

Abstract
Mutations in the Myelin Protein Zero gene (MPZ), encoding P0, the major structural glycoprotein of peripheral nerve myelin, are the cause of Charcot–Marie-Tooth (CMT) type 1B neuropathy, and most P0 mutations appear to act through gain-of-function mechanisms. Here, we investigated how misglycosylation, a pathomechanism encompassing several genetic disorders, may affect P0 function. Using in vitro assays, we showed that gain of glycosylation is more damaging for P0 trafficking and functionality as compared with a loss of glycosylation. Hence, we generated, via CRISPR/Cas9, a mouse model carrying the MPZD61N mutation, predicted to generate a new N-glycosylation site in P0. In humans, MPZD61N causes a severe early-onset form of CMT1B, suggesting that hyperglycosylation may interfere with myelin formation, leading to pathology. We show here that MPZD61N/+ mice develop a tremor as early as P15 which worsens with age and correlates with a significant motor impairment, reduced muscular strength and substantial alterations in neurophysiology. The pathological analysis confirmed a dysmyelinating phenotype characterized by diffuse hypomyelination and focal hypermyelination. We find that the mutant P0D61N does not cause significant endoplasmic reticulum stress, a common pathomechanism in CMT1B, but is properly trafficked to myelin where it causes myelin uncompaction. Finally, we show that myelinating dorsal root ganglia cultures from MPZD61N mice replicate some of the abnormalities seen in vivo, suggesting that they may represent a valuable tool to investigate therapeutic approaches. Collectively, our data indicate that the MPZD61N/+ mouse represents an authentic model of severe CMT1B affirming gain-of-glycosylation in P0 as a novel pathomechanism of disease.
Introduction
Myelin is a multilayered structure that enwraps and protects axons, acting as an insulator that facilitates the conduction of electrical impulses along nerves (1). In the peripheral nervous system, myelin is formed by the plasma membrane of Schwann cells (SCs) with each SC defining a single internode (2). The most abundant protein produced by myelinating SCs is P0, encoded by the Myelin Protein Zero (MPZ) gene; P0 accounts for up to 25–50% of the protein in peripheral nerve myelin (3) and plays a key role in myelin compaction linking and stabilizing adjacent myelin lamellae (4,5). P0 is a 29 kDa transmembrane glycoprotein, belonging to the immunoglobulin supergene family (IgCAM); it is modified as it moves through the endoplasmic reticulum (ER) to the SC plasma membrane by, among others, the addition of a single N-linked oligosaccharide at N122, which contains the HNK-1 moiety, that modulates P0–P0 interactions and myelin compaction (6).
Charcot–Marie Tooth (CMT) disease defines a large group of hereditary motor and sensor neuropathies and is the most common inherited neuromuscular disorder (7). Patients typically develop distal muscle weakness and atrophy, often associated with mild to moderate sensory loss, depressed tendon reflexes and high-arched feet (7). Despite the clinical similarities among patients with CMT, the disorder is genetically heterogeneous, resulting from mutations in more than 90 genes (8). Mutations in the MPZ gene are identified in 5% of CMT patients, representing the fourth most common cause of CMT (9). Mutations in MPZ may cause either a dys/demyelinating early-onset neuropathy with a delay in motor development (early-onset CMT1B) or an axonal neuropathy occurring in adulthood (late-onset CMT1B or CMT2I/J) (10). Finally, a minority of patients carrying MPZ mutations present an intermediate phenotype, similar to CMT1A (11). It has been shown that early-onset CMT1B is caused by multiple pathogenic mechanisms involving gain of function and dominant negative effects, amongst which protein misfolding and activation of the unfolded protein response (UPR) play a major contribution (12–15). Yet, not all P0 mutations are misfolded or activate a UPR (15), suggesting the existence of further pathomechanisms.
Recent work revealed that mutations removing constitutive or introducing new N-linkage sites are more frequent than expected (16,17), accounting for up to ~1.4% of all missense mutations in genes coding for membrane proteins (16). Protein misglycosylation has also been proposed as a pathomechanism for CMT1B; this includes mutations causing loss of glycosylation, such as the common MPZT124M mutation that causes a late-onset axonal CMT1B (18), and mutations creating a new glycosylation motif (N-X-S/T), such as the mutant MPZD61N. This mutation is associated with a severe, early-onset demyelinating CMT1B neuropathy with tomacula-like structures in human sural nerve biopsies (19,20). While misglycosylation is not rare, its role in CMT1B pathogenesis had never been thoroughly explored. Here we describe the in vitro characterization of different P0 mutants featuring the loss of the constitutive glycosylation site or the introduction of a new glycosylation motif. In addition, we report the generation and characterization of a mouse model carrying the MPZD61N mutation to investigate how peripheral myelination is affected by P0 hyperglycosylation. We show that MPZD61N mice present a severe dysmyelinating neuropathy, characterized by motor and neurophysiological defects, hypomyelination, myelin packing defects and tomacula-like structures that recapitulate the human phenotype, affirming P0 hyperglycosylation as a novel pathomechanism in CMT1B.
Results
Analysis of misglycosylating MPZ mutations in vitro
Protein misglycosylation following a missense mutation entails the introduction of a new glycosylation site or the removal of a constitutive one; this relatively frequent event (16,17) has been described also for MPZ mutations (18–21). Using NetNGlyc 1.0 and NetOGlyc 3.1 Server (22), we identified two different groups of potentially misglycosylating mutations in the MPZ gene: hyperglycosylating mutations (D61N, D109N, D118N and K138N), predicted to introduce a new glycosylation site, and hypoglycosylating mutations (T124M, T124A, T124K and N122S), predicted to remove the constitutive N122 glycosylation site of P0. Through site-directed mutagenesis, we generated constructs coding enhanced green fluorescent protein (EGFP)-tagged versions of all the presumptive hyper- and hypoglycosylated P0 proteins and expressed them in a Schwannoma cell line (RT4-D6P2T). Western blot (WB) analysis showed that all the predicted hyperglycosylated mutants displayed a molecular weight (MW) higher than P0wt, ranging from the highest MW of K138N to the lowest of D118N (Fig. 1A), whereas the predicted hypoglycosylated proteins presented a MW lower than P0wt (Fig. 1B).
Figure 1.
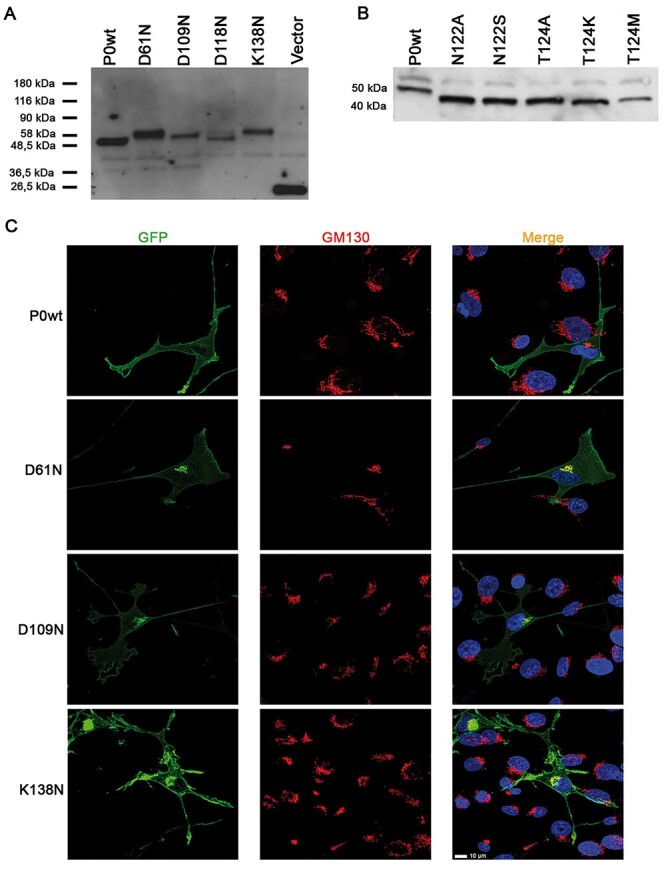
P0 mutant proteins analysis. (A) WB on extracts from Schwannoma cells transiently transfected with plasmids expressing predicted P0 hyperglycosylating mutants. (B) WB on extracts from Schwannoma cells transiently transfected with plasmids expressing predicted P0 hypoglycosylating mutants. (C) Confocal microscopy of Schwannoma cells transiently transfected with the EGFP-tagged hyperglycosylated P0 proteins D61N, D109N and K138N costained with the Golgi marker GM130; mutant proteins, but not P0wt, partially colocalize with GM130. Scale bar = 10 μm.
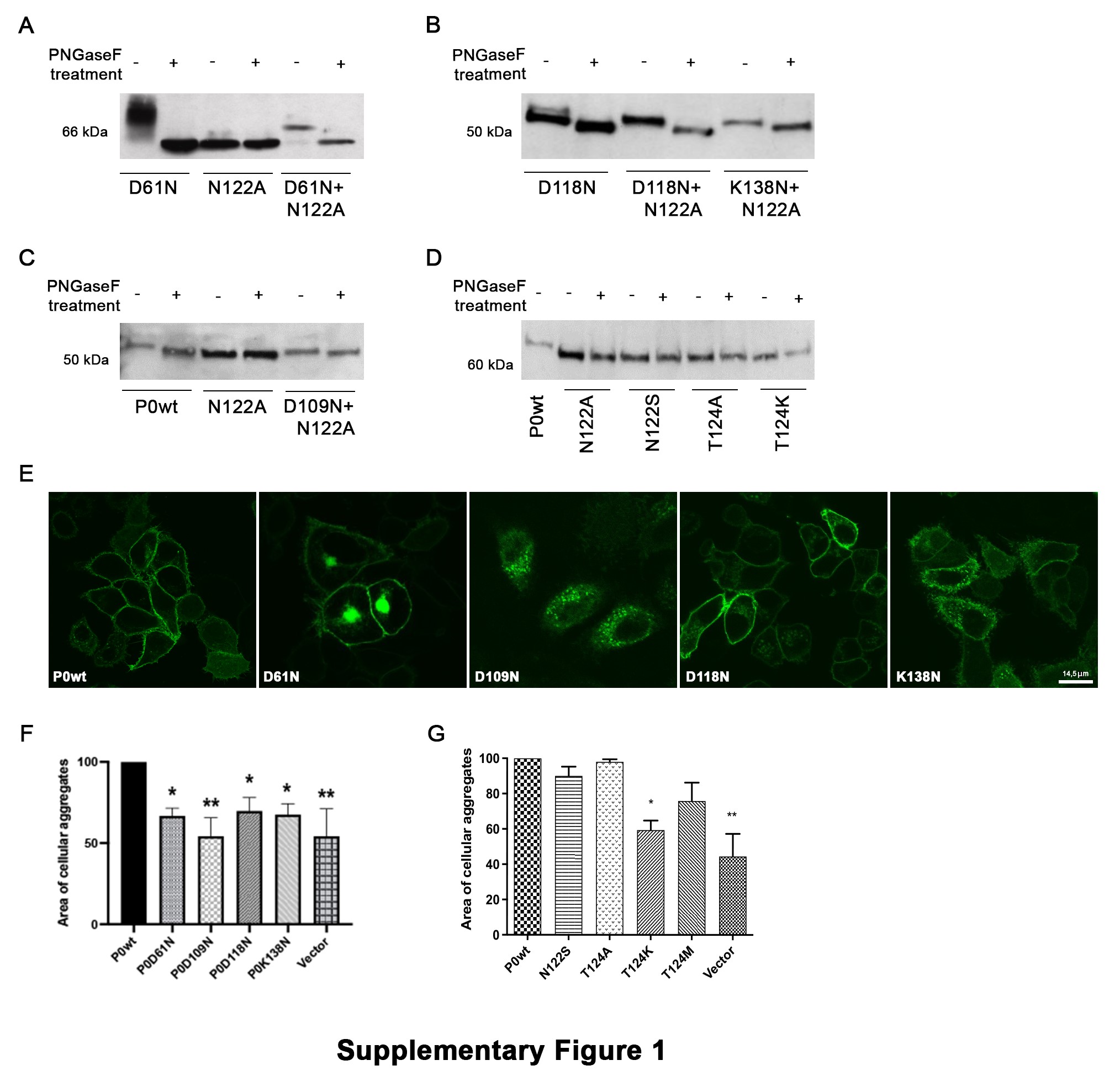
To test whether the higher MW of the hyperglycosylated P0 proteins was entirely due to extra glycans, we treated protein extracts from cells transfected with D61N and D118N mutants with PNGaseF, an amidase that cleaves N-glycans. After treatment the MW of the mutant proteins decreased to the same level as P0wt treated with the same amidase, indicating that the higher MW of these mutant proteins is due to additional oligosaccharides (Supplementary Material, Fig. S1A and B). To further demonstrate glycosylation at the new acceptor site, we generated constructs to express each gain-of-glycosylation MPZ mutation together with the N122A mutation to produce P0 variants with the unique novel glycosylation site but without the wild-type (WT) N-sequon. Again, all these mutant proteins were sensitive to PNGaseF treatment, further proving that they are all glycosylated at the novel N-sequon (Supplementary Material, Fig. S1A–C). Conversely, none of the P0 variants predicted to be nonglycosylated were sensitive to PNGaseF, confirming that they lack glycans (Supplementary Material, Fig. S1D).
