

Abstract
The unregulated activation of nuclear factor-κB (NF-κB) is a critical event in the progression of various inflammatory diseases such as ulcerative colitis, asthma, rheumatoid arthritis, bacterial induced gastritis, etc. Hence, blocking the transcriptional activity of NF-κB is a promising strategy towards the development of an anti-inflammatory agent. In this study, an integrated molecular and quantum mechanical approach was carried out to find a new potent andrographolide (AGP)-based analog that can inhibit DNA binding to NF-κB p50 and manifest anti-inflammatory activity. Our approach includes multiple sequence alignment, virtual screening, molecular docking (protein–ligand and protein-DNA), in silico site-directed mutagenesis, ADMET prediction, DFT (HOMO, LUMO, HLG, and EPM energy) analysis, MD simulation, and MM/GBSA rescoring. The virtual screening analysis of 237 AGP analogs yielded the five lead compounds based on the binding affinity. Further, molecular interactive docking and ADMET prediction of hit analogs revealed that Ana2 ((3Z,4S)-3-[2-[(4aR,6aS,7R,10aS,10bR)-3,3,6a,10b-tetramethyl-8-methylidene-1,4a,5,6,7,9,10,10a-octahydronaphtho[2,1-d][1,3]dioxin-7-yl]ethylidene]-4-hydroxyoxolan-2-one) is the most potent moiety as it displays the strongest binding affinity and better molecular/pharmacokinetic features. Moreover, DFT, MD simulation, and MM/GBSA studies corroborated the docking results and demonstrated better chemical and dynamic stability with the least binding free energy (− 29.99 kcal/mol) for the Ana2. Site-directed mutagenesis investigation (Cys62Ala) establishes the importance of the Cys62 amino acid residue towards the binding interaction and stability of Ana2 with NF-κB p50. Overall, the identified NF-κB p50 inhibitor opens up a new research horizon towards the development of plant-based anti-inflammatory drugs to combat progressive inflammatory diseases.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-022-03431-9.
Keywords: NF-κB, Andrographolide, Virtual screening, Docking, DFT, MD simulation
Introduction
Nuclear factor kappa B (NF-κB) is a pleiotropic transcription factor that regulates DNA transcription, cytokine genesis, and cell survival. It is entailed in cellular processes to inflammation, stimuli-like cytokines, and viral or bacterial antigens (Concetti and Wilson 2018). It contains a Rel homology domain (RHD) of 300-residue at its N-terminus and is synthesized as large precursors, p100, and p105 (mature p52 and p50 subunits) (Senftleben et al. 2001). The N-terminus is responsible for the DNA binding which connects to C-terminal (Rel-C) domain by a flexible 10-residue link. The NF-κB is found in dimeric form (homo and hetero of the p50 and p52 subunits) and is sequestered in the cytoplasm by inhibitors of κB (IκBs). The degradation of IκBs by IκB kinase (IKK) results in the activation of the NF-κB which triggers the expression of genes (Jacobs and Harrison 1998). The NF-κB interacts with a 10-base-pair (bp) canonical sequence of nucleotides (5′-GGGRNYYYCC-3′) at the promoter regions of numerous genes that are related to various diseases (Huxford and Ghosh 2009; Mussbacher et al. 2019). Therefore, small molecule inhibitors of NF-κB may impart a main therapeutic approach by interrupting the DNA transcription. To date, various natural compounds have been identified that can interfere with the NF-κB transcription activity (Seo et al. 2018). For example, Kumar et al. (2012) demonstrated the potential of curcumin as a potential inhibitor against NF-κB p50 by employing docking and ADME studies (Kumar and Bora 2012). Espίrito-Santo et al. (2017) reported the anti-inflammatory activity of braylin by reducing the NF-κB-dependent transcriptional activity on RAW 264.7 cells (Espírito-Santo et al. 2017). Tran et al. (2017) investigated the anti-inflammatory activity of labdane diterpenoids by inhibiting NF-κB p50 transcription activity (Tran et al. 2017). Chen et al. (2019) demonstrated the anti-inflammatory activity of some triterpenoids by attenuating the activation of NF-κB in the tetradecanoyl phorbol acetate (TPA)-treated mice model (Chen et al. 2019).
Recently, andrographolide (AGP) has garnered immense interest due to its wide spectrum of therapeutic potential. The AGP moiety has a large number of sp3 hybridized carbons, a polycyclic moiety with aliphatic side chains, rich in oxygen, and low nitrogen content which make it a potential electrophilic covalent natural drug candidate for efficient interaction with various biological proteins (Tran et al. 2020). For instance, Xia and co-workers discovered the NF-κB blocking ability of andrographolide via Cys62 covalent modification of the NF-κB p50 (Xia et al. 2004). The specific active site of the AGP at the NF-κB p50 protein involved a highly conserved sequence (RxxRxR; Arg54–Arg59) close to Cys62 (Wang et al. 2014). AGP might manifest its anti-inflammatory activity by blocking the NF-κB p50 subunit in two ways, (i) by reducing the phosphorylation of IκB, thereby hindering the nuclear transference of the NF-κB p50 and relieving inflammation (Yang et al. 2017), and (ii) by inducing the dephosphorylation of the NF-κB p50 subunit and thus leading to a reduction in inflammation; however, the exact mechanism is still unknown (Ding et al. 2017). Despite extensive efforts, poor pharmacological parameters of AGP have limited their applications towards successful drug development (Chen et al. 2010).
