

Abstract
KBG syndrome (KBGS) is characterized by distinctive facial gestalt, short stature and variable clinical findings. With ageing, some features become more recognizable, allowing a differential diagnosis. We aimed to better characterize natural history of KBGS. In the context of a European collaborative study, we collected the largest cohort of KBGS patients (49). A combined array- based Comparative Genomic Hybridization and next generation sequencing (NGS) approach investigated both genomic Copy Number Variants and SNVs. Intellectual disability (ID) (82%) ranged from mild to moderate with severe ID identified in two patients. Epilepsy was present in 26.5%. Short stature was consistent over time, while occipitofrontal circumference (median value: −0.88 SD at birth) normalized over years. Cerebral anomalies, were identified in 56% of patients and thus represented the second most relevant clinical feature reinforcing clinical suspicion in the paediatric age when short stature and vertebral/dental anomalies are vague. Macrodontia, oligodontia and dental agenesis (53%) were almost as frequent as skeletal anomalies, such as brachydactyly, short fifth finger, fifth finger clinodactyly, pectus excavatum/carinatum, delayed bone age. In 28.5% of individuals, prenatal ultrasound anomalies were reported. Except for three splicing variants, leading to a premature termination, variants were almost all frameshift. Our results, broadening the spectrum of KBGS phenotype progression, provide useful tools to facilitate differential diagnosis and improve clinical management. We suggest to consider a wider range of dental anomalies before excluding diagnosis and to perform a careful odontoiatric/ear-nose-throat (ENT) evaluation in order to look for even submucosal palate cleft given the high percentage of palate abnormalities. NGS approaches, following evidence of antenatal ultrasound anomalies, should include ANKRD11.
Introduction
KBG syndrome (KBGS) (OMIM#148050) was firstly described in 1975 by Hermann et al. (1) who reported many family members with a distinct phenotype. KBGS is a pan-ethnic syndrome presenting with variable clinical expressivity even across single families and for which an incomplete penetrance has been previously hypothesized (2–5). The prevalence of KBGS is currently unknown and about 200 patients have been described up to date, with male patients generally being more severely affected (2,3,6).
Classical clinical features include macrodontia of the upper central incisors, distinctive craniofacial findings, short stature, skeletal anomalies and neurologic involvement encompassing global developmental delay, electroencephalographic anomalies with or without seizures and intellectual disability (ID) (2). Uncommon clinical features, reported in sporadic reports, include juvenile idiopathic arthritis, dysfunctional dysphonia, multiple dental agenesis, oral frenula, idiopathic precocious thelarche, motor tics, lipoma of the corpus callosum, pilomatrixoma and endothelial corneal polymorphic dystrophy (7).
Monoallelic variants in ANKRD11 and deletions encompassing ANKRD11 cause KBGS (8,9). ANKRD11 is located on chromosome 16, has 13 exons and encodes ankyrin repeat domain 11 (10). Sequencing of ANKRD11 allows the resolution of more than half of the individuals clinically diagnosed with KBGS (5,8,11–13). Most of the pathogenic variants are truncating, whereas missense variants are rare; plenty of them cluster in exon 10, the largest exon of ANKRD11 (14). Genomic rearrangements of the 16q24.3 chromosomal region, including ANKRD11, have also been detected in KBGS patients leading to variable phenotypes depending on the contribution of other genes involved in the rearrangement (9,15).
ANKRD11 is a chromatin regulator of the neural stem cell fate through histone acetylation (16). Mice with heterozygous deletion of ANKRD11 (17) in neural crest cells present with mild midfacial hypoplasia (reduced midfacial width and a persistent open fontanelle), reproducing the human facial phenotype (16). Mice with homozygous mutations in neural crest cells are not vital at birth (18). ANKRD11 expression levels reflect the development of the bony structures during craniofacial development, strongly regulating intramembranous ossification and palate development (18). Interestingly, ANKRD11 is frequently affected by loss of heterozygosity in cancer, and it is supposed to enhance the tumour-suppressive function of TP53 oncogene (19). It has been speculated that ANKRD11 haploinsufficiency may lead to an increased cancer risk in KBGS patients but only one patient with a 16p24.3 deletion involving ANKRD11 developed a paratesticular rhabdoid tumour (12).
Although broader availability of whole exome sequencing (WES) is increasing the diagnostic rate, a high percentage of KBGS cases remain undiagnosed due to the lack of specific diagnostic clues or mild clinical presentation, except for a recurrent facial gestalt (8,20). In children, younger than 6 years of age, in particular, a clinical diagnosis of KBGS may not be obvious since facial features are subtler and part of the physical characteristics become distinguishable only with age progression. Indeed, an age-dependent evolution of the facial features from Cornelia de Lange (CdLS) to KBGS, from infancy to adolescence, has been previously highlighted (3). Nevertheless, a clinical overlap with the Aarskog, CdLS, Kabuki, Filippi, Silver-Russell and Cohen syndromes has been noted (2,21–23). In milder affected individuals, the short stature and the triangular shape of the face in late childhood could pose a differential diagnosis with Noonan syndrome (24).
In the context of a European collaborative study, we employed a combined array-based Comparative Genomic Hybridization (array-CGH), gene panel analysis and exome sequencing approach on the largest cohort of KBGS patients currently available (n = 49) to establish new genotype–phenotype correlations, define novel clinical signs and characterize the clinical evolution of KBGS with ageing.
Results
We analysed a cohort of 49 individuals affected by KBGS with an equal distribution of male and female patients (25/24) (Supplementary Material, Table S1). We divided the cohort into four age-groups (0–4, 5–10, 11–20, ≥21 years) differentiated for the gender (Supplementary Material, Fig. S1).
Molecular characterization
Detection of ANKRD11 variants was obtained by a combined exome sequencing, gene panel analyses and array-CGH. In most patients, we detected alterations only in the ANKRD11 gene. In two patients genetic defects in additional genes were detected. All patients carried constitutive variants except for patient 22, who was found to carry the variant in a mosaic state.
Five patients inherited the alteration from an affected parent, whereas in 35 patients the variants were de novo. In nine patients segregation analysis of the variants was not available. Two twins shared the same pathogenic variant. We detected 36 different variants: one missense variant, indeed lying at the −1 position of the 5′ splice site and thus able to affect splicing, two splice site variants, 29 frameshift and four nonsense variants (Supplementary Material, Table S1 and Supplementary Material, Table S2). In 35 patients out of 49, variants were located in exon 10. Among the detected variants, 17 have been previously reported, whereas 19 are unpublished (Fig. 1A). We did not detect any specific genotype–phenotype correlation. Furthermore, array-CGH analysis revealed two microdeletions extending to the neighbouring Zinc Finger 778 gene (ZNF778), one segregating in an affected mother and her three affected children (patient 8, 9, 10), and another arising de novo in patient 6 (Fig. 1B).
