

Abstract
SARS-CoV-2 and its variants cause serious health concerns throughout the world. The alarming increase in the daily number of cases has become a nightmare in many low-income countries; although some vaccines are available, their high cost and low vaccine production make them unreachable to ordinary people in developing countries. Other treatment strategies are required for novel therapeutic options. The peptide-based drug is one of the alternatives with low toxicity, more specificity, and ease of synthesis. Herein, we have applied structure-based virtual screening to identify potential peptides targeting the critical proteins of SARS-CoV-2. Non-toxic natural antiviral peptides were selected from the enormous number of peptides. Comparative modeling was applied to prepare a 3D structure of selected peptides. 3D models of the peptides were docked using the ClusPro docking server to determine their binding affinity and peptide-protein interaction. The high-scoring peptides were docked with other crucial proteins to analyze multiple targeting peptides. The two best peptides were subjected to MD simulations to validate the structure stability and evaluated RMSD, RMSF, Rg, SASA, and H-bonding from the trajectory analysis of 100 ns. The proposed lead peptides can be used as a broad-spectrum drug and potentially develop as a therapeutic to combat SARS-CoV-2, positively impacting the current pandemic.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11224-022-02113-9.
Keywords: SARS-CoV-2, Antiviral peptides, Comparative modeling, Ab initio modeling, Broad-spectrum drug
Introduction
Coronaviruses are a group of viruses consisting of RNA as genetic material. To date, a total of four subgroups of coronavirus, namely alpha, beta, gamma, and delta, have been reported, but human-affecting coronaviruses mainly belong to the first two subgroups. The severity of coronavirus infections ranged from the common cold to acute respiratory syndrome, named Middle East Respiratory Syndrome (MERS-CoV) and severe acute respiratory syndrome (SARS-CoV). As evidenced by its quick spread from Wuhan, China, to every country in the world, the 2019 novel coronavirus (SARS-CoV-2) is a new strain of the currently known coronavirus that has not been previously reported and has been found to be highly contagious [1]. The World Health Organization recorded 608 million confirmed cases of COVID-19 as of October 2, 2022, with 6.5 million fatalities (https://www.who.int/health-topics/coronavirus). COVID-19 is a zoonotic disease indicating its spread from animal to human and further from human to human. This condition’s basic signs and symptoms are similar to those of a regular cold, including fever, dry cough, and weariness. Other less prevalent symptoms include sore throat, headache, diarrhea, loss of taste and smell, and skin rash (https://www.cdc.gov/coronavirus/SARS-CoV-2/symptoms-testing/symptoms.html). In the severe stage, patients can feel shortness of breath, chest pain, and loss of speech and even movement. Based on the pandemic data and symptoms, we can easily understand the COVID-19 disease severity. On May 1, 2020, FDA recently approved the antiviral drug remdesivir to treat severely affected COVID patients. However, this drug was approved under emergency use authorization and had severe adverse reactions compared to the placebo [2]. Present COVID-19 drugs, including chloroquine, hydroxychloroquine, baricitinib, favipiravir, and umifenovir, are mainly based on the patient’s clinical symptoms [3, 4]. Nevertheless, even all these drugs are not devoid of toxicity, and there is an urgent need to work on COVID-19 drug discovery.
In recent years, peptide-based drugs have gained interest in treating various disease conditions [5–9]. The well-known examples are Lupron™ and Lantus™, approved to treat prostate cancer and diabetes, respectively [10]. As per the Pharmaceutical GlobalData, a total of 21 synthetic peptides are in the development phase for the treatment of COVID-19 (https://www.globaldata.com/industries-we-cover/pharmaceutical/). Based on all previous studies, this study was designed to identify peptides showing therapeutic potential for combating COVID-19. Peptide-based targeted therapy needs information on crucial viral protein targets to participate in viral pathogenesis and survival. A literature survey reveals the presence of multiple targets present in the SARS-CoV-2 virus. Still, the central protease (Fig. 1C) is the most crucial one, which helps in processing the SARS-CoV-2 polyprotein post-viral RNA translation [11, 12]. Its inhibition leads to the viral replication machinery’s blockade, making it a crucial drug target for drug development. The non-structural protein 9 (Nsp9) is another drug target that acts as an RNA-binding protein (Fig. 1D) and participates in both viral replication and virulence [13]. Angiotensin-converting enzyme 2 (ACE2) of human cells has so far been directly bound through the S-protein of SARS-CoV-2 (Fig. 1A) via its receptor-binding domain (RBD) [14]. This association exploited the ACE2, which leads to host infection. RBD thus supports its eligibility as a drug target for SARS-CoV-2. The approved medication remdesivir has a well-known target in the RNA-dependent RNA polymerase (Fig. 1B) of the SARS-CoV-2 virus [15, 16]. The transcription machinery and inhibition of this protein make it a perfect candidate as a target for SARS-CoV-2 medications. It serves as a crucial component of SARS-CoV-2 replication. The endoribonuclease, also known as non-structural protein 15 (Nsp15), is associated with the processing of viral RNA (Fig. 1D). This protein also protects the virus from host defense by degrading the viral RNA and interfering with the host’s innate immune system via protein interference [17, 18].
Fig. 1.
