

Abstract
Protein engineering is currently being revolutionized by deep learning applications, especially through natural language processing (NLP) techniques. It has been shown that state-of-the-art self-supervised language models trained on entire protein databases capture hidden contextual and structural information in amino acid sequences and are capable of improving sequence-to-function predictions. Yet, recent studies have reported that current compound-protein modeling approaches perform poorly on learning interactions between enzymes and substrates of interest within one protein family. We attribute this to low-grade substrate encoding methods and over-compressed sequence representations received by downstream predictive models. In this study, we propose a new substrate-encoding based on Extended Connectivity Fingerprints (ECFPs) and a convolutional-pooling of the sequence embeddings. Through testing on an activity profiling dataset of haloalkanoate dehalogenase superfamily that measures activities of 218 phosphatases against 168 substrates, we show substantial improvements in predictive performances of compound-protein interaction modeling. In addition, we also test the workflow on three other datasets from the halogenase, kinase and aminotransferase families and show that our pipeline achieves good performance on these datasets as well. We further demonstrate the utility of this downstream model architecture by showing that it achieves good performance with six different protein embeddings, including ESM-1b (Rives et al., 2021), TAPE (Rao et al., 2019), ProtBert, ProtAlbert, ProtT5, and ProtXLNet (Elnaggar et al., 2021). This study provides a new workflow for activity prediction on novel substrates that can be used to engineer new enzymes for sustainability applications.
1. Introduction
Deep learning-guided directed evolution for proteins has largely improved the protein sequence-to-function predictions and enabled the design of novel sequences with desired features. The growing High-Performance Computing (HPC) and advances in Natural Language Processing (NLP) have brought promising techniques which allow researchers to use large protein databases to enhance predictions of sequence properties (or annotations) with relatively small experimental datasets. These techniques include (1) self-supervised learning based on advanced NLP models (i.e., Transformer (Vaswani et al., 2017), BERT (Devlin et al., 2018), etc.) that can extract hidden contextual and structural patterns in amino acid sequences from a large number of unlabeled protein sequences and convert sequences to representations (often referred to as embeddings) that improve downstream prediction tasks, and (2) transfer-learning, which uses such embeddings as input for subsequent predictive models trained on a relatively smaller dataset.
Recent literature in this area has reported various approaches of generating sequence embeddings; for example, ProtTrans (Elnaggar et al., 2021) alone has presented six models trained on databases containing up to 2.1 billion sequences. Different types of embeddings have also achieved some successes in characterizing protein properties with a small amount of data. For example, the Low-N method (Biswas et al., 2021) has brought the number of experimentally characterized sequences in training set down to 24. However, a recent study (Goldman et al., 2021) showed that it is difficult to predict enzyme activity from sequence when substrates are encoded and concatenated to sequence representations as inputs to the predictive model. This leads to the objective of our study, which is to better represent the two inputs to the compound-protein interaction (CPI) model, protein sequence and substrate. Downstream supervised models for predicting properties found in current literature typically take an average over the position-wise embeddings of sequences as inputs (averaging over protein length)(Elnaggar et al., 2021). This leaves out the valuable structural information captured by the upstream self-supervised training via language models which heavily rely on the order of the amino acid sequences.
In this study, we use a “convolutional pooling” approach to utilize such information containing local sequential patterns, which should be useful in downstream prediction tasks. We also propose a novel substrate encoding method, count-encoding of extended-connectivity fingerprints (ECFPs) (Rogers and Hahn, 2010), to better characterize substrates as inputs, and compare it with the Morgan bit vector (Morgan, 1965) used in previous studies. We demonstrate the efficacy of our approach on phosphatase activities data from Huang et al. (2015), where enzyme assays were performed to measure activities of 218 phosphatases in haloalkanoate dehalogenase superfamily against 168 substrates. We also test our methods on three relatively smaller enzyme activity datasets preprocessed and analyzed in the aforementioned study by Goldman et al. (2021).
In addition to examining the basic performance of the model through randomly splitting the enzyme-substrate pairs to training, validation and test sets, we further curated each dataset into two different splits to evaluate the performance of characterizing new sequences and new substrates. The two tasks split the data in a way such that the model was trained and tested on novel sequences or substrates, not seen in the training set, respectively (hereinafter, referred to as “novel sequences characterization task” and “novel substrates characterization task”). Good prediction in such tasks implies capability of identifying enzymes that would act on novel substrates of interest (enzyme discovery) as well as identifying new sequences that are able to act on known substrates with potentially improved activity (enzyme engineering).