Next, to verify whether P0 misglycosylation alters P0 trafficking and localization, we transfected HeLa cells with the EGFP-tagged mutant proteins. Immunofluorescence analysis showed that, whereas P0wt fully reached the cell membrane, the hyperglycosylated proteins D61N, D109N and K138N were partially retained intracellularly. Conversely, the D118N hyperglycosylated mutant and all the hypoglycosylated mutant proteins mostly reached the plasma membrane (Supplementary Material, Fig. S1E, and data not shown). To further evaluate mutant protein localization, we transfected HeLa and RT4-D6P2T cells with EGFP-tagged P0D61N, P0D109N and P0K138N and costained with ER and Golgi markers. While in both cell types P0wt normally reached the plasma membrane, all mutant proteins colocalized with KDEL (ER marker) and GM130 (Golgi marker) in HeLa cells (Supplementary Material, Fig. S2A and B), whereas in RT4-D6P2T cells the mutant proteins only marginally colocalized with KDEL and appeared to mostly colocalize with GM130 (Fig. 1C, and data not shown). Overall these data suggest that the mutant hyperglycosylated P0 are partially retained in the ER–Golgi network.
In myelin, P0 has an essential role in the adhesion of adjacent membrane wraps, through the formation of functional tetramers (5). To test whether the misglycosylating mutations interfered with P0 adhesion, we performed an aggregation assay in transiently transfected HeLa cells (13). The size of aggregates formed by cells expressing wt P0 protein was used as a reference value; cells expressing hyperglycosylated P0 proteins formed aggregates that were roughly 30–40% smaller compared to P0wt, suggesting that hyperglycosylation impairs homophilic interactions (Supplementary Material, Fig. S1F). Instead, cells expressing hypoglycosylated P0 proteins formed aggregates similar to P0wt, with the exception of T124K which formed smaller aggregates (Supplementary Material, Fig. S1G).
Altogether these data indicate that the hyperglycosylated mutations, but not the hypoglycosylated ones, interfered with protein localization and protein adhesion properties, supporting the hypothesis that gain-of-glycosylation is more disruptive as compared to loss-of-glycosylation (16,17) and prompted us to further investigate P0 hyperglycosylation.
Generation of the Mpz D61N/+ mouse model
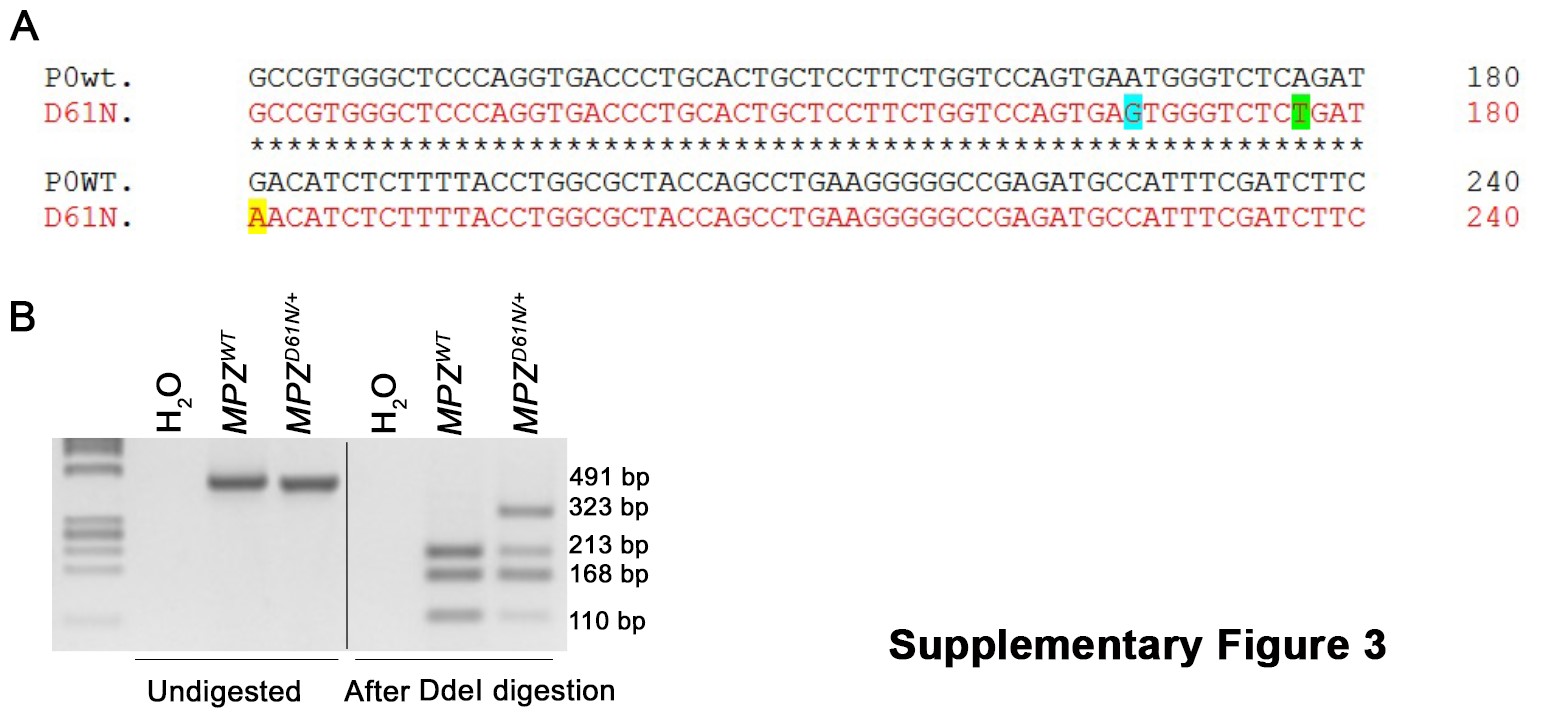
To evaluate the effects of P0 hyperglycosylation in vivo, we established a transgenic mouse model for the MpzD61N mutation using genome editing with CRISPR/CRISPR-associated protein 9 (Cas9) nuclease (23), to perfectly mimic the human mutation. In particular, for the generation of the MpzD61N model, three different single-nucleotide polymorphism (SNP) conversions were introduced: a G > A conversion (position 181) that introduced the desired D to N aminoacidic change, a silent A > G conversion (position 168) that prevented gRNA binding and recleavage and a silent A > T conversion (position 177) that eliminated the DdeI restriction enzyme site (Supplementary Material, Fig. S3A); the latter conversion allowed us to discriminate between WT and mutant mice (Supplementary Material, Fig. S3B). The A > G and A > T conversions were designed not to cause any amino acid change so that the only difference in protein sequence is the D to N in position 61.
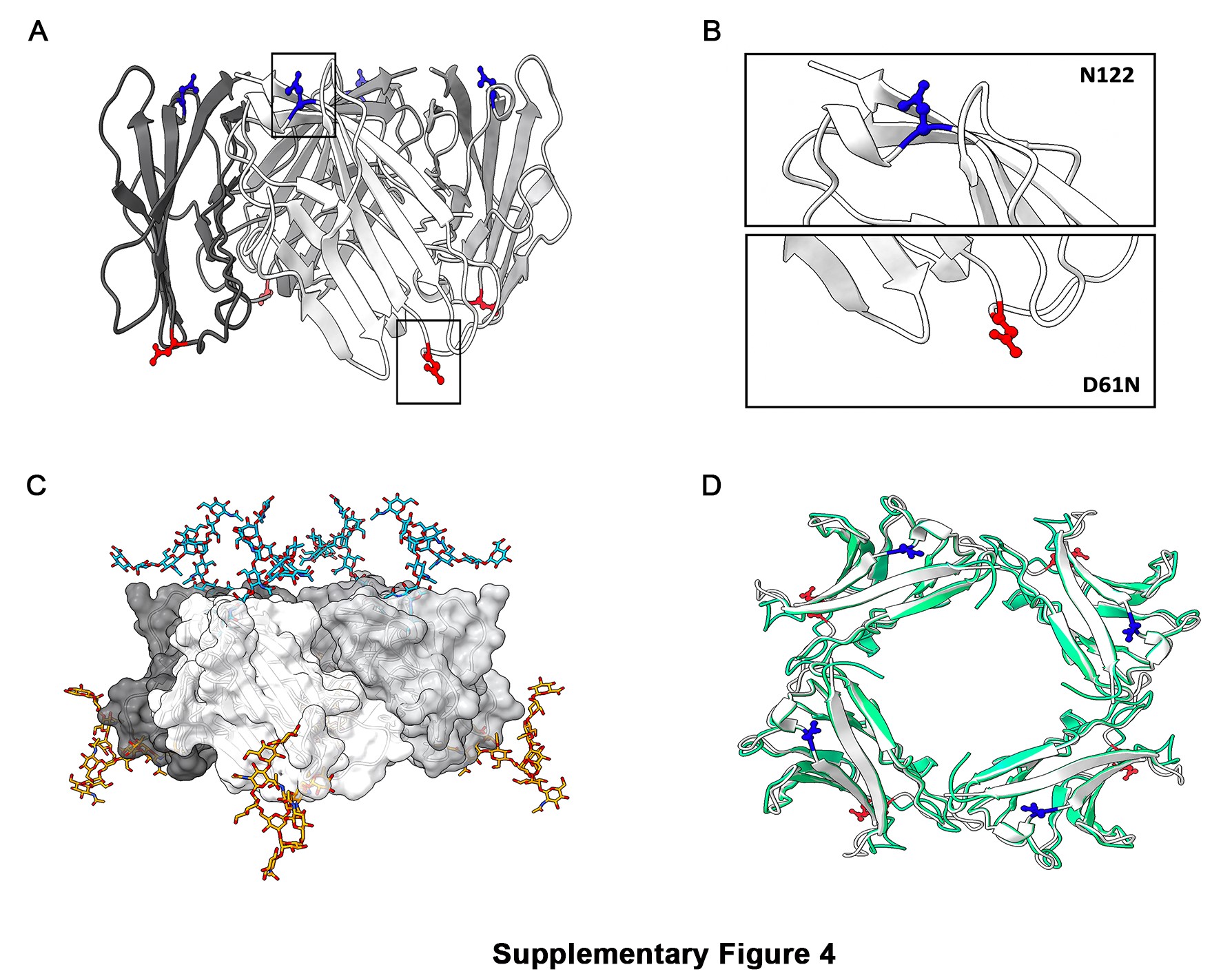
To support an investigation at a molecular level of the experimental MpzD61N/+ mouse model, the tetrameric model of the mouse variant was obtained by the superimposition of the four monomeric models of the P0D61N variants on the rat Mpz tetramer (5). Visual inspection of the tetrameric structure showed that residues D61N and N122 of each chain are accessible for attachment of N-glycan to asparagine (Supplementary Material, Fig. S4A and B). The structural compatibility of the second glycosylation site D61N, with respect to N122 residue, was evaluated by building an N-linked oligosaccharide containing the HNK-1 moiety (24). The oligosaccharide was subsequently attached to the protein glycosylation sites of the WT and of the mutated protein, to form the N-linked glycoprotein model of P0D61N.
Sugar mapping on the tetrameric structure of MpzD61N/+ showed that two glycosylation sites per tetrameric unit are allowed because there is no steric hindrance between them (Supplementary Material, Fig. S4C). The structural superimposition between the mouse and the human P0D61N tetramers (Supplementary Material, Fig. S4D) showed an identical folding of the tetrameric units as well as of the N-residues side chains involved in N-glycosylation.
To confirm that the mutant P0D61N protein was hyperglycosylated in vivo, we performed a PNGaseF assay on protein extracts from sciatic nerves of WT and MpzD61N/+ mice. The untreated WT lanes are characterized by a single band (29 kDa), while the mutant extracts present two distinct bands, one corresponding to the WT and a second one with a higher MW. After PNGaseF treatment, the MW of P0 in the D61N/+ extracts collapses to a single lower band of the same MW as the WT, demonstrating that the upper band is entirely due to extra glycan residues (Fig. 2A).
Figure 2.

Analysis of the MpzD61N mouse model. (A) WB for P0 on proteins extracted from sciatic nerves of WT and MpzD61N/+ animals treated with PNGaseF. Before PNGase digestion P0D61N/+ extracts show a double band; after treatment with PNGaseF, the MW of P0 in MpzD61N/+ samples decreases to a single band of the same MW as in the WT. (B) Table describing the tremor in WT and MpzD61N/+ mice at different time points; the scale is visual and goes from – (no tremor) to + (mild tremor) to ++++ (very severe tremor). (C) Rotarod analysis, performed at 3 months of age, shows that MpzD61N/+ have a severe reduction in motor capacity as compared with WT; ****P-value < 0.0001 by two-way ANOVA with Sidak multiple comparison post-hoc test. n = 8 for each genotype. (D) Grip strength test, performed at 3 months of age, shows that MpzD61N/+ mice have reduced strength in hind limbs as compared with WT. *P-value = 0.0246 by unpaired Student t-test. n = 13 for each genotype.
