

Abstract
The extensive characterization of glycosidic linkages in carbohydrates remains a challenge due to the lack of known standards and limitations in current analytical techniques. This study encompasses the construction of an extensive glycosidic linkage library built from synthesized standards. It includes an improved liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for the quantitation of glycosidic linkages derived from disaccharides, oligosaccharides, and polysaccharides present in complicated matrices. We present a method capable of the simultaneous identification of over 90 unique glycosidic linkages using ultrahigh-performance liquid chromatography coupled with triple quadrupole mass spectrometry (UHPLC/QqQ MS) operated in dynamic multiple reaction monitoring (dMRM) mode. To build the library, known monosaccharides commonly found in plants were subjected to partial methylation to yield partially derivatized species representing trisecting, bisecting, linear, and terminal structures. The library includes glycosidic linkage information for three hexoses (glucose, galactose, and mannose), three pentoses (xylose, arabinose, and ribose), two deoxyhexoses (fucose and rhamnose), and two hexuronic acids (glucuronic acid and galacturonic acid). The resulting partially methylated monosaccharides were then labeled with 1-phenyl-3-methyl-5-pyrazolone (PMP) followed by separation and analysis by UHPLC/dMRM MS. Validation of the synthesized standards was performed using disaccharide, oligosaccharide, and polysaccharide standards. Accuracy, reproducibility, and robustness of the method was demonstrated by analysis of xyloglucan (tamarind) and whole carrot root. The synthesized standards represent the most comprehensive group of carbohydrate linkages to date.
Keywords: Glycosidic Linkage Analysis, Glycosidic Linkage Library, Mass Spectrometry, Carbohydrates, Disaccharides, Oligosaccharides, Polysaccharides, Multiple Reaction Monitoring, Permethylation, Partial methylation, Acetylation, Deacetylation, 1-phenyl-3-methyl-5-pyrazolone, LC-MS, Tandem MS
Graphical Abstract
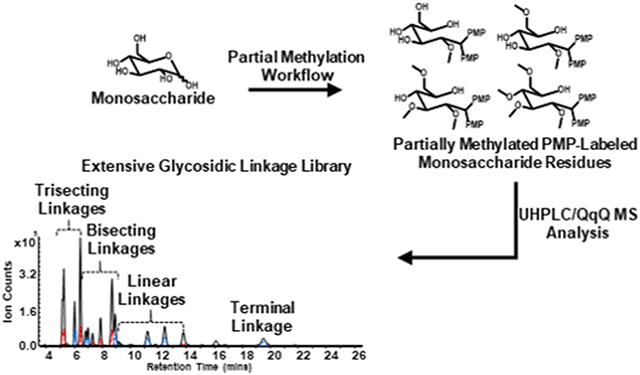
INTRODUCTION
Carbohydrates consist of monosaccharides, disaccharides, oligosaccharides, and polysaccharides, and are the most abundant biomolecules in nature. They are present in all living organisms and are known to have structural and functional biological properties that are associated with their specific compositions and structures. Carbohydrates are often found on the surface of cells as plant cell wall polysaccharides and as oligosaccharides that decorate the surfaces of animal cells that are involved in molecular trafficking, protein-folding, and biological recognition.1-3 Carbohydrates are common structural components that are degraded by enzymes in a host of biological reactions, one example being food for nutrition. The monosaccharide and glycosidic linkages of polysaccharides are specific substrates for glycosyl hydrolases present in the host or in their compliment microbes.4-7 These glycosyl hydrolases can have broad or high specificity that depend on both the monosaccharide and linkage composition. For example, in human digestion, α-amylase is responsible for the hydrolysis of α(1→4)glucose in amylose, the main component of starch. However, α-amylase is generally incapable of digesting the α(1→4,6)glucose, which is the branching component of amylopectin.8-10 Thus, characterization of the monosaccharide composition and the associated glycosidic linkages are critical to fully understand structural and functional components of carbohydrates.
The most commonly used method for linkage analysis employs the Hakomori approach using permethylation and acid hydrolysis followed by volatilization of the products for GC-MS analysis.11-16 Recently, we reported a method for linkage analysis based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) involving permethylation, acid hydrolysis, and labeling with 1-phenyl-3-methyl-5-pyrazolone (PMP) providing improvements in speed, throughput, and sensitivity over the standard GC-MS method.17 A library was also produced based on a limited set of available standards. The lack of highly pure and well characterized commercial standards severely limited this approach. Thus, the earlier compilation was created with only a fraction of the possible linkages represented. Glycosidic linkages representing many branched monomers were most notably absent.
A solution to the lack of standards is to synthesize partially methylated species from monosaccharides representing all combinations of linkages including terminal, linear, bisecting and even trisecting species. This approach has been previously used to prepare linkage standards for GC-MS analysis.18 However, some important concerns have to be addressed in order to successfully adapt this approach from gas chromatography to liquid chromatography. One is the poor potential recovery of monosaccharides with higher numbers of unmethylated sites such as those representing bisecting and trisecting species during liquid-liquid extraction.19-22 To address this issue, unmethylated hydroxyls can be acetylated using acetic anhydride.18, 23-25 A further concern with this method is that sodium hydroxide (NaOH), necessary for the methylation, will decompose acetic anhydride immediately after its addition to the solution. A more effective approach would be to use dimethyl sulfinyl anion rather than hydroxide during methylation.11, 26-29 The extent of methylation is then controlled depending on the concentration of dimethyl sulfinyl anion.18 Recovery of the desired partially methylated structures is then obtained by enrichment using liquid-liquid extraction followed by deacetylation.18, 20
In this study, we constructed a considerably more comprehensive LC-MS/MS-based glycosidic linkage library by employing a synthetic approach involving the partial methylation of monosaccharides and reducing-end labeling in combination with ultrahigh-performance liquid chromatography coupled with triple quadrupole mass spectrometry (UHPLC/QqQ MS). Tandem MS was performed in dynamic multiple reaction monitoring (dMRM) mode for the quantitation and detection of distinct glycosidic linkages derived from disaccharides, oligosaccharides, and polysaccharides present in complicated matrices. The UHPLC/dMRM MS method enabled the simultaneous analysis of an extensive group of over 90 glycosidic linkages representing terminal, linear, bisecting and even trisecting monomers of glucose, galactose, mannose, xylose, arabinose, ribose, fucose, rhamnose, glucuronic acid, and galacturonic acid. The synthetic-based library improves over a previously developed linkage library built exclusively from commercial disaccharide, oligosaccharide, and polysaccharide standards.17 To build the extended library, partial methylation of monosaccharide standards was performed with iodomethane using various concentrations of dimethyl sulfinyl anion followed by acetylation using acetic anhydride, deacetylation using trifluoroacetic acid (TFA), and reducing-end labeling with PMP. Optimal chromatographic separation accompanied by optimized MS parameters allowed for separation and analysis in 30 minutes. Validation of the synthesized linkage standards was performed by analysis of disaccharide, oligosaccharide, and polysaccharide standards. The method was applied and further validated by the analysis of xyloglucan (tamarind) and whole carrot root. The accuracy, robustness, and reproducibility of this method makes it significantly useful for the elucidation of glycosidic linkages in disaccharides, oligosaccharides, and polysaccharides derived from foods and biological samples.