Computer-aided drug design (CADD) is an indispensable tool to discover, develop, and analyze drugs and similar biologically active molecules (Singh et al. 2022a). The drug design techniques expedite the drug discovery process by applying either structure-based (target-based) or ligand-based (analog-based) approaches. The principal stages of CADD include target identification, validation, lead screening, optimization, and pre-clinical assessment (Singh et al. 2022b). CADD has greatly influenced the development of numerous therapeutic drug leads for different diseases (Kumar Bhardwaj and Purohit 2021; Kumar et al. 2021; Singh et al. 2022c). The present in silico work was envisaged to find out a novel natural potent NF-κB p50 transcription inhibitor with enhanced interaction ability and improved pharmacokinetics parameters. At first, the full amino acid sequence of NF-κB p50 protein was examined by multiple sequence alignment (MSA). Then, the hit AGP analogs obtained from virtual screening were investigated further by employing molecular docking, absorption–distribution–metabolism–excretion–toxicity (ADMET) prediction, density-functional-theory (DFT) analysis, molecular-dynamics (MD) simulation, and molecular mechanics with generalized born and surface area solvation (MM/GBSA) studies. Site-directed mutagenesis was also carried out for one selected amino acid important for the binding of DNA, AGP, and hit analog with NF-κB p50. Our study suggests one potential AGP analog that can be developed as a new promising anti-inflammatory drug candidate for inhibition of DNA binding with NF-κB p50.
Materials and methods
Retrieval, preparation, and binding site prediction of protein and DNA
The 3D PDB structure of the NF-κB p50 subunit (PDB ID: 1SVC; resolution 2.60 Å), which is a homodimer coupled to DNA, was obtained from the RCSB PDB database (Müller et al. 1995; Berman et al. 2000). This PDB structure consists of 364 amino acid length, from 2 to 365 amino acids out of 968 amino acid sequence of NF-κB1_HUMAN (Bateman et al. 2021). Amongst the 968 amino acid residues, the domain from 42 to 367 amino acids is called RHD which grips the DNA in the major groove and is responsible for the transcriptional activity [UniProtKB—P19838]. Therefore, this region is a potential site for the binding of small molecules to inhibit DNA transcription. Hence, this PDB structure consisting of RHD was selected for this study to find out potential transcriptional inhibitors against the NF-κB p50 protein. ModRefiner tool was used to clean and improve the protein structure by refining C-alpha traces based on a two-step energy minimization at the atomic level (Xu and Zhang 2011). Thereafter, Chimera molecular visualizer was utilized to visualize the NF-κB p50 structure by removing heteroatoms, and water molecules and by adding hydrogen atoms to the protein structure (Pettersen et al. 2004). Subsequently, binding pockets and active site residues prediction of NF-κB p50 and their visualization were performed by CASTp 3.0, HotSpot Wizard v3.1, and Chimera, respectively (Sumbalova et al. 2018; Tian et al. 2018). The 3D structure of the NF-κB p50 subunit, which was acquired from the PDB database, comprises double-stranded (ds) DNA with 19 base pairs, while, the extracted PDB structure of protein consists of single-stranded (ss) DNA which has the base sequence length 5′-AGATGGGGAATCCCCTAGA-3′. Computationally, ssDNA was retrieved from the NF-κBp50-DNA complex structure by Chimera and visualized in Chimera as well as Swiss-PdbViewer (Guex and Peitsch 1997).
Collections and preparation of andrographolide analogs
The chemical structure of AGP and its 237 analogs were retrieved from the PubChem database (Kim et al. 2021). OpenBabel was used, wherever needed, to convert the file formats (e.g..pdb,.pdbqt,.sdf, and smiles) of the analogs for virtual screening, molecular docking, ADMET, and MD simulation studies (O’Boyle et al. 2011).
Multiple sequence alignment
The positions of conserved amino acid residues in the NF-κB p50 subunit were determined by multiple sequence alignment analysis. To perform MSA, the amino acid sequences of the NF-κB p50 subunit of different eleven mammal species (Homo sapiens, Rattus norvegicus, Mus musculus, Bos Taurus, Equus caballus, Sus scrofa, Mesocricetus auratus, Ovis aries, Pan troglodytes, Oryctolagus cuniculus, and Cavia porcellus) were taken from UniProt database in the.fasta format. PRALINE web server was used to perform MSA of NF-κB p50 subunit of different species (Simossis and Heringa 2005). The amino acid residues were shaded in MSA based on the conservation scoring scheme that works from conservation index 0 (the least conserved alignment position) to 10 (the most conserved alignment position). As there are few or no mutations in the conserved amino acid sites in the protein, these sites are thought to be structurally or functionally important for biological action. Furthermore, these conserved amino acids may also have significance to find out residues involved in the binding of the ligand with protein (Capra and Singh 2007).
Virtual screening
Molecular docking is commonly used for virtual screening to systematically search the drug lead for a specific known protein structure (Bhardwaj and Purohit 2021). Docking-based virtual screening was performed for 237 AGP analogs against the NF-κB p50 protein to find out the hit AGP analogs. PyRx (AutoDock Vina) was used to screen the AGP analogs (Dallakyan and Olson 2015). First, AGP analogs were converted into AutoDock Vina input file format (.pdbqt) using Open Babel. The amino acid residues Arg57, Arg59, Tyr60, Cys62, Glu63, His67, His144, Lys147, Lys244, Ala245, Pro246, Asn247, Lys275, Gln277, Arg308, and Gln309 of the NF-κB p50 protein were used as the binding site for docking based virtual screening. This was followed by AutoDock Vina run by selecting the grid box covering all the binding site residues with search space as center X:46.1169, Y:20.9609, Z:42.1540, and dimensions (Å) x:46.8342, y:39.9117, z:34.5820 with 8 exhaustiveness. The binding energy (BE) cutoff value was set to < − 7.0 kcal/mol for the screening of AGP analogs.