Mpz D61N/+ mice present severe impairment in motor capacity and neurophysiology
Mice heterozygous for the Mpz variant D61N (MpzD61N/+) were viable, bred normally and pups were born with the expected Mendelian ratio and were initially indistinguishable from WT littermates. However, MpzD61N/+ mice started manifesting a tremor around postnatal day 15 (P15). The tremor was scored on a + to ++++ scale and it worsened with ageing (Fig. 2B) but without impairing life expectancy at least up to 6 months of age. To test effects on motor capacity we evaluated the latency to fall on the accelerating Rotarod (14) in mice at 3 months of age. The mean latency to fall off the rotarod over the 3-day trial period was 184.7 ± 14.21 s for MpzD61N/+ mice, compared with 329.7 ± 23.24 s for WT (P-value < 0.0001) (Fig. 2C). At 3 months we also performed grip strength analysis to evaluate the force and hind limb strength of MpzD61N/+ mice compared with WT mice. We measured the maximal force produced over six different trials. The mean force in the WT animals was 97.19 ± 4.4 gr, while in MpzD61N/+ mice the mean force was 82.68 ± 4.1 gr (P-value 0.0246) (Fig. 2D). Altogether these data indicate that MpzD61N/+ mice present motor impairment and reduced muscular strength.
Next, we performed electrophysiological analysis on mutant and control animals at 1- and 3 months of age; we evaluated motor nerve conduction velocity (MNCV), F-wave latency (FWL), distal and proximal latency and distal and proximal amplitude. At 1 month of age, all the parameters were significantly altered in MpzD61N/+ as compared with WT. MNCV was significantly lower (12.05 ± 1.22 m/s) as compared with controls (33.02 ± 1.622 m/s) (Fig. 3Aand E), with prolonged latencies and a strong decrease of cMAPs (Fig. 3B and E). By 3 months of age, all the electrophysiological parameters were unrecordable in most mutant mice or dramatically impaired in the few cases where we could record them (Fig. 3C–F), suggesting a severe neuropathy.
Figure 3.

Mpz D61N/+ mice present altered electrophysiological parameters. (A) MNCVs and (B) CMAP amplitudes recorded at 1 month in WT and MpzD61N/+ mice; ****P-value < 0.0001 by unpaired Student t-test. n = 8 for each genotype. (C) MNCVs and (D) CMAP amplitudes recorded at 3 months in WT and MpzD61N/+ mice; ****P-value < 0.0001 by unpaired Student t-test. n = 8 for each genotype. (E) Original recordings in WT and MpzD61N/+ at 1 and 3 months. Flags indicate the onset and end of the CMAPs; MpzD61N/+ mice showed a significant reduction of nerve conduction velocities, a prolonged latency of both distal and proximal CMAP as well as a reduced CMAP amplitude compared with controls. (F) Table representing the analysis of different electrophysiological parameters in WT and MpzD61N/+ animals at 3 months. In the MpzD61N/+ column, we indicated the percentage of animals in which the different parameters were unrecordable.
Mpz D61N/+ show a severe de/dysmyelinating phenotype and defects in myelin compaction
The behavioral and neurophysiological analysis outlined earlier suggested that the MpzD61N/+ mutation determined a dramatic impairment of nerve functionality. To further corroborate this, we performed morphological analysis on a semithin section from 1-month-old sciatic nerves stained with toluidine blue (Fig. 4A). Mutant fibers showed a decrease of myelin thickness, typical of a de/dysmyelinating phenotype, as measured by g-ratio analysis, (g-ratio: 0.72 ± 0.01 in MpzD61N/+ mice vs 0.65 ± 0.01 in controls; P-value 0.0027) (Fig. 4B); moreover, we also noticed a reduction in myelinated axon diameter in MpzD61N/+ nerves as compared with WT control (Fig. 4C) and, in 6-month-old sciatic nerves, a reduction in the overall number of properly myelinated fibers (Fig. 4D and E). Of note, MpzD61N/+ nerves were also characterized by the presence of tomacula-like structures, basically absent in WT controls, that increased with time, from 80 ± 24.19 per nerve section at 1 month to 222 ± 12.49 at 6 months (Fig. 4F; Supplementary Material, Fig. S5A and B), supporting the progressive worsening of the phenotype already noted in neurophysiological tests. The analysis of osmicated teased fibers from 1-month-old WT and MpzD61N/+ sciatic nerve revealed that these myelin abnormalities (tomacula/focal hypermyelination) are unique to mutant nerve and they are not restricted to paranodal areas but are also found along the entire internode (Fig. 4G–L).
Figure 4.

Morphological analysis of WT and MpzD61N/+ sciatic nerves. (A) Images of semithin section of sciatic nerves from WT and MpzD61N/+ mice at 1 month. Magnification: 100X; Scale bar = 5 μm. (B) G-ratio of WT and MpzD61N/+ fibers. Error bars, SEM; **P = 0.0027 by unpaired, two-tailed, Student t-test. n = 4 for each genotype. (C) Comparison between axon diameter of WT and MpzD61N/+ fibers. Unpaired, two-tailed, Student t-test; ****P-value < 0.0001. (D) Number of fibers in WT and MpzD61N/+ sciatic nerves at 1 month and (E) 6 months (n = 3 mice at both time points). Unpaired, two-tails, Student t-test; P-value 0.93 and 0.08, respectively. (F) Number of myelin abnormalities in MpzD61N/+ sciatic nerves at 1 month and 6 months (n = 4 mice at both time points). Error bars, SEM. P = 0.0055 by unpaired, two-tails, Student t-test. (G–L) Osmicated teased nerve fibers from WT and MpzD61N/+ mice. Focal myelin thickenings, absent in the WT, originate both at paranodal regions (magnified in K) and at internodal regions (magnified in L). Magnification 20X in G-I, 40X in J–L. Arrows in G–I indicate nodes of Ranvier.
Consistent with the hypomyelinating phenotype, WB analysis showed that the levels of P0, as well as of MBP and PMP22, two important components of peripheral nerve myelin (25,26), were decreased in mutant nerves (Fig. 5A–E). However, there were no significant differences in mRNA levels (Fig. 5F–H). The reduction in MBP and PMP22 protein levels was confirmed at 6 months of age (Fig. 5I–K). Of note, PMP22 has been shown to play an important role in regulating myelin thickness and compaction, and PMP22 haploinsufficiency causes hereditary neuropathy with liability to pressure palsy (HNPP), a neuropathy characterized by florid tomacula formation, while MBP participates in the maintenance of the major dense line (27).
Figure 5.

Analysis of myelin proteins. (A) WB and protein quantification of (B) MBP and (C) P0 in sciatic nerves of 1-month WT and MpzD61N/+ animals. Error bars, SEM; unpaired, two-tails, Student t-test; P-value 0.1357 and 0.2702, respectively. One representative blot of three is shown. (D) WB and (E) protein quantification of PMP22 in sciatic nerves of 1-month WT and MpzD61N/+ animals. Error bars, SEM; unpaired, two-tails, Student t-test; P-value = 0.0055. One representative blot of three is shown. mRNA level of (F) MBP, (G) MPZ and (H) PMP22 in sciatic nerves of 1-month WT and MpzD61N/+ animals. Error bars, SEM; unpaired, two-tails, Student t-test; P-values of 0.2779, 0.3723 and 0.5991, respectively. Each experiment was repeated five times. Protein quantification for (I) MPB, (J) P0 and (K) PMP22 in sciatic nerves of 6-month WT and MpzD61N/+ animals. Error bars, SEM; unpaired, two-tails, Student t-test; P-values of 0.0041, 0.0555 and 0.0412, respectively. β-tubulin was used as loading control.
Electron microscopy analysis on transversal and longitudinal sections of WT (Fig. 6A and B) and MpzD61N/+ sciatic nerves showed the presence of several folding and hypermyelinating figures that appeared to compress the axons (Fig. 6C and D); in sciatic nerves of 6-month-old mice, we also observed signs of axonal degeneration (Fig. 6E), macrophages engulfing myelin debris (Fig. 6F and G), rare cases of onion bulbs (Fig. 6G and H) and an increase in mast cell numbers (Fig. 6H and K), all features which are absent in WT nerves.
Figure 6.

Electron microscopy of WT and MpzD61N/+ sciatic nerves. (A) Transverse and (B) longitudinal section of WT and (C, D) MpzD61N/+ sciatic nerves. Scale bar: 5 μm in the transverse sections, 2 μm in the longitudinal sections. (E) In MpzD61N/+ sciatic nerve signs of axonal degeneration (star), (F, G) macrophages (arrowheads), (H) inflammatory infiltrate (thick arrow) and (G, H) rare cases of onion bulbs (thin arrows) are shown. Scale bar= 5 μm. (I) Transverse section of a WT and (J) MpzD61N/+ sciatic nerve myelin. The black bar indicates normally compacted myelin lamellae, whereas white bars indicate myelin uncompaction and widening of the IPL. Scale bar = 500 nm. (K) Quantification of mast cells per nerve section in WT and MpzD61N/+ reconstructed sciatic nerves. Error bars, SEM; unpaired, two-tails, Student t-test; P-value = 0.0352.
P0 is implicated in the formation of the intracellular ‘major dense line’ as well as of the extracellular intraperiod line (IPL) in myelin (4,28). The extracellular domain contains a conserved sequence that has a role in mediating homophilic P0 interaction; crystal structure analysis of P0 indicated that this region is also implicated in the formation of P0 homotetramers in trans, keeping adjacent wraps of myelin together (4). The presence of an extra glycan in the extracellular domain of P0 could therefore interfere with myelin compaction. High magnification analysis revealed that myelin periodicity was altered in many mutant fibers. In fact, whereas WT fibers showed a consistent frequency in the major dense line and IPL (Fig. 6I), in MpzD61N/+ nerves some myelin lamellae looked normally compacted while others showed decompaction and widening of the IPL (Fig. 6J).
The phenotype was strongly suggestive of a gain-of-function mechanism since morphology was very different from the mild hypomyelination observed in Mpz+/− mice (26). The phenotype was also distinct from the severe dysmyelinating phenotype, but without tomacula formation or alterations in myelin periodicity, seen in MpzR98C/+ or MpzS63del nerves in which the mutant P0 is fully retained in the ER and does not reach myelin (14,29,30).
To directly evaluate whether the presence of the extra glycan interfered with the ability of P0D61N to reach the membrane in vivo, we generated mice carrying the MpzD61N mutation in a P0 null background (MpzD61N/−), so that only the mutant P0 protein is present. Morphological analysis performed on 1-month-old sciatic nerves from WT (Fig. 7A), MpzD61N/+ (Fig. 7B) and MpzD61N/− (Fig. 7C) showed that MpzD61N/− nerves are characterized by thin myelin sheaths and a much larger number of fibers with tomacula-like structures as compared with MpzD61N/+. However, as shown by the EM images (Fig. 7D–F), the presence of myelinated fibers in MpzD61N/− sciatic nerves indicate that the mutation did not fully prevent the transport of the protein to the membrane, differently from what happens for example in MpzS63del//−/− (expressing the P0S63del transgene in an Mpz null background) nerves, in which P0S63del is retained in the ER and nerves are basically devoid of properly compact myelin (14).
Figure 7.
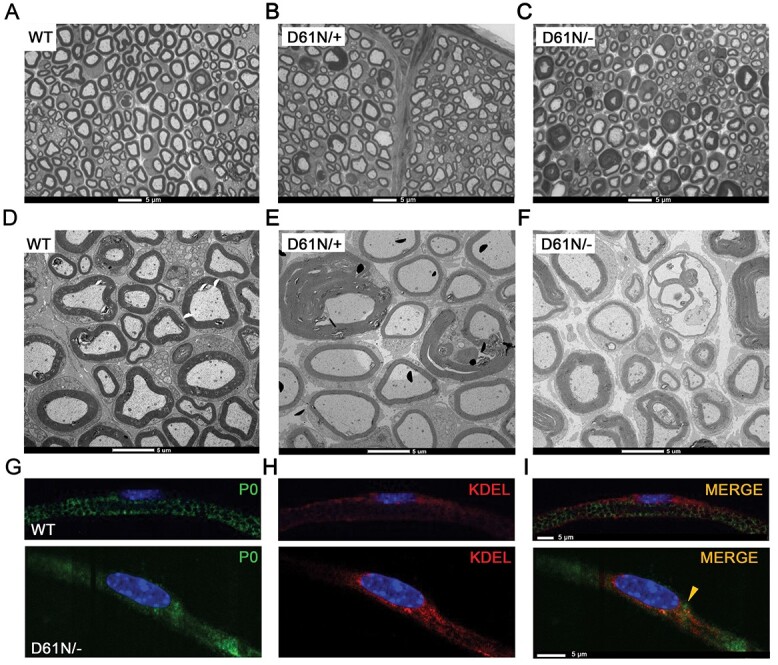
Analysis of WT, MpzD61N/+ and MpzD61N/− mice. Semithin section of sciatic nerves from (A) WT, (B) MpzD61N/+ and (C) MpzD61N/− mice at 1 month. Magnification: 100X; Scale bar = 5 μm. Electron microscopy images of transverse sections of sciatic nerves from (D) WT, (E) MpzD61N/+ and (F) MpzD61N/− mice. Scale bar = 5 μm. Immunofluorescence for (G) P0 (green) and (H) KDEL (red) in teased fibers from 1-month sciatic nerves of WT and MpzD61N/− mice. The third panel (I) represents the merge between P0 and KDEL. Nuclei were stained in blue (DAPI). Scale bar = 5 μm.