2. Method
2.1. Overview
Our proposed prediction pipeline starts with extracting “embeddings” (values in latent space) from large language models pre-trained on massive protein sequence data. Four protein language models provided by Elnaggar et al. (2021), as well as ESM-1b (Rives et al., 2021) and TAPE (Rao et al., 2019) models, are utilized in this study. Predictive performance of the pipeline is mainly evaluated with ESM-1b (Rives et al., 2021) representations, while embeddings generated by other language models are also used to validate the proposed methods.
2.2. Count-Encoder of Extended-Connectivity Fingerprints
Extended-connectivity fingerprints (ECFPs) (Rogers and Hahn, 2010) are circular topological descriptors derived using a variant of Morgan algorithm, which was proposed to identify molecular isomorphism. ECFPs equally takes into account all molecular fragments and encode all groups of neighboring atoms connected. As shown in Figure 1A, the first level ECFP identifiers (i.e., ECFP2) simply represent each non-hydrogen atom in the molecule with bonds connected. The substructures of each successive level are concatenations of the previous level substructures and their immediate neighboring atoms, which corresponds to higher levels of ECFPs (ECFP4, ECFP6, etc.). The advantage of using ECFP encoding is that all substructures of a molecule are represented directly in a vector. This feature distinguishes the ECFP and Morgan algorithm. The ECFP algorithm collects all identifiers after each iteration, in addition to the initial atom identifiers, into a set and retains the intermediate atom identifiers that are discarded in Morgan algorithm. The list of ECFP identifiers are then converted to a vector containing the counts of each distinct ECFPs, as shown in Figure 1B. Although the structural similarities of substructures are not reflected by ECFP identifiers themselves, identifiers of all shared substructures representing their common features can always be found in the count encodings. Different levels of ECFPs are obtained using Chemistry Development Kit (CDK) (Willighagen et al., 2017), an open source modular Java libraries for Cheminformatics.
