNA inhibition | |
| Use more than one siRNA to target the BSJ and at least one non-targeting control siRNA. | Reduce the risk of off-target effects (miRNA-like effects on linear form) and non-specific effects (on unrelated transcripts). |
| Consider regulatory regions in the host gene when designing guide RNAs for Cas9-dependent knockout. | Large-scale genomic deletions can affect host gene transcription and processing in a circRNA-independent manner. |
| Validate that the observed effects are circRNA-dependent, e.g., by expressing an siRNA-resistant form of the circRNA from a plasmid. | A partial overlap between a BSJ-targeting siRNA and the linear form of the RNA may lead to miRNA-like repressive effects that are independent of the circRNA (Kjems lab, unpublished observation). |
| CircRNA over-expression | |
Validate that in vitro transcribed circRNAs
|
In vitro transcription can lead to formation of dsRNA and linear RNA carrying 5’ triphosphates70. Both RNA types are known to trigger an innate immune response that may confound the effects seen in cells transfected with circRNA. |
Validate circRNA expression from plasmid
|
CircRNA-expressing vectors often produce both linear and circular forms. Many circRNAs are expressed at low levels and expression from plasmid may reach unphysiological amounts. Read-through transcription may lead to concatemer formation and generate BSJ-like sequences (false positive)96. |
| Determining the regulatory mechanism of circRNAs | |
| Quantify IP-efficiency as % of total input, not as fold-enrichment relative to a non-specific IP control. | This approach allows an estimate of the fraction of a particular circRNA that associates with a given RBP82. |
| For IP-MS and RAP-MS: make sure the circRNA of interest is expressed at sufficiently high levels to support detection of interaction partners. | The extensive sequence overlap between circular and linear RNA means that many RBPs will bind both. IP-MS of a circRNA with low expression will therefore be prone to contamination from proteins binding the linear form. |
| Include negative controls, e.g., cell line/tissue without expression of the circRNA of interest. | RNA and proteins are prone to non-specific interactions due to their charged surfaces. Pulldown studies should therefore ideally include a comparison to a sample that lacks the circRNA or protein of interest. |
| Consider endogenous expression levels when testing a circRNA sponge function (miRNA and protein). | Over-expression of circRNA or putative sponge targets can lead to non-physiological effects88,106. It is therefore important to show that a model for sponge-based regulation is relevant at endogenous expression levels. See Box for more information about circRNA sponging. |
For circRNA translation, it is important to include the following controls:
|
CircRNA expression from plasmids is an error-prone process and it is therefore important to rule out translation from contaminating linear RNA. In addition, MS is a very sensitive technique that can lead to false-positive validations unless a False-Discovery Rate is set in advance100. See Table 2 for more information about assays for circRNA translation. |
circRNA enrichment
The most widely used strategy for circRNA enrichment is RNase R treatment (Table 1). This 3’–5’ exonuclease preferentially degrades linear RNAs while leaving most circRNAs intact28. However, highly structured RNA (e.g., G-quadruplexes) or lack of 3’ overhangs can block RNase R activity, meaning that exonuclease resistance cannot unequivocally establish circularity. This problem can be reduced by including Li+ in the RNase R reaction buffer, since this destabilises RNA secondary structure and increases the clearance of linear RNA29. Furthermore, including a polyadenylation step prior to RNase R treatment stimulates degradation of structured RNAs by providing a 3’-overhang for the RNase to attack29. In addition to the risk of false positives from undegraded linear RNAs, false negatives can arise if circRNAs are nicked during purification. This seems to be particularly relevant for larger circRNAs (such as circZNF9130 and CDR1as18) and upon prolonged incubation times. Using different concentrations or batches of RNase R may also affect the number of false positive and negatives. Another way to enrich the circRNA content in a sample is to perform poly(A) counter-selection (RPAD) using immobilised oligo(dT)-primers that deplete linear, polyadenylated mRNAs from a sample of interest31; however, this may affect circRNAs containing internal A-stretches such as CDR1as32.
Global circRNA detection
CircRNA profiling is typically based on high throughput methods such as RNA-seq or microarrays using total or rRNA-depleted RNA as input since circRNAs are depleted in poly-A enriched fractions. Microarrays for circRNAs are designed with BSJ-specific probes; however, it is important to confer with the manufacturer for the extent and identity of the circRNAs detected since database annotations change frequently. Moreover, only a single probe evaluates the expression of individual circRNAs and therefore microarray-based discoveries require stringent validation.
For RNA-seq, downstream computational analysis identifies and counts the BSJ-spanning reads. Template switching and ligation artefacts produced during library preparation12 as well as gene duplications and trans-splicing33,34 have the potential to generate false positive BSJs and validation using complementary techniques is therefore important, particularly for novel circRNA candidates (Table 1). One recommended method is to perform RNA-seq with or without RNase R treatment and look for an enrichment in BSJ-reads per million total reads as well as an enrichment of the circRNA of interest. In addition, the relatively low abundance of many circRNAs means that quantification based on RNA-seq data becomes unreliable for small or very dilute samples. This is particularly important when comparing circRNA expression across samples as a low signal-to-noise ratio and variation in cell populations can strongly affect the identification of differentially expressed circRNAs. For accurate quantification of circRNA expression levels by RNA-seq, beyond the subset of the most abundant circRNAs, it is important to perform a relatively deep sequencing. Using longer reads (>100 nt) will also increase the total number of BSJ-mapping reads (Table 1).
CircRNA identification requires sequencing reads to overlap the BSJ and it is therefore difficult to define the internal composition of multi-exon circRNAs using short-read next-generation sequencing (NGS), although some studies have attempted to do this using paired-end sequencing to cover larger parts of the circRNA in BSJ-spanning reads35,36. More recently, nanopore-based long-read sequencing been used to reveal the full determination of internal circRNA complexity32,37,38. These studies use different approaches to enrich and linearise the circRNAs to allow passage through the nanopore and find extensive alternative splicing in circRNA-producing genes, leading to numerous circRNA isoforms with different exon composition and occasionally retained introns. For more detail about circRNA sequencing using long-read sequencing, we refer the reader to a recent review39. The existence of circRNA isoforms should be considered when designing probes or primers for validation of multi-exon circRNAs.
Bioinformatic analysis
Bioinformatic pipelines for annotating circRNAs from short-read RNA-seq data rely on mapping reads that overlap the unique BSJ40. Some pipelines (e.g. find_circ) split non-continuously mapping reads and align the two anchor sequences independently to the genome9. Others (e.g., circexplorer and DCC) use mapping algorithms like hisat2 and STAR to align subregions of the reads and subsequently retrieve the sequence across the BSJ41. Yet others (e.g., CIRI2) use multiple seed matches to derive circRNA candidates42. In some cases, circRNA detection pipelines require existing gene annotation to guide true BSJ-spanning reads, while others are fully de novo. The former approach typically performs with higher accuracy, while the latter can detect circRNAs derived from unannotated splice sites, such as ciRS-7/CDR1as. Prediction pipelines with de novo gene annotation may be particularly helpful when studying circRNAs in organisms with poorly annotated genomes, while this is less of a concern for human and mouse circRNAs. The different approaches employed by circRNA detection pipelines mean that many circRNA candidates are only called by a single pipeline. To reduce the number of false positive candidates we suggest to use a combination of at least two detection pipelines (e.g., CIRI2 and find_circ) when identifying circRNAs from sequencing data40 (Table 1). Comparing the number of BSJ-containing reads with reads spanning the adjacent splice junctions in linear RNA provides a circ-to-linear RNA ratio that can be used for quantitative comparisons.
3. CircRNA validation and quantification
PCR-based methods
The extensive similarity between circRNAs and their linear counterparts makes experimental validation of circRNA expression crucial. Among the most popular methods are reverse transcription quantitative PCR (RT-qPCR) and RT-PCR followed by Sanger sequencing and/or gel electrophoresis using a divergent primer pair to generate BSJ-spanning amplicons (Fig. 2A). RT-qPCR requires only small amounts of input RNA and analysis of linear splice junctions can be run in parallel to determine the circ-to-linear ratio. Alternatively, semi-quantitative RT-PCR can be used to directly amplify the linear and circular isoforms independently in a single reaction, as widely done in the field of alternative splicing43. This involves the design of three primers: a pair to amplify the BSJ and an additional primer in one of the neighbouring exons, which will serve to amplify the linear form (Fig. 2A). For loci that produce more than one unique circRNA, multiple amplicons may be produced from a single primer pair and it is therefore important to pay attention to the melting curves and ideally run the RT-qPCR product on a gel (Table 1). Placing one of the primers on the BSJ may help to prevent this problem. For circRNAs that undergo alternative splicing, it may be necessary to design additional primer pairs to differentiate between circRNA isoforms that share a BSJ.