To further evaluate the trafficking of the mutant P0D61N protein, we performed immunofluorescence staining on teased fibers from WT and MpzD61N/− sciatic nerves. As shown in Fig. 7G–I, although most P0D61N appears to reach myelin, there is a partial colocalization between P0 and KDEL, a marker of the ER, in MpzD61N/− nerves suggesting some retention of the mutant protein in the ER. In contrast, as expected (31), in WT nerves there is no colocalization between P0 and KDEL.
Mpz D61N/+ activates only a transient UPR but affects SC development and interactions with axons and the extracellular matrix
The partial colocalization of P0D61N with KDEL prompted us to evaluate if this would cause ER stress and activate a canonical UPR (14,30,32,33). We measured, via WB, the expression of ER stress/UPR markers, such as BiP and phosphorylated eukaryotic initiation factor 2α (P-eIF2α) in sciatic nerves at 1 month. Both BiP and P-eIF2α levels showed a small but significant increase in MpzD61N/+ nerves as compared with WT nerves, suggesting a mild activation of the UPR (Supplementary Material, Fig. S6A–C). Next, we measured via real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) the expression of BiP, C/EBP-homologous protein (CHOP) and spliced X-box binding protein 1 in sciatic nerves of WT and MpzD61N/+ mice; MpzS63del nerves were used as a control for UPR activation. In MpzD61N/+ nerves there is a slight, not statistically significant increase in the expression of these UPR factors (Supplementary Material, Fig. S6D–F), that is, however, not comparable with the increase that occurs in the case of MpzS63del nerves, in which the activation of the UPR is well documented (12,14). Accordingly, by 6 months of age, a time point where the UPR is still active in MpzS63del mice (12,33), we could no longer detect any increase in BiP and P-eIF2α proteins in MpzD61N/+ nerves (Supplementary Material, Fig. S6G and H). All these data indicated that a mild UPR activation is a transient event present only during the active phase of myelination in MpzD61N/+ nerves, suggesting that ER stress is unlikely to play a significant role in the pathogenesis.
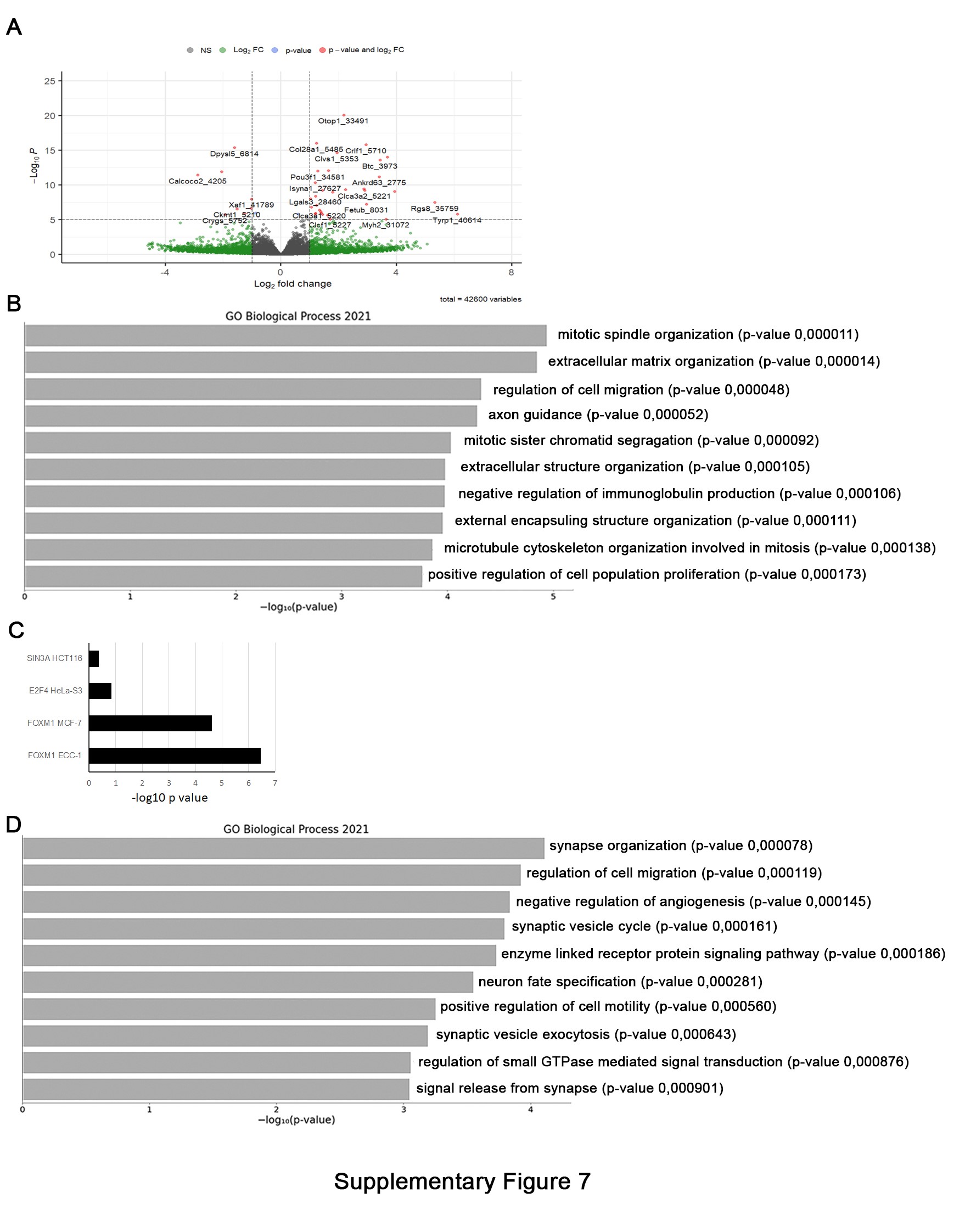
To further explore the molecular signature of D61N nerves we performed RNAseq transcriptome profiling of 2-month-old sciatic nerves in MpzD61N/+ and WT control mice. We identified 797 differentially expressed transcripts in mutant nerves, of which 446 were increased and 351 were decreased (Supplementary Material, Fig. S7A). We first examined for any changes in macrophage transcripts (e.g., Cd68) but did not observe any significant change that would reflect macrophage infiltration.
Gene ontology (GO) annotation (Supplementary Material, Fig. S7B) identified amongst the biological processes most upregulated in MpzD61N/+ nerves genes related to the mitotic spindle organization and mitosis, followed by genes related to extracellular matrix organization and extracellular structure organization, such as collagens, laminins and cadherins; these are an important component of the basal lamina and are important for SCs development and myelination (34–36). Genes belonging to the regulation of cell migration and axon guidance were also upregulated, as well as the GO category related to positive regulation of cell proliferation. In this category, we identified genes encoding transcription factors or enzymes that are normally expressed in pre- and promyelinating SCs, such as Pou3f2, Sox4, Adam17 and Id2, suggesting a developmental defect in MpzD61N/+ SCs, as already observed in other CMT1B models, such as MpzR98C/+ or MpzS63del (14,30). An analysis of induced genes with respect to transcription factor pathways (from ENCODE TF Chip-seq) identified enrichment of the FOXM1 pathway, which is involved in promoting the G2/M transition of the cell cycle. Foxm1 itself is induced ~2-fold, accompanied by induction of several FOXM1 target genes, such as Top2a, Ccnb1 and Mki67 (Supplementary Material, Fig. S7C). Another pathway evident in upregulated genes is the c-Jun pathway, which has been characterized in SC responses to nerve injury (37). This pathway is also induced in some peripheral neuropathy models (38). While the c-Jun transcript itself is not significantly changed, JUN target genes have been defined in both loss-of-function and overexpression models. Several JUN target genes are elevated, including Shh (>10-fold), Btc, Tnc, Rgs8, Fgf5, Sema4 and Gdnf and these have been shown to be expressed in SCs in recent single-cell RNA-sequencing studies (39). This likely reflects the post-transcriptional elevation of JUN or other AP-1 family members.
While there is some overlap with the expression profile of S63del with regard to JUN target gene activation, there are several distinct changes that distinguish the D61N model. At this time point, several target genes related to UPR are not significantly elevated (Ddit3/CHOP, DNAjb9 as an XBP1 target gene, and Nqo1 as an NRF2 target gene) although these remain elevated in mature S63del nerve. In addition, the observed depression of lipid biosynthetic genes in S63del (12) is not evident in the D61N model (see later).
To validate the MpzD61N/+ RNAseq data, we performed WB analysis for proteins related to the extracellular matrix. We confirmed a significant increase of E-cadherin (Fig. 8A and B) and laminin (Fig. 8C and D) in MpzD61N/+ nerves compared with WT, whereas Cyclin B1, a marker cell proliferation, only showed a small, not significant increase in mutant nerves (Fig. 8C and E). However, through qRT-PCR, we confirmed the increase in mRNA levels of the immature SC transcription factor Id2 (40), known to regulate the cell cycle, in MpzD61N/+ nerves (Fig. 8F). Of note, qRT-PCR experiments also detected a small but significant increase in the levels of JUN (Fig. 8G) that was not detected by the RNAseq analysis, further supporting the activation of the pathways in MpzD61N/+ SCs.
Figure 8.

Validation of a small selection of genes altered in the RNA-seq data. (A) WB and protein quantification (B) for E-Cadherin. (C) WB and protein quantification (D) for Laminin and (E) Cyclin B-1 in sciatic nerves of 2-month WT and MpzD61N/+ animals. Error bars, SEM; unpaired, two-tails, Student t-test; P-values of 0.0313, 0.0328 and 0.3044, respectively. One representative blot of three is shown. Quantitative RT-PCR analysis for (F) Id2 and (G) c-Jun on mRNA extracted form WT and MpzD61N/+ sciatic nerves. Error bars, SEM; unpaired, two-tails, Student t-test; P-values of 0.0202 and 0.0402, respectively; n = 5 RT from independent nerves.
Amongst the downregulated genes (Supplementary Material, Fig. S7D) we identified GO categories related to synapse organization, synaptic vesicle cycle, synaptic vesicle exocytosis and signal release from synapse; additional GO categories that appeared significantly downregulated were related to the regulation of cell migration and positive regulation of cell motility, whereas lipid biosynthetic pathways, often downregulated in demyelinating neuropathies (12), did not appear to be altered. Overall, this analysis suggests that the mutant D61N protein impacts mostly on SC proliferation, migration and in SC-axon and SC–extracellular matrix interactions.
N-butyl-deoxynojirimycin: A possible benefit for hyperglycosylated mutations in both in vitro and ex vivo models
Recent works (16,41) have suggested that gain-of-glycosylation mutations may benefit from imino-sugar administration, a class of compound potentially able to modify the N-glycosylation of proteins. We treated HeLa cells expressing the D61N mutation with 750 μM N-butyl-deoxynojirimycin (NB-DNJ) to assess if this compound was able to complement the phenotype in vitro, by improving the trafficking of the mutant protein and cell adhesion. Immunofluorescence analysis suggested that the treatment ameliorated P0D61N protein trafficking (Fig. 9A), reduced its intracellular retention and improved adhesion (Fig. 9B), without affecting WT protein trafficking; interestingly, the treatment of cells with the compound also appeared to slightly reduce the MW of the mutant protein, suggesting a potential modification in glycosylation (Fig. 9C).
Figure 9.