Figure 1:
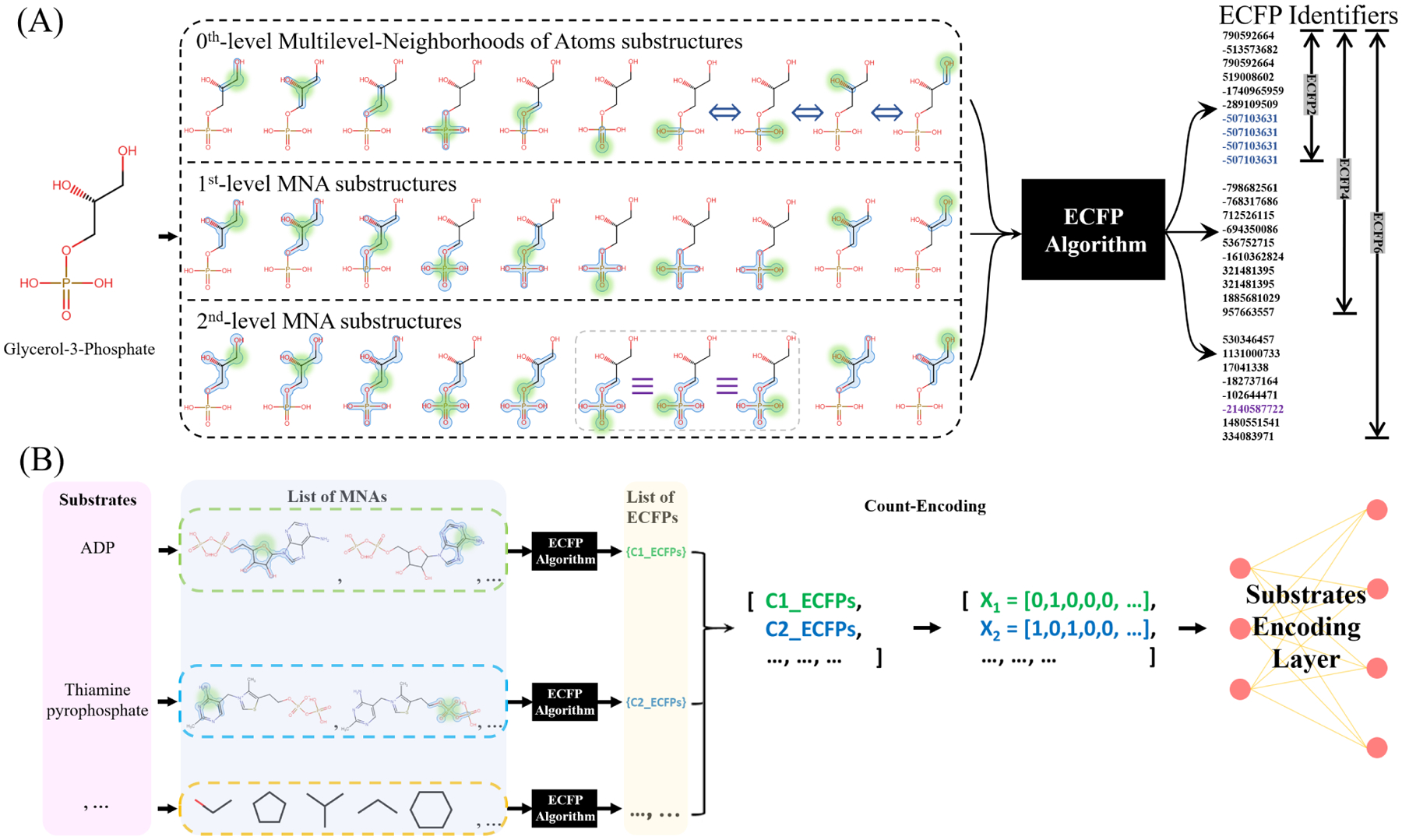
(A) Extended Connectivity Fingerprints of Glycerol-3-Phosphate. Multilevel neighborhoods of atoms (MNAs) substructures are identified surrounding each non-hydrogen atom before being converted to a set of ECFP identifiers through a numeric identifier conversion algorithm. (B) Count encoding of ECFP identifiers, each dimension in the vector representation represents the count of occurrence of a different ECFP identifier.
2.3. Pooling the Bulky Sequence Embeddings with Convolutional Neural Networks
Current protein-level predictions typically use a global pooling that averages over the sequence length in the last hidden state of pretrained language models (Elnaggar et al., 2021). As shown in Figure 2, this results in a fixed-size vector for each protein of different length. Using average pooling effectively simplifies the downstream predictive model, while at the same time impairs informative local patterns and leaves out token-level variance. In this work, we use convolutional neural networks (CNNs) to leverage the complete position-wise embeddings of amino acids. This allows the representation to preserve the sequential information of the original protein sequence as well as the structural patterns extracted by the pretrained language model.
Figure 2:
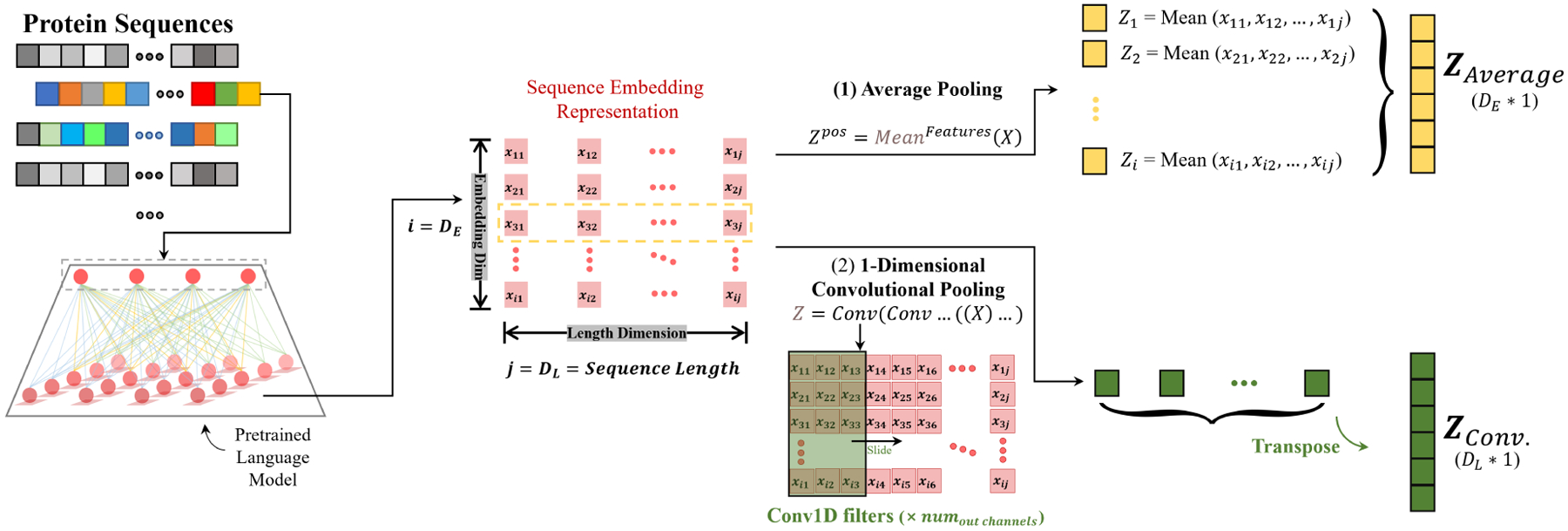
Comparison between average pooling and the idea of convolutional pooling. Global average pooling, which is used in current top models, simply averages over the entire length-dimension of the last layer of pretrained language models. It converts the representation to a fixed-length vector with dimension equals to the pretrained language model’s embedding dimension. The idea of convolutional pooling is to apply convolutional filters along the sequence length direction in order to obtain a same length sequential output as the protein sequences (padded to the same length).