Figure 2: CircRNA detection, validation and quantification.
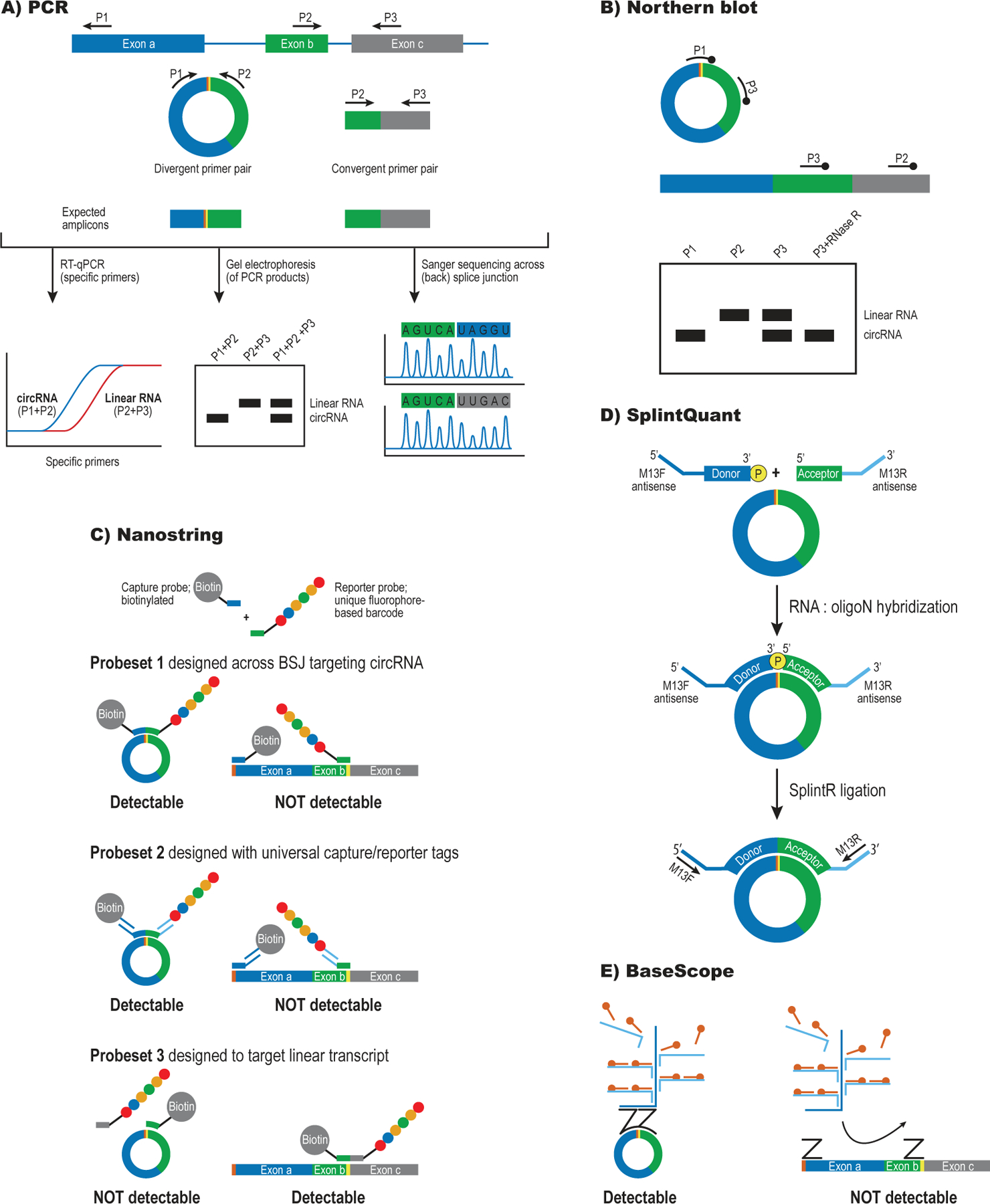
A) RT-PCR-based strategies can detect circular and linear RNAs from the same locus using divergent and convergent primer pairs. Sanger sequencing of the PCR product can confirm BSJ formation (marked in yellow and orange)
B) Hybridization-based detection of circRNA by Northern blot. Probes recognize only the circRNA (P1), only the linear RNA (P2) or both forms simultaneously (P3). The inclusion of an RNase R-treated sample can help identify the band that corresponds to the circRNA.
C) Direct circRNA detection using nanoString. This technique uses a capture probe and reporter probe that jointly recognize the BSJ to detect individual circRNAs. The amount of reporter probe is quantified using digital counting, thus avoiding an RT-step that may introduce bias. Probe sets can be designed for the circular (1,2) or linear form (3) of a given RNA.
D) Splint-Quant is an RT-free technique to quantify circRNAs. The two halves of the BSJ are recognized by donor and acceptor probes that can only be ligated and amplified upon binding to the correct target sequence.
E) BaseScope uses Z probes that bind on the two halves of the BSJ. The probes need to bind their target in close proximity to each other in order to form a docking site for a branched DNA strand that can amplify the signal to the levels required for detection.
An important concern for circRNA detection is that RNA-seq and PCR-based methods include a reverse transcription step. Most Reverse Transcriptases (RTases) exhibit template-switching activity and this is most likely the biggest contributor to false positive identification of circRNAs44 (Table 1). The RTase may also form cDNA concatemers via rolling circle amplification, especially for small circRNAs, and thus over-estimate circRNA abundance30,34. This can be avoided by supplementing PCR-based detection with methods based on direct hybridization (see below). Another possible source of false positives is trans-splicing, which may create linear mRNAs carrying concatemeric exon repeats that form junctions resembling those formed by back-splicing45. Such events are rare for endogenously expressed genes in mammals46, though, and including an RNase R treatment step can help exclude detection of false BSJs in linear RNA (Table 1).
Hybridization-based methods
Hybridization-based methods have the advantage of detecting circRNAs without an intervening RT step but often require high circRNA expression or large amounts of starting material. Northern blots can detect the circular and linear forms simultaneously due to their different migration on a gel and provide direct visualisation of circRNA resistance to RNase R (Fig. 2B). The probes used should target the BSJ and/or an internal exon sequence (shared between circular and linear RNA). Further circRNA validation can be done by incubating the RNA sample with RNase H in the presence of a DNA probe complementary to the circularised exon(s). This reaction cleaves the RNA specifically within the DNA-RNA hybrid region and shifts the mobility of the circRNA on a gel.
The nanoString nCounter technology47 offers a sensitive way to quantify circRNAs by direct hybridization and performs better on low quality RNA samples, e.g., RNA derived from formalin-fixed, paraffin-embedded (FFPE) samples30,48. The method is based on single-molecule counting of fluorescently labelled probes designed to bind unique sequences (the BSJ for circRNAs) (Fig. 2C). The linear RNA form can be quantified simultaneously if additional probes are designed to match splice sites from flanking exons. Another RT-free method is SplintQuant (Fig. 2D), where custom-made DNA probes are ligated by DNA ligase when simultaneously bound to the target circRNA. The ligated DNA product can subsequently be quantified by qPCR34. One thing to bear in mind when using hybridization-based techniques is that they give rise to a hybridization bias, which may be influenced by factors such as GC content and RNA secondary structure.
Microarrays can be designed to include probes overlapping BSJs, thereby allowing the simultaneous profiling of a large panel of circRNAs. However, not all circRNAs on currently available microarrays have been independently validated and results should therefore be interpreted with care and substantiated using other experimental systems. Similar to the RT-qPCR and sequencing approaches described above, microarrays include an RT-step that may introduce bias.
4. Detecting circRNAs by microscopy
Microscopy-based approaches such as in situ hybridization (ISH) can detect specific RNAs and co-factors in fixed cells or tissues, albeit in a low-throughput manner compared to RNA-seq or microarrays49. The detection of circRNAs by single molecule (sm)ISH is more challenging than for mRNAs since probe design is restricted to the region spanning the BSJ. Furthermore, probe binding to the BSJ may be hindered by RNA degradation, competing secondary structures, association with RBPs or RNA sequestering into granules. This can be prevented by protease treatment of fixed cells prior to probe hybridization (Table 1). In addition, the lower sensitivity of circRNA detection by ISH compared to mRNAs means that circRNAs expressed at low levels may be difficult to detect. Several steps can be taken to optimize circRNA detection, including a) chemically modified probes with increased affinity (e.g., locked nucleic acids), b) signal amplification via enzymatic reactions or branched DNA (bDNA), and c) enzymatic pre-treatment of tissues to expose the BSJ. Detection of single circRNAs can be done using two different assay types: 1) Chemical amplification of a probe set binding on either side of the BSJ (e.g., BaseScope)22,50,51 or 2) Multiple fluorophore-coupled probes (e.g., Stellaris, RNAscope) that cover the full length of the RNA. Basescope allows specific detection of the circRNA in the presence of the linear RNA, while Stellaris and RNAscope are unable to differentiate between a circRNA and its co-expressed linear counterpart.