Treatment with NB-DNJ in vitro and ex vivo. (A) Confocal images of HeLa cells expressing the mutant protein P0D61N before and after the treatment with NB-DNJ; scale bar = 14.5 μm. (B) Adhesion assay performed on HeLa cells treated with NB-DNJ, the nontreated cells have been used as control. **P < 0.01 and ***P < 0.001 by one-way ANOVA with Tukey post-hoc test. (C) WB analysis on extracts from HeLa cells expressing P0wt or P0D61N treated with NB-DNJ. (D) Images of WT (upper panel) and MpzD61N/+ (lower panel) DRGs stained for MBP. In mutant cultures, there are fewer and often abnormal myelinated internodes. Scale bars 20 and 10 μm in the left and right panels, respectively. (E) Quantification of MBP+ segments in WT and MpzD61N/+ DRGs. ****P < 0.0001 by one-way ANOVA with Tukey post-hoc test. Three different dissections were performed. (F) DRG cultures from MpzD61N/+ embryos treated with 50 or 100 μM NB-DNJ and (G) quantification of myelin abnormalities before and after the treatment. ****P < 0.0001 by one-way ANOVA followed by Holm–Sidak test. Scale bar in (F), 20 μm. WB analysis for P0 and MBP on protein extracts from DRG from (H) WT and (I) MpzD61N/+ embryos treated with ascorbic acid only (AA) or with AA + NB-DNJ for 14 days. β-tubulin was used as loading control. Quantification of the amount of (J) P0 and (K) MBP proteins in MpzD61N/+ DRGs after the treatment with NB-DNJ. *P < 0.05 by one-way ANOVA with Tukey post-hoc test.
Based on these results, we tested NB-DNJ in an ex vivo system, by establishing myelinating organotypic dorsal root ganglia (DRG) cultures from E13,5 WT and MpzD61N/+ embryos (Fig. 9D). In mutant MpzD61N/+ DRGs cultures, the number of myelinated internodes was slightly reduced (Fig. 9E) and myelin presented frequent swellings reminiscent of the tomacula-like structures we saw in nerves. We reasoned that NB-DNJ, by modulating N-glycosylation, could ameliorate the morphology of myelinated internodes; we treated WT and MpzD61N/+ DRGs with 50 μM and 100 μM NB-DNJ; DRGs treated only with ascorbic acid were used as control. Two weeks of treatment with NB-DNJ appeared to reduce myelin abnormalities in a dose-dependent fashion: myelin defects were reduced by roughly 25% after the treatment with 50 μM, and by roughly 50% with the higher dosage (Fig. 9F and G).
To investigate if the treatment modified the glycosylation of the P0D61N mutant protein, we performed a WB analysis on protein extracts from DRGs at the end of treatment (Fig. 9H and I). Surprisingly, however, the glycosylation of P0D61N did not appear to change after NB-DNJ treatment, as evidenced by the lack of any shift in MW and by the unaltered P0D61N/P0wt ratio (Fig. 9I, and data not shown). Rather, we observed a dose-dependent reduction in the amount of P0 and MBP (Fig. 9J and K), which may suggest that the decrease in myelin abnormalities could be related to an overall reduction in myelin protein synthesis after NB-DNJ treatment rather than to a direct effect of the compound on P0D61N glycosylation. Further studies will be required to elucidate the effects of imino-sugars on protein glycosylation and myelination in SCs.
Discussion
MPZ-related neuropathies are very heterogeneous, suggesting that multiple pathogenic mechanisms are involved (11). ER stress and the activation of the UPR is the most extensively studied pathomechanism, shown to be involved in a significant number of CMT1B neuropathies (7,15). Among other disease mechanisms, misglycosylation was also inferred to play a pathogenic role (17,19). The loss of the native glycosylation site or the gain of a new glycosylation site has been demonstrated in several diseases (13,25,42–47). While peripheral myelin is largely composed of glycoproteins, misglycosylation had never been investigated as a potential cause of peripheral neuropathies.
Here, we extensively investigated how peripheral myelination may be affected by P0 misglycosylation using in vitro and in vivo models. Our results suggested that hyperglycosylation is more damaging for P0 trafficking and function as compared with hypoglycosylation. By establishing the first knock-in mouse expressing an authentic gain-of-glycosylation mutation in P0, we provided evidence that the resulting severe CMT1B neuropathy is likely due to a gain-of-function in myelin, with important implications for future therapeutic approaches.
P0 gain-of-glycosylation is more disruptive than loss of glycosylation
P0 glycosylation is important for the homophilic interactions between P0 proteins and, through the adhesion-related HNK-1/L2 epitope, protein–carbohydrate interactions between adjacent P0 molecules may also be involved in the homophilic binding of P0, both in trans and cis (48). The P0 protein has a single N-glycosylation site at position 122 within the Asp122-X-Thr124 sequon. One of the most important functions of the N-glycosidic links is the promotion of the correct folding of newly synthesized polypeptides (49), and it has been demonstrated that N-glycans are efficient signals for cell surface targeting (50). By in silico prediction, we identified two sets of mutations potentially able to cause gain (D61N, D109N, D118N, K138N) or loss (N122S, T124K, T124A, T124M) of glycosylation in P0, and biochemical analysis of the mutant P0 confirmed the acquisition or loss of oligosaccharides, respectively. In humans, the two groups of mutations result in different phenotypes: the hyperglycosylating mutations correlate with severe early-onset dysmyelinating neuropathy, whereas the mutations that abolish P0 glycosylation are associated with a late-onset neuropathy with a clear axonal involvement (late-onset CMT1B or CMT2J/I) (18–20,51–56). In transfected cells, the trafficking pattern of the mutant proteins was different among the two groups: not-glycosylated P0 proteins reached the plasma membrane as the P0wt protein; instead, the hyperglycosylated proteins were partially retained intracellularly in the ER and Golgi compartments. It has been recently shown that glycosylation, also in the WT sequon, could slow down the trafficking of PMP22 (57). Whether this applies also to P0wt is currently unknown, but it is conceivable that the hyperglycosylated P0 proteins, with their additional glycan, could be slowed in the trafficking to the plasma membrane. Correct folding and trafficking to the membrane are crucial to allow the adhesive function of P0, and accordingly, the aggregation tests showed that hyperglycosylated proteins were less efficient in promoting cell adhesion as compared with unglycosylated variants. It is tempting to speculate that the presence of the second oligosaccharide can impair P0–P0 interaction (see later). Overall, our in vitro results indicate that mutations causing loss of glycosylation in P0 determine a milder cellular phenotype as compared with mutations associated with a gain of glycosylation. This confirms previous observations made in other pathologies, such as antithrombin deficiency and dysfibrinogenemia that suggested that losing a glycosylation site is less harmful than acquiring a new one (17) and establishes a clear genotype–phenotype correlation for misglycosylating mutations in CMT1B.
The MPZD61N mouse recapitulates human pathology
Despite the development of many animal models of CMT (58), there were none for mutations causing a gain-of-glycosylation; to fill this gap, we generated a knock-in mouse model carrying the MPZD61N mutation that in humans causes a severe, early onset, dysmyelinating neuropathy (19,20). Thanks to CRISPR/Cas9 technology, only the desired amino acidic substitution D61→N has been introduced, thus completely reproducing the patients’ genotype, without the insertion of LoxP sites that have been shown to cause a reduction in P0 transcription, complicating interpretation (59). Biochemical analysis confirmed that the amino acid substitution introduced the second glycosylation site, as expected, and that the mutant P0D61N is expressed at similar levels as P0wt.
MPZ D61N/+ mice manifested a severe neuropathic phenotype, characterized by extensive motor and neurophysiological impairment. Sciatic nerves of mutant mice presented widespread dysmyelination, with reduced myelin thickness, signs of myelin uncompaction, diffused tomacula-like structures and, at later stages, axonal degeneration and loss of myelinated fibers. This phenotype appears to faithfully recapitulate the human pathology: in fact, patients carrying the D61N substitution show ambulatory impairment, severely reduced NCVs and sural nerves biopsies revealed fibers surrounded by thin myelin sheaths but also tomacula and fibers with abnormally compacted myelin (19,20). Similar pathological findings have been reported in two subgroups of patients affected by CMT1B due to mutations in the P0 extracellular domains (P0ECD). However, nerve biopsies of these patients usually manifest either defects in myelin compaction, mostly affecting the IPL, or tomacula, but normally not both (60–62). How can we explain this peculiar phenotype? The presence of myelin in MpzD61N/− nerves suggested that the mutant P0D61N protein can reach the plasma membrane, and be at least partially adhesive, although less than P0WT, since in MpzD61N/− mice myelin is more compacted than in Mpz−/− mice (63). Normally P0ECD on the SC membrane form tetramers that link with tetramers emanating from the opposing membrane surface. Each P0ECD has a structure similar to the variable domain of immunoglobulins, with 10 β strands arranged as 2 β sheets. D61N is positioned in the loop between strand B of one β sheet and strand C on the other β sheet. This B-C loop is highly extended and, together with tryptophan 28, is thought to be directly involved in the interactions with the opposing myelin membrane, determining the spacing of the IPL (5). It seems therefore reasonable to speculate that the presence of extra-glycans in this loop may interfere with such interactions, causing the disruption of the IPL spacing. Very similar pathological abnormalities have been seen in another CMT1B mouse model, the P0Myc mouse, which is characterized by tremor, reduced nerve conduction velocity, dysmyelination with a widening of myelin lamellae at the IPL and tomacula (64); it has been postulated that the N-terminal 13 amino acid Myc tag could disrupt interactions of multiple domains of P0 (64). It is conceivable that the extra glycans of P0D61N may exert a similar disruptive effect at multiple structural levels in myelin resulting in both tomacula and packing defects.
While relatively rare in CMT1B, tomacula formation is the hallmark of other neuropathies, such as HNPP due to a 1.5 Mb deletion on chromosome 17 encompassing the PMP22 gene, which is modeled by PMP22 deficient mice (65). Despite no differences in mRNA levels, the overall amount of PMP22 protein appeared reduced in mutant MpzD61N/+ nerves as compared to WT nerves. The morphological similarities between PMP22/+ mice and MpzD61N/+ mice raise the intriguing possibility that the decrease in PMP22 protein levels in sciatic nerves of MpzD61N/+ mice could be one of the contributing factors to the myelin defects described earlier.
P0D61N interferes with SC differentiation and extracellular matrix organization
Besides myelin packing defects and tomacula, MPZD61N nerves are characterized by diffuse hypomyelination. How the D61N mutation in P0 interferes with myelination from a molecular point of view is still unclear. We detected a partial ER retention for the P0 D61N protein in vivo. This, however, was not sufficient to elicit a chronic UPR, like those seen in other CMT1B models, such as the MpzS63del and MpzR98C mice (12,30,33). In fact, while at 1 month old, the UPR appeared marginally increased, by 6 months of age UPR markers’ levels were comparable with WT, indicating that this pathway is only transiently activated and unlikely to be significantly involved in the pathomechanism.
RNAseq analysis confirmed the absence of a significant stress response in MpzD61N/+ nerves but rather a striking dysregulation of pathways involved in SC differentiation and proliferation and of pathways related to extracellular matrix structure and organization. In these categories, there were genes belonging to the laminin, collagen and cadherin families. Laminins and collagens are proteins of the extracellular matrix important for SC proliferation, survival, radial sorting and myelination (34); similarly, cadherins are necessary for SC development, their proliferation, migration and early SC precursor–axon interactions (66). Laminin α2 chain (merosin) deficiency causes congenital muscular dystrophy. In patients with LAMA2 mutation, sural nerve biopsies show focally thickened myelin (tomacula-like) and thinner and uncompacted myelin (67). The similarity between the LAMA2 mutant nerve phenotype and our mouse model could suggest that the dysregulation of pathways important for extracellular matrix organization may contribute to the observed myelin defects.
The RNA-seq analysis also identified upregulation of several JUN target genes that have been described in the context of peripheral nerve injury, and JUN upregulation has been described in several neuropathy models. Models of JUN overexpression show hypomyelination (68), but JUN overexpression per se would not account for tomacula formation. Moreover, there is an enrichment of genes activated by the FOXM1 transcription factor that is involved in G2/M cell cycle regulation. Identification of these pathways offers some potential therapeutic targets that will be investigated further.
Therapies for hyperglycosylating CMT1B
The use of a class of compounds belonging to the imino-sugar category was proposed as a potential treatment for a gain of glycosylation mutations (41,69). These molecules can interfere with specific enzymes involved in the protein glycosylation chain, such as Golgi and ER glycosidases (70). NB-DNJ, commercially known as miglustat, has already been used for the treatment of cystic fibrosis (71), Gaucher disease (72) and Niemann–Pick disease (73) with a good safety profile in humans.
After promising preliminary experiments on cells expressing P0D61N-EGFP we tested this drug on DRG explants, a system that better models human disease. Intriguingly, mutant DRGs were characterized by the presence of many myelin abnormalities, that appeared like a ‘string of pearls’, due to the presence of focal enlargements of the myelin, reminiscent of the tomacula-like structures seen in nerves. The treatment with miglustat ameliorated the morphology of mutant DRGs but without significantly modifying the glycosylation of P0. The reduction in P0 and MBP proteins in the treated DRGs suggests that in SCs miglustat may interfere with myelin protein synthesis, indicating that drugs with a broad effect on cellular glycosylation may not be appropriate for treating hyperglycosylating CMT1B. Mass spectrometry analysis would be required to characterize the nature of the extra glycan introduced with the MpzD61N mutation and help us to act in a more targeted manner. Alternatively, therapy approaches based on allele-specific knockdown may be necessary when dealing with mutations that affect such crucial cellular processes.