Protein–ligand docking
AutoDock 4.2 was used for docking analysis to ascertain the selectivity of the AGP and five hit AGP analogs identified via virtual screening towards the inhibition of NF-κB p50 (Morris et al. 2009). The grid was set to ensure to cover all the active site residues (Arg57, Arg59, Tyr60, Cys62, Glu63, His67, His144, Lys147, Lys244, Ala245, Pro246, Lys275, Gln277, Arg308, and Gln309). Further, the grid box dimensions used along the x, y, and z-axis were 94 Å, 66 Å, and 76 Å, respectively, with a grid spacing of 0.375 Å and the grid center 32.42, 22.93, and 33.44 at xyz-coordinates. Lamarckian genetic algorithm (GA) was used for ligand conformational search. Other docking parameters were used as: GA population size = 150, maximum number of energy evaluations = 2,500,000, GA run = 10, maximum number of generations = 27,000, and the root mean square (RMS) cluster tolerance = 2.0 Å. Further, the docking analysis in terms of binding energy, total interacting amino acid residues, number and types of both hydrogen bonds (H-bonds), and hydrophobic (HYD) interactions was computed for the best conformation of the molecules. The output files obtained from the docking study were visualized and analyzed by Chimera, Accelrys discovery studio, and Maestro 12.0.
Protein-DNA docking
NF-κB p50 and DNA docking was performed using NPDock (Nucleic acid–Protein Dock) which utilizes poses scoring, cluster of the top-scored models, and refinement of the most potential solutions to provide the best results (Tuszynska et al. 2015). The top-scoring complex of NF-κB p50 and DNA was recovered from NPDock and studied further for interaction analysis to extract structural features from DNA–nucleic acid complexes by DNAproDB (Sagendorf et al. 2017).
Site-directed mutagenesis and molecular docking with the mutated protein
There are seven key mutations (at 62, 678, 903, 907, 921, 923, and 932 residue positions of NF-kB p50) reported for NF-kB p50 protein in the UniProt database and amongst these, the mutation at the 62 residue position in the RHD domain of the protein is of main importance in the DNA binding activity (Heissmeyer et al. 1999; Cernuda-Morollón et al. 2001; Demarchi et al. 2003; Cockman et al. 2006). Therefore, in this study amino acid Cys62 was selected for the mutational analysis. The site-directed mutagenesis technique was used to identify the significance of Cys62 in the formation of a stable complex of NF-κB p50 and AGP analogs. Cys62 amino acid residue of the NF-κB p50 protein structure was mutated to alanine (Ala) residue with help of spdv followed by energy minimization and secondary structure refinement using ModRefiner (Morrison and Weiss 2001). Further, this mutated NF-κB p50 protein structure was used for docking and simulation studies of AGP and selected analogs to assess the importance of the mutated residue in the molecular interactions and protein stability. All the docking parameters were kept the same as followed for the docking studies of the native protein with AGA and selected analogs (Section: Protein–ligand docking).
DFT analysis
Quantum–mechanical (QM) descriptors especially highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and electrostatic potential map (EPM) be of high importance in controlling several chemical reactions and also have a vital role in the pharmacological effects (Zheng et al. 2013). Hit AGP analogs and AGP were submitted for electronic structure calculations using DFT analysis carried out on Spartan 20 (www.Wavefun.com) in order to evaluate QM parameters. First, the chemicals under study were presented for use in the density functional calculation of equilibrium geometric variables in a gas state at the ground level. Later vibrational wavenumbers are calculated by optimizing the geometry of the analogs using the three-parameter exchange potential of Becke (Berman et al. 2015) and Lee Yang Parr correlation functional (B3LYP) (Gill et al. 1992) method with 6-31G* basis set (Stephens et al. 1994). The combination of B3LYP and 6-31G* (B3LYP/6-31G*) constitutes a theoretical model and provides a good account to calculate all types of reaction energies. It is also good for the organic molecules and gives better electronic and spectroscopic results closer to the experimental values. Therefore, the combination of B3LYP and 6-31G* was taken for the calculations of the DFT analysis (Chaudhary et al. 2020). Further, to calculate the DFT, the data related to B3LYP/6-31G* basis set were taken as the number of unpaired electrons (multiplicity) = 0, total charge = neutral coupling, and coupling constant = empirical. Thereafter, analogs’ chemical surfaces were observed to evaluate the HOMO, LUMO, and EPM. Electron-rich outermost orbitals that can donate electrons to the paired protein are referred to as HOMO, moreover, the lowest energy innermost orbitals which receive electrons from paired protein are defined as LUMO. Further, HOMO, LUMO, HOMO–LUMO gap (HLG), and Electrostatic Potential Energy (EPE) were computed for examining the kinetic stability and chemical reactivity of the AGP and selected AGP analogs (Zhan et al. 2003).
ADMET parameters assessment
In silico prediction of pharmacological properties and evaluation of drug-like features of AGP and the hit AGP analogs was performed with admetSAR, SwissADME, ProTox-II, and ADMETlab 2 (Cheng et al. 2012; Daina et al. 2017; Banerjee et al. 2018; Xiong et al. 2021). Computational analyses were performed to anticipate the pharmacokinetic parameters, i.e. bioavailability, distribution volume, plasma protein binding, elimination, and toxicity. Further, the drug-like potential of hit AGP analogs was assessed according to Lipinski’s rule of five (Lipinski 2004).