Detection using BSJ-specific probes
BaseScope is a chromogenic ISH assay that allows detection of single circRNA molecules52,53. It uses two adjacent Z-probes with 18–23 nt sequence complementary to the BSJ (Fig. 2E) and can therefore be used to detect circRNAs in the presence of the linear counterpart. Following probe binding, bDNA amplifies the signal, while an alkaline phosphatase-labelled probe converts Fast Red into a precipitate that marks the spatial localization of the circRNA. Chromogenic assays offer high sensitivity but do not support extensive multiplexing, meaning that co-localization with several other RNAs can only be determined in adjacent tissue sections. On the other hand, BaseScope technology is reliable and can be applied to a wide range of tissues following treatment with different fixatives49.
Detection using general probe sets
Single-molecule Fluorescent ISH (smFISH) allows multiplex detection of different RNAs in the same sample. Stellaris RNA FISH uses DNA probes54 and a probe set contains 30 to 50 20-nt fluorophore-labelled oligos that bind different parts of the target RNA. Because of possible off-target effects, Stellaris probes can only be used for circRNAs that lack linear counterparts or on RNase R-treated tissue. Stellaris assays detect RNA molecules with a high signal-to-noise ratio but require high-resolution microscopy to capture the signal produced by the probes. This also holds true for other smFISH approaches that employ fluorescent or DIG-labelled oligos50,51.
5. CircRNA inhibition
RNAi-dependent depletion
A major challenge for functional studies of circRNAs is to prove that the observed effects are specifically caused by the circRNA and not the linear isoforms that frequently co-exist in cells. The most common way to deplete a circRNA is to design siRNAs against the BSJ as this should degrade the circRNA and leave the co-existing linear RNA from the same locus intact (Fig. 3A). All RNAi experiments come with a risk of off-target effects and it is important to design 2–3 siRNA sequences, off-set from each other but still targeting the BSJ, to test the knockdown efficiency (circ-to-linear ratio) and to include scrambled siRNA control sequences (Table 1). When designing siRNAs, sequence matches between positions 2–7 and the linear mRNA should be avoided as this could trigger miRNA-like repression, leading to an overestimate of the regulatory effect conferred by the circRNA. In systems where siRNA transfection efficiency is low (e.g., neurons), we suggest using viral delivery of short hairpin RNA (shRNA) vectors. This strategy also creates a more stable circRNA knock-down compared to the transient depletion achieved with transfected siRNAs55.
Figure 3: Strategies for circRNA depletion.
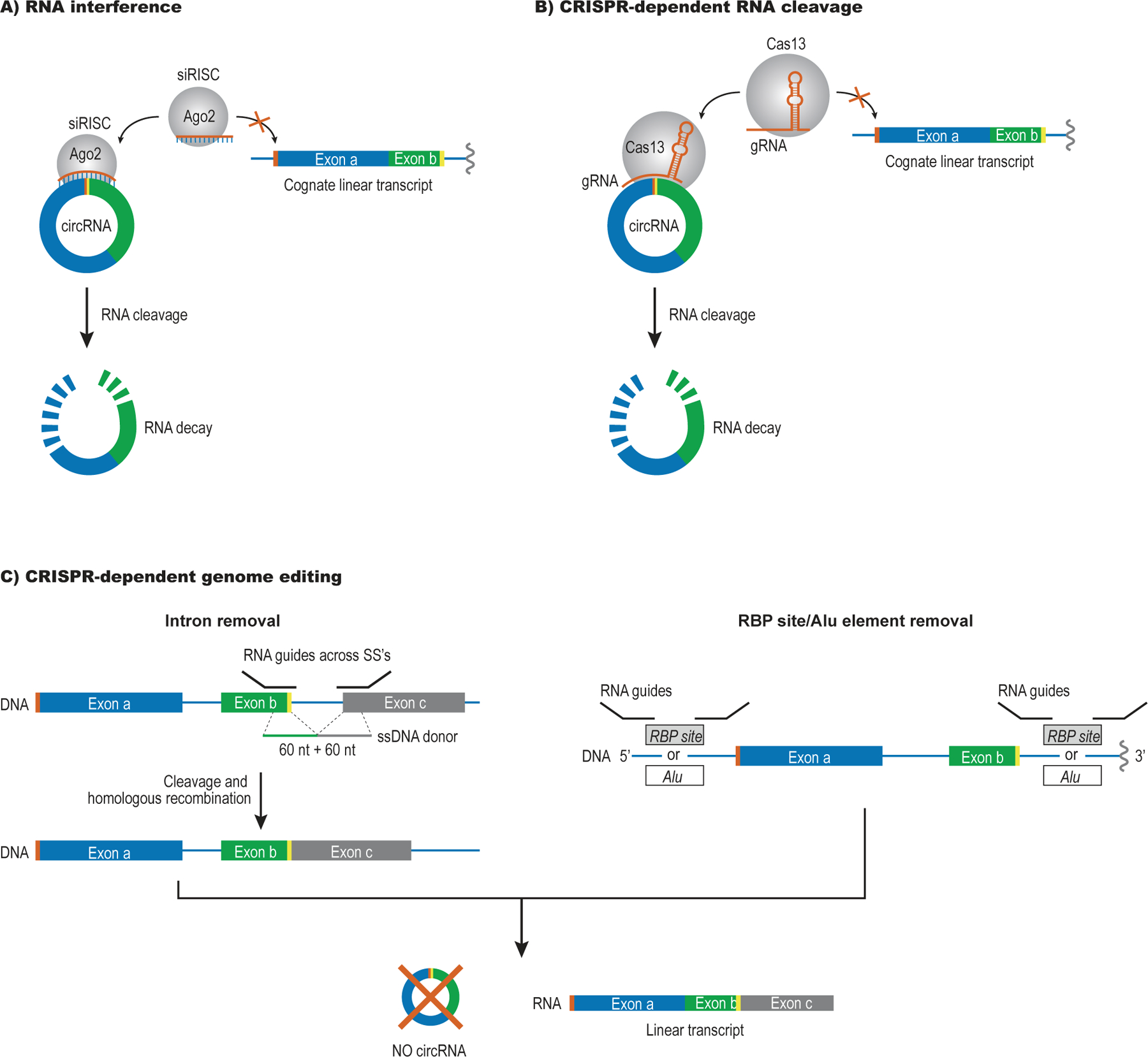
A) Short interfering (si)RNAs designed against the BSJ (yellow and orange) can drive selective cleavage of circRNAs by RNA interference (RNAi), leaving the linearized RNA subject to degradation by exonucleases (RNA decay). A note of caution for RNAi is that a partial overlap with the siRNA can drive miRNA-like repression of the linear form, thus creating off-target effects.
B) Guide (g) RNAs targeting the BSJ can be used for selective degradation of the circRNA form via Cas13, while leaving the linear RNA intact. gRNAs require a more extensive sequence overlap with their target than siRNAs and are therefore less prone to off-target effects.
C) CircRNA biogenesis can be disrupted by Cas9-dependent genome editing, e.g., by using gRNAs to remove an intron flanking a circularizing exon or delete sequence elements that bring the two splice sites together (e.g., Alu elements and/or RBP sites). In either case, back-splicing is prevented on the edited allele.
Another depletion strategy is to target the BSJ of a circRNA using antisense oligonucleotides (ASOs) or gapmers containing modified nucleotides, such as LNA (locked nucleic acids), since they show high sequence specificity56.
Post-transcriptional circRNA depletion using CRISPR/Cas13
RNA-guided RNA-targeting systems (CRISPR/Cas13) provide another way to selectively degrade circRNAs as they trigger RNA cleavage in a sequence-specific manner (Fig. 3B)57,58. This strategy is less invasive than genome editing (see below) since the gene of interest remains intact; however, the guide RNA needs to overlap the BSJ and optimisation may be needed to avoid cleavage of linear RNA. Two recent studies have used Cas13 to specifically target the BSJ of circRNAs and demonstrated increased specificity and reduced off-target effects compared to shRNA-mediated depletion59,60.