Figure 1.

ANKRD11 variants localization and genomic rearrangements (A) Genetic pathogenic variants identified in our study in the ANKRD11 protein, with a schematic representation of its domains (ANK: Ankyrin repeats (p.126–295); RD1 Repression domain 1 (p.318–611); ad: Activation domain (p.2076–2145); RD2: Repression domain 2 (p.2369–2663)). The novel mutations identified in the patients described in this study are indicated in red. (B) Genomic coordinates of the microdeletions (highlighted with red dashed lines) as identified by array-CGH in two KBGS patients.
Clinical features prevalence
The prevalence of the main clinical features is summarized in Figure 2. As expected, the most prevalent feature was mild ID. However, cerebral anomalies were found in 56% of patients (18/32) who underwent brain magnetic resonance imaging (MRI). Skeletal and dental anomalies were also frequent. Congenital heart defects were reported in 16.3% of cases.
Figure 2.
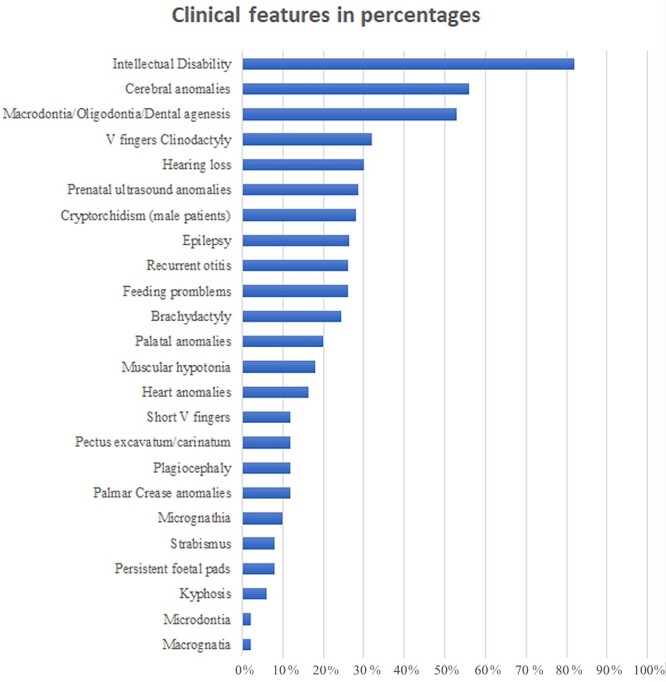
Clinical features distribution: The horizontal bars represent the different percentages (x axis) of each clinical feature (y axis) as observed in our KBGS patient cohort.
Growth
At birth, all growth parameters tended to be lower than average; weight at birth had a median of −0.81 SD, length had a median of −0.65 SD and occipitofrontal circumference (OFC) had a median of −0.88 SD. With ageing, all auxological parameters, except for height, tended to normalize (Fig. 3). Weight reached a median of −0.645 SD between 11 and 21 years of age, and − 0.48 SD above 20 years. OFC also increased to a median of +0.03 SD and + 0.62 SD over time, with values in the range of normality above 21 years of age (P-value = 0.04 in the range ≥ 21 years versus 5–10 years). Persistent microcephaly was detected in 12% (6/49) of patients. Height did not improve with ageing with a deflection in the growth curve around the age of 10 and a median of −1.54 SD in the cohort above 21 years of age. The difference of the median height SD for patients ≥21 years versus the median length SD at birth was statistically significantly different (P-value = 0.0268). Delayed bone maturation was reported in five cases (patients 5, 17, 41, 48 and 49).
Figure 3.
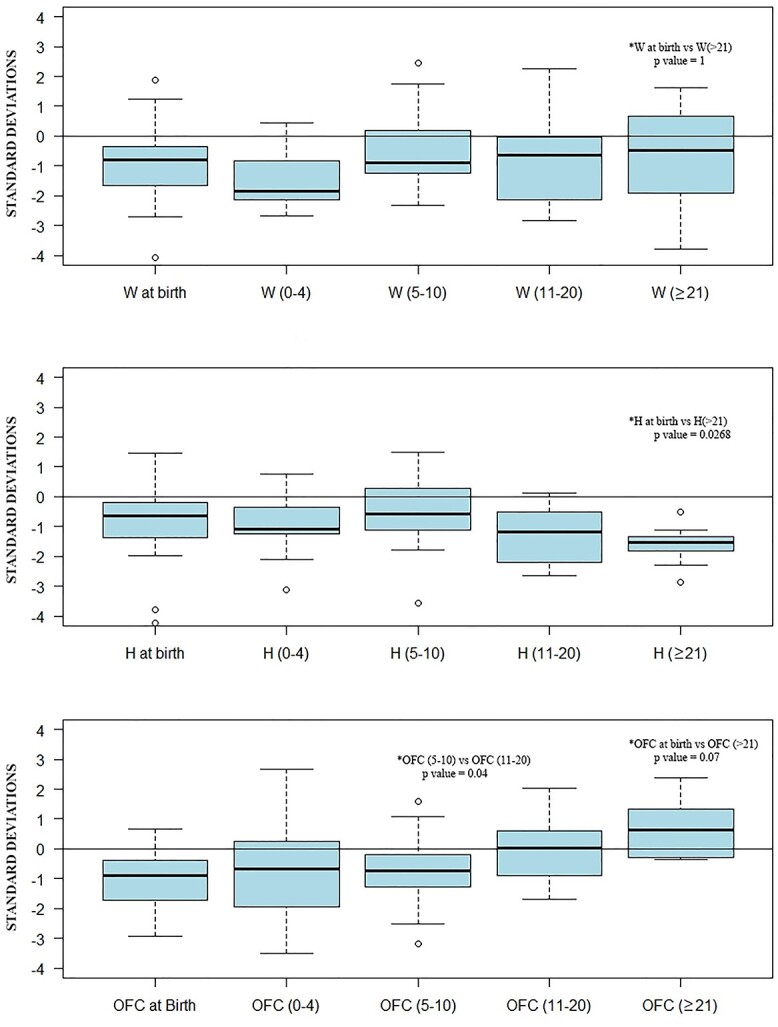
Auxological parameters distribution over time: Standard deviations for auxological parameters divided into age groups (0–4 years old, 5–10 years old, 11–20 years old and ≥21 years old). The statistically significant differences (P-value < 0.05) of the median standard deviations for the auxological parameters for the different demographic classes are indicated on the box plots.