Figure 3 illustrates “convolutional pooling” in a downstream predictive model. Convolutional pooling utilizes a combined structure of convolutional layers and a residual block to improve the flexibility of the model to adapt to the true number of degrees of freedom in the original problem without significant prior knowledge (He et al., 2016). Embedding dimension (DE) in the architecture is determined by the language model used, which is 1280 for ESM-1b. Substrate encoding dimension (DS) in Figure 3(A) is 1024 as Morgan bit vector is used, while the value of DS in Figure 3(B) is 1413 for ECFP6 encoder. Maximum sequence length (DL) ranges from ~300 to ~600 across the four datasets analyzed in this work.
Figure 3:
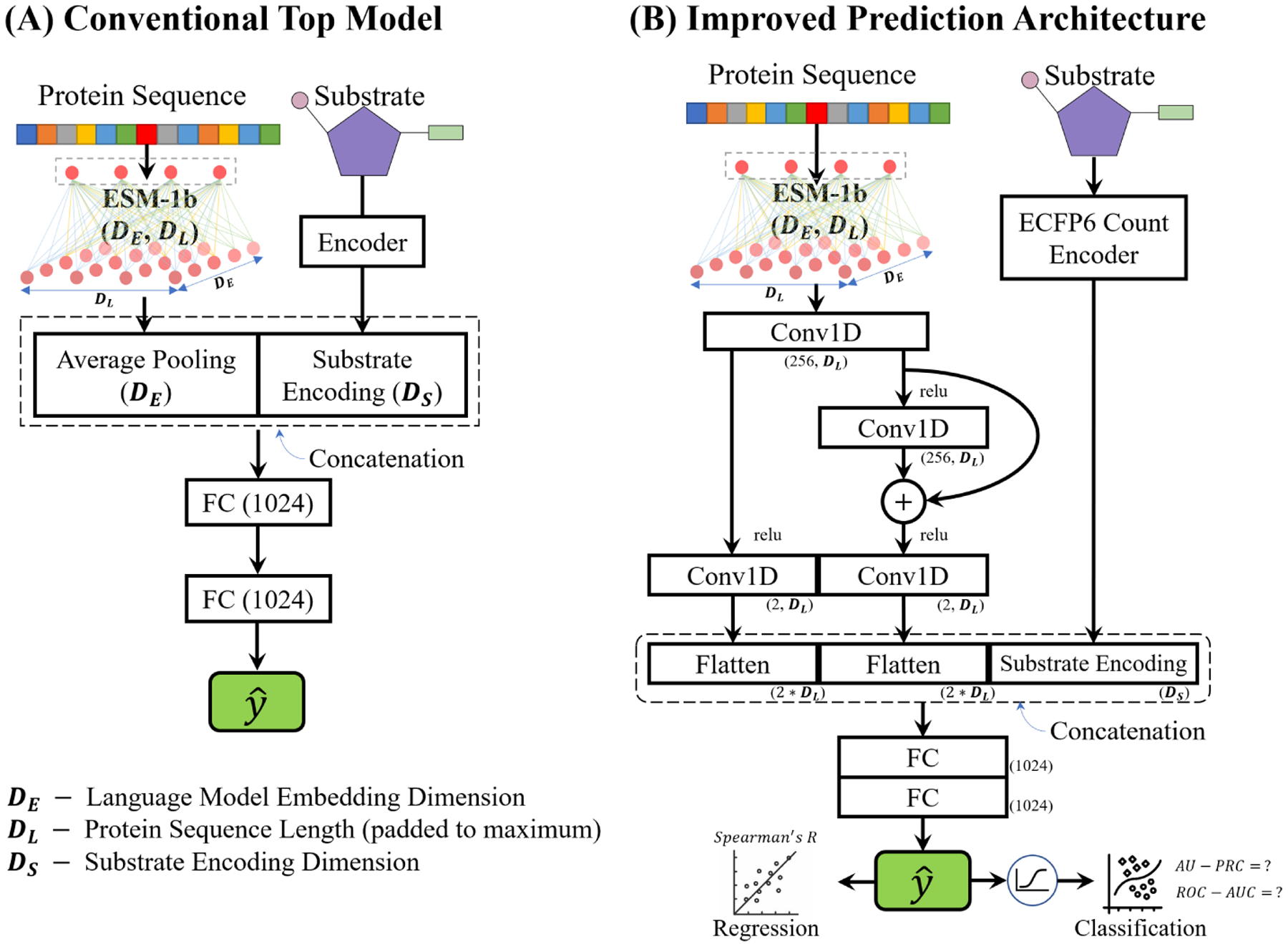
(A) A conventional model for downstream predictions (used as baseline for validating the performance). The baseline model uses average pooling and Morgan bit vector substrates encoding. (B) Architecture of the proposed predictive model. One-dimensional convolutional layers together with a residual block were used to parse sequence embeddings. Two detectors sharing the same initial convolutional layer are used to extract local information and global features in parallel: a basal detector that contains two 1-D convolutional layers with a kernel size of 3 captures local patterns of amino acids, while a superior detector composed of 3 convolutional layers (using the same kernel size) with a residual connection effectively formulates global patterns from panoramic view of the sequence embeddings. The filtered outputs from both detectors were then flattened and concatenated with substrate encodings to generate single vector for each sequence-compound pair, followed by two fully-connected (FC) layers. Dimensions of each block’s output are shown with ESM-1b embeddings and ECFP6 encoder.
3. Results
3.1. Outline