EXPERIMENTAL PROCEDURES
Samples and materials.
Iodomethane (contains copper stabilizer, 99.5%), TFA, NaOH pellets (semi-conductor grade, 99.99% trace metals basis), sodium hydride (NaH) (dry, 90%), hydrochloric acid (HCl) (ACS reagent, 37%), phenolphthalein (ACS reagent), ammonium acetate (NH4Ac) (99.99% trace metals basis), ammonium hydroxide solution (NH4OH) (28-30%, NH3 basis), PMP, anhydrous dimethyl sulfoxide (DMSO), dichloromethane (DCM), methanol (MeOH) (HPLC grade), stachyose, D-glucose, D-galactose, D-mannose, L-fucose, L-rhamnose, D-arabinose, D-xylose, D-ribose, D-glucuronic acid, and D-galacturonic acid were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN) (HPLC grade) was purchased from Honeywell (Muskegon, MI). 2-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-D-xylobiose, 1,5-a-L-arabinotriose, 1,3-a-1,6-a-D-mannotriose, isomaltotriose, 4-O-(b-D-galactopyranosyl)-D-galactopyranose, lactose, 2'-fucosyllactose (synthetic), nigerose, 3-O-(b-D-galactopyranosyl)-D-galactopyranose, 3-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-b-D-mannotriose, maltohexaose, 1,6-a-D-mannotriose, and amylopectin all were obtained from Carbosynth (Compton, UK). Xyloglucan (tamarind), 33-α-L-arabinofuranosyl-xylotetraose, and sophorose were acquired from Megazyme (Chicago, IL). Whole carrots were purchased from Whole Foods (Davis, CA). Nanopure water was used for all experiments.
Optimization of deacetylation reaction.
A stock solution of α-D-glucose pentaacetate was prepared at a concentration of 2 μg/μL in MeOH. Aliquots containing 25 μL of this stock solution was transferred to micro-tubes with screw caps and dried by vacuum centrifugation. Next, the samples were hydrolyzed using 100 μL of 4 M TFA and by incubation at 100 °C for 0, 15, 30, 60, and 90 minutes, respectively. Three experimental replicates were performed for each time-point. After each time-point, 900 μL of ice-cold water was added to the samples followed by vortexing. An aliquot containing 50 μL of this solution was immediately subjected to PMP-labeling following a previously developed procedure.30
Optimization of acetylation reaction.
A stock solution of D-glucose was prepared in water at a concentration of 1 μg/μL. Aliquots containing 50 μL of this solution were transferred to cryovials (5 mL) and dried by vacuum centrifugation. Samples were purged with argon followed by the addition of 150 μL of anhydrous DMSO. Next, 75 μL of 1-methyl-imidazole was added followed by 750 μL of anhydrous acetic anhydride. Samples were reacted on a shaker at room temperature for 0, 5, 10, and 20 minutes, respectively. Three experimental replicates were performed for each time-point. After each time point, the samples were placed in an ice-bath followed by the addition of 1.5 mL ice-cold water and vortexed to quench the excess acetic anhydride in each sample. After 10 minutes, the samples were removed from the ice-bath and enriched by liquid-liquid extraction using 500 μL DCM and subsequent washes with 4 x 750 μL ice-cold water. The aqueous top layer was discarded and the bottom organic layer containing products was collected and dried by vacuum centrifugation. Samples were hydrolyzed using the optimized time of 30 minutes followed by PMP-labeling using the previously described method.
Preparation of dimethyl sulfinyl anion.
Anhydrous DMSO (20 mL, 280.0 mmol) pre-heated to 70 °C was added drop-wise via addition funnel to solid NaH (1.5 g, 62.5 mmol) in a two-neck argon purged round-bottomed flask. The round-bottomed flask was fitted with an entrance and exit gas port to allow fresh argon to enter and hydrogen gas by-product to escape. The reaction mixture was vigorously stirred using a magnetic stir bar and reacted at 70 °C for 1 hour in an oil bath. The solution was flushed with argon throughout the reaction. After 1 hour, no further bubbling was observed, indicating completion of the reaction. The reaction solution was cooled to room temperature and the resulting color of the final solution was dark green. The solution was centrifuged at 4k rpm for 30 minutes and the supernatant was filtered using a 0.2 μm syringe filter and collected. The concentration of dimethyl sulfinyl anion in the filtrate was determined by titration with 0.1 M HCl using phenolphthalein as an indicator. Final concentration of the anion was determined to be 1.2 M.
Partial methylation of monosaccharides.
Standard solutions of monosaccharides were prepared in water at a concentration of 10 μg/μL. Aliquots containing 5 μL of solution were transferred to cryovials (5 mL) and dried by vacuum centrifugation. Dimethyl sulfinyl anion was prepared at concentrations of 0.01, 0.1, and 1.0 M in anhydrous DMSO. Each of the monosaccharide samples were reconstituted in 150 μL of each dimethyl sulfinyl anion concentration, flushed with argon, and placed on a shaker at for 30 minutes at room temperature. Next, 40 μL of iodomethane was added followed by reacting on a shaker for 50 minutes. Residual iodomethane was then removed by vacuum centrifugation for 30 minutes. Acetylation of remaining free hydroxyl groups was carried out using the optimal reaction time of 20 minutes using the method previously described. Subsequent liquid-liquid extractions, hydrolysis, and PMP-labeling were then performed following the methods previously mentioned. The samples were reconstituted in 100 μL of 70% MeOH/water (v/v) and an aliquot containing 20 μL of partially methylated PMP-labeled monosaccharides produced by each concentration of dimethyl sulfinyl anion filtrate were pooled.
Preparation of polysaccharide standards and whole food samples for derivatization and analysis.
A stock solution of xyloglucan polysaccharide standard was prepared in water at a concentration of 10 μg/μL. Whole carrot root was chopped, lyophilized, ground to a fine powder using a mortar and pestle, then a stock solution of this processed material was prepared at a concentration of 10 μg/μL in water. Further homogenization of sample was accomplished by bead blasting for 3 minutes using 1.4 mm stainless steel magnetic beads and a Next Advance Bullet Blender Storm 24 (Next Advance, Troy, NY). The resulting sample solutions were incubated at 100 °C for 60 minutes followed by a second round of bead blasting.
UHPLC/QqQ MS Analysis.
The partially methylated PMP-labeled monosaccharides were separated and analyzed on an Agilent 1290 Infinity II UHPLC system coupled to an Agilent 6495A triple quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA.). The analysis utilizes 1 μL of sample injected onto a Agilent Poroshell 120 EC-C18 column (2.1 mm x 150 mm i.d., 1.9 μm particle size) equipped with an Agilent Poroshell 120 EC-C18 guard cartridge (2.1 mm × 5 mm i.d., 1.9 μm particle size) and separated using a 26-minute isocratic method followed by a 4-minute wash and re-equilibration step at a constant flow rate of 0.35 mL/min: Mobile phase A: 25 mM NH4Ac in 5% ACN/water (v/v), adjusted to pH 8.2 using NH4OH; Mobile phase B: 95% ACN/water (v/v). The following elution gradient was used: 0.00-26.00 mins, 17.00% B; 26.00-26.01 mins, 17.00-99.00% B; 26.01-28.00 mins, 99.00% B; 28.00-28.01 mins, 99.00-17.00% B; 28.01-30.00 mins, 17.00% B.