CircRNA knockout by genome editing
Genome editing by CRISPR/Cas9 can be used to prevent or reduce back-splicing. For circRNAs that exhibit little or no linear RNA co-expression, this can be done by deleting the entire locus22 but care should be taken to avoid long-range regulatory elements such as enhancers or insulators regulating nearby genes; however, for the majority of circRNAs this is not an option. Instead, CRISPR/Cas9 can be used to remove either entire introns or sequence elements in them that drive circRNA formation61,62. This principle is shown in Fig. 3C for a gene that produces a circRNA from two neighbouring exons: Guide RNAs targeting the splice junctions of the distal intron are combined with a single-stranded DNA donor spanning the exon-a/exon-b-junction to prevent back-splicing. Alternatively, gRNAs can be used to remove a specific repeat element or an RBP-binding site that drives circRNA formation (Fig. 3C). RT-qPCR or equivalent methods should be used to confirm that circRNA expression is reduced by 50% (corresponding to the loss of one allele) after genome editing, while the linear counterpart is not affected.
For all circRNA depletion strategies it is important to consider specificity, side-effects and validation throughout the process (Table 1). Ideally, such studies should be supplemented with rescue experiments, such as expressing an siRNA or gRNA-resistant form of the depleted circRNA; however, care should be taken not to interfere with the back splicing reaction and resistance to RNAi/Cas13 should be validated. Another way to conduct rescue experiments is to express a circRNA from a plasmid (see next section) after endogenous circRNA formation has been disrupted via genome-editing63.
6. CircRNA over-expression
Transfection of circRNAs generated by in vitro transcription
A circRNA of interest can be introduced by transfecting cells with in vitro transcribed, ligated RNAs (Fig. 4A). These artificial circRNAs are often generated by GMP-primed transcription by T7 RNA polymerase, followed by ligation by T4 RNA ligase64. This strategy works well for relatively short circRNAs (up to ~300 nt) while longer circRNAs (up to a few kb) can be generated by using Group I permuted-intron-exon (PIE) systems65 or chemical ligation to generate circRNAs in vitro64. However, the self-splicing process in PIE systems requires specific sequences to flank the BSJ and it will therefore not fully recreate the endogenous BSJ sequence. For all in vitro synthesis techniques, we highly recommend to include subsequent RNase R treatment and gel purification steps to obtain pure circRNA.
Figure 4: Over-expression of circRNA.
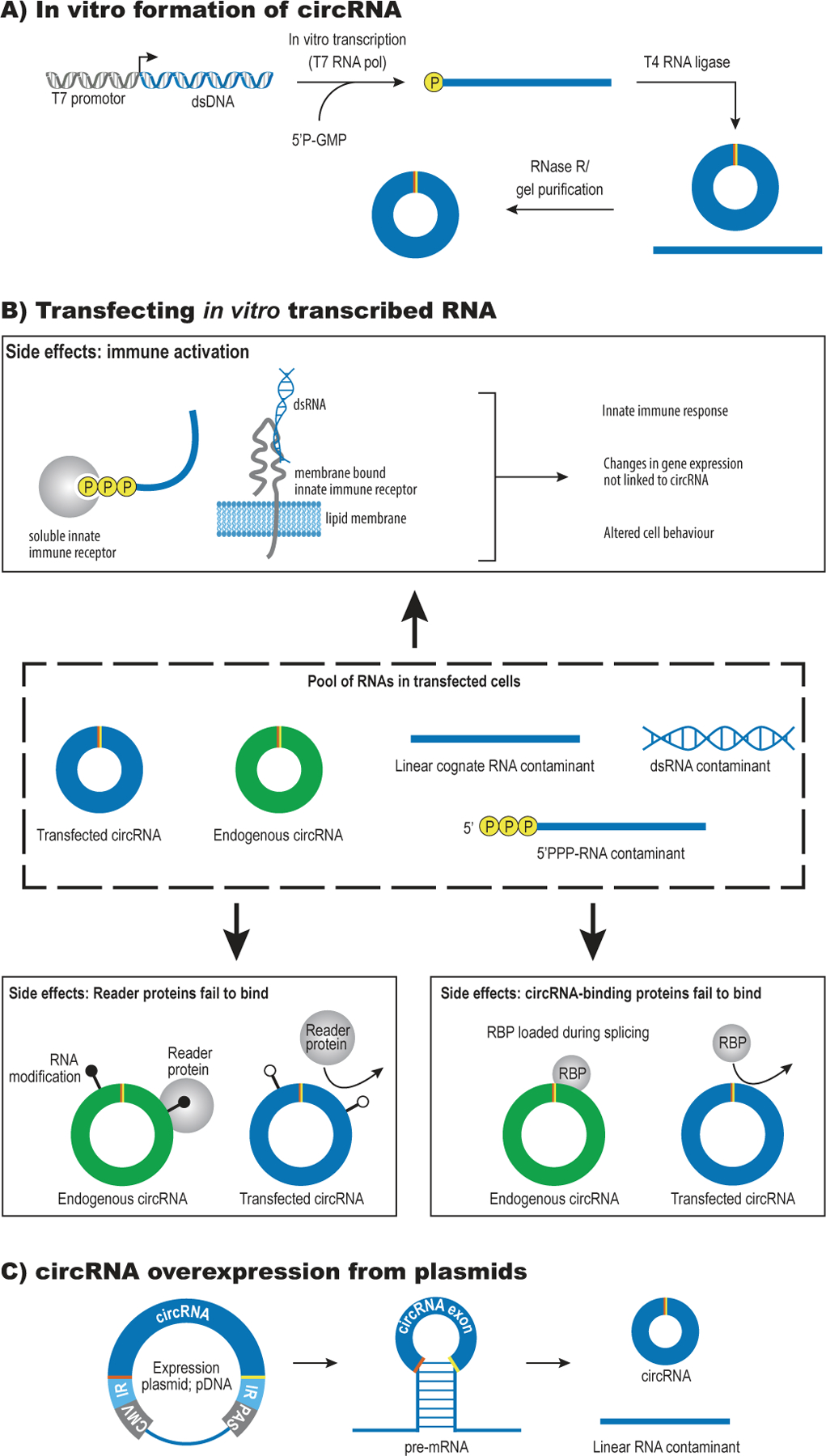
A) In vitro formation of circRNAs: The circRNA sequence is transcribed by T7 RNA polymerase using 5’P-GMP to avoid the presence of a triphosphate on the 5’ end of the RNA. The linear RNA is then circularized using T4 RNA ligase and any remaining, unligated RNA is removed by RNase R treatment or gel purification.
B) Possible side-effects triggered by transfection of in vitro generated circRNAs:
Immune activation: 5’triphosphates, extended dsRNA regions and circRNAs that lack m6A marks may be recognized by innate immune receptors in the cytoplasm and trigger a signaling cascade that confounds the biological effect of the introduced circRNA.
Reader proteins fail to bind: Endogenous circRNA function may depend on RNA modifications that serve as docking sites for specific reader proteins.
CircRNA-binding proteins fail to bind: Endogenous circRNAs may function as circRNPs and depend on proteins that are loaded co-transcriptionally (e.g., the exon-junction complex).
C) A common way to over-express circRNAs is by inserting the exon of interest after a strong promoter (CMV) and between a set of inverted repeats (IR) in a plasmid.
Transfection of in vitro-generated circRNAs allows full control of the RNA form (length, amount, circ-to-linear ratio) and amount but may not recapitulate endogenous circRNA function (Fig. 4B). In addition to possible co-transcriptional effects, circRNAs can carry RNA modifications such as m6A66 and associate with RNA-binding proteins, and it is unknown if their functional role can be recapitulated by in vitro-generated transcripts. The introduction of foreign RNA to cells may also have unwanted effects such as innate immune activation that can confound functional conclusions67. It has been suggested that non-self sequences in the flanking introns may trigger RIG-I signalling68 and that m6A modifications can counteract this immunogenicity69. However, these findings are contrasted by a study that found a limited immune response to non-modified circRNAs and instead suggested that these effects may be avoided or reduced by careful purification of the in vitro-generated circRNAs prior to transfection70. As an example, T7 transcription is known to produce small amounts of dsRNA that can trigger an innate immune response. Furthermore, the stability of transfected circRNAs needs to be monitored throughout the experiment to ensure that effects attributed to circRNAs are not driven by linear forms (Table 1).