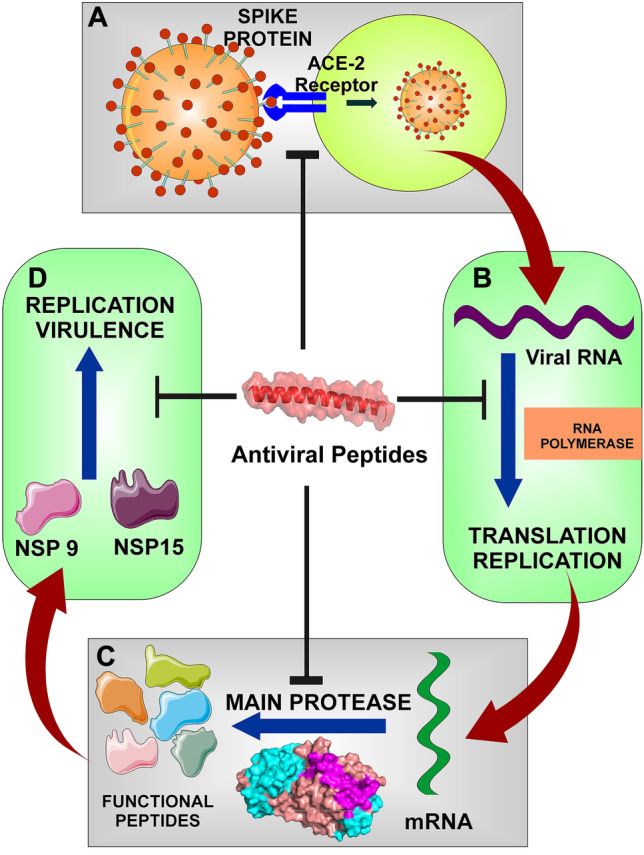
A The spike protein binds to the ACE2 receptor of the host cell and directs its entry into the cell. B RNA polymerase is involved in both translation and replication of the viral RNA. C The main protease is required for the polyprotein processing to produce functional peptides. D The Nsp9 and Nsp15 are crucial for the virulence and replication of the virus
In this study, natural antiviral peptides were virtually screened based on their structural properties against the critical SARS-CoV-2 targets. Screening the peptide library was carried out based on different parameters like toxicity, affinity, and stability to identify a suitable candidate to target the SARS-CoV-2 proteins. These peptides could emerge as promising therapeutics to tackle this deadly virus.
Results
Sorting of antiviral peptides from the AVP database (AVPdb)
To find the therapeutic antiviral peptides against the multiple targets of the novel coronavirus (SARS-CoV-2), the whole library with a total number of 2060 natural AVPs was analyzed. Analyzing each AVP with all five targets was challenging and generated extensive data. So to reduce the complexity of the data, AVPs were narrowed down through three different criteria. The first criterion was that AVPs previously reported against viruses in the coronaviridae family (104 AVPs) were deleted because we intended to find other AVPs that could efficiently target SARS-CoV-2. The second criterion was to remove the AVPs that were toxic, and the ToxinPred web server was used for the prediction of the toxic or non-toxic behavior of the AVPs. We found 69 AVPs having toxic nature. So, we have removed all those toxic AVPs. The third and last criteria were that those AVPs with less than 27 amino acids had been removed. This was done because the Robetta web server selected for modeling these AVPs did not allow the modeling of those peptides with at least 27 amino acids. So, we have removed 1453 AVPs from the library with this criterion. From the compilation of all these three criteria, we have removed a total of 1626 AVPs (104 coronaviridae family, 69 toxic, and 1453 < 27 amino acids) from the 2060 AVPs (Fig. 2).
Fig. 2.
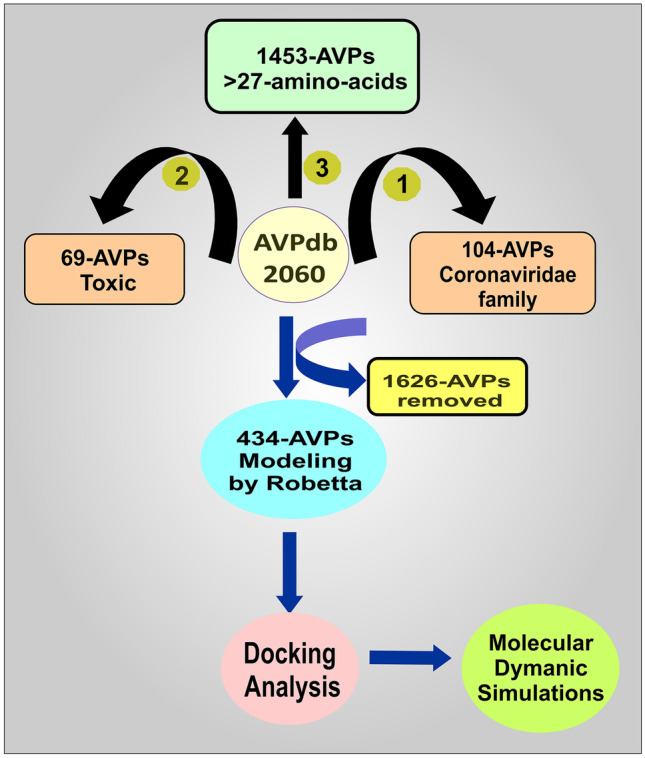
Flow chart for the structure-based screening of the peptides and identification of potential lead AVP for targeting the SARS-CoV-2
Now, 434 AVPs remained to analyze for their binding affinities with the targets. We have modeled all these 434 AVPs using the Robetta web server to obtain this purpose.
Evaluation of the AVPs’ affinity with the molecular targets of SARS-CoV-2
For binding affinity assessment of the selected AVPs (434) with the targets, main protease, and non-structural protein 9 (Nsp9), the ClusPro web server was used. The docking screening of all those 434 AVPs was performed with both targets. With the results of this docking screening, we have selected only the top-scoring AVPs (the most negative free energy > − 1000) among all the 434 AVPs, and the score is also compared with a reference peptide (IEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEIWDLGKGDFR) [19]. The top-scoring AVPs include 11 AVPs (AVP1155, AVP1235, AVP1757, AVP1758, AVP1760, AVP1788, AVP1789, AVP1791, AVP1792, AVP1794, and AVP1801) with both targets. The source and nomenclature of these peptides are presented in Table 1.
Table 1.