An electrospray ionization (ESI) source operated in positive ion mode was used to introduce sample into the QqQ. Capillary and fragmentor voltages were set at 1800 V and 380 V, respectively. Nebulizer pressure was set to 30 psi. Drying and sheath gas flow rates were set at 11 L/min and 12 L/min, respectively. Nitrogen drying and sheath gas temperatures were set at 290 °C and 300 °C, respectively. Collision induced dissociation (CID) was performed by setting the collision energy to 35 V. Acquired UHPLC/QqQ MS data was retrieved using Agilent MassHunter Workstation Data Acquisition Version B.06.01 and analyzed with Agilent MassHunter Quantitative Analysis software Version B.06.00.
RESULTS AND DISCUSSION
A synthesis-based methodology was used to develop an extensive library for the analysis of linkage residues derived from disaccharides, oligosaccharides, and polysaccharides existing in complicated matrices. The method is comprised of partial methylation, acetylation, deacetylation, and PMP-labeling followed by UHPLC/QqQ MS analysis (Scheme 1). First, partial methylation was performed using varying concentrations of prepared dimethyl sulfinyl anion. Remaining free hydroxyls were then acetylated to improve enrichment efficiency during liquid-liquid extraction following partial methylation. Next, deacetylation by acid was performed to reestablish the free hydroxyl groups that mimic positions of putative glycosidic bonds. The partially methylated monosaccharides were labeled further to improve chromatographic separation and ionization in the LC-MS/MS analysis.
Scheme 1.

Workflow for the development of the extended linkage library. The library is built by sequential partial methylation, acetylation, hydrolysis, and PMP-labeling using monosaccharide standards as building blocks.
Construction of the synthetic glycosidic linkage library
Glycosidic linkage reference compounds representing over 90 terminal, linear, bisecting, and trisecting neutral and acidic monosaccharide species were produced by the partial methylation and PMP-labeling of D-glucose, D-galactose, D-mannose, L-fucose, L-rhamnose, D-xylose, D-arabinose, D-ribose, D-glucuronic acid, and D-galacturonic acid. Validation of the synthetic linkages was performed using commercial disaccharide, oligosaccharide, and polysaccharide standards prepared using a previously established procedure.17 Disaccharide, oligosaccharide, and polysaccharide standards used for the validation include stachyose, sophorose, isomaltotriose, lactose, nigerose, 2'-fucosyllactose, 1,5-a-L-arabinotriose, 1,4-D-xylobiose, 33-α-L-arabinofuranosyl-xylotetraose, 1,6-a-D-mannotriose, 1,4-b-D-mannotriose, 1,3-a-1,6-a-D-mannotriose, 3-O-(a-D-mannopyranosyl)-D-mannopyranose, 2-O-(a-D-mannopyranosyl)-D-mannopyranose, 3-O-(b-D-galactopyranosyl)-D-galactopyranose, 4-O-(b-D-galactopyranosyl)-D-galactopyranose, and xyloglucan. Table 1 displays the synthesis-based LC-MS/MS linkage library that lists 92 neutral and acidic linkage residues with their corresponding retention times, degree of permethylation (DoPe), precursor ions, and product ions.
Table 1.
Library containing 92 unique glycosidic linkages obtained from the partial methylation and PMP-labeling of monosaccharide standards.
| Linkage Residue | Retention Time (mins) |
Degree of Permethylation (DoPe) |
MRM Transition Pair | |
|---|---|---|---|---|
| Precursor Ion (m/z) |
Product Ions (m/z) |
|||
| 3,4-Glucuronic Acid | 2.70 | 1 | 553.3 | 175.1, 231.2 |
| 2,X,X-Mannose | 2.80 | 1 | 525.2 | 175.1, 217.1 |
| X-Glucuronic Acid | 2.82 | 2 | 567.6 | 175.1, 231.2 |
| 2,X-Glucuronic Acid | 2.90 | 1 | 553.3 | 175.1, 217.1 |
| 2,X-Glucuronic Acid | 3.10 | 1 | 553.3 | 175.1, 217.1 |
| 2,X,X-Mannose | 3.16 | 1 | 525.2 | 175.1, 217.1 |
| 2,X-Galacturonic Acid | 3.30 | 1 | 553.3 | 175.1, 217.1 |
| 2,X-Mannose | 3.30 | 2 | 539.2 | 175.1, 217.1 |
| X-Glucuronic Acid | 3.32 | 2 | 567.6 | 175.1, 231.2 |
| 3,4-Galacturonic Acid | 3.72 | 1 | 553.3 | 175.1, 231.2 |
| 2,X,X-Mannose | 3.80 | 1 | 525.2 | 175.1, 217.1 |
| X-Galacturonic Acid | 3.84 | 2 | 567.6 | 175.1, 231.2 |
| 2,X-F-Ribose | 3.88 | 1 | 495.2 | 175.1, 217.1 |
| X-Galacturonic Acid | 4.04 | 2 | 567.6 | 175.1, 231.2 |
| 2,X-Mannose | 4.14 | 2 | 539.2 | 175.1, 217.1 |
| 2,X-Rhamnose | 4.16 | 1 | 509.2 | 175.1, 217.1 |
| 2,X,X-Glucose | 5.12 | 1 | 525.2 | 175.1, 217.1 |
| 2,X-Mannose | 5.15 | 2 | 539.2 | 175.1, 217.1 |
| 2,X-Rhamnose | 5.24 | 1 | 509.2 | 175.1, 217.1 |
| 2,X,X-Glucose | 5.24 | 1 | 525.2 | 175.1, 217.1 |
| 2-Mannose* | 5.25 | 3 | 553.3 | 175.1, 217.1 |
| 2-Rhamnose | 5.46 | 2 | 523.2 | 175.1, 217.1 |
| 2,X,X-Galactose | 5.66 | 1 | 525.2 | 175.1, 217.1 |
| 2,X-F-Ribose | 5.68 | 1 | 495.2 | 175.1, 217.1 |
| 2,X,X-Galactose | 5.88 | 1 | 525.2 | 175.1, 217.1 |
| 3,4,6-Glucose | 6.00 | 1 | 525.2 | 175.1, 231.2 |
| 2,X-P-Xylose | 6.02 | 1 | 495.2 | 175.1, 217.1 |