Expressing circRNAs from plasmids
A common way to over-express circRNAs is using minigene-containing plasmids that drive back-splicing using natural or artificial inverted repeats (Fig. 4C)8,71. Generally, the circularizing exon(s) are inserted after a strong promoter and flanked by introns carrying inverted repeat sequences. Initial analysis found that functional splice sites and splice signals (branch point, pyrimidine tract) are necessary for circRNA formation, whereas the exonic sequence itself can be replaced by other sequences72. Removing the poly(A) signal, present in many standard vectors, may improve circularization efficiency for some circRNAs (Bindereif lab, personal observation), but also increases the risk of polymerase read-through. Another strategy to express circRNAs from a plasmid is to insert the circRNA of interest within an autocatalytic ribozyme that generates free 5’ and 3’ ends in close proximity73. This method was shown to produce circRNAs through the activity of endogenous RNA ligase RtcB but it adds additional sequence next to the BSJ, which may change the properties of the circRNA generated. In addition, the RtcB system is mainly efficient for smaller circRNAs (<500nt).
Back-splicing is generally an inefficient process and while circRNA-expressing vectors are closer to the natural biogenesis pathway than in vitro transcribed circRNAs they often co-produce linear and circular RNAs (Fig. 4C). We therefore recommend that all plasmid-based circRNA expression involves a quantitative comparison of the circ-to-linear ratio of the RNAs formed, the formation of alternative splice products and a confirmation that the correct BSJ has been created (Table 1). Such analysis can be done by RT-qPCR, Northern blot (+/− RNase R treatment) or equivalent methods. Another concern with plasmid-based expression is the formation of linear read-through transcripts that extend around the whole plasmid. This phenomenon can potentially form concatemers of linear spliced exons with splice junctions indistinguishable from the BSJs in the circRNA, resulting in skewed quantification and functional misinterpretation. This may be avoided by transfecting the circRNA expression cassette as a linear DNA fragment that is chemically protected at the ends to avoid ligation or to insert the expression cassette into the genome. Finally, circularisation efficiency differs widely between circRNAs - likely dependent on sequence, length, and RNA structure - and this should be considered when interpreting phenotypes arising from circRNA overexpression. Mini-gene cassettes can be inserted into viral vectors for stable circRNA over-expression. This is commonly done using a targeted recombinase (e.g., Flip-In), retroviral delivery or adenovirus-associated virus (AAV) vectors74.
7. Determining the regulatory mechanism for circRNAs
If depletion or over-expression of a circRNA of interest (without simultaneous changes in the linear RNA) produces a phenotype, the typical next step is to investigate the underlying mechanism. A number of different mechanisms have been proposed for circRNAs and we will only cover the most frequently reported ones here. For all assays used to investigate circRNA mechanism and function it is important to determine endogenous expression levels of the components involved and use these as reference points for further investigations in order to draw physiologically relevant conclusions.
CircRNA-protein interactions
As with most other RNAs, circRNAs do not work alone and a starting point for delineating their mechanism of action is to identify protein binding partners. Some circRNAs act as protein sponges as for instance demonstrated for Mbl in flies and PKR and AUF1 in human75–77. Alternatively, circRNAs may serve as templates for complex assembly78 or co-factor recruitment79. Many experimental challenges for studying circRNA-protein interactions are shared with the general RNA-protein interaction field and we therefore refer the readers to other reviews for a detailed description80,81. It is important to consider that most of a circRNA sequence matches the linear RNA, meaning that circRNA-protein complexes (circRNPs) may not have a fundamentally different composition than linear mRNPs, although differences in biogenesis, nuclear export, secondary structure, translational activity and stability between linear and circular RNAs could lead to formation of distinct RNP complexes68.
A general strategy to identify circRNPs is to fractionate cell or tissue lysates by glycerol or sucrose gradient centrifugation. Comparing the sedimentation profiles of circRNAs present in lysate versus RNA prepared from the same lysate provides an estimate of the total molecular weight of their protein components82,83. Gradient sedimentation is also useful as an initial purification step in antisense pulldown strategies, thereby enriching for a certain circRNP complex size.
Immuno-Precipitation (IP) from cell lysates can be used to identify circRNAs associated with an RBP of interest. The RBP-associated circRNAs are identified by RNA-seq and ranked by BSJ read counts, followed by individual validation. To assess the biological relevance of the interaction, we recommend quantifying IP-efficiency as a percentage relative to total input, instead of measuring fold-enrichment relative to a non-specific IP control. This allows an estimate of the fraction of a particular circRNA that associates with a given RBP82 (Table 1). The stringency of the RNA-protein interaction can be increased by crosslinking the sample before performing the IP step (PAR-CLIP, HITS-CLIP, iCLIP)84,85. For circRNAs that contain sequences not found in linear RNA, an IP enrichment of circRNA-specific sequences (and a reduction in mRNA-specific sequences) further indicates a specific interaction of the circRNA to the RBP.
Pulldown experiments using circRNA-specific antisense oligonucleotides followed by mass spectrometry (MS) analysis can be used to identify unknown protein binding partners for a circRNA of interest8,86. The targeted circRNA should be expressed at sufficiently high level to support detection of interaction partners by MS87 (Table 1) and the RBPs identified need to be carefully validated as RNP pull-downs are prone to unspecific binding. Protein-interaction candidates based on circRNA pulldown should be validated by quantitative IP of the endogenous circRNP from cell lysates. A useful control is to perform antisense pulldown and MS in cells that do not express the circRNA of interest. CircRNA-pulldowns are typically performed using anti-sense oligos overlapping the BSJ to avoid binding to the linear form. To further minimize co-purification of the linear isoforms, cell lysates can be pre-treated with RNase R or subjected to other enrichment protocols. Circ-to-linear ratios should be closely monitored throughout the experiment.
CircRNAs as miRNA sponges
Two of the papers that helped reignite the circRNA field found that CDR1as functions as a sponge for miR-7 through more than 60 highly conserved binding sites and suggested that miRNA sponging could be a general role for circRNAs8,9. This has inspired a large number of studies claiming that individual circRNAs exert important regulatory roles via their miRNA sponging capabilities. However, there are many methodological challenges associated with studying miRNA sponging and the field is associated with a number of controversies88 (see Box). The main concern is that the circRNA-miRNA-mRNA stoichiometry required for efficiency competition for miRNA binding (the competitive endogenous RNA model) is rarely seen under physiological conditions89. Similar discussions have been ongoing in the lncRNA field for several years and they are equally relevant for circRNA research90.
Box: Controversies of the miRNA sponge theory.
The idea that circRNAs act via miRNA sponging was questioned early on by the finding that most circRNAs do not carry more miRNA binding sites than would be expected by chance107. Moreover, while some circRNAs can accumulate to high levels, most are expressed at relatively low levels compared to miRNAs and mRNAs. It is therefore important to consider the stoichiometry between the number of miRNA binding sites on the circRNA of interest relative to the total number of cellular binding sites for a given miRNA (including pseudogenes, lncRNAs and mRNAs), when making conclusions about biologically relevant miRNA sponging. CircRNAs could potentially bind the miRNA with higher affinity than other target transcripts if additional complementarity is present outside of the miRNA seed sequence (as seen for circZNF91 and miR-23b33). However, this is rarely investigated and not likely to apply to many of the circRNAs that have been proposed to function as miRNA sponges. Another source of controversy is that negative correlations between circRNA and miRNA as well positive correlations between circRNA and miRNA target genes are often interpreted as evidence for a miRNA sponging. However, correlations between circRNAs and miRNAs target genes may be explained by different cellular compositions across patient samples91 and most circRNAs do probably not degrade the miRNAs, as target RNA-directed miRNA degradation (TDMD) requires extensive complementarity outside of the seed sequence108. Indeed, deletion of the CDR1as locus in mouse was found to destabilize miR-7, suggesting that miRNA-binding circRNAs may have other functions than repressing miRNA levels and activity22.
We suggest that any study on miRNA sponging first conduct a thorough bioinformatics characterization of the circRNA in question to determine the abundance of miRNA binding sites and potential AGO binding. Next, in situ analyses of the circRNA and miRNA should confirm that they co-localize within the same cells in the tissue under investigation91. However, co-localization does not prove a sponging function and it is important to provide direct evidence of the circRNA-miRNA interaction by RIP or pulldown experiments92. Luciferase reporters with miRNA binding sites in the 3’UTR are often used to test a direct interaction with a miRNA but do not prove biological function at endogenous expression levels. Likewise, phenotypic effects associated with circRNA overexpression should be sensitive to mutagenesis of the miRNA binding sites and also reflect the stoichiometry of endogenous expression to avoid over-estimating the extent of biologically relevant sponging89. Finally, a strong indication of de facto miRNA association can be obtained from sequencing of the miRNAs recovered upon endogenous circRNA pulldown or from CLIP-seq data if Ago2 associates with the putative sponge sequences on a circRNA8,62. This strategy should ideally be combined with circRNA perturbation (preferably depletion) and show a concomitant downstream effect on other miRNA targets.