The source and nomenclature of the selected AVPs
| Id | Length | Virus | Family | Nomenclature | Source | Reference |
|---|---|---|---|---|---|---|
| AVP1155 | 41 | Influenza A virus | Orthomyxoviridae | PB1 4–1 | Conotoxin | [20] |
| AVP1235 | 33 | Dengue 2 virus | Flaviviridae | DN59 | DENV envelope glycoprotein (gpE) | [21] |
| AVP1757 | 29 | Dengue 1 virus | Flaviviridae | DV2 | DENV2 envelope protein (stem peptide) | [22] |
| AVP1758 | 29 | Dengue 1 virus | Flaviviridae | DV3 | DENV3 envelope protein (stem peptide) | [22] |
| AVP1760 | 29 | Dengue 2 virus | Flaviviridae | DV2 | DENV2 envelope protein (stem peptide) | [22] |
| AVP1788 | 29 | Dengue 4 virus | Flaviviridae | DV2 | DENV2 envelope protein (stem peptide) | [22] |
| AVP1789 | 29 | Dengue 4 virus | Flaviviridae | DV3 | DENV3 envelope protein (stem peptide) | [22] |
| AVP1791 | 29 | Dengue 3 virus | Flaviviridae | DV2 | DENV2 envelope protein (stem peptide) | [22] |
| AVP1792 | 29 | Dengue 2 virus | Flaviviridae | DV3 | DENV3 envelope protein (stem peptide) | [22] |
| AVP1794 | 29 | Dengue 3 virus | Flaviviridae | DV3 | DENV3 envelope protein (stem peptide) | [22] |
| AVP1801 | 34 | Marek’s disease virus | Herpesviridae | gHH3 | MDV H glycoprotein (gH) | [23] |
These docking scores exhibit an excellent binding affinity for both the main protease and the non-structural protein 9 (Nsp9) targets, according to our analysis. We are curious about the possibility that these preselected AVPs could also target other SARS-CoV-2 targets due to their high docking scores. So, with this curiosity, we have found another three targets (RBD/ACE2-B0AT1 complex, SARS-Cov-2 RNA-dependent RNA polymerase in complex with cofactors, and Nsp15 endoribonuclease from SARS-CoV-2) and performed their docking analysis with the selected AVPs by using the same web server (ClusPro). Fortunately, the docking scores of these three targets with the selected AVPs are also as good as the initial targets (main protease and non-structural protein 9 (Nsp9)) (Table 2). Obtained docking scores of peptides were compared with a reference peptide. Although most of the peptides have shown a better affinity for the targets than the reference, we determined the top 4 scoring peptides for each target (Fig. 3). From this, we found AVP1155 was among the top four highest scorings for the targets 6M03, 6W4B, 6M17, and 6VWW, whereas AVP1235 and AVP1788 in targets 6M03, 6M17, and 6VWW. Therefore AVP1155, AVP1235, and AVP1788 can be used as potential lead peptides to target most coronavirus proteins with greater affinity.
Table 2.
The ClusPro docking scores of 11 selected AVPs with all five targets
| AVPs | Therapeutic targets of SARS-CoV-2 | ||||
|---|---|---|---|---|---|
| 6M03 | 6W4B | 6M17 | 6M71 | 6VWW | |
| Reference peptide | −895 | −869 | −911.3 | −915.1 | −998.1 |
| AVP1155 | −1078.5 | −1162.0 | −1669 | −1262.4 | −1599.2 |
| AVP1235 | −1202.0 | −1024.0 | −1452 | −1191 | −1346.1 |
| AVP1757 | −1030.8 | −1116.6 | −1347.8 | −1272.4 | −1209.5 |
| AVP1758 | −1026.3 | −1035.6 | −1257.3 | −1212.3 | −1300.6 |
| AVP1760 | −1029.3 | −1050.9 | −1303.2 | −1275 | −1176.7 |
| AVP1788 | −1080.9 | −1017.8 | −1460.7 | −1155.1 | −1303.1 |
| AVP1789 | −1047.9 | −1030.9 | −1276.8 | −1197.8 | −1279 |
| AVP1791 | −1060.8 | −1053.7 | −1346.2 | −1302.9 | −1213.1 |
| AVP1792 | −1066.8 | −1033.4 | −1248.2 | −1150.2 | −1256.7 |
| AVP1794 | −1024.1 | −1047.5 | −1260.6 | −1264.4 | −1278.1 |
| AVP1801 | −1078.4 | −1014.0 | −1312 | −1139.5 | −1334.9 |
Fig. 3.

Plot representing the binding affinity of the peptides with the different protein targets of the SARS-CoV-2
Physicochemical characterization
The physiochemical parameters of the selected AVPs have been estimated with the ProtParam tool’s utilization. Theoretical pI, half-life, stability index, aliphatic index, and GRAVY scores are among the physiochemical parameters involved (Table 3).
Table 3.
Physiochemical characteristics of the AVPs
| Id | Molecular weight | Theoretical pI | Estimated half-life (hour) | Instability index | Aliphatic index | GRAVY |
|---|---|---|---|---|---|---|
| AVP1155 | 4948.02 | 6.25 | 1.2 | 68.69 | 125.85 | 0.89 |
| AVP1235 | 3443.96 | 5.19 | 30 | 8.74 | 100.61 | 0.776 |
| AVP1757 | 2941.34 | 6.79 | 4.4 | −3 | 91.03 | 0.738 |
| AVP1758 | 3026.46 | 6.79 | 4.4 | 13.77 | 94.14 | 0.569 |
| AVP1760 | 2941.34 | 6.79 | 4.4 | −3 | 91.03 | 0.738 |
| AVP1788 | 2941.34 | 6.79 | 4.4 | −3 | 91.03 | 0.738 |
| AVP1789 | 3026.46 | 6.79 | 4.4 | 13.77 | 94.14 | 0.569 |
| AVP1791 | 2941.34 | 6.79 | 4.4 | −3 | 91.03 | 0.738 |
| AVP1792 | 3026.46 | 6.79 | 4.4 | 13.77 | 94.14 | 0.569 |
| AVP1794 | 3026.46 | 6.79 | 4.4 | 13.77 | 94.14 | 0.569 |
| AVP1801 | 3795.48 | 10.45 | 1.4 | 36.06 | 143.53 | 0.221 |
Among the peptides selected for the multiple targeting, AVP1155 has shown a more excellent score (68.69) and half-life of 1.2 h, indicating its low stability. Besides the high affinity, it cannot be used for therapeutics at present, so it may require further development to improve its stability. However, now it is possible to increase the peptide’s stability due to significant advancements in science. The basic processes for continuing to increase the stability of therapeutic peptides include N- and/or C-terminal reconfiguration (N-acylation or N-formylation at the N-terminal end, C-termination amidation) and even partial replacement, reconfiguration of the backbone, conversion of peptides with the d-amino acid or unnatural amino acid, crosslinking, nanoparticle formulations of the peptides, and/or by increasing the molecular mass. Conjugating the peptide with polymer also increases the stability of the peptide; for example, polyethylene glycol (PEG) is the most promising polymer [24–26]. On the other hand, AVP1235 and AVP1788 are both stable in nature, but AVP 1235 has a half-life of 30 h compared to the 4.4 h of AVP 1788. So, based on these parameters, AVP 1235 has been selected for further stability evaluation at the microscopic level.