| 2,X,X-Galactose | 6.15 | 1 | 525.2 | 175.1, 217.1 |
| 3,4,6-Galactose | 6.40 | 1 | 525.2 | 175.1, 231.2 |
| 2,X,X-Glucose | 6.41 | 1 | 525.2 | 175.1, 217.1 |
| 2,X-Galactose | 6.54 | 2 | 539.2 | 175.1, 217.1 |
| 2,X-F-Arabinose | 6.55 | 1 | 495.2 | 175.1, 217.1 |
| 3,5-F-Arabinose | 6.78 | 1 | 495.2 | 175.1, 231.2 |
| X,X-Glucose | 6.79 | 2 | 539.2 | 175.1, 231.2 |
| 3,4-P-Xylose* | 6.82 | 1 | 495.2 | 175.1, 231.2 |
| 2,X-F-Arabinose | 6.92 | 1 | 495.2 | 175.1, 217.1 |
| 4,6-Glucose | 6.98 | 2 | 539.2 | 175.1, 231.2 |
| 2,X-P-Xylose | 7.00 | 1 | 495.2 | 175.1, 217.1 |
| 3,4,6-Mannose | 7.15 | 1 | 525.2 | 175.1, 231.2 |
| X,X-Galactose | 7.18 | 2 | 539.2 | 175.1, 231.2 |
| 2,X-Glucose | 7.30 | 2 | 539.2 | 175.1, 217.1 |
| X,X-Galactose | 7.40 | 2 | 539.2 | 175.1, 231.2 |
| 3,4-Fucose | 7.44 | 1 | 509.2 | 175.1, 231.2 |
| 3,5-F-Ribose | 7.45 | 1 | 495.2 | 175.1, 231.2 |
| 2,X-Glucose | 7.85 | 2 | 539.2 | 175.1, 217.1 |
| 2,X-Galactose | 7.89 | 2 | 539.2 | 175.1, 217.1 |
| 4-P-Xylose* | 8.05 | 2 | 509.2 | 175.1, 231.2 |
| 5-F-Arabinose* | 8.18 | 2 | 509.2 | 175.1, 231.2 |
| 3,6-Mannose* | 8.20 | 2 | 539.2 | 175.1, 231.2 |
| X,X-Galactose | 8.20 | 2 | 539.2 | 175.1, 231.2 |
| 2,X-Fucose | 8.25 | 1 | 509.2 | 175.1, 217.1 |
| 2-F-Arabinose | 8.32 | 2 | 509.2 | 175.1, 217.1 |
| 3-P-Xylose | 8.34 | 2 | 509.2 | 175.1, 231.2 |
| 2-P-Xylose | 8.52 | 2 | 509.2 | 175.1, 217.1 |
| 2,X-Glucose | 8.68 | 2 | 539.2 | 175.1, 217.1 |
| 2-F-Ribose | 8.70 | 2 | 509.2 | 175.1, 217.1 |
| 2,X-Galactose | 8.79 | 2 | 539.2 | 175.1, 217.1 |
| X,X-Glucose | 8.92 | 2 | 539.2 | 175.1, 231.2 |
| 2,X-Fucose | 9.00 | 1 | 509.2 | 175.1, 217.1 |
| X-F-Arabinose | 9.10 | 2 | 509.2 | 175.1, 231.2 |
| 3,4-Rhamnose | 9.10 | 1 | 509.2 | 175.1, 231.2 |
| 6-Glucose* | 9.14 | 3 | 553.3 | 175.1, 231.2 |
| X-F-Ribose | 9.25 | 2 | 509.2 | 175.1, 231.2 |
| 6-Galactose* | 9.38 | 3 | 553.3 | 175.1, 231.2 |
| X-Fucose | 9.48 | 2 | 523.2 | 175.1, 231.2 |
| X,X-Mannose | 9.85 | 2 | 539.2 | 175.1, 231.2 |
| X,X-Mannose | 10.02 | 2 | 539.2 | 175.1, 231.2 |
| 6-Mannose* | 10.10 | 3 | 553.3 | 175.1, 231.2 |
| 4-Galactose* | 10.26 | 3 | 553.3 | 175.1, 231.2 |
| 2-Fucose | 10.36 | 2 | 523.2 | 175.1, 217.1 |
| X-F-Ribose | 10.42 | 2 | 509.2 | 175.1, 231.2 |
| X-F-Arabinose | 10.65 | 2 | 509.2 | 175.1, 231.2 |
| 4-Glucose* | 11.20 | 3 | 553.3 | 175.1, 231.2 |
| T-P-Xylose* | 11.25 | 3 | 523.2 | 175.1, 231.2 |
| 2-Galactose* | 11.28 | 3 | 553.3 | 175.1, 217.1 |
| X-Fucose | 11.50 | 2 | 523.2 | 175.1, 231.2 |
| 3-Galactose* | 11.81 | 3 | 553.3 | 175.1, 231.2 |
| X-Rhamnose | 12.35 | 2 | 523.2 | 175.1, 231.2 |
| 3-Glucose* | 12.45 | 3 | 553.3 | 175.1, 231.2 |
| X-Rhamnose | 13.80 | 2 | 523.2 | 175.1, 231.2 |
| 2-Glucose* | 13.80 | 3 | 553.3 | 175.1, 217.1 |
| T-F-Arabinose* | 13.92 | 3 | 523.2 | 175.1, 231.2 |
| 3-Mannose* | 14.70 | 3 | 553.3 | 175.1, 231.2 |
| 4-Mannose* | 15.10 | 3 | 553.3 | 175.1, 231.2 |
| T-Fucose* | 16.55 | 3 | 537.2 | 175.1, 231.2 |
| T-Rhamnose | 16.55 | 3 | 537.2 | 175.1, 231.2 |
| T-Galactose* | 17.84 | 4 | 567.6 | 175.1, 231.2 |
| T-F-Ribose | 18.40 | 3 | 523.2 | 175.1, 231.2 |
| T-Mannose* | 19.00 | 4 | 567.6 | 175.1, 231.2 |
| T-Galacturonic Acid | 19.10 | 3 | 581.2 | 175.1, 231.2 |
| T-Glucose* | 19.50 | 4 | 567.6 | 175.1, 231.2 |
| T-Glucuronic Acid | 23.10 | 3 | 581.2 | 175.1, 231.2 |
Validated with a commercial disaccharide, oligosaccharide, or polysaccharide standard
The retention time, DoPe value, and corresponding MRM transitions are used to identify each linkage.17 For MRM, the precursor ion mass is equal to the sum of the native monosaccharide mass, the mass of a methyl group multiplied by the DoPe value, the mass of two PMP molecules, and a proton ([M + 14*DoPe value + 330 + H]+). The DoPe value provides the number of methyl groups associated with each monosaccharide class, gives an indication of the linkages, and can be used to calculate the precursor ion mass. For example, terminal hexoses are represented by a DoPe value of 4 which corresponds to a precursor ion mass of m/z 567.6. Linear, bisecting, and trisecting hexoses are represented by DoPe values of 3, 2, or 1, respectively, which correspond to a precursor ion mass of m/z 553.3, m/z 539.2, or m/z 525.2, respectively. However, terminal pentoses, deoxyhexoses, and hexuronic acids are represented by a DoPe value of 3, thus, their respective precursor ion masses are m/z 523.2, m/z 537.2, or m/z 581.2, respectively. Linear pentoses, deoxyhexoses, and hexuronic acids are represented by a DoPe value of 2 which corresponds to a precursor ion mass of m/z 509.2, m/z 523.2, or m/z 567.6, respectively. Bisecting species of pentoses, deoxyhexoses, and hexuronic acids are represented by a DoPe value of 1 and correspond to a precursor ion mass of m/z 495.2, m/z 509.2, or m/z 553.3, respectively. Due to their inherent structures, pentoses, deoxyhexoses, and hexuronic acids cannot possess a trisecting linkage.