MD simulation study
MD simulation is an important study to produce more reliable interactive binding affinity results and to validate the docking results (Sabe et al. 2021; Bhardwaj et al. 2022; Kumar et al. 2022). MD simulation was executed to evaluate the protein stability, conformation, and interaction modes between the AGP/hit AGP analogs and NF-κB p50 [both native and mutated (Cys62Ala)] protein complex. The simulation studies were performed for NF-κB protein (both mutated and non-mutated) alone (control) and with its complexes with AGP and its hit analogs using GROMACS 2019.2 (Abraham et al. 2015). The topology for the AGP and AGP analogs was computed from PRODRG server (Schüttelkopf and Van Aalten 2004) in the framework of GROMOS96 54a7 force field (Schmid et al. 2011). The complex structure was solvated by a simple point charge (SPC) water model in a dodecahedron box type and neutralized by adding 0.15 M NaCl salt. Thereafter, the steepest descent algorithm was used to minimize the system for 50,000 steps. Then, simulations were carried out by NVT/NPT equilibration type using the constant temperature (300 K) and pressure (1.0 bar) with a leap-frog MD integrator. Finally, the MD simulation was run for 100 ns with 5000 frames per simulation. The MD simulation trajectory was analyzed in terms of root means square deviation (RMSD), root mean square fluctuation (RMSF), solvent-accessible surface area (SASA), the radius of gyration (Rg), and the number of H-bonds using GROMACS simulation software “WebGRO for Macromolecular Simulations (https://simlab.uams.edu/)”. In addition to this, principal component analysis (PCA), calculating projection (PC1 and PC2) and eigenvector, was also carried out to assess the obtained conformational space and Cα atomic motion pattern for all the native protein–ligand complexes and selective mutated protein–ligand complexes along with the protein (native and mutated) alone (Amadei et al. 1993). Besides, the dynamic behavior of protein was also calculated employing atomic coordinates which are generated from MD simulation.
MM/GBSA rescoring study
Fast Amber Rescoring (far) was utilized for rescoring of AGP/hit analogs’ docking pose using MM/GBSA methods (Wang et al. 2019b). The combination of Generalized Amber Force Field2 (GAFF2) and ff14SB force field was set for ligand, and receptor, respectively (Maier et al. 2015). Further, the partial charge of analog was assigned by the AM1-BCC method via the antechamber module of Amber (Jakalian et al. 2002). Then, free energy was computed for each analog complex (Wang et al. 2019a).
Results and discussion
Protein binding site prediction and validation
The refined structure of NF-κB p50 protein consists of 311 amino acid residues that are from Pro43 to Glu353 of the complete protein sequence. Further, CASTp 3.0 predicted 63 binding pockets while HotSpot Wizard v3.1 computed 26 binding pockets for the NF-κB p50 protein (Tables S1, S2). Based on the area, shape, volume, and internal cavities of the binding pocket obtained from the CASTp results, second binding pocket residues (Arg57, Arg59, Tyr60, Cys62, Glu63, His67, His144, Lys147) were selected as contributing amino acids at binding pocket. Similarly, based on the druggability score obtained from the HotSpot Wizard v3.1, residues Lys244, Ala245, Pro246, Asn247, Lys275, Gln277, Arg308, and Gln309 were selected as binding site residues. Thereafter, the active binding site comprising of 16 amino acid residues (acquired from both CASTp and HotSopt Wizard), i.e. Arg57, Arg59, Tyr60, Cys62, Glu63, His67, His144, Lys147, Lys244, Ala245, Pro246, Asn247, Lys275, Gln277, Arg308, and Gln309, was selected. This binding site includes the conserved RxxRxR (Arg54, Gly55, Phe56, Arg57, Phe58, Arg59) motif that has positively charged Arg residues which is responsible for making Cys62 active for covalent adduct with small molecules and leads to the inhibition of DNA binding (Xia et al. 2004; Nguyen et al. 2015). Therefore, the binding site including these conserved residues was taken into consideration for further analyses (Fig. S1).
Multiple sequence alignment
Since there is no co-crystallized ligand in the PDB structure of the NF-κB p50 subunit, MSA analysis was carried out to find out the conserved amino acid residues in the NF-κB p50 protein that are functionally responsible for ligand binding. BLOSUM62 exchange weight matrix was used for scoring the MSA. Further, the percent sequence identity for MSA was found to be 0.89 (Table S3). It was observed from the MSA that the amino acid sequence from 534 to 928 was most conserved in all the selected species followed by the amino acid sequence from 74 to 409 which was moderately conserved. Additionally, the amino acid sequence length from 1 to 73, 410–533, and 929–1059 were found to be less conserved (Fig. S2a). Interestingly, the predicted binding site residues in NF-κB p50 (Arg57, Arg59, Tyr60, Cys62, Glu63, His67, His144, Lys147, Lys244, Ala245, Pro246, Asn247, Lys275, Gln277, Arg308, and Gln309) were identified as conserved in MSA among all the species (Fig. S2b). Therefore, we considered these amino acid residues as functionally important for the ligand binding for AGP and hit analogs’ anti-inflammatory activity.
Virtual screening analysis
Virtual screening of 237 AGP analogs revealed that the binding energies were found to be in the range of − 7.4 kcal/mol (PubChem ID: 101362374) to − 5.1 kcal/mol (PubChem ID: 46907192), whereas the binding energy for AGP was found to be − 6.4 kcal/mol (Table S4). Since no co-crystallized structure is reported for the selected NF-κB p50 protein, hence we selected the top five analogs [PubChem IDs: 132210373 (Ana1), 101362374 (Ana2), 132210388 (Ana3), 132210390 (Ana4), and 132223040 (Ana5)], having binding energy < − 7.0 kcal/mol for further studies. Furthermore, visualization of the docked pose of all 237 AGP analogs revealed that these five selected hit analogs interact at the binding pocket like the DNA (Fig. S3). Structurally, these five screened analogs share the common oxolane and ethylidene moiety with the andrographolide structure, while these vary with the naphthalene moiety. Ana1 has the octahydronaphtho-dioxin ring, Ana2 possesses octahydronaphtho-dioxin, and Ana3-5 contains substituted hexahydro-naphthalene moiety in place of naphthalene moiety as in andrographolide. In terms of binding energy, naphthol groups containing analogs show more binding affinity towards NF-κB p50 protein compared to AGP and the rest of the analogs. Therefore, these five selected analogs were used for subsequent studies to find out the most efficient compound for NF-κB inhibition (Tables 1, S5).