Analysis of ΔG and Kd between the peptide and SARS-CoV-2 proteins
Finally, the binding affinity (ΔG) and the Kd (dissociation constant) were calculated using the PRODIGY web server. The tool predicts the binding affinity between the antiviral peptides (AVP1155 and AVP1235) with all the important proteins of SARS-CoV-2 (main protease (3C-like proteinase), spike protein, RNA-directed RNA polymerase, endoribonuclease, and Nsp9 (RNA-binding protein)) (Table 4).
Table 4.
AVPs and proteins from SARS-CoV-2 with predicted values for binding affinities (G) and dissociation constants (Kd)
| S. no. | Protein-antiviral peptide complex | ΔG (kcal mol−1) | Kd (M) at 25.0 ℃ |
|---|---|---|---|
| 1 | 6M03_AVP1155 | −10.2 | 3.2e−08 |
| 2 | 6M03_AVP1235 | −11.7 | 2.6e−09 |
| 3 | 6M17_AVP1155 | −11.3 | 5.4e−09 |
| 4 | 6M17_AVP1235 | −11.3 | 4.8e−09 |
| 5 | 6M71_AVP1155 | −13.0 | 2.8e−10 |
| 6 | 6M71_AVP1235 | −14.0 | 5.4e−11 |
| 7 | 6VWW_AVP1155 | −8.6 | 5.2e−07 |
| 8 | 6VWW_AVP1235 | −7.8 | 2.0e−06 |
| 9 | 6W4B_AVP1155 | −5.1 | 1.8e−04 |
| 10 | 6W4B_AVP1235 | −6.1 | 3.3e−05 |
Molecular dynamics simulations
Molecular dynamics simulation was performed for 100 ns using the GROMACS v5.0 for ten complexes: 6M03_AVP1155, 6M03_AVP1235, 6M17_AVP1155, 6M17_AVP1235, 6M71_AVP1155, 6M71_AVP1235, 6VWW_AVP1155, 6VWW_AVP1235, 6W4B_AVP1155, 6W4B_AVP1235. The root-mean-square deviation (RMSD) reveals the average deviation between the two proteins, i.e., the initially submitted structure and the dynamics nature of the protein under the physiological state during the molecular dynamics simulation. The calculated RMSD value for all the complexes is in the acceptable range. The average RMSD values for seven complexes (6M03_AVP1155, 6M03_AVP1235, 6M17_AVP1155, 6M17_AVP1235, 6M71_AVP1155, 6M71_AVP1235, and 6W4B_AVP1235) were less than 0.5 nm, whereas three complexes (6VWW_AVP1155, 6VWW_AVP1235, and 6W4B_AVP1155) have average RMSD of more than 0.5 nm. The stable RMSD was obtained for 6M03_AVP1155, 6M03_AVP1235, 6M17AP_1235, 6M71_AVP1155, and 6M71_AVP1235, whereas the remaining complexes were stable with little deviation (Fig. 4). The root-mean-square fluctuation then characterizes the movement of each residue involved in molecular dynamics. For each of the complexes described above, RMSF was computed. The average RMSF score for all the complexes was below 0.2 nm except for the 6VWW_AVP1155, 6VWW_AVP1235, 6W4B_AVP1155, and 6W4B_AVP1235 (Fig. 5). The gyration radius indicates the structural compactness of the questioned protein. Proteins with the smallest gyration radius represent the tightest packing and the better protein structure. Here, we have calculated the radius of gyration for all the selected protein-peptide complexes. The Rg score for all the complexes was less than 3.0 nm. Six complexes show the Rg between 1.5 and 1.9 nm, whereas in four complexes, Rg scores were between 2.4 and 2.9 nm (Fig. 6). Solvent-accessible surface area (SASA) of a protein refers to the surface area of the protein that is accessible to the available solvent in the virtual physiological condition. Except for complexes 6M71_AVP1235 (258.64 nm2), 6VWW_AVP1155 (358.98 nm2), and 6VWW_AVP1235 (356.56 nm2), all the complexes have SASA scores between 118.56 and 167.82 nm2, which shows the low accessibility for the solvent (Fig. 7). Finally, the hydrogen bond was analyzed that is the formation and the breakage of the hydrogen bond during the molecular dynamics simulation. Complexes 6M03_AVP1155 and 6M03_AVP1235 have an average hydrogen bonding of ~10, whereas 6M71_AVP1155 and 6M71_AVP1235 with ~ 5 hydrogen bonds during the simulation period. The lowest number of hydrogen bonding was found for complexes 6VWW_AVP1155 and 6VWW_AVP1235, i.e., between 1 and 1.5 (Fig. 8).
Fig. 4.

RMSD plot of selected complexes of antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9)
Fig. 5.

Graphical representation of RMS fluctuation plot of selected complexes of antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9)
Fig. 6.

Radius of gyration of antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9)
Fig. 7.

Solvent accessibility surface area plot of selected complexes of antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9)
Fig. 8.