The precursor ion is fragmented by CID to yield a fragment ion common to all, m/z 175.1, and is used as the quantifying ion. Some monosaccharides yield unique fragments that readily identify the linkage. For example, a 2-linked hexose (glucose, galactose, mannose) yield unique fragments corresponding to both m/z 217.1 and m/z 175.1. However, structures lacking linkage at the 2-position such as a 3-, 4-, or 6-linked hexose yield unique fragments corresponding to both m/z 231.2 and m/z 175.1.
The synthesis produced a stochastic set of partially methylated species whose identity was assigned using a combination of known standards and process of deduction. The possible combinations for each linkage species (terminal, linear, bisecting, & trisecting) must first be understood. For example, for hexoses such as glucose, galactose, and mannose, there are three possible combinations for trisecting species: (1→2,3,6)-, (1→2,4,6)-, or (1→3,4,6)-positions. Bisecting hexose species have six possible combinations, which include linkages at the (1→2,3)-, (1→2,4)-, (1→2,6)-, (1→3,4)-, (1→3,6)-, or (1→4,6)-positions. Linear hexose species can exist in four possible combinations with linkages at the (1→2)-, (1→3)-, (1→4)-, or (1→6)-positions. The terminal position yields a single product and was readily identified.
Deoxyhexoses such as fucose and rhamnose lack trisecting species, however, they have three possible combinations for bisecting species with linkages at the (1→2,3)-, (1→2,4)-, or (1→3,4)-positions. Linear deoxyhexose species possess three possible linkage combinations with linkages at the (1→2)-, (1→3)-, or (1→4)-positions. With the addition of a terminal deoxyhexose species, a total of seven linkage combinations are possible for a single deoxyhexose monosaccharide. Pentoses such as xylose, which exists typically in the pyranose form in plant carbohydrates,31 share the same linkage combinations for bisecting, linear and terminal species.
Pentoses in the furanose form, such as arabinose and ribose, also lack a trisecting species. However, they have three possible combinations for bisecting species with linkages at the (1→2,3)-, (1→2,5)-, or (1→3,5)-positions. The linear species of pentoses in the furanose form can have three possible combinations with linkages at the (1→2)-, (1→3)-, or (1→5)-positions. Seven possible linkage combinations, including the terminal species, are possible for pentoses existing in the furanose form. For the remainder of this article, the short-hand notation described in previous work for the linkages was used.17 For example, the notation 2,3,6-glucose is used to represent glucose linked at the (1→2,3,6)-positions while 4-P-xylose is used to represent xylose in the pyranose form linked at the (1→4)-positions.
To improve enrichment of partially methylated structures during liquid-liquid extraction, general conditions for complete deacetylation and acetylation were first optimized. After partial methylation, remaining hydroxyls are acetylated to maximize partitioning of the compounds into the organic layer during liquid-liquid extraction. The acetyl groups are then removed by acid hydrolysis to obtain the desired partially methylated structures. α-D-Glucose pentaacetate and D-glucose were used as models for fully acetylated and minimally methylated monosaccharides, respectively. After each condition, the monosaccharides were derivatized with PMP and their corresponding absolute ion peak area abundances were compared. Deacetylation was optimized by hydrolysis of α-D-glucose pentaacetate with TFA at varying timepoints (Figure S-1A). Next, acetylation was optimized with D-glucose at varying time points followed by complete deacetylation after enrichment by liquid-liquid extraction (Figure S-1B). Complete deacetylation and acetylation were observed at 30 and 20 minutes, respectively, and were applied to construct the library.
To ensure the generation of all linkage types possible for each monosaccharide, partial methylation was separately performed using three dimethyl sulfonyl anion concentrations spanning three orders of magnitude. Validation of the partial methylation workflow was demonstrated by analysis of linear and terminal hexose residues produced by partial methylation of glucose and by comparison with linkage residues derived from sophorose, nigerose, lactose, and isomaltotriose. MRM transitions developed in previous work were used to monitor linear hexose species (m/z 553.3 → m/z 175.1, m/z 217.1, m/z 231.2) and terminal hexose species (m/z 567.6 → m/z 175.1, m/z 231.2).17 Figure 1A-E displays MRM chromatograms for linear and terminal glucose residues obtained by analysis of sophorose, nigerose, lactose, isomaltotriose, and glucose linkages generated by the partial methylation workflow, respectively. As shown by Figure 1E, partially methylated glucose residues corresponding to 6-, 4-, 3-, 2-, and T-glucose were produced and observed at 9.14, 11.20,12.45, 13.80, and 19.50 minutes, respectively. These signals match the retention times and transitions observed by analysis of the standards (Figure 1A-D) thereby validating the utility of the partial methylation method. Additional signals for 4-mannose and T-mannose were observed for the lactose sample (Figure 1C) due to contamination of a mannose-containing disaccharide or oligosaccharide present in the stock reagent bottle. The contaminants were identified and confirmed by analysis of 1,4-b-D-mannotriose.
Figure 1.

MRM chromatograms illustrating linkage linear and terminal residues obtained from (A) sophorose, (B) nigerose, (C) lactose, (D) isomaltotriose, and (E) glucose linkage standards generated using the partial methylation workflow. The solid black trace represents transitions including the quantifying ion m/z 175.1, while the dashed blue and red traces represent transitions include qualifying ions m/z 231.2 and m/z 217.1, respectively. Additional 4-mannose and T-mannose peaks were observed for lactose and are contaminants present in the stock reagent bottle. The contaminants were assigned by analysis of 1,4-b-D-mannotriose standard.
Additional peaks that were identified as linkage residues using both quantifying and qualifying transitions, but were not assignable using standards, were tentatively assigned into the linkage library for later evaluation. Examples include bisecting species obtained by partial methylation of arabinose, xylose, and ribose (Figure S-2). It was expected that three bisecting species each for arabinose, xylose, and ribose would be observed, respectively, when monitoring the transitions m/z 495.2 → m/z 175.1, m/z 217.1, m/z 231.2. Because there can only be linkages at the 3,5-positions for arabinose and ribose in the furanose form and 3,4-positions for xylose in the pyranose form, peaks corresponding to transitions of m/z 495.2 → m/z 175.1, m/z 231.2 were observed at retention times 6.78, 6.82, and 7.45 minutes for arabinose, xylose, and ribose, respectively. Additional bisecting species identified at retention times 6.55 minutes and 6.92 minutes for arabinose, 6.02 minutes and 7.00 minutes for xylose, and 3.88 minutes and 5.68 minutes for ribose, were observed with transitions of m/z 495.2 → m/z 175.1, m/z 217.1 indicating linkage at the 2-position. Because the two possible linkage combinations for the trisecting species containing a linkage at the 2-position are not distinguishable, these bisecting species were tentatively assigned as “2,X-” followed by the corresponding monosaccharide.