CircRNA translation
Early reports proposed that viral circRNAs could be templates for translation2, but this was questioned by the finding that mammalian circRNAs, like circSry, are not associated with polysomes5. When circRNAs were rediscovered in 2012–2013, they were assumed to be non-coding, given the absence of a cap and a poly(A)-tail, but recent work has shown that some circRNAs may undergo translation93–95. One of these circRNAs, circZNF609, contains an open reading frame that runs over the BSJ, adding a C-terminus specific for the circRNA-encoded protein isoform. Since circRNAs rely on cap-independent translation, their protein products could be subject to different translational control than linear mRNAs. In addition, the high stability of circRNAs could facilitate translation in specific subcellular compartments (e.g., synapses) where mRNA supply is particularly limited.
The lack of known consensus motifs for IRES elements makes the identification of translatable circRNAs difficult and experimental validation is therefore a must. A commonly used method is to express circRNAs from plasmids and look for a protein product unique to the circular form94,95; however, care must be taken when interpreting the results (Table 1). As discussed above, plasmids can produce linear concatemeric transcripts that produce the same protein product as the circRNA, albeit in a cap- and poly(A)-dependent manner96,97. It is therefore important to include a control vector without the flanking elements required for back-splicing in order to delineate contributions from circRNA and linear concatemers. The detection of protein that can only be expressed from the endogenously produced circRNA would be the ideal demonstration of translation but this is rarely seen, presumably due to low abundance (Table 2).
Table 2:
Evidence for circRNA translation
| Available assays | Level of support | Rationale |
|---|---|---|
| BSJ detected in Riboseq data | Low | Binding may reflect background signal |
| Selective conservation of circRNA-specific ORF | Medium | May be related to regulatory elements (e.g., uORFs) in the linear form |
| CircRNA associated with polysomes AND sensitive to puromycin treatment | Medium | Suggests proximity to active translation |
| Protein product detected upon expression of the circRNA | Medium | Rule out linear contamination and concatemer formation from vector |
| Endogenous protein isoform encoded by circRNA detected by mass spectrometry | High | The bulk FDR does not apply to circRNAs – this has to be determined specifically to avoid false positives |
| Detection of endogenous circRNA-derived peptides sensitive to circRNA depletion | High | Peptide levels may often be below detection limits |
Other approaches for studying potential circRNA translation include: 1) Determining circRNA association to polysomes and the response to puromycin treatment94, 2) Using the Ribotag method (cells expressing HA-tagged Rpl22) to purify ribosome-associated RNAs in a tissue- or cell-type specific manner98, 3) Using ribosome footprinting followed by RNA sequencing (Riboseq) to search for translation of endogenous circRNAs in a genome-wide manner95,99. For all strategies, it is important to remember that ribosome association does not prove translation, even if a triplet-phased signal is observed in Riboseq data97. On the other hand, negative findings in vitro do not disprove translation in vivo. Cap-independent translation may occur in response to specific forms of stimuli or stress, suggesting that the right conditions for circRNA translation could have been missed in some cases.
A direct and more rigorous method to test the existence of circRNA-derived proteins is mass spectrometry. Whole-cell extracts or size-selected protein fractions can be analyzed for the presence of peptides derived from the BSJ or downstream of it. Here, it is essential to compute a False Discovery Rate (FDR) specifically for the putative circRNA-derived peptides and not simply accept all peptides passing a full proteome FDR cutoff, as this may lead to the detection of false positives100 (Table 2). Targeted mass spectrometry methods such as parallel reaction monitoring may facilitate detection of peptides expressed at very low levels but to our knowledge this has not yet been applied in circRNA studies. Antibodies raised against the predicted new amino acid sequence offer a specific tool for studying a putative circRNA-derived protein. Alternatively, genome editing can be used to insert specific tags in the endogenous locus, downstream of the BSJ, allowing detection of the putative protein product94.
Based on published MS data, it appears that there may be a very low level of pervasive translation for many circRNAs101; however, whether and to what extent the huge repertoire of natural circRNAs provides widespread and significant translation potential, and whether any novel biological functions rely on circRNA-encoded protein isoforms, remain important open questions.
8. Conclusion and outlook
The first wave of research in a new field is filled with excitement but also inevitably confusion due to contradictory conclusions and unclear experiments. The last 7–8 years have seen the circRNA field explode and diversify and we hope that this will continue in the future. One area where we see great potential is in the development of techniques for circRNA profiling at single-cell level since many circRNAs are expressed in a cell type-specific manner. A few studies have detected circRNAs in single cells using random primers coupled to fixed anchor sequences102 or by combining microfluidics with strand-specific total RNA sequencing103; however, these methods only work for highly abundant circRNAs and will need further development to be relevant for general circRNA analysis. The development of spatial transcriptomics offers another way to study RNA expression at single-cell resolution in complex tissues. Similar to the techniques mentioned above, applying this technique to circRNAs will require careful optimisation for recognition of the BSJ. Another area that will be interesting to follow in the circRNA field is single-molecule imaging in fixed and live cells or tissue. Recent techniques enable the imaging of single RNAs in large, cleared and expanded samples using light sheet microscopy104 and the detection of smFISH spots in large volumes or many images can be facilitated by the recently developed Radial Symmetry FISH (RS-FISH)105.
When it comes to the biogenesis and mechanism of action for circRNAs, a technique that holds great potential is to use catalytically inactive versions of Cas13 to target the BSJ. Fluorescently labelled gRNAs will enable tracking of circRNAs in live cells and tethering of RNA-binding proteins or RNA-modifying enzymes can block binding sites or provide new functions. The site-specific tethering of RNA-modifying enzymes will also be a helpful tool to further delineate how RNA modifications such as m6A affect circRNA stability and function.
We invite labs across the world to join us in developing the circRNA field further in the coming years and hope that this review can become a reference point for technical guidelines in the field.
Acknowledgements:
This work was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 721890 (circRTrain). AFN and JK are funded by the Danish National Research Foundation grant #135 (CellPAT), MJO is funded by the Danish Cancer society grant R269-A15768, LSK is funded by the Lundbeck Foundation (R307-2018-3433). IB acknowledges funding from ERC-2019-SyG 855923-ASTRA and from AIRC-Progetto IG 2019 Id. 23053. AB is funded by the Deutsche Forschungsgemeinschaft via grants RTG 2355, SPP 1935 and RU5116. SK acknowledges funding from the National Institute of Health (R01GM122406 and R01AG057700)
Glossary
- AGO
Argonaute (Ago) proteins are the effectors in RNA interference. Ago proteins bind miRNAs and siRNAs and trigger degradation of target transcripts (via deadenylation or direct cleavage)
- Anchor sequences
The two halves of a non-continuously mapping read that are used to ‘anchor’ the exons that have been joined across the BSJ.
- Back-splicing
A splicing event where the 5’ splice site is spliced to an upstream 3’ splice site, resulting in the formation of one or more covalently joined circular exons
- bDNA technology
The use of branched DNA oligos as signal amplifiers in hybridisation-based imaging
- BSJ
Back-Splice Junction; the unique sequence element created when 5’ and 3’ splice sites are joined in the reverse order to create a circRNA.
- CiRS-7/CDR1as
One of the best characterised circRNAs so far, mainly expressed in neurons and affects stability and function of miR-7 via direct base-pairing
- (sm)ISH
Single-molecule In Situ Hybridisation
- Long-Read Sequencing
A single molecule, real-time sequencing technique that can read long sequences of 500-50000 bps.
- RNase R
An exonuclease that degrades any RNA containing free ends, often used to enrich the circRNA content of a given sample
- nanoString
A RT-free technique for quantifying RNAs with a known sequence. Uses fluorescently labelled probes and a single-molecule counting device.
- Ribosome profiling
A deep-sequencing-based technique that facilitates the detailed measurement of global ribosome occupancy on mRNA
- RT
Reverse transcription, creating a cDNA form of the RNA of interest prior to PCR- or microarray-based analyses
- RTase
Reverse Transcriptase enzyme
- Trans-splicing
A splicing event that occurs between two independent pre-mRNA transcripts
- Z probe
A structured oligonucleotide probe that recognizes a target sequence in one end while the other end forms part of a composite binding surface for an amplification probe (often bDNA). Illustrated in Fig 2E.