Dysmorphic features
In all patients, a typical facial gestalt was detected (Fig. 4). The main features include prominent forehead, brachycephaly, triangular face, synophrys, hypertelorism, long philtrum, thin upper lip, depressed and wide nasal bridge, anteverted nares and hypertrichosis. Facial dysmorphic features are summarized in Supplementary Material, Table S1 and illustrated in Figure 4.
Figure 4.

Patient’s clinical features: Clinical features of patients at different ages (A:18, B:15, C:42, D:13, E:48, F–G:9, H–I:14, J–K:44, L:40, M–N:33, O–P:36). Common clinical features are triangular face, long philtrum, depressed and wide nasal bridge, anteverted nares.
Neurodevelopmental problems
In our cohort of patients, 82% (36/44) presented with ID which mostly ranged from mild to moderate (72.2% and 19.4%, respectively) in line with previous reports (25). Seven patients (patients 7, 8, 9, 13, 20, 26, 38 in Supplementary Material, Table S1) out of the 44 evaluable individuals (15,6%), did not display ID. Severe ID was identified only in two patients with concomitant genetic conditions (12,16), namely maternal 15q11.2 deletion and paternal inherited exostosis due to EXT1 mutation, respectively. About 18% of patients presented hypotonia at birth or at the first evaluation. Epilepsy was present in 26.5% of patients (13/49) and mostly correlated with cerebral anomalies at the MRI. Indeed, out of 13 patients who developed seizures, eight presented brain anomalies, in three of them (12,25,26) no cerebral alteration was detected by brain imaging and in two of them no MRI was performed. Electroencephalography (EEG) often revealed mild non-specific background abnormalities with modest voltage asymmetry, slow background activity (patient 2, 8, 47) or an irregular brain activity during sleeping (patient 41). Generalized slow spikes and waves, generalized polyspikes and generalized paroxysmal fast activity during sleep were only registered in patient 12, a 34-year-old male, who displayed a refractory Lennox–Gastaut syndrome with severe ID and who indeed harboured a deletion of the 15q11.2 region including TUBGCP5, CYFIP1, NIPA1, NIPA2. In patient 45 slight pointed plurifocal anomalies in the left hemispheric area were already registered at 1 month of age. Behavioural problems were detected in 65% of individuals (32/49) with Autism spectrum disorder (ASD) or attention deficit hyperactivity disorder (ADHD) reported in 30% of patients (15/49), tantrums and aggressive behaviour in 20% of patients (10/49), anxiety in 6% (3/49), emotional lability and shyness in 8.1% of individuals (4/48).
Cerebral anomalies
Cerebral abnormalities were identified in 56% of patients (18/32) who underwent brain MRI. Enlarged cisterna magna (including mega cisterna magna) was the most shared clinical finding among affected individuals being detected by MRI in 23% of patients (6/32). Common clinical findings were also arachnoid cysts. Other brain anomalies include periventricular nodular heterotopia (PNH) (patients 10 and 13), short and thin corpus callosum (patients 26, 27 and 45), microcephaly (22% of patients, 11/49), cortical atrophy and dysplasia (patients 2 and 10), trigonocephaly (patient 17) (Fig. 6).
Figure 6.

Brain MRI images. (A–B) Megacisterna Magna (patient 46); (C–E) Frontal heterotopy in the white matter near the lateral ventricles and in the temporo-basal region. T2 signal intensity suggestive of cortical dysplasia (patient 10); (F): Subarachnoid cyst (patient 16); (G) Retro-cerebellar cisterna expansion (patient 15).
Dental and skeletal anomalies
A wide range of dental defects were observed in 53% (26/49) of cases: macrodontia, oligodontia, dental fusion, dental crowding and dental agenesis. Delayed dental eruption was observed in two patients. Patient 16 displayed macrodontia with a fusion of the maxillary lateral. Microdontia was observed in one patient. In 10% of patients, micrognathia was present, while only one patient presented macrognathia. Palatal anomalies were detected in 20% of cases (10/49) with 12% of patients (6/49) presenting with a highly arched palate. Cleft soft palate was described in one patient (patient 14) and velopharyngeal dysfunction in another patient (patient 19).
Skeletal anomalies including pectus excavatum, scoliosis, fifth finger clinodactyly, brachydactyly, vertebral malformations such as malformation of fifth and sixth thoracic vertebrae, were detected in 89% of cases (44/49). About 32% of patients (16/49) presented with fifth finger clinodactyly. Short fifth finger was also common (12%, 6/49), along with brachydactyly (24.4%, 12/49) and pectus excavatum/carinatum (12%, 6/49). Kyphotic posture was described in three patients (6%). Delayed bone maturation was reported in five cases (patients 5, 17, 41, 48 and 49). In patient 45 a reduced development of the growth nuclei of the bones and absence of the growth nucleus of the radius was observed. Large anterior fontanelle were detected in 10% of cases.
Hearing loss and recurrent otitis
Hearing loss was found in 30% (15/49) of patients in accordance with previous reports (5). Conductive hearing loss was present in nine of them, mixed hearing loss was found in two, one patient didn’t pass otoacoustic emissions (OAE) and only in one case sensorineural hearing loss was reported (patient 2). In patient 29 conductive hearing loss in his left ear and sensorineural hearing loss in his right ear coexisted. Only three patients presented unilateral hearing loss (patient 6, 25 and 35).
Recurrent otitis were found in 26% (13/49) of patients. Of the 15 patients that presented hearing loss only half of them presented recurrent otitis (7/15, 46%).
Additional congenital anomalies
Anomalies of palmar creases were found in 12% of patients and they included decreased palmar creases in two cases (patients 2 and 6), deeper palmar crease in one case (patient 4) and a single palmar fold in three cases (patients 5, 40 and 45). Persistence of foetal pads was noted in 8% of cases (patient 4, 14, 16 and 28).