Graphical representation of hydrogen bond formation and breakage during the molecular dynamics simulation between the selected complexes of antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9)
Discussion
Peptides can antagonize the protein–protein interactions, making them critical in the therapeutics research of various deadly pathogens. Currently, around 140 peptide therapies are under clinical evaluation, and among them, 21 peptide-based therapies, such as plitidepsin, aviptadil, BIO-11006, and solnatide, are under development against COVID-19 [27, 28]. Fewer side effects, better tolerance, minimal interference, and greater specificity are the main benefits of peptides over chemical drugs [29]. In the future, after further research, peptide-based drugs can have a promising impact on the progression of COVID-19. Here, we have evaluated five attractive targets for determining the potential peptides that can be used to develop anti-nCoV therapeutic drugs. The main protease is crucial for the virus as it processes the replicase protein required to replicate the coronavirus [30]. Nsp9 is an RNA-binding replicase protein encoded by the ORF1 and is an essential protein for the RNA synthesis of the virus [31]. As the primary inducer of the pathogenicity of this virus, the ACE-2 receptor-binding domain is the most common target of SARS-CoV-2. Extreme affinity exists between the RBD in the spike protein and the ACE-2 receptors expressed in human cells, and this interaction facilitates viral fusion and entry into host cells [32, 33]. RNA-dependent RNA polymerase, also known as Nsp12, plays a pivotal role in the replication and transcription of the virus. Therefore, it is one of the most attractive targets of SARS-CoV-2 [34]. The Nsp15 endoribonuclease aids in the evasion of the virus from the immune response by preventing its detection by the dsRNA sensors [18].
Here, we have used the structure-based virtual screening to identify stable, non-toxic, and strongly interacting peptides for targeting the SARS-CoV-2, leading to pandemic and mortality worldwide. There are currently around 21 peptide-based drugs under clinical evaluation, but more alternatives and therapies must be identified to tackle the current situation. The selection of peptides from a massive number of natural antiviral peptides has been made using three criteria. From the library of 2060 antiviral peptides, we removed those peptides that are already under clinical trials against SARS as we want to identify some novel therapeutic peptides against SARS-CoV-2. The second criterion used to narrow down the list was toxicity, using the ToxinPred web server and selecting those peptides which were non-toxic [35, 36]. The third criterion we used was peptide length, and we selected 434 peptides having a minimum of 27 peptides as Robetta cannot model the peptides of small length [37]. The other reason to eliminate the small peptide is their low specificity and less stability, as they cannot maintain their secondary structure and can be easily degraded [38, 39]. The interaction between the peptide structure and the two targets (main protease and Nsp9) was evaluated by docking them in the ClusPro server, predicting the affinity based on minimum energy between the protein and peptide [40, 41]. The 11 peptides with lower scores for the minimum energy compared to the reference peptide were selected and docked with three more essential targets (ACE-2 receptor-binding domain, RNA-dependent RNA polymerase, and Nsp15 endoribonuclease) of the SARS-CoV-2 to determine whether it can target the multiple sites of the pathogen.
Interestingly, these 11 peptides have shown a strong affinity with the main protease and Nsp9 and the other three targets. Among them, we selected two peptides (1155 and 1235) which showed overall better interaction with all the five targets, and these peptides were further verified for their structural stability at the microscopic level by molecular dynamics simulations for 100 ns. We also determined the physicochemical characteristics, such as aliphatic index, hydrophobicity, and half-life, using the ProtParam [42, 43].
Moreover, this study has identified 11 potential therapeutic peptides that can target multiple sites of the SARS-CoV-2 to obstruct the protein–protein interaction of the virus and host, which are critical for the fusion, replication, and virulence of the novel coronavirus. Our study provides new alternatives in the direction of peptide-based drug development to treat COVID-19. Further research to determine their efficacy and safety is required to acknowledge their impact on the pandemic caused by SARS-CoV-2 [44]. The post-MD analysis of all the aforementioned SARS-CoV-2 proteins and antiviral peptides calculated the electrostatic energy contribution, van der Waals energy contribution, and total energy contribution (Fig. 9). Changes in the energy profile for 100 ns of the molecular dynamics simulation were studied, and it was discovered that all of these energy charts are stable throughout the dynamics [33, 43, 44].
Fig. 9.

Energy contribution for the antiviral peptides (AVP1155 and AVP1235) and SARS-CoV-2 proteins (main protease, RBD with ACE2, RdRp, Nsp15, and Nsp9). A Electrostatic energy pattern from an MD simulation plotted. The description depicts how electrostatic forces changed throughout the MD simulation. B van der Waals (VDW) energy contribution and changes in the VDW energy during the MD simulation. C Contributions to the total energy and changes in the total energy over the duration of the 100 ns MD simulation
Conclusion
As essential proteins for the replication and virulence of the SARS-CoV-2 mediated disease progression, the main protease and Nsp9 are potential targets to combat COVID-19. This study has identified the natural antiviral peptides which are non-toxic and have a strong affinity for target proteins by structural-based virtual screening. These selected peptides can be used as potential therapeutics to block the protein–protein interaction of the virus with the host organism. In addition to the targets mentioned above, these peptides have shown strong affinity in three more targets of the SARS-CoV-2, which indicates their multiple targeting properties. The trajectory analysis for 100 ns during MDS has indicated their structure stability at the microscopic level. The screened peptides can be further evaluated for their efficacy and safety by in vitro and in vivo experiments. Our findings can aid in the acceleration of the development of therapeutics against SARS-CoV-2.
Methodology
Screening of antiviral peptide library
To identify a therapeutic peptide that can hit the multiple targets of the novel coronavirus (SARS-CoV-2), we have screened a library of natural antiviral peptides (AVPs) taken from a database of antiviral peptides (AVPdb) (http://crdd.osdd.net/servers/avpdb/dd.php). This AVPdb is an inclusive collection of antiviral peptides targeting 60 pathologically highlighted viruses [45]. This library involves 2060 natural antiviral peptides from various families of different viruses. Hence, we have removed the antiviral peptides reported against the coronaviridae family because we wanted to identify other AVPs that can potentially target the significant hits of the novel coronavirus.