Table 1.
Virtual screening and docking result of AGP and selected analogs against NF-κB p50

Molecular interactive docking, binding mode, and site-directed mutagenesis studies
The five hit AGP analogs and AGP were submitted for docking studies to carry out the molecular interactive binding analysis with NF-κB p50 protein and also to find out the accurate binding confirmation, precise interactive residues, and binding energy. The binding energy values were found in the range of − 4.92 kcal/mol (Ana5) to − 6.66 kcal/mol (Ana1), while for AGP it was found to be − 4.70 kcal/mol. The order of binding affinity was found to be Ana1 (BE = − 6.66 kcal/mol) > Ana2 (BE = − 6.15 kcal/mol) > Ana3 (BE = − 5.24 kcal/mol) > Ana4 (BE = − 5.21 kcal/mol) > Ana5 (BE = − 4.92 kcal/mol) > AGP (BE = − 4.70 kcal/mol) (Table 1). It is evident from the comparative interaction studies that AGP and hit analogs bind at the common binding pocket of NF-κB p50 which consists of the RxxRxR motif (Arg54 to Arg59) next to Cys62 (Fig. 1a) and hydrophobic (HYD) residues such as Val, Leu, Pro, and Ala. This is in good agreement with the literature that the RxxRxR motif, which leads up Cys62 in NF-κB, is important for DNA interaction and redox homeostasis (Hopkins and Neumann 2019), thereby inhibition of NF-κB and DNA binding (Pande et al. 2009). The 2D binding poses of all the hit molecules are depicted in Fig. 1c–h, whereas a detailed molecular interactive analysis of AGP, shortlisted AGP analogs, and DNA with native NF-κB p50 protein are provided in Table 2 with total interacting residues, types of H-bonds, and HYD interaction along with their distance with the interacting site name of the chemical moiety.
Fig. 1.

a 3D spatial orientation of AGP, selected analogs, and DNA in the substrate binding site of NF-κB p50 after docking. Color code: light grey = protein, green surface = binding pocket, dark blue = ssDNA helix, ball and stick representation = AGP/analogs. b The 2D interactive plot of DNA-NF-κB p50 docking. c–h Pictorial representation of 2D interaction of AGP and Ana1–5, respectively; Residue color red = acidic amino acid, green = hydrophobic amino acids, purple = basic amino acid, blue = polar amino acid; Hydrogen bonds = dashed pink lines, SASA = gray atom background.
Table 2.
Molecular interactive analysis of AGP, shortlisted AGP analogs, and DNA with native NF-κB p50 protein
| Compound name | Total interacting residues | HYD bond | H-bond | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Name of residue | Distance | Analog’s interacting site | Type | Name of residue | Distance | Analog’s interacting site | Type | ||
|
DNA 5′-AGATGGGGAATCCCCTAGA-3′ |
Tyr60, Cys62, His144, Thr146, Asp209, Ser211, Tyr241, Lys244, Ala245, Pro246, Asn247, Ser338, Asp339, Leu340 | – | – | – | – | – | – | – | – |
| AGP | Tyr60, His144, Val145, Thr146, Lys147, Lys148, Leu210, Asp242, Lys244, Ala245, Pro246, Lys275 |
His144 Ala245 Pro246 Lys244 |
6.46 4.77; 5.97 4.64; 5.80 4.54; 6.95 |
2-methylidene 2-methylidene; Naphthalene 8a-methyl; 4a-8a bond 5-methyl; Naphthalene |
Alkyl |
His144, Lys244, Thr146, Lys275 |
5.04 5.90 3.94 5.52 |
2-one 5-hydroxymethyl 2-one 5-hydroxymethyl |
C-H bond H-bond C-H bond H-bond |
| Ana1 | Tyr60, His144, Leu210, Ser211, Asp242, Lys244, Ala245, Pro246, Lys275 |
Tyr60 His144 Leu210 Lys244 Ala245 Pro246 |
5.67 6.69 4.88 3.92; 5.54 4.22; 4.75; 4.90 5.21; 5.48 |
2-methylidene 10b-methyl 10b-methyl 2-methylidene; Naphthol 4a-methyl; Naphthol; 10b-methyl 4a-methyl; Naphthol |
Alkyl | Lys244 | 3.89 | 4-hydroxy | H-bond |
| Ana2 | Arg57, Tyr60, Ser243, Ala245, Pro246, Ser249, Lys244, Lys275 |
Lys244 Lys275 |
4.28; 5.04 3.62 |
Naphthol; Naphthol 8-mehylidene |
Alkyl |
Arg57 Tyr60 Lys275 |
4.23 6.28 5.53 |
4-O (dioxin) 4-hydroxy 2-one |
H-bond H bond |
| Ana3 | Tyr60, His144, Leu210, Ser211, Asp242, Lys244, Ala245, Pro246, Asn247, Lys275 |
Tyr60 His144 Leu210 Lys244 Ala245 Pro246 |
5.86 7.82 4.49 3.76 4.35; 5.03 5.58 |
2-methyl Naphthalene 5-methyl 2-methyl Naphthalene; 4a-8a bond 4a-8a bond |
Alkyl |
Lys244 Pro246 |
3.88 4.30; 4.64 |
4-hydroxy 6-fluoromethyl; 6-fluoromethyl |
H-bond C-H bond C-H bond |
| Ana4 | Arg57, Tyr60, Ser243, Lys244, Ala245, Pro246, Ser249, Asn250, Lys275 |
Pro246 Lys244 Lys275 |
4.84 5.27 4.15; 4.84; 5.16 |
2-methyl 8a-methyl 2-methyl; 8a-methyl; Naphthalene |
Alkyl |
Arg57 Tyr60 Ser243 Lys244 |
3.73 6.20 4.29 5.48 |
6-fluoromethyl 1-oxo 6-fluoromethyl 4-carbon |
H-bond H-bond H-bond C-H bond |
| Ana5 | Ser243, Lys244, Ser249, Asn250, Cys273, Asp274, Lys275, Phe310 |
Lys244 Lys275 |
4.37 3.62; 4.70; 4.98 |