Footnotes
Competing Interests statement:
The authors have no competing interests
Reference list
- 1.Sanger HL, Klotz G, Riesner D, Gross HJ & Kleinschmidt AK Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 73, 3852–3856, doi: 10.1073/pnas.73.11.3852 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsu MT & Coca-Prados M Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 280, 339–340, doi: 10.1038/280339a0 (1979). [DOI] [PubMed] [Google Scholar]
- 3.Zaug AJ, Grabowski PJ & Cech TR Autocatalytic cyclization of an excised intervening sequence RNA is a cleavage-ligation reaction. Nature 301, 578–583, doi: 10.1038/301578a0 (1983). [DOI] [PubMed] [Google Scholar]
- 4.Kjems J & Garrett RA Novel splicing mechanism for the ribosomal RNA intron in the archaebacterium Desulfurococcus mobilis. Cell 54, 693–703, doi: 10.1016/s0092-8674(88)80014-x (1988). [DOI] [PubMed] [Google Scholar]
- 5.Capel B et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73, 1019–1030, doi: 10.1016/0092-8674(93)90279-y (1993). [DOI] [PubMed] [Google Scholar]
- 6.Nigro JM et al. Scrambled exons. Cell 64, 607–613, doi: 10.1016/0092-8674(91)90244-s (1991). [DOI] [PubMed] [Google Scholar]
- 7.Cocquerelle C, Mascrez B, Hetuin D & Bailleul B Missplicing Yields Circular Rna Molecules. FASEB J 7, 155–160 (1993). [DOI] [PubMed] [Google Scholar]
- 8. Hansen TB et al. Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388, doi: 10.1038/nature11993 (2013). Reported that circRNA ciRS-7/CDR1as carries a large number of binding sites for miR7 and regulates availability of the miRNA.
- 9. Memczak S et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338, doi: 10.1038/nature11928 (2013). Reported that circRNA ciRS-7/CDR1as carries a large number of binding sites for miR7 and regulates availability of the miRNA.
- 10. Salzman J, Gawad C, Wang PL, Lacayo N & Brown PO Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. Plos One 7, e30733, doi: 10.1371/journal.pone.0030733 (2012). One of the first studies to report widespread production of circular RNA isoforms from a range of human genes.
- 11.Patop IL, Wust S & Kadener S Past, present, and future of circRNAs. EMBO J 38, e100836, doi: 10.15252/embj.2018100836 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo L & Salzman J Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17, 679–692, doi: 10.1038/nrg.2016.114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kristensen LS et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20, 675–691, doi: 10.1038/s41576-019-0158-7 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Li X, Yang L & Chen LL The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell 71, 428–442, doi: 10.1016/j.molcel.2018.06.034 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Kristensen LS, Jakobsen T, Hager H & Kjems J The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol, doi: 10.1038/s41571-021-00585-y (2021). [DOI] [PubMed] [Google Scholar]
- 16.Dodbele S, Mutlu N & Wilusz JE Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep 22, e52072, doi: 10.15252/embr.202052072 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsitsipatis D & Gorospe M Practical guide for circular RNA analysis: Steps, tips, and resources. Wiley Interdiscip Rev RNA 12, e1633, doi: 10.1002/wrna.1633 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Jeck WR et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157, doi: 10.1261/rna.035667.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enuka Y et al. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 44, 1370–1383, doi: 10.1093/nar/gkv1367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen TB et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30, 4414–4422, doi: 10.1038/emboj.2011.359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett SP, Parker KR, Horn C, Mata M & Salzman J ciRS-7 exonic sequence is embedded in a long non-coding RNA locus. PLoS Genet 13, e1007114, doi: 10.1371/journal.pgen.1007114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piwecka M et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357, doi: 10.1126/science.aam8526 (2017). The first circRNA knockout mouse showed ciRS-7/CDR1as to play a role in brain development via its ability to bind miR-7.
- 23.Seal RL et al. A guide to naming human non-coding RNA genes. EMBO J 39, e103777, doi: 10.15252/embj.2019103777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glazar P, Papavasileiou P & Rajewsky N circBase: a database for circular RNAs. RNA 20, 1666–1670, doi: 10.1261/rna.043687.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Wang Q, Shen J, Yang BB & Ding X Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol 16, 899–905, doi: 10.1080/15476286.2019.1600395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong R, Ma XK, Li GW & Yang L CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genomics Proteomics Bioinformatics 16, 226–233, doi: 10.1016/j.gpb.2018.08.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachmayr-Heyda A et al. Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5, 8057, doi: 10.1038/srep08057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34, e63, doi: 10.1093/nar/gkl151 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao MS & Wilusz JE An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends. Nucleic Acids Res 47, 8755–8769, doi: 10.1093/nar/gkz576 (2019). A modified protocol for circRNA enrichment that facilitates a better separation between circular and linear RNAs.
- 30.Dahl M et al. Enzyme-free digital counting of endogenous circular RNA molecules in B-cell malignancies. Lab Invest 98, 1657–1669, doi: 10.1038/s41374-018-0108-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panda AC et al. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 45, e116, doi: 10.1093/nar/gkx297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rahimi K, Veno MT, Dupont DM & Kjems J Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun 12, 4825, doi: 10.1038/s41467-021-24975-z (2021). Developed a long-read sequencing protocol that reveals the complexity and internal composition of large, multi-exon circRNAs.
- 33.Kristensen LS, Okholm TLH, Veno MT & Kjems J Circular RNAs are abundantly expressed and upregulated during human epidermal stem cell differentiation. RNA Biol 15, 280–291, doi: 10.1080/15476286.2017.1409931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conn V & Conn SJ SplintQuant: a method for accurately quantifying circular RNA transcript abundance without reverse transcription bias. RNA 25, 1202–1210, doi: 10.1261/rna.070953.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 7, 12060, doi: 10.1038/ncomms12060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y, Ji P, Chen S, Hou L & Zhao F Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 11, 2, doi: 10.1186/s13073-019-0614-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J et al. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol 39, 836–845, doi: 10.1038/s41587-021-00842-6 (2021). Developed a long-read sequencing protocol that reveals the complexity and internal composition of large, multi-exon circRNAs.
- 38. Xin R et al. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat Commun 12, 266, doi: 10.1038/s41467-020-20459-8 (2021). Developed a long-read sequencing protocol that reveals the complexity and internal composition of large, multi-exon circRNAs.
- 39.Rahimi K, Faerch Nielsen A, Veno MT & Kjems J Nanopore long-read sequencing of circRNAs. Methods, doi: 10.1016/j.ymeth.2021.09.010 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Hansen TB Improved circRNA Identification by Combining Prediction Algorithms. Front Cell Dev Biol 6, 20, doi: 10.3389/fcell.2018.00020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng J, Metge F & Dieterich C Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 32, 1094–1096, doi: 10.1093/bioinformatics/btv656 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Gao Y, Zhang J & Zhao F Circular RNA identification based on multiple seed matching. Brief Bioinform 19, 803–810, doi: 10.1093/bib/bbx014 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Tapial J et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res 27, 1759–1768, doi: 10.1101/gr.220962.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang C et al. Template switching causes artificial junction formation and false identification of circular RNAs. bioRxiv, 259556, doi: 10.1101/259556 (2018). [DOI] [Google Scholar]
- 45.Yu CY, Liu HJ, Hung LY, Kuo HC & Chuang TJ Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro? Nucleic Acids Res 42, 9410–9423, doi: 10.1093/nar/gku643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuang TJ et al. Integrative transcriptome sequencing reveals extensive alternative trans-splicing and cis-backsplicing in human cells. Nucleic Acids Res 46, 3671–3691, doi: 10.1093/nar/gky032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geiss GK et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26, 317–325, doi: 10.1038/nbt1385 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Kristensen LS Profiling of circRNAs using an enzyme-free digital counting method. Methods, doi: 10.1016/j.ymeth.2021.02.004 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Bejugam PR, Das A & Panda AC Seeing Is Believing: Visualizing Circular RNAs. Noncoding RNA 6, doi: 10.3390/ncrna6040045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veno MT et al. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol 16, 245, doi: 10.1186/s13059-015-0801-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You X et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18, 603–610, doi: 10.1038/nn.3975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suenkel C, Cavalli D, Massalini S, Calegari F & Rajewsky N A Highly Conserved Circular RNA Is Required to Keep Neural Cells in a Progenitor State in the Mammalian Brain. Cell Rep 30, 2170–2179 e2175, doi: 10.1016/j.celrep.2020.01.083 (2020). [DOI] [PubMed] [Google Scholar]
- 53.D’Ambra E et al. Circ-Hdgfrp3 shuttles along neurites and is trapped in aggregates formed by ALS-associated mutant FUS. iScience 24, 103504, doi: 10.1016/j.isci.2021.103504 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879, doi: 10.1038/nmeth.1253 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pamudurti NR et al. An in vivo strategy for knockdown of circular RNAs. Cell Discov 6, 52, doi: 10.1038/s41421-020-0182-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marrosu E, Ala P, Muntoni F & Zhou H Gapmer Antisense Oligonucleotides Suppress the Mutant Allele of COL6A3 and Restore Functional Protein in Ullrich Muscular Dystrophy. Mol Ther Nucleic Acids 8, 416–427, doi: 10.1016/j.omtn.2017.07.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614, doi: 10.1016/j.cell.2018.02.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abudayyeh OO et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284, doi: 10.1038/nature24049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S et al. Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat Methods 18, 51–59, doi: 10.1038/s41592-020-01011-4 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y et al. Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol 22, 41, doi: 10.1186/s13059-021-02263-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Y et al. The Biogenesis of Nascent Circular RNAs. Cell Rep 15, 611–624, doi: 10.1016/j.celrep.2016.03.058 (2016). Established the role of flanking sequence elements in driving circRNA formation by back-splicing.