An overall good predictive capability of the count-encoding of ECFPs can be observed as it is tested by the “simple task” of random splitting of the ~35,000 sequence-substrate pairs. The performance is compared with that of using Morgan bit vector substrates representations examined by Goldman et al. (2021) on the same dataset. We tested another encoding approach, Junction-Tree Variational Auto Encoder (JT-VAE) (Jin et al., 2018) mentioned in the same literature and found that this approach was outperformed by the two former encoding methods. Figure 4 presents a comparison of Spearman’s correlation (R), area under Precision-Recall curve (AU-PRC), and area under receiver operating characteristic curve (ROC-AUC) on the simple task of activities regression and classification using different substrate encodings.
Figure 4:
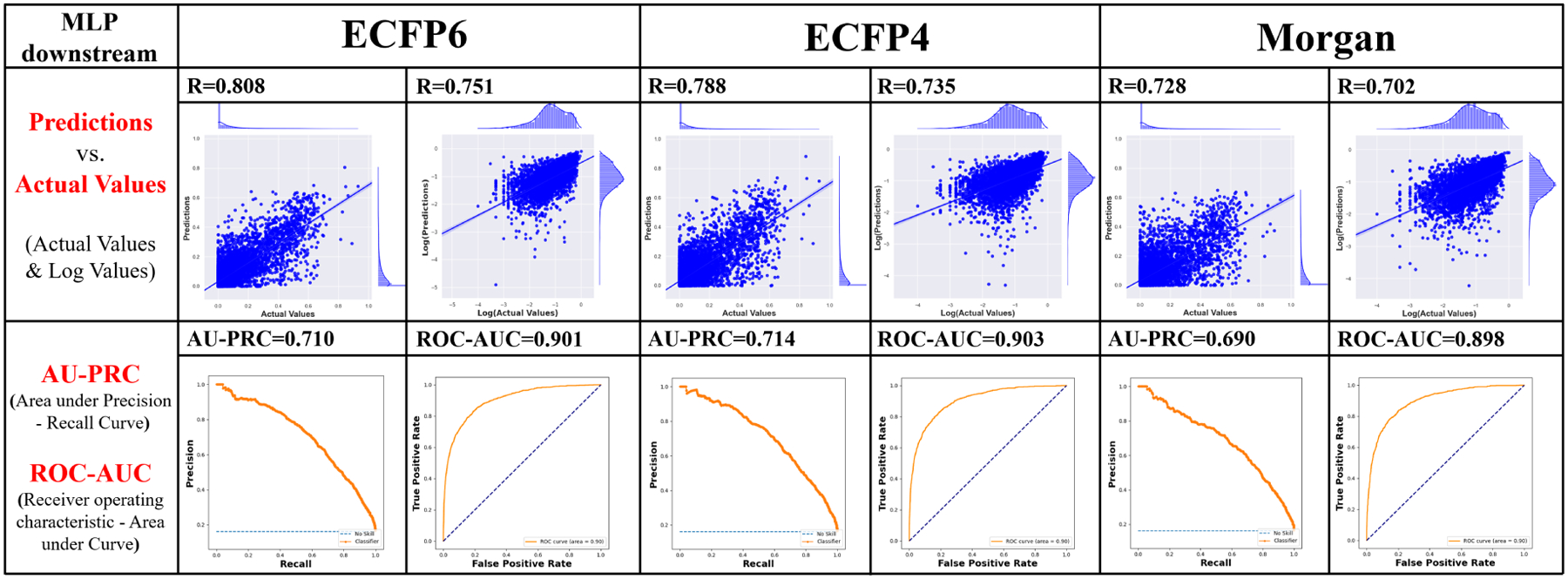
A simple comparison of different substrates encodings, tested on multilayer perceptron (MLP) top models using ESM-1b embeddings with average pooling. Figures in the first row show results of regression models. Scatter plots were used to correlate predicted values against actual values. A stronger correlation can be observed in both the logarithmic values plots (where zeros are removed) and actual values plots for ECFP6 as compared with the other two encodings (ECFP4, and Morgan bit vector). Binary classification based on different chemical encodings approaches were also tested. The same classification labels as Goldman et al. (2021) were used directly. The comparison between AU-PRC as well as ROC-AUC value also shows the two ECFP’s achieve better predictions than Morgan bit vector.
Figure 4 and Table 1 illustrate that, compared to average pooling, convolutional pooling consistently improves the CPI prediction with different substrate encodings and sequence embeddings. Table 1 shows embedding dimensions and Spearman’s correlations for results on 6 different pre-trained protein language models (without tuning the predictive model): ESM-1b (Rives et al., 2021), TAPE (Rao et al., 2019), ProtBert (Elnaggar et al., 2021), ProtT5 (Elnaggar et al., 2021), ProtXLNet (Elnaggar et al., 2021) and ProtAlbert (Elnaggar et al., 2021). ECFP6 encoding is used for substrates.
Table 1:
Results of using different sequence embeddings on downstream predictions (ECFP6 encoding used for substrates). R denotes Spearman’s correlation and the best performing result is highlighted in bold.
| Language Models | ||||||
|---|---|---|---|---|---|---|
| ESM-1b | TAPE | ProtBert | ProtT5 | ProtXLNet | ProtAlbert | |
| Embedding Dimension | 1280 | 768 | 1024 | 1024 | 1024 | 4096 |
| R conv. pooling | 0.816 | 0.813 | 0.810 | 0.818 | 0.810 | 0.820 |
| R avg. pooling | 0.788 | 0.786 | 0.763 | 0.763 | 0.777 | 0.795 |
3.2. Substrate Discovery: Prediction of Activity on Novel Substrates
We also evaluated our models on the “novel substrates characterization task”, which examined, using a different split of the dataset, the potential of applying our workflow to predict activities on novel substrates. Table 2 reports the performance comparison between ECFP6 and baseline Morgan encoding algorithms on predicting the enzyme activities on novel substrates. ECFP6 was found to offer superior robustness of characterizing unseen substrates over Morgan fingerprint due to its ability to interpret all substructures of the substrates. The classification results also confirms the robustness of the ECFP6 and convolutional pooling pipeline in identifying the presence of enzyme activity on novel substrates, as shown by an increased AU-PRC value. Table 3 shows extended results of the “novel substrates characterization task” covering the performance of this pipeline on three other datasets presented in Goldman et al. (2021). These datasets include activity data from halogenase family (62 substrates on 42 sequences) (Fisher et al., 2019), kinase family (72 substrates on 318 sequences) (Davis et al., 2011) and the aminotransferase family (18 substrates on 25 sequences) (Li et al., 2020). Due to fewer substrates presented, the regression results for these three datasets were not comparable to that for the phosphatases dataset, but the classification models resulted in fairly good metric scores (i.e., AU-PRC and ROC-AUC), which are higher than that reported in Goldman et al. (2021).