5-methyl 8a-methyl; Naphthalene; 2-methyl |
Alkyl |
Asn250 Ser243 |
2.98; 3.90 3.08; 4.70 |
6-fluoromethyl; 6-hydroxy 6-hydroxy; 6-fluoromethyl |
H-bond H-bond H-bond H-bond |
HYD hydrophobic interaction, H-bond Hydrogen bond
DNA-NF-κB p50 protein docking studies revealed that the hit AGP analogs bind at the same binding pocket where DNA binds with the only exception of Ana5 (Fig. 1a, b and Table 2). The Ana2 and Ana4 bind close to the Cys62 containing RxxRxR motif, indicating their potential towards NF-κB p50 inhibition (Fig. 1e, g). Further, to confirm the importance of Cys62 residues in the molecular interaction, a site-directed mutagenesis study was carried out on Cys62 (mutated to Ala62) residue which showed a reduction in the binding affinity of the AGP and hit analog with the mutated protein. The binding energy was found to be − 4.53 kcal/mol (AGP), − 5.51 kcal/mol (Ana1), − 5.67 kcal/mol (Ana2), − 4.81 kcal/mol (Ana3), − 4.85 kcal/mol (Ana4), and − 4.30 kcal/mol (Ana5), respectively (Table S6). This indicates that Cys62 residue plays an important role in the interaction of AGP and hit AGP analog with NF-κB p50. As shown in Fig. 2a, b, the superimposition of AGP and hit analogs with non-mutated and mutated NF-κB p50 protein displayed that in the case of non-mutated protein binding pocket for the hit AGP analogs is nearer to the Cys62 as compared to mutated protein. These results infer that AGP and hit AGP analogs strongly interact with non-mutated NF-κB p50 and bind to the RxxRxR motif next to Cys62 which is a key residue for the small molecule binding and subsequent inhibitory action for NF-κB p50 protein.
Fig. 2.

Overlay of the docked pose of AGP and selected analogs with binding pocket residues in a NF-κB p50 (Cys62) and b mutated NF-κB p50 (Cys62Ala). Color code: AGP = Red, Ana1 = Orange, Ana2 = Green, Ana3 = Brown, Ana4 = Blue, Ana5 = Grey; Binding pocket for native NF-κB p50 (Cys62) and mutated NF-κB p50 (Cys62Ala) = Light Blue; Representation of atoms/bonds of AGP and analogs = Stick
DFT analysis
The DFT analysis was computed to identify the nature of the electronic and energetic states of AGP and its screened analogs. Figure 3 displays the contour map of the molecular HOMO, LUMO, and EPM of AGP and its selected analogs. The DFT analysis calculated the values of HOMO, LUMO, HLG, and EPE in the range of − 6.31 to − 7.09 eV, − 1.28 to − 1.37 eV, 4.94 to 5.78 eV, and − 48.07 to − 49.39 (min)/62.89 to 65.86 kcal/mol (max), respectively (Table 3). Additionally, the lowest HLG value was found to be for Ana3 (4.94 eV), while the highest value was observed for Ana5 (5.78 eV). Ana2 (HLG = 5.18 eV) and Ana4 (HLG = 5.17 eV) displayed the HLG value close to Ana3, indicating that the Ana2, Ana3, and Ana4 are relatively more chemically reactive than the AGP, Ana4, and Ana5 and form a stable complex with NF-κB p50 protein (Zheng et al. 2013; Panwar and Singh 2021). The HOMO and LUMO positions of the analogs are used to locate the nucleophilic and electrophilic sites based on the charge distribution, respectively. As AGP is an electrophilic covalent binding inhibitor, analyzing the QM parameters is of importance in assessing the reactivity of the hit AGP analogs toward the NF-κB p50 inhibition (Singh et al. 2011; Tran et al. 2020). Furthermore, the HOMO is mostly distributed close to the naphthalene/Naphthol moiety except for the Ana5, while the LUMO is dispersed around the oxolane moiety for all analogs (Fig. 3). Thereafter, the EPM was computed to illustrate the map of the electrostatic potential region, H-bond interaction site, physicochemical properties, and molecular size and shape representation. It is evident from Fig. 3 that in all the analogs the negative potential (red color) lies near oxolane moiety as well as near the naphthol ring in the case of Ana1 and Ana2, while the positive potential (blue color) lies near the aliphatic chain of the analogs. Negative potential sites of the analogs act as electrophilic attack and positive potential sites act as the nucleophilic attack in case of biological interaction and the more negative value of electrostatic potential energy (EPE) suggests a more stable configuration and binding. It was observed from the EPE analysis that there is a subtle difference in the negative potential energy amongst all the analogs signifying their nearly same type of configuration and binding with the protein. Overall, from the DFT analysis, frontier orbital energies (HOMO and LUMO) along with EPM establish the direct inhibitory activity of AGP and analogs against NF-κB p50 interaction.