Materials and Methods
Plasmids generation and site-directed mutagenesis
The full-length human cDNA of WT MPZ was cloned into the pEGFP–N1 vector as previously described (13). Plasmids coding for all the mutations in fusion with EGFP vector (pEGFP from Clonetech) has been generated through site-directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit, Stratagene), using specific primers containing the nucleotide changes (Table S1), according to manufacturer’s protocol. The mutant cDNAs were amplified by PCR using primers containing restriction enzyme sites for EcoRI and BamHI and then fused in-frame into the expression vectors for mammalian cells pEGFP–N1 (Clontech, Mountain View).
Cellular cultures
HeLa and Schwannoma RT4-D6P2T cell lines were maintained in Dulbecco modified eagle medium (DMEM, Gibco, Invitrogen) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin/streptomycin at 37°C, in a humidified incubator with 5% CO2.
Transient transfection and protein localization
HeLa and Schwannoma cells were transiently transfected by single transfection using Lipofectamine 2000 according to manufacturer’s protocol (Invitrogen).
To study the expression of P0 on the cell surface, HeLa and Schwannoma cells were transfected with both P0wt and mutant P0 linked to EGFP, allowed to grow for 24 h in a chamber slide (Nalge Nunc International), washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 10 min, washed and mounted. To evaluate protein localization, transfected cells were fixed with 4% PFA for 15 min at room temperature (RT) and then washed with PBS 1X. Cells were permeabilized with cold methanol for 5 min at RT, washed with PBS 1X and then blocked for 1 h at RT with blocking solution [3% bovine serum albumin (BSA), 1% goat serum and 0.2% Triton X in PBS 1X]. Primary antibodies diluted 1:100 in blocking solution were added overnight at 4°C. The following morning the cells were washed thrice with PBS 1X and secondary antibodies, diluted 1:200 in blocking solution, were added for 45 min at RT. The cells were washed, stained with Hoechst solution for 5 min at RT and mounted.
Adhesion test
After 24 h from transient transfection, HeLa cells were washed with PBS and incubated with Cell Dissociation Buffer (Invitrogen, San Giuliano Milanese, MI, Italy), for 3 min at RT, counted in Bürker chambers and resuspended to a final concentration of 1.5 × 106 cells/ml in DMEM containing 10% fetal bovine serum (FBS) and 25 mM of Hepes (Sigma) to maintain the cellular pH. By three passages through an 18-gauge syringe, we obtained a single-cell suspension. Suspensions, containing a minimum of 95% single cells, were allowed to aggregate at 37°C with gentle rocking at 5 rpm. After 3 h, the tubes were gently inverted and aliquots of 10 μl were removed, put on a microscope slide without using fixatives and immediately examined under the microscope (Olympus AX60). From each slide, 30 frames, randomly selected at 20X magnification, were digitalized and stored, using the Pro Plus Imaging System (Immagini e Computer). The area of cellular aggregates (>4 cells) was measured with Pro Plus Imaging System (Immagini e Computer) and the average area of aggregates formed by HeLa cells transfected with each mutant was calculated and normalized to the average area of aggregates produced by cells expressing P0wt.
Transgenic mice
All experiments involving animals were performed in accordance with Italian national regulations and in accord with experimental protocols approved by the San Raffaele Scientific Institute Animal Care and Use Committees.
MPZ D61N mice were generated by the Core Facility for Conditional Mutagenesis using CRISPR/Cas9 technology and kept on a C57BL/6 genetic background. The generation of the MPZD61N mouse model was performed in collaboration with Sigma-Aldrich which designed the guide RNA.
The donor oligo contains:
1. G > A SNP conversion that introduce the desired mutation;
2. A > G SNP conversion that is necessary to prevent gRNA binding and recleavage possibility and
3. A > T SNP conversion that eliminates the DdeI restriction enzyme site.
For genotyping, genomic DNA was extracted from tail biopsies. All PCR products were stained with SYBR Safe DNA Gel Stain (Invitrogen), run in 3% agarose gels and detected with UVP GelDOC-It Imaging System.
D61N PCR primers:
D61N sense: 5′ TGCTCTCTCCAGCCCTGG 3′
D61N antisense: 5′ GGAGCCATCCTTCCAGCGA 3′
PCR program:
95°C 10′
 30°cycles
30°cycles
72°C 10′
4°C
The PCR product (491 bp) was then digested (37°C, 30′) with DdeI. Three bands (110, 168 and 213 bp) were detected in WT and four (110, 168, 213 and 323 bp) in D61N/+ samples.
Morphological and morphometric analysis
Sciatic nerves were freshly dissected, fixed in 2% glutaraldehyde in phosphate buffer, osmicated in 1% OsO4, alcohol dehydrated, infiltrated with propylene oxide and embedded in Epon. Transverse semithin sections and ultrathin sections were cut with an ultracut microtome. Semithin sections were stained with toluidine blue and acquired with a Leica DM5000 microscope equipped with a DFC480 digital camera, whereas ultrathin sections were stained with lead citrate and photographed with a Zeiss (Oberkochen, Germany) EM10 electron microscope. G-ratio (axon diameter/fiber diameter) was measured on semithin sections using ImageJ software; four-six images per nerve were acquired with a 100X objective; ~800–2000 fibers per condition were measured. The proportion of fibers with myelin outfoldings was also determined (74).
Electrophysiology
As many as eight MPZD61N/+ and eight control mice were analyzed at 1 month and 3 months old. Electrophysiological tests were performed using an EMG system (NeuroMep Micro, Neurosoft, Russia). Mice were anesthetized with trichloroethanol, 0.3 mg/g of body weight, and placed under a heating lamp to maintain a constant body temperature. Monopolar needle electrodes were inserted subcutaneously to stimulate the tibial nerve at the ankle and, subsequently, the sciatic nerve at the sciatic notch; the cathode was placed close to the nerve and the anode was inserted proximally to the cathode. The stimulation consisted of single 100 μs, 1 Hz supramaximal pulses. The muscular response was recorded by inserting the active electrode into the muscles in the middle of the paw and the reference electrode in the skin between the first and second digit. NCV (m/s), peak-to-peak cMAP amplitude (mV) and FWL (ms) were measured. FWL measurement was obtained by stimulating the tibial nerve at the ankle and recording the responses in the paw muscles, using the same pair of needle electrodes used for the nerve conduction study.
Behavioral analysis
Tremor was estimated visually on a + to ++++ scale.
Rotarod test. Motor ability was assessed using the accelerating Rotarod (Ugo Basile, Comerio, Italy). Groups of 3=month-old transgenic and control littermates were tested in two sessions of three trials each per day (6 h rest between the two daily sessions) for three consecutive days. During the test, the rod accelerated from 4 to 40 rotations per minute, and the time that the animal remained on the rod (maximum 900 s) was measured.
Grip strength test. A grip strength test is a simple noninvasive method designed to evaluate mouse muscle force and hind limb strength. The grip strength test was performed on a group of 9 MPZD61N/+ and 10 WT mice at 3 months of age using a grip strength meter (Grip-Strength Meter, 47 200, Ugo Basile, Italy) to determine hind limb muscle strength. The grip strength meter was arranged horizontally on the table. While holding the tail, the mouse was lowered toward the grip strength meter to enable it to grasp the T-shaped bar with the hind paws. After grasping the bar, the mice were pulled backward till the grasp was released. The test was repeated six consecutive times. For each session, the average value of the peak force (in gf) was calculated (75).
TaqMan quantitative PCR analysis
Total RNA was extracted with Trizol (Roche Diagnostic GmbH, Germany) and retrotranscribed as previously described (14). TaqMan assays were performed following the manufacturer’s instructions (TaqMan, PE Applied Biosystems Instruments) on an ABI PRISM 7700 sequence detection system (Applied Biosystems Instruments). Normalization was performed using 18S rRNA as a reference gene. Target and reference genes PCR amplification were performed in separate tubes with Assay on Demand (Applied Biosystems Instruments): 18S assay, Hs99999901_s1; Ddit3/CHOP assay, Mm00492097_m1; Xbp-1 s assay, Mm03464496_m1; Hspa5/BiP assay, Mm00517691_m1; MPZ assay, Mm00485141_g1; MBP assay, Mm01266402_m1; PMP22 assay, Mm01333393_m1; Id2 assay, Mm007011781_m; and c-Jun assay, Mm004950 62_s1.
WB analysis
Sciatic nerves were dissected and frozen in liquid nitrogen. Frozen nerves were pulverized on dry ice and proteins were extracted in denaturing lysis buffer [Tris–HCl 50 mM pH 7,5, NaCl 150 mM, ethylenediaminetetraacetic acid (EDTA) 10 mM, 2% sodium dodecyl-sulfate (SDS)] containing protease inhibitor cocktail (PIC 100X, Roche) and phosphatase inhibitors (Roche). Total protein concentration was determined by bicinchoninic acid assay (Pierce) following the manufacturer’s instructions. Equal amounts of proteins were separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Biorad) and gels were transferred onto nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% milk (milk powder/1X PBS-Tween 0.1%) or 5% BSA (BSA powder/1X PBS-Tween 0.1%) and incubated with primary antibodies diluted in 1% milk or 1% BSA/1X PBS-Tween 0.1% at 4°C overnight. Horseradish peroxidase (HRP)-conjugated antibodies were diluted in 1% milk or 1% BSA/1X PBS-Tween 0.1% and incubated for 1 h at RT. Signals were detected by the ECL method. Densitometric analysis was performed with NIH-Image-J software.
PNGaseF treatment
For the digestion with PNGaseF, an amidase that cleaves N-glycans, proteins were extracted from sciatic nerves or from Hela cells as previously described for the WB analysis, but using the RIPA buffer, digested for 1 h at 37°C with PNGaseF, according to the manufacturer’s protocol (NEB), separated by SDS-PAGE and immunoblotted.
Teased fibers preparation
Sciatic nerves were freshly dissected and fixed in 2% glutaraldehyde in phosphate buffer. Some nerves were osmicated in 1% OsO4 in triethyl phosphate, placed in glycerol and single fibers were placed on slides and separated with fine needles; osmicated fibers were analyzed with a light microscope (Leica DM5000). For immunofluorescence on teased fibers, slides were coated with 5% of 3-(aminopropyl) triethoxysilane (TESPA) (A3648; Sigma-Aldrich), and single fibers were separated with fine needles; slides were analyzed with Leica TCS SP8 SMD FLIM laser scanning confocal.
Myelinating DRG explant cultures
DRG explants were isolated from E13.5 WT and MpzD61N/+ embryos, seeded on rat collagen I-coated coverslip and maintained in cultures as previously described (76). Myelination was induced by supplementing media with 50 μg/ml ascorbic acid (Sigma-Aldrich) for 14 days. The culture medium was refreshed every 2 days. Samples were fixed and prepared for immunofluorescence.
Immunohistochemistry and analysis of myelination
SC cell/DRG neuron cocultures were fixed for 15 min in 4% paraformaldehyde, permeabilized for 3 min in ice-cold methanol, blocked for 1 h with 1% BSA + 10% NGS in PBS and incubated at 4°C overnight with primary antibody. After washing, the coverslips were incubated for 1 h at room temperature in the dark with secondary antibody, washed, incubated for 5 min in the dark with Hoechst solution for nuclei staining, washed and mounted with Vectashield. To quantify the amount of myelin, eight nonoverlapping images per DRG were acquired with a Leica DM5000 microscope (10X objective) equipped with a Leica DFC480 digital camera and the number of MBP-positive myelinated fibers was counted. The percentage of MBP-positive fibers showing myelin abnormalities among the total number of MBP-positive fibers was indicated. At least three independent dissections were performed.
RNA-sequencing
For D61N and control mice, at least 1000 ng total RNA was purified and sent to Genewiz (South Plainfield, NJ) for library preparation after PolyA selection and Illumina sequencing (Illumina HiSeq 2x150bp). Illumina sequencing data were mapped to the GRCm38/mm10 genome using the STAR aligner (77). Data were analyzed using DESeq2 to determine differentially regulated genes (P-value < 0.5) (78). RNA-seq data are deposited in National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE196979.
Antibodies
For WB, the following primary antibodies were used: chicken anti-MPZ (P0) (PZO; Aves); mouse anti-myelin basic protein (MAB 382; Chemicon); rabbit anti-PMP22 (AB211052; ABCAM); goat anti-Cyclin B1 (AF6000; R&D System); rabbit anti-Laminin (L9393; Sigma); mouse anti-E Cadherin (610 181 BD Biosciences); rabbit anti-eIF2α (D7D3) XP (5324; Cell Signaling); rabbit anti-phospho-eIF2α (Ser51) (D9G8) XP (3398; Cell Signaling); rabbit anti-GRP78 (BiP) (NB 300520; Novus Biological); mouse anti-β-tubulin (T4026; Sigma); rabbit anti-Calnexin (C4731; Sigma) and mouse anti-GFP tag (GF28R) (MA515256; Thermo Fisher).