Table 2:
Results of ECFP6 and Morgan substrate encoders on “novel substrates characterization task”.
| ECFP6 | Morgan | |
|---|---|---|
| R conv. pooling | 0.681 | 0.658 |
| R avg. pooling | 0.654 | 0.649 |
| AU-PRC (convolutional pooling) | 0.588 | 0.562 |
| ROC-AUC (convolutional pooling) | 0.858 | 0.853 |
Table 3:
Full regression and classification results of three characterization tasks on the four enzyme datasets. Baseline model here used Morgan bit vector encoding and two layers of fully connected MLPs.
| Tasks | Dataset | Phosphatase | Halogenase | Kinasea | Aminotransferase |
|---|---|---|---|---|---|
| #Seq. × #Subs. | 218 × 168 | 42 × 62 | 318 × 72 | 25 × 18 | |
| Simple task | R Conv+ECFP6 | 0.816 | 0.892 | 0.845 | 0.838 |
| R Baseline | 0.728 | 0.838 | 0.805 | 0.808 | |
| AU-PRC | 0.710 | 0.732 | 0.809 | 0.867 | |
| ROC-AUC | 0.901 | 0.937 | 0.905 | 0.905 | |
| Substrates task | R Conv+ECFP6 | 0.681 | 0.545 | 0.335 | 0.470 |
| R Baseline | 0.649 | 0.521 | 0.205 | 0.322 | |
| AU-PRC | 0.588 | 0.606 | 0.403 | 0.756 | |
| ROC-AUC | 0.858 | 0.931 | 0.730 | 0.697 | |
| Sequence task | R Conv+ECFP6 | 0.465 | 0.673 | 0.735 | 0.790 |
| R Baseline | 0.422 | 0.581 | 0.716 | 0.796 | |
| AU-PRC | 0.418 | 0.743 | 0.745 | 0.790 | |
| ROC-AUC | 0.695 | 0.909 | 0.889 | 0.842 |
Classification performed to Kinase dataset uses self-defined labels.
3.3. Enzyme Discovery: Prediction of Activity for Novel Sequences
The “novel sequences characterization task” was also performed on all four datasets mentioned in the previous section, with results shown in Table 3. The use of convolutional pooling and ECFP encoding consistently outperformed the baseline model on the first three enzyme datasets. However, the proposed pipeline seemed not able to earn a better result than the baseline in the aminotransferase case, likely due to the limited number of substrates and sequences.
For the phosphatase dataset, while we ensured that the same sequences do not appear in both the training and the test sets, we did not exclude sequences in the training set that may potentially be similar to sequences in the test set. The phosphatase data was from the original paper (Huang et al., 2015), where enzyme sequences with greater than 40 percent identity were clustered and different sequences from diverse clusters were experimentally characterized. We note that several enzymes from the same cluster were similar in sequence and eliminating them would have resulted in a smaller training set. Further characterizing phosphatases to obtain additional sequences will be valuable to explore the diversity of this family and such a dataset would offer a more comprehensive test of our workflow.
4. Conclusion
In this study, we encode the substrates using a new approach that enables a better characterization of substructural information. We also mitigate the problem of information loss resulting from averaging the position-wise embeddings, by learning a convolutional pooling of those embeddings to retain the information captured by language models. The proposed new approaches of encoding substrates and pooling the sequence embeddings are tested on four high-quality enzyme activity datasets and compared with recent studies. Taken together, the results suggest that our proposed compound-protein interaction modeling pipeline achieves better predictions compared to current methods. These results also set the stage for using our proposed workflows for enzyme discovery by identifying potential novel substrates for existing sequences in these families and also for identifying new sequence variants that can have improved activity through generative modeling, paving the way for enzyme engineering for these families. Finally, our results also demonstrate the potential for applying these methods to other enzyme families that have been characterized to the same extent as the datasets studied here. All our models and results are available through https://github.com/LMSE/CmpdEnzymPred.
Acknowledgments
We acknowledge Dr. Antoine Koehl for helpful discussions and the Miller Institute for enabling this research. This work was supported in part by the Natural Sciences and Engineering Research Council (NSERC), the NSERC Industrial Biocatalysis Network (IBN), Biochemicals from Cellulosic Biomass (BioCeB) grant from the Ontario Research Fund (Research Excellence), a grant from the Genome Canada Genomics Applied Partnership Program (GAPP), and a grant R35-GM134922 from the National Institute of Health (NIH). YSS is a Chan Zuckerberg Biohub Investigator.
Contributor Information
Zhiqing Xu, University of Toronto.
Jinghao Wu, University of Toronto.
Yun S. Song, University of California, Berkeley
Radhakrishnan Mahadevan, University of Toronto.