We have reduced this library based on toxicity and the number of amino acids with the intention of modeling to achieve the goal of potential therapeutic peptides. The first criterion was the toxic or non-toxic nature of the AVPs; the ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/) has been utilized to predict the toxic or non-toxic nature of the AVPs. This server scans the dataset of toxic peptides and their single mutant analogs from Swiss-Prot and TrEMBLE to evaluate the toxicity of all potentially overlapping peptides [46].
Tertiary structure prediction of AVPs
To model AVPs, we have introduced these AVPs to the Robetta server (http://robetta.bakerlab.org). This server allows predicting the automated protein structure with the utilization of genomic data (Peptide/protein sequences). It generates the models of the provided sequences via the homology modeling method if the sequence matches any known protein sequence. This server analyzes the input sequences and pairs them into the domains for homology modeling to construct the models. The sequences lack the homology with any known protein that is structured by de novo modeling. Unfortunately, this server does not allow input sequences of less than 27 amino acids; therefore, we have removed all those AVPs that do not have 27 amino acids [47]. Removing short peptides is also a reason for stability; most of the peptides approved for clinical trials are higher than 25 amino acids, suggesting that these selected AVPs can be stable and require less conjugation for therapeutic purposes [48].
Assessment of binding affinity of AVPs with the targets of the novel coronavirus
From the screening of the whole antiviral peptide database (AVPdb), we have selected 434 AVPs to evaluate the binding affinity with the targets of the novel coronavirus. Initially, we selected only two targets, the main protease and Nsp9. The protein data bank (https://www.rcsb.org/structure/) has provided the crystal structures of the main protease (PDB-6M03) and Nsp9 (PDB-6W4B). To perform the docking analysis of all 434 AVPs with the targets (main protease and Nsp9), the ClusPro web server v2.0 (https://cluspro.bu.edu/login.php) has been utilized. The PDB files for the receptor and ligand are input into this server, which is an entirely automated docking server. It utilizes the PIPER algorithm to predict the rank for the stability level, interaction potential, semi-definite programming-based underestimation (SDU) energy, and cluster size [49, 50].
Binding affinity analysis of the top-scoring AVPs with other potential targets
With the ClusPro web server results, we selected the AVPs with the most negative weighted scores with both targets (main protease and Nsp9) to further assess the binding affinity. A reference peptide made up of two sequential helices taken from the protease domain (PD) of angiotensin-converting enzyme 2 (ACE2), which only targets the SARS-CoV-2 receptor, was used to compare the docking scores of the chosen AVPs [51]. Surprisingly, we found excellent docking scores of the selected AVPs compared to the reference peptide. The good docking scores generated a new curiosity in our mind that the selected peptides may also have good binding efficacy with other targets of the novel coronavirus. With this curiosity, we performed a literature survey again and found some other crucial targets of SARS-CoV-2. From this literature survey, we have selected another three targets of the SARS-CoV-2, which involves RBD/ACE2-B0AT1 complex (PDB-6M17), SARS-Cov-2 RNA-dependent RNA polymerase in complex with cofactors (PDB-6M71), and Nsp15 endoribonuclease from SARS-CoV-2 (PDB-6VWW) [17, 18, 34]. The crystal structure of these three targets was extracted from the protein data bank. The ClusPro web server has been utilized again to analyze the binding affinity of the selected AVPs with these three targets (6M17, 6M71, and 6VWW).
Characterization of the AVPs through physicochemistry
To further examine the physiochemical characteristics of selected AVPs, we have utilized the ProtParam tool (https://web.expasy.org/protparam/), which predicts different physiochemical parameters of the protein (AVPs) [52]. The parameters include molecular weight, stability index, aliphatic index, theoretical Pi, and in vivo half-life based on the “N-end rule,” which talks about how proteins degrade based on N-terminal amino acids [53]. Additionally, it was predicted that if a protein’s instability index is less than 40, it will be stable, and vice versa [42, 54].
Determination of ΔG (binding affinity) and dissociation constant (Kd)
The PROtein binDIng enerGY (PRODIGY) prediction tool (https://wenmr.science.uu.nl/prodigy/) was utilized before performing molecular dynamics to estimate the binding energy and dissociation constant between the antiviral peptide and targets (complexes). The protein-peptide complexes (chains) were renamed using the PyMol standalone tool, and the complexes were saved in pdb format once again. These complexes were required by the PRODIGY software for binding energy prediction, and the protein was defined in interactor 1, while the peptide chain name was added in interactor 2.
Molecular dynamics simulations to ensure the structural stability of the AVP target complexes
The structural stability and equilibration state of the selected lead AVPs (AVP1155 and AVP1235) were ensured with the therapeutic targets of SARS-CoV-2 (main protease (3C-like proteinase), spike protein, RNA-directed RNA polymerase, endoribonuclease, and Nsp9 (RNA-binding protein)) in the virtual in vivo environment. Molecular dynamics simulation was performed using the GROMACS v5.0 standalone tool [55, 56].
Only the chains that interact with peptides and antiviral peptides, as well as the chains that do not interact with peptides, were removed for PDB ID: 6M17 and 6M71. Six chains, A, B, C, D, E, and F, are present in PDB ID: 6M17, but only chain E interacted with the antiviral peptides. The protein-peptide complex had all its chains removed except for chain E. There were four chains in PDB ID: 6M71, but only chain A interacted with peptides; therefore, all other chains (chains B, C, and D) were eliminated using the PyMol standalone tool. The whole protein opted for simulation in the cases of PDB IDs 6M03, 6VWW, and 6W4B.
To evaluate the interactions between the AVPs and their prospective molecular targets at the microscopic level, GROMOS96 43a1 force field and the particle mesh Ewald summation method were used to run the MD simulation of the complexes mentioned above [57, 58]. For protein topology creation and characterization of bonded and non-bonded regions, the pdb2gmx command was given. Na+ and Cl− ions were used for system charge neutralization. Energy minimization was performed that utilizes the steepest descent algorithm to confirm the appropriate geometry of the system without any steric clashes [59]. For system equilibration, the leapfrog algorithm was used with an increase in pressure of the system up to 1 bar and 300 K temperature [60]. The trajectory of the MD simulation was analyzed for 100 ns.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
SS, PC, VS, and AR are thankful to the Central University of Rajasthan for the fellowship. BVK is thankful to NMIMS (deemed-to-be) University, Mumbai, for providing a computational facility. VKP is thankful to the Central University of Rajasthan for providing a lab facility.