Linkage analysis of polysaccharides
The ability to perform linkage analysis on both soluble and insoluble branched polysaccharides is necessary to accurately characterize samples in complicated matrices such as whole food. Method validation and reproducibility were evaluated using xyloglucan (tamarind), a branched hemicellulose polysaccharide with low solubility. Using retention time information and transitions corresponding to the linkage residues listed in Table 1, the quadrupoles were operated in dMRM mode to improve signal intensities while lowering the duty cycle. A representative dMRM chromatogram for xyloglucan is shown in Figure 2A. The results obtained from the analysis qualitatively match the theoretical structure of xyloglucan.32 A total of 20 linkage residues were identified and a list containing linkages with relative peak area abundances ≥0.1% are shown in Table 2. Standard deviation values of ≤0.8% were determined for each linkage residue, indicating robustness and reproducibility of the method for analysis of branched insoluble polysaccharides. Abundant signals for T-P-xylose, 2-P-xylose, and T-galactose were expected and observed with relative abundances of 45.8 ± 0.6%, 26.8 ± 0.5%, and 12.6 ± 0.1%, respectively. A signal corresponding to the co-eluting T-F-arabinose/X-rhamnose peak with a relative abundance of 4.9 ± 0.3% was additionally observed. Based on a previous monosaccharide analysis, the composition of T-F-arabinose/X-rhamnose is solely due to the presence of T-F-arabinose monosaccharide rather than X-rhamnose.33 Representing the backbone and branched points, 4-glucose and 4,6-glucose were observed with relative abundances of 3.5 ± 0.8% and 4.6 ± 0.4%, respectively.
Figure 2.
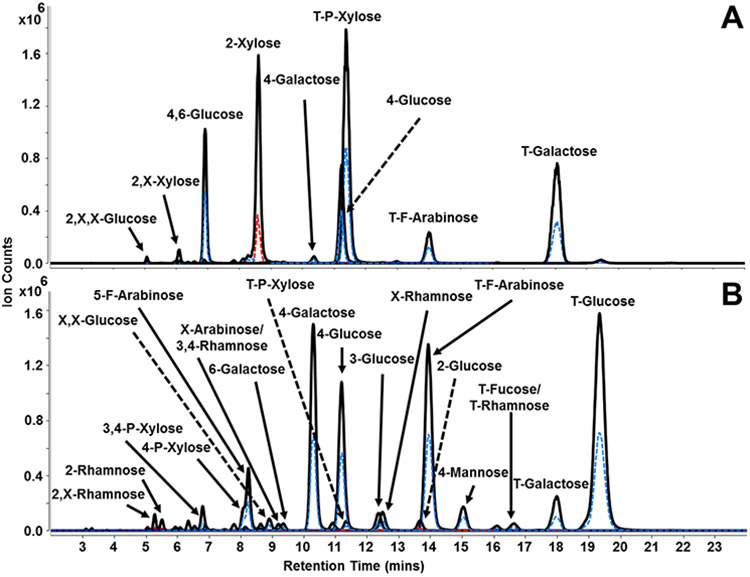
Representative dynamic MRM chromatograms for the linkage analysis of (A) xyloglucan and (B) whole carrot root. The solid black trace represents transitions with the quantifying ion m/z 175.1, while the dashed blue and red traces represent transitions of qualifying ions m/z 231.2 and m/z 217.1, respectively.
Table 2.
Relative peak area linkage composition of xyloglucan (tamarind) (n=3).
| Linkage Residue | RT (mins) |
Relative Abundance (%) |
|---|---|---|
| T-P-Xylose | 11.25 | 45.8 ± 0.6 |
| 2-P-Xylose | 8.52 | 26.8 ± 0.5 |
| T-Galactose | 17.84 | 12.6 ± 0.1 |
| T-F-Arabinose | 13.92 | 4.9 ± 0.3 |
| 4,6-Glucose | 6.98 | 4.6 ± 0.4 |
| 4-Glucose | 11.2 | 3.5 ± 0.8 |
| 2,X-P-Xylose | 6.02 | 0.4 ± 0.1 |
| 4-Galactose | 10.26 | 0.3 ± 0.0 |
| 3,4-P-Xylose | 6.82 | 0.1 ± 0.0 |
| 2,X,X-Glucose | 5.12 | 0.1 ± 0.0 |
| 2,X,X-Galactose | 6.41 | 0.1 ± 0.0 |
| X,X-Glucose | 8.92 | 0.1 ± 0.0 |
| 2,X-F-Arabinose | 6.55 | 0.1 ± 0.0 |
| 2,X-Mannose | 5.15 | 0.1 ± 0.0 |
| 2,X-Glucose/2,X-Galactose | 7.85 | 0.1 ± 0.0 |
| 2,X-Glucose | 8.68 | 0.1 ± 0.0 |
The overall method was used to analyze carbohydrates in a complicated matrix such as whole carrot root. In a previous GC-MS study, a total of 13 linkages were determined.17 In that study, we also described a more limited LC-MS/MS workflow which identified 24 linkages with relative abundances of ≥0.1%. The presented workflow in this study is an improvement over the previous methods facilitated by improved LC-MS/MS conditions and the use of a synthetic-based library. Figure 2B displays a representative annotated dMRM chromatogram for whole carrot root with over 39 linkages having relative abundances of ≥0.1% (Table 3). The relative standard deviations were determined to be ≤3.9%, demonstrating reproducibility of the method for analysis of linkage residues derived from disaccharides, oligosaccharides, and polysaccharides existing in complicated matrices. Slightly lower relative T-glucose (28.1 ± 2.4%) and 4-glucose (9.4 ± 2.0%) abundances were observed and expected in comparison to the more limited LC-MS/MS study and is a result of the increase in the number of linkages that can be monitored.17
Table 3.
Relative peak area linkage composition of whole carrot root (n=3).