About 16% of patients (8/49) displayed heart defects including ASD, ventricular septal defects, atrioventricular defect and valve insufficiency. Bilateral or unilateral cryptorchidism was detected in 28% of patients (7/25). Vesicoureteral reflux was reported in patients 31, 32 and 48. About 8% of patients had strabismus (4/49). Hydrocele and double spleen were found, respectively, in patients 29 and 42. Anteriorly placed anus and anal atresia were detected in patient 9, two fistulas in the anal region emerged in patient 36 (Fig. 5). Lingual frenulum alterations were observed in four patients with one of them presenting longer than normal frenulum and the other three presenting shorter frenulum (two of them needed surgery). Feeding difficulties were present in 13/49 patients (26%) mostly consisting of low feeding activity, unable to breastfeed, discoordinated sucking-swallowing-breathing rhythm, difficulty in chewing or swallowing.
Figure 5.

Other Clinical features: Brachydactyly patients 48 (A), 42 (B–C), 13 (D) and 44 (E), dental anomalies patients 16 (F) and 44 (G), mild eruption delay of permanent first molars and small size of the inferior lateral left incisor and of the superior lateral right incisor in patient 49 (H) and anal fistula patient 36 (I).
Antenatal ultrasound anomalies
In 28.5% of individuals (14/49), antenatal ultrasound anomalies were reported namely increased nuchal translucency, polyhydramnios and intrauterine growth restriction (IUGR).
Coexistence of two conditions
In patient 12, a maternal 15q11.2 microdeletion including TUBGCP5, CYFIP1, NIPA1 and NIPA2 genes was also detected. He presented with severe ID and major behavioural disorder. He also suffered from Lennox–Gastaut syndrome from age 5. At 33 years of age, language was limited to a few words.
In patient 16 a previously described pathogenic variant in EXT1 gene was also identified. His skeletal phenotype was indeed more complex. He presented multiple exostoses in the context of a Madelung deformity, curved forearms and short upper limbs. He also presented with a severe ID. Considering that mutations of EXT1 gene are not associated with ID, his more severe cognitive phenotype is probably a random association. In conclusion, the only two severe ID individuals collected in this study had a coexistence of KBGS with a second unrelated condition.
Discussion
Short stature, macrodontia and distinctive facial features are the most common clinical traits in KBGS (2). Even if none of the individual symptoms identified so far is a KBGS diagnostic requisite, the combination of them can lead to disease suspicion. In line with previous reports, our study reinforces the concept that general clinical features, which justify genetic investigation for KBGS, are developmental delay, recognizable facial gestalt (becoming more apparent with ageing) and skeletal/dental anomalies. In accordance with previous studies (5) hearing loss was present in about 30% of patients and feeding difficulties in about 26% (5). Height below average was present in 20/49 patients, while short stature (height < −2SD) was detected in 5/49 individuals. ID generally ranges from mild to moderate in line with previous reports (27) and seizures generally correlated with cerebral anomalies. The occurrence of cerebral anomalies suggests that performing a brain MRI might help reaching a diagnosis of KBGS. In contrast with previous reports (13), we did not find a correlation between ID severity and location of the ANKRD11 variants because both mild and moderate ID were observed in patients with variants in the RD2 domain as well as in patients with variants lying in the region between RD1 and ad domains. We found only two individuals with severe ID that both presented with a concomitant genetic condition. Most of the adult patients with mild ID (26/49, 53% of cases) were able to attend high school with teaching aid. One of them is a kindergarten teacher and one patient is currently attending university. Behavioural issues were present in 65% of individuals with ASD and ADHD being the most prevalent finding followed by aggressive behaviour.
The finding of distinctive dysmorphisms is generally suggestive of a KBGS even if making a definitive diagnosis early in life is challenging because of milder features and the lack of some clinical signs such as dental anomalies and short stature (2). In order to provide useful criteria for clinical diagnosis, a presumptive scheme of KBGS natural history, showing the natural evolution of the major clinical findings over time, is provided in Figure 7. As recently underlined, atypical findings such as a wide anterior fontanelle accompanied by minor facial features, can be the only presenting features of KBGS in children (28). Enlarged fontanelles are mostly caused by a slow or incomplete closure of the skull bones (29). This finding, which we detected in about 10% of patients, is in line with the reports about ANKRD11 role in mice in which heterozygous deletion of ANKRD11 in neural crest cells leads to reduced midfacial width and persistent open fontanelle (16).
Figure 7.

Natural history of KBGS phenotype: Graphic representation of the major clinical findings over time. Our cohort was divided into age groups. Clinical findings spanning all age groups are listed below the main figure. Clinical signs typical of each age group are indicated in red above the main figure.
Various brain abnormalities including cerebellar vermis hypoplasia (30), enlarged cisterna magna, Chiari I malformation, PNH, pineal cyst, dysgenesis of the corpus callosum, arachnoid cysts have been previously reported as single case reports. However, the frequency of brain malformations was not known because brain MRI has not been previously performed in large cohorts of affected individuals. Notably, being detected in more than 50% of the 32 patients who underwent brain MRI, our cohort suggests that cerebral anomalies are far more frequent in KBGS than previously estimated. We found an enlarged or a mega cisterna magna (greater than 10 mm) in 23% of patients. Furthermore, we report a series of unique clinical features such as trigonocephaly, small hypophysis and hypotrophy of the optic nerve, broadening the cerebral phenotypic spectrum of KBGS. Nevertheless, according to our data, we suggest considering a wider range of dental anomalies including microdontia before ruling out KBGS diagnosis. This clinical finding has been previously reported in another patient displaying maxillary canine microdontia (31). We also highlight the importance of performing a careful dental/ENT evaluation to look for even submucosal cleft palate given the high frequency of palate abnormalities.
In conclusion, broadening the diagnostic criteria can help the ascertainment of KBGS with an improvement of the clinical management and can facilitate differential diagnosis (25). In some individuals, KBGS symptoms overlap with other syndromes leading to a misdiagnosis. In patients 11 and 16, CdLS was suspected in the first years of life. A clinical diagnosis of Nicolaides–Baraitser syndrome was proposed for patient 14. Thus, consistent with previous reports, our study supports an age-dependent phenotypic evolution going from Coffin-Siris/Nicolaides Baraitser or CdLS to KBGS from infancy to adolescence. This also highlights the importance of clinical follow-ups to achieve the proper diagnosis (3). Differential diagnosis with RASopathies spectrum disorders is often raised as well, due to some common features, namely short stature and triangular shape of the face. Notably, an increased nuchal translucency, often observed in RASopathies, has been reported in the prenatal era in some of our KBGS patients, thus highlighting the importance of a wider targeted panel approach which should include ANKRD11 in the context of prenatal diagnosis.