For immunofluorescence, the following primary antibodies were used: rat anti-MBP; chicken anti-neurofilament M (822 701; Biolegend); chicken anti-MPZ (P0) (PZO; Aves); mouse anti-KDEL (ADI-SPA-827; Enzo) and mouse anti-GM130 (610 823; BD Biosciences).
Secondary antibodies included donkey anti-chicken-HRP (AB 16349; ABCAM); HRP-conjugated goat anti-rabbit (P0448; Dako); anti-mouse IgG peroxidase (A3682; Sigma Aldrich) and mouse IgG HRP-linked whole Ab (10 196 124; Fisher Scientific); donkey anti goat-HRP (sc-2020; Santa Cruz Biotechnology). For immunofluorescence, secondary antibodies included Cy3-conjugated goat anti-rat (112 166 062; Jackson ImmunoResearch); Alexa Fluor 546 goat anti-mouse IgG (A11030; Thermo Fisher); Rhodamine (TRITC)-conjugated donkey anti-mouse IgG (715–025-150; Jackson ImmunoResearch) and fluorescein-labeled goat anti-chicken IgY (F1005; Aves). Nuclei were marked using Hoechst or DAPI. Samples were mounted onto slides with Vectashield mounting medium (Vector Laboratories).
Protein variants molecular modeling
Mouse and human Mpz monomeric models of the D61N variant were modeled with the SwissModel online web server (79) using as template the X-rays deposited in PDB, code 1neu (5) and 3oai (46), respectively. Mouse and human sequences were retrieved from UniProt, entry codes P27573 and P25189. The complete tetrameric structure for rats was derived using symmetry information from the input coordinate file PDB 1neu. Human and mouse tetrameric variants were built superimposing the single units on the rat tetrameric structure. Glycoprotein web tool was used to build oligosaccharides and attach them to the protein to form N-linked glycoproteins.
Experimental design and statistical analysis
Experiments were not randomized, but data collection and analysis were performed blindly to the conditions of the experiments. Researchers blinded to the genotype performed behavioral analyses, NCVs and morphometric analyses. Most experiments were performed with the same number of male and female animals. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally used in the field. Graphs and data were analyzed using GraphPad Prism Software. Data show the mean ± standard error of mean (SEM). One-way or two-way ANOVA with post hoc analysis (when specified in the text) or Student t test was used. Significance levels were marked on figures as follows: P-values (P), *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Maria Carla Panzeri, Valeria Berno and the Alembic facility at the San Raffaele Scientific Institute for excellent technical support, and Dr Angelo Quattrini for help in the morphological analysis. We are also grateful to Dr Paola Fossa and Dr Pasqualina D’Ursi for their help with the in silico molecular modeling of P0D61N.
Conflict of Interest statement. None declared.
Contributor Information
Francesca A Veneri, Biology of Myelin Unit, Division of Genetics and Cell Biology, IRCCS Ospedale San Raffaele, 20132 Milan, Italy; Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genova, IRCCS AOU San Martino-IST, 16132 Genova, Italy.
Valeria Prada, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genova, IRCCS AOU San Martino-IST, 16132 Genova, Italy.
Rosa Mastrangelo, Biology of Myelin Unit, Division of Genetics and Cell Biology, IRCCS Ospedale San Raffaele, 20132 Milan, Italy.
Cinzia Ferri, Biology of Myelin Unit, Division of Genetics and Cell Biology, IRCCS Ospedale San Raffaele, 20132 Milan, Italy.
Lucilla Nobbio, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genova, IRCCS AOU San Martino-IST, 16132 Genova, Italy.
Mario Passalacqua, Department of Experimental Medicine, University of Genova, 16132 Genova, Italy.
Maria Milanesi, Experimental Oncology and Immunology, Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy.
Francesca Bianchi, Movement Disorders Unit, Division of Neuroscience, IRCCS Ospedale San Raffaele, 20132 Milan, Italy.
Ubaldo Del Carro, Movement Disorders Unit, Division of Neuroscience, IRCCS Ospedale San Raffaele, 20132 Milan, Italy.
Jean-Michel Vallat, Department and Laboratory of Neurology, National Reference Center for ‘Rare Peripheral Neuropathies’, University Hospital of Limoges (CHU Limoges), Dupuytren Hospital, 87000 Limoges, France.
Phu Duong, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin–Madison, Madison, WI 53706, USA.
John Svaren, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin–Madison, Madison, WI 53706, USA.
Angelo Schenone, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genova, IRCCS AOU San Martino-IST, 16132 Genova, Italy; Department of Neurology, IRCCS Ospedale Policlinico San Martino, 16132 Genova, Italy.
Marina Grandis, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genova, IRCCS AOU San Martino-IST, 16132 Genova, Italy; Department of Neurology, IRCCS Ospedale Policlinico San Martino, 16132 Genova, Italy.
Maurizio D’Antonio, Biology of Myelin Unit, Division of Genetics and Cell Biology, IRCCS Ospedale San Raffaele, 20132 Milan, Italy.
Funding
Research funded by AFM-Téléthon (grant #20572 to M.G. and M.D.) and by a Fronzaroli Fellowship from Fondazione Centro San Raffaele to F.V.
References
- 1. Sherman, D.L. and Brophy, P.J. (2005) Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci., 6, 683–690. [DOI] [PubMed] [Google Scholar]
- 2. Jessen, K.R. and Mirsky, R. (2005) The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci., 6, 671–682. [DOI] [PubMed] [Google Scholar]
- 3. Patzig, J., Jahn, O., Tenzer, S., Wichert, S.P., de Monasterio-Schrader, P., Rosfa, S., Kuharev, J., Yan, K., Bormuth, I., Bremer, J. et al. (2011) Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J. Neurosci., 31, 16369–16386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Filbin, M.T., Walsh, F.S., Trapp, B.D., Pizzey, J.A. and Tennekoon, G.I. (1990) Role of myelin P0 protein as a homophilic adhesion molecule. Nature, 344, 871–872. [DOI] [PubMed] [Google Scholar]
- 5. Shapiro, L., Doyle, J.P., Hensley, P., Colman, D.R. and Hendrickson, W.A. (1996) Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron, 17, 435–449. [DOI] [PubMed] [Google Scholar]
- 6. Griffith, L.S., Schmitz, B. and Schachner, M. (1992) L2/HNK-1 carbohydrate and protein-protein interactions mediate the homophilic binding of the neural adhesion molecule P0. J. Neurosci. Res., 33, 639–648. [DOI] [PubMed] [Google Scholar]
- 7. Brennan, K.M., Bai, Y. and Shy, M.E. (2015) Demyelinating CMT--what's known, what's new and what's in store. Neurosci. Lett., 596, 14–26. [DOI] [PubMed] [Google Scholar]
- 8. Pareyson, D., Saveri, P. and Pisciotta, C. (2017) New developments in Charcot-Marie-tooth neuropathy and related diseases. Curr. Opin. Neurol., 30, 471–480. [DOI] [PubMed] [Google Scholar]
- 9. Saporta, A.S., Sottile, S.L., Miller, L.J., Feely, S.M., Siskind, C.E. and Shy, M.E. (2011) Charcot-Marie-tooth disease subtypes and genetic testing strategies. Ann. Neurol., 69, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shy, M.E., Jani, A., Krajewski, K., Grandis, M., Lewis, R.A., Li, J., Shy, R.R., Balsamo, J., Lilien, J., Garbern, J.Y. et al. (2004) Phenotypic clustering in MPZ mutations. Brain, 127, 371–384. [DOI] [PubMed] [Google Scholar]
- 11. Callegari, I., Gemelli, C., Geroldi, A., Veneri, F., Mandich, P., D'Antonio, M., Pareyson, D., Shy, M.E., Schenone, A., Prada, V. et al. (2019) Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J. Neurol., 266, 2629–2645. [DOI] [PubMed] [Google Scholar]
- 12. D'Antonio, M., Musner, N., Scapin, C., Ungaro, D., Del Carro, U., Ron, D., Feltri, M.L. and Wrabetz, L. (2013) Resetting translational homeostasis restores myelination in Charcot-Marie-tooth disease type 1B mice. J. Exp. Med., 210, 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grandis, M., Vigo, T., Passalacqua, M., Jain, M., Scazzola, S., La Padula, V., Brucal, M., Benvenuto, F., Nobbio, L., Cadoni, A. et al. (2008) Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum. Mol. Genet., 17, 1877–1889. [DOI] [PubMed] [Google Scholar]
- 14. Wrabetz, L., D'Antonio, M., Pennuto, M., Dati, G., Tinelli, E., Fratta, P., Previtali, S., Imperiale, D., Zielasek, J., Toyka, K. et al. (2006) Different intracellular pathomechanisms produce diverse myelin protein zero neuropathies in transgenic mice. J. Neurosci., 26, 2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai, Y., Wu, X., Brennan, K.M., Wang, D.S., D'Antonio, M., Moran, J., Svaren, J. and Shy, M.E. (2018) Myelin protein zero mutations and the unfolded protein response in Charcot Marie tooth disease type 1B. Ann. Clin. Transl. Neurol., 5, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vogt, G., Chapgier, A., Yang, K., Chuzhanova, N., Feinberg, J., Fieschi, C., Boisson-Dupuis, S., Alcais, A., Filipe-Santos, O., Bustamante, J. et al. (2005) Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat. Genet., 37, 692–700. [DOI] [PubMed] [Google Scholar]
- 17. Vogt, G., Vogt, B., Chuzhanova, N., Julenius, K., Cooper, D.N. and Casanova, J.L. (2007) Gain-of-glycosylation mutations. Curr. Opin. Genet. Dev., 17, 245–251. [DOI] [PubMed] [Google Scholar]
- 18. De Jonghe, P., Timmerman, V., Ceuterick, C., Nelis, E., De Vriendt, E., Lofgren, A., Vercruyssen, A., Verellen, C., Van Maldergem, L., Martin, J.J. et al. (1999) The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-tooth phenotype. Brain, 122(Pt 2), 281–290. [DOI] [PubMed] [Google Scholar]
- 19. Prada, V., Passalacqua, M., Bono, M., Luzzi, P., Scazzola, S., Nobbio, L.A., Capponi, S., Bellone, E., Mandich, P., Mancardi, G. et al. (2012) Gain of glycosylation: a new pathomechanism of myelin protein zero mutations. Ann. Neurol., 71, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yonekawa, T., Komaki, H., Saito, Y., Takashima, H. and Sasaki, M. (2013) Congenital hypomyelinating neuropathy attributable to a de novo p.Asp61Asn mutation of the myelin protein zero gene. Pediatr. Neurol., 48, 59–62. [DOI] [PubMed] [Google Scholar]
- 21. Sedzik, J., Jastrzebski, J.P. and Grandis, M. (2015) Glycans of myelin proteins. J. Neurosci. Res., 93, 1–18. [DOI] [PubMed] [Google Scholar]
- 22. Julenius, K., Molgaard, A., Gupta, R. and Brunak, S. (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology, 15, 153–164. [DOI] [PubMed] [Google Scholar]
- 23. Wang, H., Yang, H., Shivalila, C.S., Dawlaty, M.M., Cheng, A.W., Zhang, F. and Jaenisch, R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voshol, H., van Zuylen, C.W., Orberger, G., Vliegenthart, J.F. and Schachner, M. (1996) Structure of the HNK-1 carbohydrate epitope on bovine peripheral myelin glycoprotein P0. J. Biol. Chem., 271, 22957–22960. [DOI] [PubMed] [Google Scholar]
- 25. Li, J., Parker, B., Martyn, C., Natarajan, C. and Guo, J. (2013) The PMP22 gene and its related diseases. Mol. Neurobiol., 47, 673–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martini, R. and Schachner, M. (1997) Molecular bases of myelin formation as revealed by investigations on mice deficient in glial cell surface molecules. Glia, 19, 298–310. [PubMed] [Google Scholar]
- 27. Garbay, B., Heape, A.M., Sargueil, F. and Cassagne, C. (2000) Myelin synthesis in the peripheral nervous system. Prog. Neurobiol., 61, 267–304. [DOI] [PubMed] [Google Scholar]
- 28. D'Urso, D., Brophy, P.J., Staugaitis, S.M., Gillespie, C.S., Frey, A.B., Stempak, J.G. and Colman, D.R. (1990) Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron, 4, 449–460. [DOI] [PubMed] [Google Scholar]