| Linkage Residue | RT (mins) |
Relative Abundance (%) |
|---|---|---|
| T-F-Arabinose | 13.92 | 28.4 ± 3.9 |
| T-Glucose | 19.50 | 28.1 ± 2.4 |
| 4-Galactose | 10.26 | 12.5 ± 0.8 |
| 4-Glucose | 11.20 | 9.4 ± 2.0 |
| 5-F-Arabinose | 8.18 | 5.6 ± 0.1 |
| T-Galactose | 17.84 | 3.0 ± 0.1 |
| 4-Mannose | 15.10 | 1.8 ± 0.2 |
| 4-P-Xylose | 8.05 | 1.5 ± 0.8 |
| 3-Glucose | 12.45 | 1.4 ± 0.3 |
| 3,4-P-Xylose | 6.82 | 1.0 ± 0.1 |
| T-P-Xylose | 11.25 | 0.9 ± 0.1 |
| T-Fucose/T-Rhamnose | 16.55 | 0.7 ± 0.1 |
| X,X-Glucose | 8.92 | 0.6 ± 0.0 |
| 2-Glucose | 13.8 | 0.6 ± 0.0 |
| X-F-Arabinose/3,4-Rhamnose | 9.10 | 0.6 ± 0.1 |
| 2,X-Rhamnose | 5.24 | 0.4 ± 0.1 |
| 6-Galactose | 9.38 | 0.4 ± 0.0 |
| 2-Rhamnose | 5.46 | 0.4 ± 0.1 |
| 2,X-Glucose | 8.68 | 0.3 ± 0.0 |
| 2,X-Glucose | 7.85 | 0.2 ± 0.1 |
| 3.4.6-Galactose | 6.40 | 0.2 ± 0.1 |
| 2,X,X-Glucose | 6.41 | 0.2 ± 0.1 |
| 4,6-Glucose | 6.98 | 0.2 ± 0.0 |
| 2,X-F-Arabinose | 6.55 | 0.2 ± 0.0 |
| X,X-Galactose/X,X-Mannose | 8.20 | 0.1 ± 0.0 |
| 2-Xylose | 8.52 | 0.1 ± 0.2 |
| 6-Glucose | 9.14 | 0.1 ± 0.0 |
| 2,X,X-Galactose | 6.15 | 0.1 ± 0.0 |
| 3,4-Fucose | 7.44 | 0.1 ± 0.0 |
| 3,4,6-Glucose | 6.00 | 0.1 ± 0.1 |
| 2,X,X-Glucose | 5.12 | 0.1 ± 0.1 |
The linkage composition further pointed to the presence of polysaccharides other than starch and cellulose, both of which are known to be significant carbohydrate components in carrots.34 For example, the identification of T-F-arabinose (28.4 ± 3.9%), 4-galactose (12.5 ± 0.8%), and T-galactose (3.0 ±0.1%) suggested the presence of type I arabinogalactans, which typically possess a β(1→4)-galactose backbone with T-F-arabinose capping, rather than type II arabinogalactans, which consist of repeating β(1→3)-galactose monosaccharides.35 Other pentoses such as 4-P-xylose (1.5 ± 0.8%), 3,4-P-xylose (1.0 ± 0.1%), and 5-F-arabinose (5.6 ± 0.1%) were also identified and suggested the additional presence of xylans and arabinans.36, 37
Quantitative information was obtained using this method, however, there are certain caveats that should be considered in the LC-MS platform which are also common for GC-MS. Although the PMP labels produced equal ionization between the different linkage representations of a given species, changes in the effluent compositions and/or the simultaneous elution of two or more species could have yielded competitive conditions where the signals of the different species were attenuated relative to the single eluting species. Because of the vast number of linkage possibilities, it is difficult to achieve complete isomeric separation. For example, there were 3 pairs of co-eluting isomeric compounds unable to be chromatographically resolved by the method: 3,6-mannose/X,X-galactose (8.20 mins), X-F-arabinose/3,4-rhamnose (9.10 mins), and T-fucose/T-rhamnose (16.55 mins). However, a complimentary monosaccharide analysis may provide additional information to accurately identify the correct associated monosaccharide. Additionally, the permethylation reaction could be affected by the availability of the polymer to the reaction. Similarly, acid hydrolysis could be affected by the secondary and tertiary structures of the polymer in the matrix. Lastly, in-depth characterization of individual carbohydrate structures existing in a complicated mixture would require isolation of the components prior to application of this analytical method.
CONCLUSIONS
In this study, a synthetic-based linkage library in addition to an improved UHPLC/QqQ MS method employing dynamic multiple reaction monitoring was developed for the accurate simultaneous profiling of 92 glycosidic linkages derived from disaccharides, oligosaccharides, and polysaccharides existing in complicated mixtures. While an extended analysis time is needed to simultaneously and accurately profile 92 linkages, the presented method provides a more substantially improved comprehensive analysis over the previous LC-MS/MS and GC-MS methods.17 Optimized partial methylation conditions were used to synthesize trisecting, bisecting, linear, and terminal species for hexoses, pentoses, deoxyhexoses, and hexuronic acids using commercial monosaccharides as building blocks. The synthesized linkages were validated by deductive reasoning through analysis of residues derived from commercial disaccharide, oligosaccharide, and polysaccharide standards. Additional signals positively identified as synthesized glycosidic linkages were tentatively assigned for later evaluation pending the availability of commercial standards. The method’s ability to accurately characterize a highly branched polysaccharide was validated by comparison to the known structure. Further validation for the ability to analyze polysaccharides within a complicated matrix was performed by comparison with our previous rapid linkage analysis method and GC-MS. Glycosidic linkages derived from fructose, N-acetylglucosamine, and N-acetylgalactosamine require a different synthetic approach, and methods for the creation of standards derived from these monosaccharides are currently being developed and will be the topic of future reports. The combined library and UHPLC/QqQ MS method developed in this work will allow for the accurate profiling and relative quantitation of linkages derived from disaccharides, oligosaccharides, and polysaccharides present in complicated matrices. Additionally, the concept of this work may provide as an orthogonal approach for the linkage profiling of N- and O- glycosylation, an analysis typically reliant on the availability of enzymes, and will be a topic of future reports.
Supplementary Material
Funding Sources
Funding provided by the Mars Corp (MDCF001) is gratefully acknowledged.
Footnotes
Supporting Information.
Supporting figures include the effect of hydrolysis time on the deacetylation of α-D-glucose pentaacetate and the effect of acetylation time for 50 μg of D-glucose (Figure S-1) along with MRM chromatograms for bisecting species obtained by the partial methylation and PMP-labeling of arabinose, xylose, and ribose monosaccharides (Figure S-2).