None of our patients developed cancer and thus we were not able to establish a correlation between ANKRD11 loss of function variants and cancer predisposition, as postulated in previous studies (12). However, as the eldest patient in our cohort is 41 years old, we cannot exclude that KBGS patients have an increased cancer risk in adulthood. Notably, in patient 16 we were not able to establish the maternal origin for the pathogenic variant since the mother died in her forties due to myeloid acute leukaemia, a kind of tumour often related to TP53 pathway dysregulation. However, available iconographic documents strongly suggest that she also carried a pathogenic variant in ANKRD11. Thus, although at the moment no specific measures for cancer surveillance need to be recommended for KBGS patients, a systematic follow-up is necessary to collect additional useful information to clarify if there is a cancer risk in KBGS.
According to previous studies, pathogenic ANKRD11 variants were almost all exclusively frameshift, followed by rare reports of more rarely nonsense variants, confirming the haploinsufficiency as disease mechanism. Interestingly, in our cohort, the only apparent missense mutation lies at −1 position of the 5′ splice site that can affect mRNA processing (14). This finding emphasizes the importance of careful evaluation of ANKRD11 missense variants as causative of KBGS and highlights that a splicing effect should be considered when clinical diagnosis is convincing. We cannot indeed exclude though that ANKRD11 missense variants could be responsible for a distinct phenotype. In our cohort, we detected a 251 kb microdeletion involving the ANKRD11 and ZNF778 genes, segregating in a family with a phenotype partially distinct from KBGS. Recently, a microdeletion, causing haploinsufficiency of the ANKRD11-flanking genes, has been found in patients with a 16q24.3 microdeletion syndrome (32,33). Our patients presented KBGS-like features that overlapped with those of the 16q24.3 microdeletion syndrome (dyslalia, strabismus, mild ID and heterotopy), defining a ‘KBGS plus phenotype’. We identified a mosaic ANKRD11 variant in patient 22. A mosaic state has been established for a previously reported patient with a mild phenotype (26). Our patient with mosaic KBGS presented mild ID, ASD and minor facial dysmorphisms confirming that a milder phenotype is likely associated with ANKRD11 mosaic variants.
In conclusion, our results demonstrate that the combined use of exome sequencing analysis and array-CGH can result in the diagnosis of KBGS, particularly in paediatric patients with immature clinical findings (34,35). Furthermore, our data underline the importance of considering additional signs, such as cerebral anomalies and suggest that anamnestic obstetric history, positive for IUGR and/or increased nuchal translucency, supports KBGS diagnosis in the paediatric age.
Materials and Methods
Selection of patients and DNA samples’ preparation
For each patient clinical evaluation and genetic counselling was performed in order to collect family history and better define facial and physical characteristics. In all patients audiological examination was performed to check for hearing loss as well as EEG to look for parossistic activities. ENT evaluation followed the suspicion of palatal anomalies and/or velopharyngeal dysfunction, brain MRI was performed in 32/49 patients (65%). All patients or their parents gave their written informed consent to the study that was carried out according to the Declaration of Helsinki.
Genomic DNA was extracted, for some samples, from EDTA peripheral blood samples using MagCore HF16 (Diatech Lab Line), according to the manufacturer’s instructions. The DNA quantity was estimated using the NanoDropTM 2000/2000c Spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA).
For other samples DNA was extracted from peripheral blood using the standard salting out method or the QiaSymphony DSP DNA midi kit. The DNA quantity was estimated using the NanoDropTM 1000 Spectrophotometer (Thermo Fisher Scientific).
Whole-Genome array-CGH
High-resolution whole-genome (array-CGH) analysis was performed on genomic DNA of patients 6, 8, 9, 10 in Supplementary Material, Table S1, using the SurePrint G3 Human CGH Microarray 8 × 60 k (Agilent Technologies, Santa Clara, CA, USA), a dual-colour array containing 60-mer high-quality probes with 41 Kb genome-wide median probe spacing. Copy Number Variants (CNVs) were analysed and mapped using the Human Genome Assembly GRCh37/hg19.
Slides were scanned using an Agilent G2565CA Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA) and processed using Feature Extraction software (v10.5.1.1). Results were analysed using Agilent CytoGenomics software (v5.1) with default settings. The results included imbalances with at least three consecutive probes with abnormal log2 ratios. The Database of Genome Variants (DGV- http://dgv.tcag.ca/dgv/app/home;), DECIPHER (DatabasE of Chromosomal Imbalances and Phenotypes using Ensembl Resources- https://www.deciphergenomics.org/), PubMed (https://pubmed.ncbi.nlm.nih.gov;), UCSC genome browser (https://genome.ucsc.edu;), Database of Human CNVs (http://gvarianti.homelinux.net/gvariantib37/index.php;), SFARI (Simon’s Foundation Autism Research Initiative) Gene Database (https://gene.sfari.org;) and OMIM (Online Mendelian Inheritance in Man- https://www.omim.org/;) databases were used for the interpretation of the results. Each DNA sample was analysed twice through array-CGH, in order to confirm the result.
Exome sequencing and data analysis
For most patients, exome sequencing was performed using the Life Technologies Ion Proton sequencer (Life Technologies, Carlsbad, CA, USA) on genomic DNA samples of proband and parents, when available. This system enables >92% of bases covered 20X. Sample preparation and sequencing were performed with AmpliSeq™Exome strategy, following the manufacturer’s protocol (Life Technologies). The library preparation was performed using the Ion AmpliSeq™Exome Kit (Life Technologies), which allows us to target ~33 Mb of coding exons plus 15 Mb of flanking regions for a total of ~58 Mb, in total more than 97% of the coding regions described by Consensus Coding Sequences (CCDS) annotation. In total 12 primer pools for highly specific enrichment of exons within the human genome were used. Taking advantage of a barcode system, three samples were loaded together in a single run and sequenced. Data analysis was performed with Torrent Suite™ Software v3.6.2 (Life Technologies). The provider generated at least 30 effective mean depths per sample. Using specific parameters, we were able to remove the adaptors’ contamination and low-quality sequences, so the total amount of clean data was mapped to the UCSC/hg19 reference genome. Indel and variant calls were made using GATK version 2.7 (Broad Institute, Cambridge, MA, USA) (and its recommended parameters) and then the variants were also annotated against external datasets, including 1000 genomes and dbSNP (36).