- 62.Zheng Q et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7, 11215, doi: 10.1038/ncomms11215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guarnerio J et al. Intragenic antagonistic roles of protein and circRNA in tumorigenesis. Cell Res 29, 628–640, doi: 10.1038/s41422-019-0192-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petkovic S & Muller S RNA circularization strategies in vivo and in vitro. Nucleic Acids Res 43, 2454–2465, doi: 10.1093/nar/gkv045 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puttaraju M & Been MD Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res 20, 5357–5364, doi: 10.1093/nar/20.20.5357 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou C et al. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep 20, 2262–2276, doi: 10.1016/j.celrep.2017.08.027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kariko K, Muramatsu H, Ludwig J & Weissman D Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39, e142, doi: 10.1093/nar/gkr695 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen YG et al. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell 67, 228-+, doi: 10.1016/j.molcel.2017.05.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen YG et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell 76, 96–109 e109, doi: 10.1016/j.molcel.2019.07.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wesselhoeft RA et al. RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol Cell 74, 508–520 e504, doi: 10.1016/j.molcel.2019.02.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liang D & Wilusz JE Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28, 2233–2247, doi: 10.1101/gad.251926.114 (2014). Showed that base-pairing between inverted repeat sequences in introns flanking the circularizing exons can drive circRNA formation.
- 72.Starke S et al. Exon circularization requires canonical splice signals. Cell Rep 10, 103–111, doi: 10.1016/j.celrep.2014.12.002 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Litke JL & Jaffrey SR Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat Biotechnol 37, 667–675, doi: 10.1038/s41587-019-0090-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meganck RM et al. Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo. Mol Ther Nucleic Acids 13, 89–98, doi: 10.1016/j.omtn.2018.08.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashwal-Fluss R et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56, 55–66, doi: 10.1016/j.molcel.2014.08.019 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Liu CX et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 177, 865–880 e821, doi: 10.1016/j.cell.2019.03.046 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Tsitsipatis D et al. AUF1 ligand circPCNX reduces cell proliferation by competing with p21 mRNA to increase p21 production. Nucleic Acids Res 49, 1631–1646, doi: 10.1093/nar/gkaa1246 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Q et al. CircACC1 Regulates Assembly and Activation of AMPK Complex under Metabolic Stress. Cell Metab 30, 157–173 e157, doi: 10.1016/j.cmet.2019.05.009 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Chen N et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol 19, 218, doi: 10.1186/s13059-018-1594-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramanathan M, Porter DF & Khavari PA Methods to study RNA-protein interactions. Nat Methods 16, 225–234, doi: 10.1038/s41592-019-0330-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Das A, Sinha T, Shyamal S & Panda AC Emerging Role of Circular RNA-Protein Interactions. Noncoding RNA 7, doi: 10.3390/ncrna7030048 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schneider T et al. CircRNA-protein complexes: IMP3 protein component defines subfamily of circRNPs. Sci Rep 6, 31313, doi: 10.1038/srep31313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Preusser C et al. Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles 7, 1424473, doi: 10.1080/20013078.2018.1424473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okholm TLH et al. Transcriptome-wide profiles of circular RNA and RNA-binding protein interactions reveal effects on circular RNA biogenesis and cancer pathway expression. Genome Med 12, 112, doi: 10.1186/s13073-020-00812-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee FCY & Ule J Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol Cell 69, 354–369, doi: 10.1016/j.molcel.2018.01.005 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Pandey PR et al. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res 48, 3789–3805, doi: 10.1093/nar/gkaa035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chu C et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416, doi: 10.1016/j.cell.2015.03.025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jarlstad Olesen MT & S. K. Circular L RNAs as microRNA sponges: evidence and controversies. Essays Biochem, doi: 10.1042/EBC20200060 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Denzler R, Agarwal V, Stefano J, Bartel DP & Stoffel M Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54, 766–776, doi: 10.1016/j.molcel.2014.03.045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomson DW & Dinger ME Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17, 272–283, doi: 10.1038/nrg.2016.20 (2016). [DOI] [PubMed] [Google Scholar]
- 91. Kristensen LS et al. Spatial expression analyses of the putative oncogene ciRS-7 in cancer reshape the microRNA sponge theory. Nat Commun 11, 4551, doi: 10.1038/s41467-020-18355-2 (2020). This study found that a presumably oncogenic circRNA is only expressed in stromal cells, not in cancer cells, thereby highlighting the limitations of bulk RNA-seq analysis for circRNA profiling of tissue.
- 92.Li M et al. A circular transcript of ncx1 gene mediates ischemic myocardial injury by targeting miR-133a-3p. Theranostics 8, 5855–5869, doi: 10.7150/thno.27285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y & Wang Z Efficient backsplicing produces translatable circular mRNAs. RNA 21, 172–179, doi: 10.1261/rna.048272.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Legnini I et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell 66, 22–37 e29, doi: 10.1016/j.molcel.2017.02.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pamudurti NR et al. Translation of CircRNAs. Mol Cell 66, 9–21 e27, doi: 10.1016/j.molcel.2017.02.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ho-Xuan H et al. Comprehensive analysis of translation from overexpressed circular RNAs reveals pervasive translation from linear transcripts. Nucleic Acids Res 48, 10368–10382, doi: 10.1093/nar/gkaa704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stagsted LV, Nielsen KM, Daugaard I & Hansen TB Noncoding AUG circRNAs constitute an abundant and conserved subclass of circles. Life Sci Alliance 2, doi: 10.26508/lsa.201900398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanz E et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106, 13939–13944, doi: 10.1073/pnas.0907143106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Heesch S et al. The Translational Landscape of the Human Heart. Cell 178, 242–260 e229, doi: 10.1016/j.cell.2019.05.010 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Hansen TB Signal and noise in circRNA translation. Methods, doi: 10.1016/j.ymeth.2021.02.007 (2021). [DOI] [PubMed] [Google Scholar]
- 101.Chen CK et al. Structured elements drive extensive circular RNA translation. Mol Cell 81, 4300–4318 e4313, doi: 10.1016/j.molcel.2021.07.042 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fan X et al. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol 16, 148, doi: 10.1186/s13059-015-0706-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Verboom K et al. SMARTer single cell total RNA sequencing. Nucleic Acids Res 47, e93, doi: 10.1093/nar/gkz535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y et al. Expansion-Assisted Iterative-FISH defines lateral hypothalamus spatio-molecular organization. bioRxiv, 2021.2003.2008.434304, doi: 10.1101/2021.03.08.434304 (2021). [DOI] [PubMed] [Google Scholar]
- 105.Bahry E et al. RS-FISH: Precise, interactive and scalable smFISH spot detection using Radial Symmetry. bioRxiv, 2021.2003.2009.434205, doi: 10.1101/2021.03.09.434205 (2021). [DOI] [Google Scholar]
- 106.Denzler R et al. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol Cell 64, 565–579, doi: 10.1016/j.molcel.2016.09.027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo JU, Agarwal V, Guo H & Bartel DP Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15, 409, doi: 10.1186/s13059-014-0409-z (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghini F et al. Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9, 3119, doi: 10.1038/s41467-018-05182-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]