REFERENCES
- 1.Dwek RA, Glycobiology: Toward Understanding the Function of Sugars. Chemical Reviews 1996, 96 (2), 683–720. [DOI] [PubMed] [Google Scholar]
- 2.Kailemia MJ; Park D; Lebrilla CB, Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem 2017, 409 (2), 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moremen KW; Tiemeyer M; Nairn AV, Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012, 13 (7), 448–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lovegrove A; Edwards CH; De Noni I; Patel H; El SN; Grassby T; Zielke C; Ulmius M; Nilsson L; Butterworth PJ; Ellis PR; Shewry PR, Role of polysaccharides in food, digestion, and health. Crit Rev Food Sci Nutr 2017, 57 (2), 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benbettaïeb N; Gay J-P; Karbowiak T; Debeaufort F, Tuning the Functional Properties of Polysaccharide-Protein Bio-Based Edible Films by Chemical, Enzymatic, and Physical Cross-Linking. Comprehensive Reviews in Food Science and Food Safety 2016, 15 (4), 739–752. [DOI] [PubMed] [Google Scholar]
- 6.Karaki N; Aljawish A; Humeau C; Muniglia L; Jasniewski J, Enzymatic modification of polysaccharides: Mechanisms, properties, and potential applications: A review. Enzyme Microb Technol 2016, 90, 1–18. [DOI] [PubMed] [Google Scholar]
- 7.Nieto B, M., Gum Polysaccharides as Biopolymers - Structure, Properties and Applications. Journal of Chemical Engineering & Process Technology 2018, 09. [Google Scholar]
- 8.Tester RF; Qi X; Karkalas J, Hydrolysis of native starches with amylases. Animal Feed Science and Technology 2006, 130 (1-2), 39–54. [Google Scholar]
- 9.Butterworth PJ; Warren FJ; Ellis PR, Human α-amylase and starch digestion: An interesting marriage. Starch - Stärke 2011, 63 (7), 395–405. [Google Scholar]
- 10.Singh J; Dartois A; Kaur L, Starch digestibility in food matrix: a review. Trends in Food Science & Technology 2010, 21 (4), 168–180. [Google Scholar]
- 11.Hakomori S, A Rapid Permethylation of Glycolipid, and Polysaccharide Catalyzed by Methylsulfinyl Carbanion in Dimethyl Sulfoxide. J Biochem 1964, 55, 205–8. [PubMed] [Google Scholar]
- 12.Waeghe TJ; Darvill AG; McNeil M; Albersheim P, Determination, by methylation analysis, of the glycosyl-linkage compositions of microgram quantities of complex carbohydrates. Carbohydrate Research 1983, 123 (2), 281–304. [Google Scholar]
- 13.Sims IM; Carnachan SM; Bell TJ; Hinkley SFR, Methylation analysis of polysaccharides: Technical advice. Carbohydr Polym 2018, 188, 1–7. [DOI] [PubMed] [Google Scholar]
- 14.Lim SJ; Wan Aida WM; Schiehser S; Rosenau T; Böhmdorfer S, Structural elucidation of fucoidan from Cladosiphon okamuranus (Okinawa mozuku). Food Chemistry 2019, 272, 222–226. [DOI] [PubMed] [Google Scholar]
- 15.Huang W; Deng H; Jin S; Ma X; Zha K; Xie M, The isolation, structural characterization and anti-osteosarcoma activity of a water soluble polysaccharide from Agrimonia pilosa. Carbohydr Polym 2018, 187, 19–25. [DOI] [PubMed] [Google Scholar]
- 16.Dong CX; Liu L; Wang CY; Fu Z; Zhang Y; Hou X; Peng C; Ran RX; Yao Z, Structural characterization of polysaccharides from Saposhnikovia divaricata and their antagonistic effects against the immunosuppression by the culture supernatants of melanoma cells on RAW264.7 macrophages. Int J Biol Macromol 2018, 113, 748–756. [DOI] [PubMed] [Google Scholar]
- 17.Galermo AG; Nandita E; Barboza M; Amicucci MJ; Vo TT; Lebrilla CB, Liquid Chromatography-Tandem Mass Spectrometry Approach for Determining Glycosidic Linkages. Anal Chem 2018, 90 (21), 13073–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doares SH; Albersheim P; Darvill AG, An Improved Method for the Preparation of Standards for Glycosyl-Linkage Analysis of Complex Carbohydrates. Carbohydrate Research 1991, 210, 311–317. [Google Scholar]
- 19.Heiss C; Wang Z; Azadi P, Sodium hydroxide permethylation of heparin disaccharides. Rapid Commun Mass Spectrom 2011, 25 (6), 774–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biermann CJ; McGinnis GD, Analysis of carbohydrates by GLC and MS. CRC Press: Boca Raton, Fla., 1989. [Google Scholar]
- 21.Dell A, Preparation and desorption mass spectrometry of permethyl and peracetyl derivatives of oligosaccharides. Methods Enzymol 1990, 193, 647–60. [DOI] [PubMed] [Google Scholar]
- 22.Ruhaak LR; Zauner G; Huhn C; Bruggink C; Deelder AM; Wuhrer M, Glycan labeling strategies and their use in identification and quantification. Anal Bioanal Chem 2010, 397 (8), 3457–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geyer R; Geyer H, Saccharide Linkage Analysis Using Methylation and Other Techniques. Methods Enzymol 1994, 230, 86–108. [DOI] [PubMed] [Google Scholar]
- 24.Connors KA; Pandit NK, N-Methylimidazole as a catalyst for analytical acetylations of hydroxy compounds. Analytical Chemistry 1978, 50 (11), 1542–1545. [Google Scholar]
- 25.Pandit NK; Connors KA, Kinetics and Mechanism of Hydroxy Group Acetylations Catalyzed by N-Methylimidazole. Journal of Pharmaceutical Sciences 1982, 71 (5), 485–491. [DOI] [PubMed] [Google Scholar]
- 26.Ciucanu I; Kerek F, A simple and rapid method for the permethylation of carbohydrates. Carbohydrate Research 1984, 131 (2), 209–217. [Google Scholar]
- 27.Price NP, Permethylation linkage analysis techniques for residual carbohydrates. Appl Biochem Biotechnol 2008, 148 (1-3), 271–6. [DOI] [PubMed] [Google Scholar]
- 28.Corey EJ; Chaykovsky M, Dimethylsulfoxonium Methylide. Journal of the American Chemical Society 1962, 84 (5), 867–868. [Google Scholar]
- 29.Corey EJ; Chaykovsky M, Methylsulfinyl Carbanion (CH3-SO-CH2-). Formation and Applications to Organic Synthesis. Journal of the American Chemical Society 1965, 87 (6), 1345–1353. [Google Scholar]
- 30.Xu G; Amicucci MJ; Cheng Z; Galermo AG; Lebrilla CB, Revisiting monosaccharide analysis - quantitation of a comprehensive set of monosaccharides using dynamic multiple reaction monitoring. Analyst 2017, 143 (1), 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorsheim K; Siegbahn A; Johnsson RE; Stalbrand H; Manner S; Widmalm G; Ellervik U, Chemistry of xylopyranosides. Carbohydr Res 2015, 418, 65–88. [DOI] [PubMed] [Google Scholar]
- 32.Fry SC, The Structure and Functions of Xyloglucan. Journal of Experimental Botany 1989, 40 (1), 1–11. [Google Scholar]
- 33.Amicucci MJ; Galermo AG; Nandita E; Vo T-TT; Liu Y; Lee M; Xu G; Lebrilla CB, A rapid-throughput adaptable method for determining the monosaccharide composition of polysaccharides. International Journal of Mass Spectrometry 2019, 438, 22–28. [Google Scholar]
- 34.Theander O; Åman P, Studies on dietary fibre. A method for the analysis and chemical characterisation of total dietary fibre. Journal of the Science of Food and Agriculture 1982, 33 (4), 340–344. [Google Scholar]
- 35.Serpe MD; Nothnagel EA, Arabinogalactan-Proteins in the Multiple Domains of the Plant Cell Surface. Adv Bot Res 1999, 30, 207–289. [Google Scholar]
- 36.Bastawde KB, Xylan structure, microbial xylanases, and their mode of action. World J Microbiol Biotechnol 1992, 8 (4), 353–68. [DOI] [PubMed] [Google Scholar]
- 37.Kikuchi A; Edashige Y; Ishii T; Fujii T; Satoh S, Variations in the structure of neutral sugar chains in the pectic polysaccharides of morphologically different carrot calli and correlations with the size of cell clusters. Planta 2017, 198 (4), 634–639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.