For Finnish patients an NGS-based clinical trio exome was performed with Sophia Genetics custom clinical exome solution and Illumina sequencing. Variant annotation and filtering were performed with Sophia DDM™ and visualized with IGV (Broad Institute).
Prioritization of the variants was obtained excluding polymorphisms (minor allele frequency, MAF < 0,01), synonymous variants, variants classified as benign or likely benign. Frameshift, stopgain and splice site variants were prioritized as pathogenic. The potential impact of variants on splicing was evaluated using Alamut® Visual software—version 2.11–0 (Interactive Biosoftware, France), which employs five different algorithms: SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer and HumanSplicingFinder.
The following public databases were used for the interpretation of the variants: ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), LOVD (https://databases.lovd.nl/shared/genes), the Human Genome Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac). Each disease-candidate variant was confirmed by Sanger sequencing.
Additional genetic tests
Estonian patients (Supplementary Material, Table S1 patients 29–40) were investigated by TruSight One (TSO) and TruSight One Expanded (TSOE) panels (Illumina Inc., San Diego, California). These panels cover ~4800 and ~6700 genes associated with known genetic disorders or clinical phenotypes. A detailed description of the method was published earlier (37). Detected variants were validated by Sanger sequencing.
In patients 17 and 18 in Supplementary Material, Table S1, a targeted panel approach including ANKRD11 was employed to ascertain the disease. The targeted panel was realized via Next Generation Sequencing on the 454 Junior platform (Roche) with specific amplification. Analysis was carried out using the GS Amplicon Variant Analyzer (AVA) application version 2.9 (Roche). In alternative amplification and high-throughput sequencing of ANKRD11 coding regions and exon-intron junctions was obtained by the NimbleGen SeqCapEZ Custom Enrichment Kit (Roche) and Illumina platform Nextseq550 (sensitivity >99%). Results interpretation focused on exonic regions with a read depth >20X.
Informed Consent Statement
The patients/participants or their parents provided their written informed consent to participate in this study.
Supplementary Material
{kind=link}
Acknowledgements
We are grateful to our patients for their cooperation. This work is generated within the European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders (ERN ITHACA). ‘Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases’, member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, and of the EuroBioBank network, provided us with specimens. This work has also been partially supported by grant-RC Linea 1‘Studio fenotipo-genotipo delle malattie genetiche rare ad espressione neuropsichiatrica in età evolutive’ and the 5x 1000 voluntary contributions, Italian Ministry of Health (R.B.).
Conflict of Interest statement. The authors declare that they have no conflict of interest.
Contributor Information
Lorenzo Loberti, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Lucia Pia Bruno, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy.
Stefania Granata, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Gabriella Doddato, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy.
Sara Resciniti, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy.
Francesca Fava, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Michele Carullo, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy.
Elisa Rahikkala, Department of Clinical Genetics, PEDEGO Research Unit, and Medical Research Center Oulu, University of Oulu and Oulu University Hospital, Oulu 90014, Finland.
Guillaume Jouret, National Center of Genetics (NCG), Laboratoire national de santé (LNS), L-3555 Dudelange, Luxembourg.
Leonie A Menke, Amsterdam UMC location University of Amsterdam, Department of Pediatrics, Amsterdam 1100, The Netherlands.
Damien Lederer, Institut de Pathologie et de Génétique; Centre de Génétique Humaine, Gosselies 6041, Belgium.
Pascal Vrielynck, William Lennox Neurological Hospital, Reference Center for Refractory Epilepsy UCLouvain, Ottignies 1340, Belgium.
Lukáš Ryba, Department of Biology and Medical Genetics, Charles University – 2nd Faculty of Medicine and University Hospital Motol, Prague 150 00, Czech Republic.
Nicola Brunetti-Pierri, Department of Translational Medicine, University of Naples "Federico II", Naples 80125, Italy.
Amaia Lasa-Aranzasti, Area of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcellona 08035, Spain.
Anna Maria Cueto-González, Area of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcellona 08035, Spain.
Laura Trujillano, Area of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcellona 08035, Spain.
Irene Valenzuela, Area of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcellona 08035, Spain.
Eduardo F Tizzano, Area of Clinical and Molecular Genetics, Vall d’Hebron University Hospital, Barcellona 08035, Spain.
Alessandro Mauro Spinelli, Regional Coordinating Center for Rare Diseases, Udine 33100, Italy.
Irene Bruno, Institute for Maternal and Child Health, Trieste 34100, Italy.
Aurora Currò, Genetic Counseling Service, Department of Pediatrics, Regional Hospital of Bolzano, Bolzano 39100, Italy.
Franco Stanzial, Genetic Counseling Service, Department of Pediatrics, Regional Hospital of Bolzano, Bolzano 39100, Italy.
Francesco Benedicenti, Genetic Counseling Service, Department of Pediatrics, Regional Hospital of Bolzano, Bolzano 39100, Italy.
Diego Lopergolo, IRCCS Stella Maris Foundation, Molecular Medicine for Neurodegenerative and Neuromuscular Disease Unit, Pisa 98125, Italy.
Filippo Maria Santorelli, IRCCS Stella Maris Foundation, Molecular Medicine for Neurodegenerative and Neuromuscular Disease Unit, Pisa 98125, Italy.
Constantia Aristidou, Department of Clinical Genetics and Genomics, The Cyprus Institute of Neurology & Genetics, Nicosia 1683, Cyprus.
George A Tanteles, Department of Clinical Genetics and Genomics, The Cyprus Institute of Neurology & Genetics, Nicosia 1683, Cyprus.
Isabelle Maystadt, Institut de Pathologie et de Génétique; Centre de Génétique Humaine, Gosselies 6041, Belgium.
Tinatin Tkemaladze, Department of Molecular and Medical Genetics, Tbilisi State Medical University, Tbilisi 0162, Georgia.
Tiia Reimand, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia; Institute of Clinical Medicine, University of Tartu, Tartu 50406, Estonia.
Helen Lokke, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia; Institute of Clinical Medicine, University of Tartu, Tartu 50406, Estonia.
Katrin Õunap, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia; Institute of Clinical Medicine, University of Tartu, Tartu 50406, Estonia.
Maria K Haanpää, Department of Genomics and Clinical Genetics, Turku University Hospital, Turku 20500, Finland.
Andrea Holubová, Department of Biology and Medical Genetics, Charles University – 2nd Faculty of Medicine and University Hospital Motol, Prague 150 00, Czech Republic.