Fig. 3.

Depiction of Frontier orbital energies (HOMO, and LUMO) and Electrostatic Potential Map (EPM) on the surface of AGP and selected analogs; a AGP, b Ana1, c Ana2, d Ana3, e Ana4, f Ana5. Color code: Blue = Most positive potential; Red = Most negative potential; Order of Potential = Red (most − ve) < Orange < Yellow < Green < Blue (most + ve)
Table 3.
Frontier orbital energies (HOMO, and LUMO), HLG, and electrostatic potential energy (EPE) of AGP and selected AGP analogs
| Compounds | HOMO (eV) | LUMO (eV) | HLG (eV) | EPE kcal/mol | |
|---|---|---|---|---|---|
| Min | Max | ||||
| AGP | − 6.54 | − 1.28 | 5.26 | − 48.97 | 65.08 |
| Ana1 | − 6.87 | − 1.29 | 5.58 | − 48.81 | 62.89 |
| Ana2 | − 6.48 | − 1.30 | 5.18 | − 49.39 | 64.89 |
| Ana3 | − 6.31 | − 1.37 | 4.94 | − 48.07 | 65.86 |
| Ana4 | − 6.49 | − 1.32 | 5.17 | − 48.40 | 63.70 |
| Ana5 | − 7.09 | − 1.31 | 5.78 | − 48.66 | 64.28 |
EPE electrostatic potential energy, HLG HOMO–LUMO Gap, HOMO highest occupied molecular orbital, LUMO lowest unoccupied molecular orbital
ADMET analysis
Assessment of molecular and pharmacokinetic parameters is essential to get a safe and effective concentration of a chemical compound to be biologically active; therefore, the ADMET parameters were predicted for AGP and hit analogs. The acceptable range of these parameters defines the safe use and theoretical strength of the chemical compound as a drug-like candidate. According to the Lipinski’s rule of five, all the studied compounds showed the parameter within the limits and thus suitable for oral drug delivery (Lipinski et al. 2012). Further, Ana2, Ana3, and Ana4 exhibited a favorable response to blood–brain barrier (BBB) standards which predict that analogs can cross the BBB and thus can show CNS activity. In addition to this, volume distribution (VD) was also calculated as one of the distribution parameters for all the compounds, which revealed a promising range for all the compounds. The acceptable range of VD signifies their ability to bind plasma protein and the amount of distribution in the body fluid and absorption by tissue. Furthermore, all the hit analogs are P-glycoprotein and CYP (Cytochrome) non-inhibitor. As P-glycoprotein inhibition can block the absorption, permeability, and retention of the analogs (Amin 2013) and CYP family enzymes metabolize the drugs, hit AGP analogs showed good absorption and metabolic properties. Moreover, the elimination parameters were predicted in terms of clearance (CL) and half-life (T1/2) which revealed that Ana2-4 exhibited CL > 13 mL/min/kg while Ana1-5 showed T1/2 < 0.1, indicating their good clearance and safe use as drug-like candidates. Additionally, according to the globally harmonized system of classification (www.unece.org), all the hit analogs displayed predicted toxicity at IV class (300 < LD50 ≤ 2000) suggesting their potential for oral administration. Then, the toxicity assessment predicted all analogs as non-carcinogenic, non-hepatotoxic, and non-mutagenic, and thus anticipated their safe use as drug-like candidates. However, except for Ana1 and Ana2, the rest of the analogs and AGP showed the human ether-a-go-go-related gene (hERG) non-inhibitory property that can lead to long QT syndrome (Sanguinetti and Tristani-Firouzi 2006) (Table 4). Overall, all the hit analogs satisfied the predicted ADMET features and are suitable to be used as drug-like candidates after experimental validation.
Table 4.