Author contribution
The protocol was designed by SS, PC, VS, BVK, and VKP. Methodology was performed by SS, PC, VS, AR, and BVK. The manuscript was written by SS, PC, VS, AR, BVK, and VKP.
Availability of data and material
No data is available.
Code availability
N/A.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mackenzie JS, Smith DW (2020) COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don’t. Microbiol Aust MA20013-MA. 10.1071/MA20013 [DOI] [PMC free article] [PubMed]
- 2.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 — preliminary report. N Engl J Med. 2020 doi: 10.1056/NEJMoa2007764. [DOI] [PubMed] [Google Scholar]
- 3.Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug discoveries & therapeutics. 2020;14(1):58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 4.Triggle CR, Bansal D, Farag EABA, Ding H, Sultan AA (2020) COVID-19: learning from lessons to guide treatment and prevention interventions. mSphere 5(3):e00317–20. 10.1128/mSphere.00317-20 [DOI] [PMC free article] [PubMed]
- 5.Nicol MQ, Ligertwood Y, Bacon MN, Dutia BM, Nash AA. A novel family of peptides with potent activity against influenza A viruses. J Gen Virol. 2012;93(5):980–986. doi: 10.1099/vir.0.038679-0. [DOI] [PubMed] [Google Scholar]
- 6.Hurt AC, Hui DS, Hay A, Hayden FG. Overview of the 3rd isirv-Antiviral Group Conference – advances in clinical management. Influenza Other Respir Viruses. 2015;9(1):20–31. doi: 10.1111/irv.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nyanguile O (2019) Peptide antiviral strategies as an alternative to treat lower respiratory viral infections. Front Immunol 101366. 10.3389/fimmu.2019.01366 [DOI] [PMC free article] [PubMed]
- 8.Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho NN, Franco OL. Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci. 2019;76(18):3525–3542. doi: 10.1007/s00018-019-03138-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones JC, Turpin EA, Bultmann H, Brandt CR, Schultz-Cherry S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J Virol. 2006;80(24):11960–11967. doi: 10.1128/jvi.01678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18(17–18):807–817. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Littler DR, Gully BS, Colson RN, Rossjohn J (2020) Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. bioRxiv 03(28):013920. 10.1101/2020.03.28.013920 [DOI] [PMC free article] [PubMed]
- 14.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (New York, NY) 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Yan L (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. 368(6492):779–82. 10.1126/science.abb7498 [DOI] [PMC free article] [PubMed]
- 16.Panda M, Kalita E, Singh S, Kumar K, Rao A, Prajapati VK (2022) MiRNA-SARS-CoV-2 dialogue and prospective anti-COVID-19 therapies. 120761 [DOI] [PMC free article] [PubMed]
- 17.Liu X, Fang P, Fang L, Hong Y, Zhu X, Wang D, et al. Porcine deltacoronavirus Nsp15 antagonizes interferon-β production independently of its endoribonuclease activity. Mol Immunol. 2019;114:100–107. doi: 10.1016/j.molimm.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng X, Hackbart M, Mettelman RC, O’Brien A, Mielech AM, Yi G, et al. Coronavirus non-structural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci. 2017;114(21):E4251–E4260. doi: 10.1073/pnas.1618310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Y, Král P (2020) Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. 14(4):5143–7 [DOI] [PMC free article] [PubMed]
- 20.Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of flavivirus entry derived from the E protein stem. J Virol. 2010;84(24):12549–12554. doi: 10.1128/JVI.01440-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang H, Xu Y, Li L, Weng L, Wang Q, Zhang S, et al. Inhibition of influenza virus replication by constrained peptides targeting nucleoprotein. Antiviral Chem Chemother. 2011;22(3):119–130. doi: 10.3851/IMP1902. [DOI] [PubMed] [Google Scholar]
- 22.Lok S-M, Costin JM, Hrobowski YM, Hoffmann AR, Rowe DK, Kukkaro P, et al. Release of dengue virus genome induced by a peptide inhibitor. PLoS ONE. 2012;7(11):e50995. doi: 10.1371/journal.pone.0050995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X-J, Li C-G, Chi X-J, Wang M. Characterisation and evaluation of antiviral recombinant peptides based on the heptad repeat regions of NDV and IBV fusion glycoproteins. Virology. 2011;416(1–2):65–74. doi: 10.1016/j.virol.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Evans BJ, King AT, Katsifis A, Matesic L, Jamie JF (2020) Methods to enhance the metabolic stability of peptide-based PET radiopharmaceuticals. Molecules (Basel, Switzerland) 25(10). 10.3390/molecules25102314 [DOI] [PMC free article] [PubMed]
- 25.Yao JF, Yang H, Zhao YZ, Xue M. Metabolism of peptide drugs and strategies to improve their metabolic stability. Curr Drug Metab. 2018;19(11):892–901. doi: 10.2174/1389200219666180628171531. [DOI] [PubMed] [Google Scholar]
- 26.Al Musaimi O, Lombardi L, Williams DR, Albericio FJP. Strategies for improving peptide stability and delivery. 2022;15(10):1283. doi: 10.3390/ph15101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perišić O (2020) Recognition of potential COVID-19 drug treatments through the study of existing protein-drug and protein-protein structures: an analysis of kinetically active residues [DOI] [PMC free article] [PubMed]
- 28.Wu MA, Fossali T, Pandolfi L, Carsana L, Ottolina D, Frangipane V et al (2020) COVID-19: the key role of pulmonary capillary leakage. An observational cohort study. medRxiv