Veronika Zoubková, Department of Biology and Medical Genetics, Charles University – 2nd Faculty of Medicine and University Hospital Motol, Prague 150 00, Czech Republic.
Martin Schwarz, Department of Biology and Medical Genetics, Charles University – 2nd Faculty of Medicine and University Hospital Motol, Prague 150 00, Czech Republic.
Riina Žordania, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia.
Kai Muru, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia; Institute of Clinical Medicine, University of Tartu, Tartu 50406, Estonia.
Laura Roht, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia; Institute of Clinical Medicine, University of Tartu, Tartu 50406, Estonia.
Annika Tihveräinen, Department of Child Neurology, Turku University Hospital, Turku 20500, Finland.
Rita Teek, Department of Clinical Genetics, Genetic and Personalized Medicine Clinic, Tartu University Hospital, Tartu 50406, Estonia.
Ulvi Thomson, Centre for Neurological Diseases, West-Tallinn Central Hospital, Tallinn 10617, Estonia.
Isis Atallah, Division of Genetic Medicine, Lausanne University Hospital (CHUV) and University of Lausanne, 1011 Lausanne, Switzerland.
Andrea Superti-Furga, Division of Genetic Medicine, Lausanne University Hospital (CHUV) and University of Lausanne, 1011 Lausanne, Switzerland.
Sabrina Buoni, Division of Child and Adolescent Neuropsychiatry, University of Siena, Siena 53100, Italy.
Roberto Canitano, Division of Child and Adolescent Neuropsychiatry, University of Siena, Siena 53100, Italy.
Valeria Scandurra, Division of Child and Adolescent Neuropsychiatry, University of Siena, Siena 53100, Italy.
Annalisa Rossetti, Clinical Paediatrics, Department of Molecular Medicine and Development, University of Siena, Siena 53100, Italy.
Salvatore Grosso, Clinical Paediatrics, Department of Molecular Medicine and Development, University of Siena, Siena 53100, Italy.
Roberta Battini, IRCCS Stella Maris Foundation, Department of Developmental Neuroscience, Pisa 98125, Italy; Department of Clinical and Experimental Medicine, University of Pisa, Pisa 56122, Italy.
Margherita Baldassarri, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy.
Maria Antonietta Mencarelli, Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Caterina Lo Rizzo, Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Mirella Bruttini, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Francesca Mari, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Francesca Ariani, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Alessandra Renieri, Medical Genetics, University of Siena, Siena 53100, Italy; Med Biotech Hub and Competence Centre, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Anna Maria Pinto, Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Funding
K.Õ., T.R., K.M. and L.R. were supported by Estonian Research Council grant PRG471.
Authors Contribution
Author Contributions: L.B. and L.L. made important contributions in the interpretation of the molecular results and drafted the manuscript. A.M.P. and A.R. made substantial contributions to the conception and design of the study and reviewed the manuscript. All co-authors performed clinical data acquisition, clinical evaluations and variants interpretation. All authors approved the final version of the manuscript for publication.
References
- 1. Herrmann, J., Pallister, P.D., Tiddy, W. and Opitz, J.M. (1975) The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Artic. Ser., 11, 7–18. [PubMed] [Google Scholar]
- 2. Morel Swols, D., Foster, J. and Tekin, M. (2017) KBG syndrome. Orphanet J. Rare Dis., 12, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parenti, I., Mallozzi, M.B., Hüning, I., Gervasini, C., Kuechler, A., Agolini, E., Albrecht, B., Baquero-Montoya, C., Bohring, A., Bramswig, N.C. et al. (2021) ANKRD11 variants: KBG syndrome and beyond. Clin. Genet., 100, 187–200. [DOI] [PubMed] [Google Scholar]
- 4. Ropers, H.H. and Wienker, T. (2015) Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. Eur. J. Med. Genet., 58, 715–718. [DOI] [PubMed] [Google Scholar]
- 5. Low, K., Ashraf, T., Canham, N., Clayton-Smith, J., Deshpande, C., Donaldson, A., Fisher, R., Flinter, F., Foulds, N., Fryer, A. et al. (2016) Clinical and genetic aspects of KBG syndrome. Am. J. Med. Genet. A, 170, 2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wojciechowska, K., Nurzyńska-Flak, J., Styka, B., Kacprzak, M. and Lejman, M. (2021) Case Report: Two Newly Diagnosed Patients With KBG Syndrome—Two Different Molecular Changes. Front. Pediatr., 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gnazzo, M., Lepri, F.R., Dentici, M.L., Capolino, R., Pisaneschi, E., Agolini, E., Rinelli, M., Alesi, V., Versacci, P., Genovese, S. et al. (2020) KBG syndrome: Common and uncommon clinical features based on 31 new patients. Am. J. Med. Genet. A, 182, 1073–1083. [DOI] [PubMed] [Google Scholar]
- 8. Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., Bademci, G., Agolini, E., Guo, S., Konuk, B. et al. (2011) Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet., 89, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sacharow, S., Li, D., Fan, Y.S. and Tekin, M. (2012) Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am. J. Med. Genet. A, 158A, 547–552. [DOI] [PubMed] [Google Scholar]
- 10. Zhang, A., Yeung, P.L., Li, C.-W., Tsai, S.-C., Dinh, G.K., Wu, X., Li, H. and Chen, J.D. (2004) Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J. Biol. Chem., 279, 33799–33805. [DOI] [PubMed] [Google Scholar]
- 11. Goldenberg, A., Riccardi, F., Tessier, A., Pfundt, R., Busa, T., Cacciagli, P., Capri, Y., Coutton, C., Delahaye-Duriez, A., Frebourg, T. et al. (2016) Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am. J. Med. Genet. A, 170, 2847–2859. [DOI] [PubMed] [Google Scholar]
- 12. Behnert, A., Auber, B., Steinemann, D., Frühwald, M.C., Huisinga, C., Hussein, K., Kratz, C. and Ripperger, T. (2018) KBG syndrome patient due to 16q24.3 microdeletion presenting with a paratesticular rhabdoid tumor: Coincidence or cancer predisposition? Am. J. Med. Genet. A, 176, 1449–1454. [DOI] [PubMed] [Google Scholar]