References
- Biswas Surojit, Khimulya Grigory, Alley Ethan C., Esvelt Kevin M., and Church George M.. Low-n protein engineering with data-efficient deep learning. Nature Methods, 18:389–396, 2021. ISSN 1548–7105. doi: 10.1038/s41592-021-01100-y. [DOI] [PubMed] [Google Scholar]
- Davis Mindy I, Hunt Jeremy P, Herrgard Sanna, Ciceri Pietro, Wodicka Lisa M, Pallares Gabriel, Hocker Michael, Treiber Daniel K, and Zarrinkar Patrick P. Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology, 29(11):1046–1051, 2011. [DOI] [PubMed] [Google Scholar]
- Devlin Jacob, Chang Ming-Wei, Lee Kenton, and Toutanova Kristina. BERT: pre-training of deep bidirectional transformers for language understanding. CoRR, abs/1810.04805, 2018. [Google Scholar]
- Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Yu W, Jones L, Gibbs T, Feher T,Angerer C, Steinegger M, Bhowmik D, and Rost B. ProtTrans: Towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Transactions on Pattern Analysis and Machine Intelligence, 14(08):1–16, 2021. doi: 10.1109/TPAMI.2021.3095381. [DOI] [PubMed] [Google Scholar]
- Fisher Brian F., Snodgrass Harrison M., Jones Krysten A., Andorfer Mary C., and Lewis Jared C.. Site-selective c–h halogenation using flavin-dependent halogenases identified via family-wide activity profiling. ACS Central Science, 5(11):1844–1856, 2019. doi: 10.1021/acscentsci.9b00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman Samuel, Das Ria, Yang Kevin K, and Coley Connor W. Machine learning modeling of family wide enzyme-substrate specificity screens. arXiv preprint arXiv:2109.03900, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Kaiming, Zhang Xiangyu, Ren Shaoqing, and Sun Jian. Deep residual learning for image recognition. In 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), pages 770–778, 2016. doi: 10.1109/CVPR.2016.90. [DOI] [Google Scholar]
- Huang Hua, Pandya Chetanya, Liu Chunliang, Al-Obaidi Nawar F., Wang Min, Zheng Li, Keating Sarah Toews, Aono Miyuki, Love James D., Evans Brandon, Seidel Ronald D., Hillerich Brandan S., Garforth Scott J., Almo Steven C., Mariano Patrick S., Dunaway-Mariano Debra, Allen Karen N., and Farelli Jeremiah D.. Panoramic view of a superfamily of phosphatases through substrate profiling. Proceedings of the National Academy of Sciences, 112(16):E1974–E1983, 2015. ISSN 0027–8424. doi: 10.1073/pnas.1423570112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Wengong, Barzilay Regina, and Jaakkola Tommi S.. Junction tree variational autoencoder for molecular graph generation. CoRR, abs/1802.04364, 2018. [Google Scholar]
- Li Tao, Cui Xuexian, Cui Yinglu, Sun Jinyuan, Chen Yanchun, Zhu Tong, Li Chuijian, Li Ruifeng, and Wu Bian. Exploration of transaminase diversity for the oxidative conversion of natural amino acids into 2-ketoacids and high-value chemicals. ACS Catalysis, 10(14):7950–7957, 2020. doi: 10.1021/acscatal.0c01895. [DOI] [Google Scholar]
- Morgan HL. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. Journal of Chemical Documentation, 5(2):107–113, 1965. doi: 10.1021/c160017a018. [DOI] [Google Scholar]
- Rao Roshan, Bhattacharya Nicholas, Thomas Neil, Duan Yan, Chen Peter, Canny John, Abbeel Pieter, and Song Yun S.. Evaluating protein transfer learning with TAPE. In Advances in Neural Information Processing Systems 32, pages 9689–9701. 2019. [PMC free article] [PubMed] [Google Scholar]
- Rives Alexander, Meier Joshua, Sercu Tom, Goyal Siddharth, Lin Zeming, Liu Jason, Guo Demi, Ott Myle, C Lawrence Zitnick Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences, 118(15), 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers David and Hahn Mathew. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50(5):742–754, 2010. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Vaswani Ashish, Shazeer Noam, Parmar Niki, Uszkoreit Jakob, Jones Llion, Aidan N Gomez, Ł ukasz Kaiser, and Illia Polosukhin. Attention is all you need. In Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, and Garnett R, editors, Advances in Neural Information Processing Systems, volume 30. Curran Associates, Inc., 2017. [Google Scholar]
- Willighagen Egon L., Mayfield John W., Alvarsson Jonathan, Berg Arvid, Carlsson Lars, Jeliazkova Nina, Kuhn Stefan, Pluskal Tomáš, Miquel Rojas-Chertó Ola Spjuth, Torrance Gilleain, Evelo Chris T., Guha Rajarshi, and Steinbeck Christoph. The chemistry development kit (cdk) v2.0: atom typing, depiction, molecular formulas, and substructure searching. Journal of Cheminformatics, 9(33), 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]