- 29.Mustafa S, Balkhy H, Gabere MN. Current treatment options and the role of peptides as potential therapeutic components for Middle East Respiratory Syndrome (MERS): a review. J Infect Public Health. 2018;11(1):9–17. doi: 10.1016/j.jiph.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutton G, Fry E, Carter L, Sainsbury S, Walter T, Nettleship J, et al. The Nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure. 2004;12(2):341–353. doi: 10.1016/j.str.2004.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17(6):613–620. doi: 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gopi P, Gurnani M, Singh S, Sharma P, Pandya P (2022) Structural aspects of SARS-CoV-2 mutations: implications to plausible infectivity with ACE-2 using computational modeling approach. J Biomol Struct Dyn 1–16. 10.1080/07391102.2022.2108901 [DOI] [PubMed]
- 34.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma V, Singh S, Ratnakar TS, Prajapati VK. Chapter 29 - Immunoinformatics and reverse vaccinology methods to design peptide-based vaccines. In: Tripathi T, Dubey VK, editors. Advances in protein molecular and structural biology methods. Academic Press; 2022. pp. 477–487. [Google Scholar]
- 37.Song Y, DiMaio F,Wang RY-R, Kim D, Miles C, Brunette T et al (2013) High-resolution comparative modeling with RosettaCM. Structure 21(10):1735–1742 [DOI] [PMC free article] [PubMed]
- 38.Mooney C, Haslam NJ, Pollastri G, Shields DC. Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS ONE. 2012;7(10):e45012. doi: 10.1371/journal.pone.0045012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Du X, Hashim S, Shy A, Xu B. Aromatic–aromatic interactions enable α-helix to β-sheet transition of peptides to form supramolecular hydrogels. J Am Chem Soc. 2017;139(1):71–74. doi: 10.1021/jacs.6b11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12(2):255. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chouhan P, Singh S, Sharma V, Prajapati VK (2022) Therapeutics. Anti-IL-10 antibody humanization by SDR grafting with enhanced affinity to neutralize the adverse response of interleukin-10. 28(5):1–14
- 42.Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. Springer, pp 571–607
- 43.Gopi P, Singh S, Islam MM, Yadav A, Gupta N, Pandya P (2022) Thermodynamic and structural profiles of multi‐target binding of vinblastine in solution. e2989 [DOI] [PubMed]
- 44.Singh S, Gopi P. Pandya PJSAPAM, Spectroscopy B. Structural aspects of formetanate hydrochloride binding with human serum albumin using spectroscopic and molecular modeling techniques. 2022;281:121618. doi: 10.1016/j.saa.2022.121618. [DOI] [PubMed] [Google Scholar]
- 45.Qureshi A, Thakur N, Tandon H, Kumar M. AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014;42(D1):D1147–D1153. doi: 10.1093/nar/gkt1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source Drug Discovery C et al (2013) In silico approach for predicting toxicity of peptides and proteins. PloS one 8(9):e73957-e. 10.1371/journal.pone.0073957 [DOI] [PMC free article] [PubMed]
- 47.Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32(suppl_2):W526-W31 [DOI] [PMC free article] [PubMed]
- 48.Lau JL, Dunn MK. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26(10):2700–2707. doi: 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 49.Kozakov D, Hall DR, Beglov D, Brenke R, Comeau SR, Shen Y et al (2010) Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins: Struct Funct Bioinform 78(15):3124–30 [DOI] [PMC free article] [PubMed]
- 50.Narula A, Pandey RK, Khatoon N, Mishra A, Prajapati VK. Excavating chikungunya genome to design B and T cell multi-epitope subunit vaccine using comprehensive immunoinformatics approach to control chikungunya infection. Infect Genet Evol. 2018;61:4–15. doi: 10.1016/j.meegid.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 51.Han Y, Král P (2020) Computational design of ACE2-based short peptide inhibitors of SARS-CoV-2. ACS Nano [DOI] [PMC free article] [PubMed]
- 52.Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sanchez JC, et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis. 1993;14(1):1023–1031. doi: 10.1002/elps.11501401163. [DOI] [PubMed] [Google Scholar]
- 53.Zhang C, Vasmatzis G, Cornette JL, DeLisi C. Determination of atomic desolvation energies from the structures of crystallized proteins. J Mol Biol. 1997;267(3):707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 54.Ojha R, Gupta N, Naik B, Singh S, Verma VK, Prusty D, et al. High throughput and comprehensive approach to develop multiepitope vaccine against minacious COVID-19. Eur J Pharm Sci. 2020;151:105375. doi: 10.1016/j.ejps.2020.105375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pandey RK, Sharma D, Bhatt TK, Sundar S, Prajapati VK. Developing imidazole analogues as potential inhibitor for Leishmania donovani trypanothione reductase: virtual screening, molecular docking, dynamics and ADMET approach. J Biomol Struct Dyn. 2015;33(12):2541–2553. doi: 10.1080/07391102.2015.1085904. [DOI] [PubMed] [Google Scholar]
- 56.Naik B, Mattaparthi VSK, Gupta N, Ojha R, Das P, Singh S et al (2021) Chemical system biology approach to identify multi-targeting FDA inhibitors for treating COVID-19 and associated health complications. J Biomol Struct Dyn 1–25. 10.1080/07391102.2021.1931451 [DOI] [PMC free article] [PubMed]
- 57.Singh S, Prajapati VK (2022) Exploring actinomycetes natural products to identify potential multi-target inhibitors against Leishmania donovani. 3 Biotech 12(9):235. 10.1007/s13205-022-03304-1 [DOI] [PMC free article] [PubMed]
- 58.Singh S, Kumar K, Panda M, Srivastava A, Mishra A, Prajapati VK. High-throughput virtual screening of small-molecule inhibitors targeting immune cell checkpoints to discover new immunotherapeutics for human diseases. Mol Diversity. 2022 doi: 10.1007/s11030-022-10452-2. [DOI] [PubMed] [Google Scholar]
- 59.Jaidhan B, Rao PS, Apparao A. Energy minimization and conformation analysis of molecules using steepest descent method. Int J Comput Sci Inf Technol. 2014;5(3):3525–3528. [Google Scholar]
- 60.Naik B, Gupta N, Ojha R, Singh S, Prajapati VK, Prusty D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int J Biol Macromol. 2020;160:1–17. doi: 10.1016/j.ijbiomac.2020.05.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data is available.
N/A.