- 13. Li, Q., Sun, C., Yang, L., Lu, W. and Luo, F. (2021) Comprehensive analysis of clinical spectrum and genotype associations in Chinese and literature reported KBG syndrome. Transl. Pediatr., 10, 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ockeloen, C.W., Willemsen, M.H., de Munnik, S., van Bon, B.W.M., de Leeuw, N., Verrips, A., Kant, S.G., Jones, E.A., Brunner, H.G., van Loon, R.L.E. et al. (2015) Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur. J. Hum. Genet. EJHG, 23, 1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palumbo, O., Palumbo, P., Di Muro, E., Cinque, L., Petracca, A., Carella, M. and Castori, M. (2020) A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. G. E. N., 11, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roth, D.M., Baddam, P., Lin, H., Vidal-García, M., Aponte, J.D., De Souza, S.-T., Godziuk, D., Watson, A.E.S., Footz, T., Schachter, N.F. et al. (2021) The Chromatin Regulator Ankrd11 Controls Palate and Cranial Bone Development. Front. Cell Dev. Biol., 9, 645386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walz, K., Cohen, D., Neilsen, P.M., Foster, J., Brancati, F., Demir, K., Fisher, R., Moffat, M., Verbeek, N.E., Bjørgo, K. et al. (2015) Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum. Genet., 134, 181–190. [DOI] [PubMed] [Google Scholar]
- 18. Dickinson, M.E., Flenniken, A.M., Ji, X., Teboul, L., Wong, M.D., White, J.K., Meehan, T.F., Weninger, W.J., Westerberg, H., Adissu, H. et al. (2017) Correction: Corrigendum: High-throughput discovery of novel developmental phenotypes. Nature, 551, 398–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neilsen, P.M., Cheney, K.M., Li, C.-W., Chen, J.D., Cawrse, J.E., Schulz, R.B., Powell, J.A., Kumar, R. and Callen, D.F. (2008) Identification of ANKRD11 as a p53 coactivator. J. Cell Sci., 121, 3541–3552. [DOI] [PubMed] [Google Scholar]
- 20. Crippa, M., Rusconi, D., Castronovo, C., Bestetti, I., Russo, S., Cereda, A., Selicorni, A., Larizza, L. and Finelli, P. (2015) Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet., 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cucco, F., Sarogni, P., Rossato, S., Alpa, M., Patimo, A., Latorre, A., Magnani, C., Puisac, B., Ramos, F.J., Pié, J. et al. (2020) Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia de Lange syndrome. Am. J. Med. Genet. A, 182, 1690–1696. [DOI] [PubMed] [Google Scholar]
- 22. Scarano, E., Tassone, M., Graziano, C., Gibertoni, D., Tamburrino, F., Perri, A., Gnazzo, M., Severi, G., Lepri, F. and Mazzanti, L. (2019) Novel Mutations and Unreported Clinical Features in KBG Syndrome. Mol. Syndromol., 10, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahmed, A., Mufeed, A., Ramachamparambathu, A.K. and Hasoon, U. (2016) Identifying Aarskog Syndrome. J. Clin. Diagn. Res. JCDR, 10, ZD09–ZD11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sayed, I.S.M., Abdel-Hamid, M.S. and Abdel-Salam, G.M.H. (2020) KBG syndrome in two patients from Egypt. Am. J. Med. Genet. A, 182, 1309–1312. [DOI] [PubMed] [Google Scholar]
- 25. Lo-Castro, A., Brancati, F., Digilio, M.C., Garaci, F.G., Bollero, P., Alfieri, P. and Curatolo, P. (2013) Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am. J. Med. Genet. B Neuropsychiatr. Genet., 162, 17–23. [DOI] [PubMed] [Google Scholar]
- 26. Khalifa, M., Stein, J., Grau, L., Nelson, V., Meck, J., Aradhya, S. and Duby, J. (2013) Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am. J. Med. Genet. A, 161, 835–840. [DOI] [PubMed] [Google Scholar]
- 27. Alfieri, P., Caciolo, C., Lazzaro, G., Menghini, D., Cumbo, F., Dentici, M.L., Digilio, M.C., Gnazzo, M., Demaria, F., Pironi, V. et al. (2021) Cognitive and Adaptive Characterization of Children and Adolescents with KBG Syndrome: An Explorative Study. J. Clin. Med., 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kutkowska-Kaźmierczak, A., Boczar, M., Kalka, E., Castañeda, J., Klapecki, J., Pietrzyk, A., Barczyk, A., Malinowska, O., Landowska, A., Gambin, T. et al. (2021) Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3. G. E. N., 12, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kiesler, J. and Ricer, R. (2003) The Abnormal Fontanel. Am. Fam. Physician, 67, 2547–2552. [PubMed] [Google Scholar]
- 30. Zollino, M., Battaglia, A., D’Avanzo, M.G., Della Bruna, M.M., Marini, R., Scarano, G., Cappa, M. and Neri, G. (1994) Six additional cases of the KBG syndrome: clinical reports and outline of the diagnostic criteria. Am. J. Med. Genet., 52, 302–307. [DOI] [PubMed] [Google Scholar]
- 31. Parloir, C., Fryns, J.P., Deroover, J., Lebas, E., Goffaux, P. and van den Berghe, H. (1977) Short stature, craniofacial dysmorphism and dento-skeletal abnormalities in a large kindred. A variant of K.B.G. syndrome or a new mental retardation syndrome. Clin. Genet., 12, 263–266. [DOI] [PubMed] [Google Scholar]
- 32. Willemsen, M.H., Fernandez, B.A., Bacino, C.A., Gerkes, E., de Brouwer, A.P., Pfundt, R., Sikkema-Raddatz, B., Scherer, S.W., Marshall, C.R., Potocki, L. et al. (2010) Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur. J. Hum. Genet., 18, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Novara, F., Rinaldi, B., Sisodiya, S.M., Coppola, A., Giglio, S., Stanzial, F., Benedicenti, F., Donaldson, A., Andrieux, J., Stapleton, R. et al. (2017) Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases. Eur. J. Hum. Genet., 25, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark, M.M., Stark, Z., Farnaes, L., Tan, T.Y., White, S.M., Dimmock, D. and Kingsmore, S.F. (2018) Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med., 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Niguidula, N., Alamillo, C., Shahmirzadi Mowlavi, L., Powis, Z., Cohen, J.S. and Farwell Hagman, K.D. (2018) Clinical whole-exome sequencing results impact medical management. Mol. Genet. Genomic Med., 6, 1068–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M. et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pajusalu, S., Kahre, T., Roomere, H., Murumets, Ü., Roht, L., Simenson, K., Reimand, T. and Õunap, K. (2018) Large gene panel sequencing in clinical diagnostics-results from 501 consecutive cases. Clin. Genet., 93, 78–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.