- 29. Fratta, P., Saveri, P., Zambroni, D., Ferri, C., Tinelli, E., Messing, A., D'Antonio, M., Feltri, M.L. and Wrabetz, L. (2011) P0S63del impedes the arrival of wild-type P0 glycoprotein to myelin in CMT1B mice. Hum. Mol. Genet., 20, 2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saporta, M.A., Shy, B.R., Patzko, A., Bai, Y., Pennuto, M., Ferri, C., Tinelli, E., Saveri, P., Kirschner, D., Crowther, M. et al. (2012) MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-tooth disease type 1B. Brain, 135, 2032–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sidoli, M., Musner, N. and Silvestri, N. (2016) Ablation of perk in Schwann cells improves myelination in the S63del Charcot-Marie-tooth 1B mouse. J. Neurosci., 36, 11350–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. D'Antonio, M., Feltri, M.L. and Wrabetz, L. (2009) Myelin under stress. J. Neurosci. Res., 87, 3241–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pennuto, M., Tinelli, E., Malaguti, M., Del Carro, U., D'Antonio, M., Ron, D., Quattrini, A., Feltri, M.L. and Wrabetz, L. (2008) Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-tooth 1B mice. Neuron, 57, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chernousov, M.A., Yu, W.M., Chen, Z.L., Carey, D.J. and Strickland, S. (2008) Regulation of Schwann cell function by the extracellular matrix. Glia, 56, 1498–1507. [DOI] [PubMed] [Google Scholar]
- 35. Court, F.A., Wrabetz, L. and Feltri, M.L. (2006) Basal lamina: Schwann cells wrap to the rhythm of space-time. Curr. Opin. Neurobiol., 16, 501–507. [DOI] [PubMed] [Google Scholar]
- 36. Previtali, S.C., Nodari, A., Taveggia, C., Pardini, C., Dina, G., Villa, A., Wrabetz, L., Quattrini, A. and Feltri, M.L. (2003) Expression of laminin receptors in schwann cell differentiation: evidence for distinct roles. J. Neurosci., 23, 5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arthur-Farraj, P.J., Latouche, M., Wilton, D.K., Quintes, S., Chabrol, E., Banerjee, A., Woodhoo, A., Jenkins, B., Rahman, M., Turmaine, M. et al. (2012) C-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron, 75, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hantke, J., Carty, L., Wagstaff, L.J., Turmaine, M., Wilton, D.K., Quintes, S., Koltzenburg, M., Baas, F., Mirsky, R. and Jessen, K.R. (2014) C-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain, 137, 2922–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalinski, A.L., Yoon, C., Huffman, L.D., Duncker, P.C., Kohen, R., Passino, R., Hafner, H., Johnson, C., Kawaguchi, R., Carbajal, K.S. et al. (2020) Analysis of the immune response to sciatic nerve injury identifies efferocytosis as a key mechanism of nerve debridement. elife, 9, e60223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Florio, F., Ferri, C., Scapin, C. and Feltri, M.L. (2018) Sustained expression of negative regulators of myelination protects Schwann cells from Dysmyelination in a Charcot-Marie-tooth 1B mouse model. J. Neurosci., 38, 4275–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vogt, G., Bustamante, J., Chapgier, A., Feinberg, J., Boisson Dupuis, S., Picard, C., Mahlaoui, N., Gineau, L., Alcais, A., Lamaze, C. et al. (2008) Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J. Exp. Med., 205, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee, M., Brennan, A., Blanchard, A., Zoidl, G., Dong, Z., Tabernero, A., Zoidl, C., Dent, M.A., Jessen, K.R. and Mirsky, R. (1997) P0 is constitutively expressed in the rat neural crest and embryonic nerves and is negatively and positively regulated by axons to generate non-myelin-forming and myelin-forming Schwann cells, respectively. Mol. Cell. Neurosci., 8, 336–350. [DOI] [PubMed] [Google Scholar]
- 43. Lee, Y.C., Lin, K.P., Chang, M.H., Liao, Y.C., Tsai, C.P., Liao, K.K. and Soong, B.W. (2010) Cellular characterization of MPZ mutations presenting with diverse clinical phenotypes. J. Neurol., 257, 1661–1668. [DOI] [PubMed] [Google Scholar]
- 44. Lemke, G. and Chao, M. (1988) Axons regulate Schwann cell expression of the major myelin and NGF receptor genes. Development (Cambridge, England), 102, 499–504. [DOI] [PubMed] [Google Scholar]
- 45. Lemke, G., Lamar, E. and Patterson, J. (1988) Isolation and analysis of the gene encoding peripheral myelin protein zero. Neuron, 1, 73–83. [DOI] [PubMed] [Google Scholar]
- 46. Liu, Z., Wang, Y., Yedidi, R.S., Brunzelle, J.S., Kovari, I.A., Sohi, J., Kamholz, J. and Kovari, L.C. (2012) Crystal structure of the extracellular domain of human myelin protein zero. Proteins, 80, 307–313. [DOI] [PubMed] [Google Scholar]
- 47. Lobsiger, C.S., Taylor, V. and Suter, U. (2002) The early life of a Schwann cell. Biol. Chem., 383, 245–253. [DOI] [PubMed] [Google Scholar]
- 48. Quarles, R.H. (1997) Glycoproteins of myelin sheaths. J. Mol. Neurosci., 8, 1–12. [DOI] [PubMed] [Google Scholar]
- 49. Helenius, A. and Aebi, M. (2001) Intracellular functions of N-linked glycans. Science (New York, N.Y.), 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- 50. Huet, G., Gouyer, V., Delacour, D., Richet, C., Zanetta, J.P., Delannoy, P. and Degand, P. (2003) Involvement of glycosylation in the intracellular trafficking of glycoproteins in polarized epithelial cells. Biochimie, 85, 323–330. [DOI] [PubMed] [Google Scholar]
- 51. Blanquet-Grossard, F., Pham-Dinh, D., Dautigny, A., Latour, P., Bonnebouche, C., Diraison, P., Chapon, F., Chazot, G. and Vandenberghe, A. (1996) Charcot-Marie-tooth type 1B neuropathy: a mutation at the single glycosylation site in the major peripheral myelin glycoprotein Po. Hum. Mutat., 8, 185–186. [DOI] [PubMed] [Google Scholar]
- 52. Choi, B.O., Lee, M.S., Shin, S.H., Hwang, J.H., Choi, K.G., Kim, W.K., Sunwoo, I.N., Kim, N.K. and Chung, K.W. (2004) Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-tooth neuropathy patients. Hum. Mutat., 24, 185–186. [DOI] [PubMed] [Google Scholar]
- 53. Kochanski, A., Drac, H., Kabzinska, D., Ryniewicz, B., Rowinska-Marcinska, K., Nowakowski, A. and Hausmanowa-Petrusewicz, I. (2004) A novel MPZ gene mutation in congenital neuropathy with hypomyelination. Neurology, 62, 2122–2123. [DOI] [PubMed] [Google Scholar]
- 54. Lagueny, A., Latour, P., Vital, A., Rajabally, Y., Le Masson, G., Ferrer, X., Bernard, I., Julien, J., Vital, C. and Vandenberghe, A. (1999) Peripheral myelin modification in CMT1B correlates with MPZ gene mutations. Neuromuscul. Disord., 9, 361–367. [DOI] [PubMed] [Google Scholar]
- 55. Mandich, P., Fossa, P., Capponi, S., Geroldi, A., Acquaviva, M., Gulli, R., Ciotti, P., Manganelli, F., Grandis, M. and Bellone, E. (2009) Clinical features and molecular modelling of novel MPZ mutations in demyelinating and axonal neuropathies. Eur. J. Hum. Genet., 17, 1129–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mersiyanova, I.V., Ismailov, S.M., Polyakov, A.V., Dadali, E.L., Fedotov, V.P., Nelis, E., Lofgren, A., Timmerman, V., van Broeckhoven, C. and Evgrafov, O.V. (2000) Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-tooth neuropathy patients. Hum. Mutat., 15, 340–347. [DOI] [PubMed] [Google Scholar]
- 57. Marinko, J.T., Wright, M.T., Schlebach, J.P., Clowes, K.R., Heintzman, D.R., Plate, L. and Sanders, C.R. (2021) Glycosylation limits forward trafficking of the tetraspan membrane protein PMP22. J. Biol. Chem., 296, 100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hörste, M.Z. and G. and Nave, K.A. (2006) Animal models of inherited neuropathies. Curr. Opin. Neurol., 19, 464–473. [DOI] [PubMed] [Google Scholar]
- 59. Fratta, P., Ornaghi, F., Dati, G., Zambroni, D., Saveri, P., Belin, S., D'Adamo, P., Shy, M., Quattrini, A., Laura Feltri, M. et al. (2019) A nonsense mutation in myelin protein zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum. Mol. Genet., 28, 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gabreëls-Festen, A.A., Hoogendijk, J.E., Meijerink, P.H., Gabreëls, F.J., Bolhuis, P.A., van Beersum, S., Kulkens, T., Nelis, E., Jennekens, F.G., de Visser, M. et al. (1996) Two divergent types of nerve pathology in patients with different P0 mutations in Charcot-Marie-tooth disease. Neurology, 47, 761–765. [DOI] [PubMed] [Google Scholar]
- 61. Kirschner, D.A., Szumowski, K., Gabreëls-Festen, A.A., Hoogendijk, J.E. and Bolhuis, P.A. (1996) Inherited demyelinating peripheral neuropathies: relating myelin packing abnormalities to P0 molecular defects. J. Neurosci. Res., 46, 502–508. [DOI] [PubMed] [Google Scholar]
- 62. Thomas, F.P., Lebo, R.V., Rosoklija, G., Ding, X.S., Lovelace, R.E., Latov, N. and Hays, A.P. (1994) Tomaculous neuropathy in chromosome 1 Charcot-Marie-tooth syndrome. Acta Neuropathol., 87, 91–97. [DOI] [PubMed] [Google Scholar]
- 63. Wrabetz, L., Feltri, M.L., Quattrini, A., Imperiale, D., Previtali, S., D'Antonio, M., Martini, R., Yin, X., Trapp, B.D., Zhou, L. et al. (2000) P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J. Cell Biol., 148, 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Previtali, S.C., Quattrini, A., Fasolini, M., Panzeri, M.C., Villa, A., Filbin, M.T., Li, W., Chiu, S.Y., Messing, A., Wrabetz, L. et al. (2000) Epitope-tagged P(0) glycoprotein causes Charcot-Marie-tooth-like neuropathy in transgenic mice. J. Cell Biol., 151, 1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Adlkofer, K., Frei, R., Neuberg, D.H., Zielasek, J., Toyka, K.V. and Suter, U. (1997) Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating tomaculous neuropathy. J. Neurosci., 17, 4662–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lewallen, K.A., Shen, Y.A., De la Torre, A.R., Ng, B.K., Meijer, D. and Chan, J.R. (2011) Assessing the role of the cadherin/catenin complex at the Schwann cell-axon interface and in the initiation of myelination. J. Neurosci., 31, 3032–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Previtali, S.C. and Zambon, A.A. (2020) LAMA2 neuropathies: human findings and Pathomechanisms from mouse models. Front. Mol. Neurosci., 13, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fazal, S.V., Gomez-Sanchez, J.A., Wagstaff, L.J., Musner, N., Otto, G., Janz, M., Mirsky, R. and Jessen, K.R. (2017) Graded elevation of c-Jun in Schwann cells in vivo: gene dosage determines effects on development, Remyelination, tumorigenesis, and Hypomyelination. J. Neurosci., 37, 12297–12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dwek, R.A., Butters, T.D., Platt, F.M. and Zitzmann, N. (2002) Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug Discov., 1, 65–75. [DOI] [PubMed] [Google Scholar]
- 70. Elbein, A.D. (1991) Glycosidase inhibitors: inhibitors of N-linked oligosaccharide processing. FASEB J, 5, 3055–3063. [DOI] [PubMed] [Google Scholar]
- 71. Leonard, A., Lebecque, P., Dingemanse, J. and Leal, T. (2013) Miglustat effects on the basal nasal potential differences in cystic fibrosis. J. Cyst. Fibros., 12, 89. [DOI] [PubMed] [Google Scholar]
- 72. Ficicioglu, C. (2008) Review of miglustat for clinical management in Gaucher disease type 1. Ther. Clin. Risk Manag., 4, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lyseng-Williamson, K.A. (2014) Miglustat: a review of its use in Niemann-pick disease type C. Drugs, 74, 61–74. [DOI] [PubMed] [Google Scholar]
- 74. Bolino, A., Bolis, A., Previtali, S.C., Dina, G., Bussini, S., Dati, G., Amadio, S., Del Carro, U., Mruk, D.D., Feltri, M.L. et al. (2004) Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J. Cell Biol., 167, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wiesmann, M., Timmer, N.M., Zinnhardt, B., Reinhard, D., Eligehausen, S., Konigs, A., Ben Jeddi, H., Dederen, P.J., Jacobs, A.H. and Kiliaan, A.J. (2018) Effect of a multinutrient intervention after ischemic stroke in female C57Bl/6. Mice, 144, 549–564. [DOI] [PubMed] [Google Scholar]
- 76. Taveggia, C., Zanazzi, G., Petrylak, A., Yano, H., Rosenbluth, J., Einheber, S., Xu, X., Esper, R.M., Loeb, J.A., Shrager, P. et al. (2005) Neuregulin-1 type III determines the ensheathment fate of axons. Neuron, 47, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Anders, S., McCarthy, D.J., Chen, Y., Okoniewski, M., Smyth, G.K., Huber, W. and Robinson, M.D. (2013) Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc., 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- 79. Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Gallo Cassarino, T., Bertoni, M., Bordoli, L. et al. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res., 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.