Molecular and pharmacokinetic properties of AGP and hit AGP analogs
| Properties | AGP | Ana1 | Ana2 | Ana3 | Ana4 | Ana5 |
|---|---|---|---|---|---|---|
| MW | 350.45 | 410.52 | 390.51 | 388.42 | 388.42 | 404.46 |
| H-bond acceptors | 5 | 6 | 5 | 7 | 7 | 7 |
| H-bond donors | 3 | 1 | 1 | 2 | 2 | 2 |
| TPSA | 86.99 | 101.27 | 64.99 | 66.76 | 66.76 | 66.76 |
| Consensus Log P | 2.30 | 2.94 | 3.56 | 3.54 | 3.54 | 4.14 |
| Lipinski #violations | Yes; 0 | Yes; 0 | Yes; 0 | Yes; 0 | Yes; 0 | Yes; 0 |
| Water solubility | Soluble | Soluble | Moderately soluble | Soluble | Soluble | Moderately soluble |
| GI absorption | High | High | High | High | High | High |
| Human oral bioavailability | No | No | No | Yes | Yes | Yes |
| P-glycoprotein inhibitor | No | No | No | No | No | No |
| BBB permeation | No | No | Yes | Yes | Yes | No |
| VD (0.04–20 L/kg: excellent) | 0.612 | 0.884 | 1.457 | 0.784 | 0.784 | 0.87 |
| CYP inhibitor | No | No | No | No | No | No |
|
CL (≥ 5: excellent) ml/min/kg |
8.907 | 9.827 | 13.293 | 13.682 | 13.682 | 10.480 |
| T1/2 (0–0.3: excellent) | 0.249 | 0.082 | 0.096 | 0.041 | 0.041 | 0.034 |
| Acute oral Toxicity (c) | III | III | III | III | III | III |
| Carcinogenicity | No | No | No | No | No | No |
| Hepatotoxicity | No | No | No | No | No | No |
| Human either-a-go-go inhibition | No | Yes | Yes | No | No | No |
| Ames mutagenesis | No | No | No | No | No | No |
| Avian toxicity | No | No | No | No | No | No |
| Predicted LD50 | 1890 mg/kg | 400 mg/kg | 590 mg/kg | 400 mg/kg | 400 mg/kg | 1890 mg/kg |
| Predicted Toxicity Class | 4 | 4 | 4 | 4 | 4 | 4 |
CL clearance of a drug, Consensus Log P arithmetic mean of the five predictive values (XLOGP3, WLOGP, MLOGP Silicos-IT Log P, and iLOGP), CYP cytochrome, GI gastro intestinal, H-bond hydrogen bond, LD50 lethal dose at which 50% of test subjects die upon exposure to a compound, MW molecular weight, TPSA topological polar surface area, T1/2 half-life, VD volume distribution
Chemical structurality of the hit analogs
Structurally, all the hit analogs share the basic chemical scaffold that contains two cyclic functional groups joined with ethyledine moiety. Literature suggests that chemical compounds having bicyclic groups possesses myriad biological activities as anticancer, anti-inflammatory, antirheumatic, antifungal, and antihypertensive (Horton et al. 2003). The bicyclic moiety-containing drugs usually interact with respective molecular targets’ polar amino acid groups including hydroxyl, and amides (Hajduk et al. 2000). In the present study, among the hit analogs, 4-hydroxyoxolan-2-one and substituted naphthol/naphthalene moieties are linked with ethyledine. Structurally, Ana3 and Ana4 are stereoisomers whereas Ana1, Ana2, and Ana5 are distinct and have different substituted naphthol/naphthalene groups. Compared to the AGP, hit analogs have different groups at 2, 5, and 6 positions of naphthalene moiety (Table 1). In terms of molecular interactive analysis, Ana1 and Ana2 showed better binding energy and interaction than other hit analogs (Fig. 1, Tables 1, 2) indicating the importance of naphthol groups containing AGP analog for the better binding with the protein. DFT analysis revealed that Ana2, Ana3, and Ana4 have closely similar values in term of HLG demonstrating their almost similar chemical reactivity (Table 3). Moreover, in ADMET prediction, all the hit analogs passed the Lipinski rule of five and proved their safe use as a drug like candidate (Table 4).
MD simulation analysis
The atomistic MD simulation approach can be used to investigate the various structural information, conformational behaviors, stability, and dynamic characteristics of the protein–ligand complex. Therefore, AGP/hit AGP analogs—NF-κB p50 (native and mutated) complexes together with NF-κB p50 (native and mutated) protein alone (control) simulated for 100 ns of the time period. Generated outcomes analyzed by RMSD, RMSF, H-bond, Rg, SASA, and PCA to validate the constancy of simulated complexes. The average RMSD values for the NF-κB p50 (native) protein, AGP, and Ana (1–5)-NF-κB p50 (native) protein complex were found to be 0.60 nm, 0.42 nm, 0.75 nm, 0.50 nm, 0.50 nm, 0.53 nm, and 0.60 nm, respectively (Table S7). All the analogs, except Ana1, showed better stability and lower fluctuation than the NF-κB p50 protein which indicates that these AGP analogs are dynamically active and more efficient towards stabilizing the protein (Fig. 4a).
Fig. 4.

Graphical depiction of MD simulation studies of native NF-κB p50 Protein, AGP/selected analogs-native NF-κB p50 complex for 100 ns. a RMSD of backbone, b RMSF profiling, c Rg curve, d SASA graph, e number of H-bonds in AGP/Ana1-5—NF-κB p50 protein complex, and f depiction of Cα motion in term of PC1 and PC2
Figure 4b shows the RMSF values of NF-κB p50 (native) protein and AGP/Ana1-5 complexes with native protein which indicates the flexibility of residual motility during the binding of the ligand. It is evident that except for Ana5 rest of the Analogs complexes with native protein showed a similar pattern of fluctuation but lesser in value than the native NF-κB p50 protein. Moreover, the active site residues (50–70; 240–250; 270–280; 300–320) possessed relatively lesser fluctuations indicating the stability of these regions (Fig. S4). This may be ascribed to the formation of strong H-bonds and Van der Waal interactions between amino acid residue and ligand in the NF-κB p50-AGP/analogs complex. Since the stability of the native NF-κB p50-AGP/analogs complex is mainly ascertained by H-bond analysis, the obtained results indicate that the protein complex formed with Ana1, Ana2, and Ana4 displayed 4 H-bonds, while the AGP, Ana3, and Ana5 exhibited the maximum number of 3 H-bonds through the MD trajectory (Table S7). Therefore, the native NF-κB p50 complex with Ana1, Ana2, and Ana4 might have better binding stability and a more potent effect (Fig. 4e).
The compactness of the native NF-κB p50 protein and its complex with AGP/analogs was determined by Rg analysis. All the protein systems (both protein and complex) were found to be compact and none of the complexes showed any noticeable fluctuation during the simulation period. Further, the compactness of all the systems was calculated to be in the range of 2.32–2.74 nm, and amongst these, native NF-κB p50-Ana1 displayed the minimum deviation (2.36–2.57 nm), whereas the protein-Ana5 complex showed the maximum variation (2.32–2.74 nm) (Fig. 4c and Table S7). Overall, the complex structure was found to be undisturbed throughout the trajectory time and this is due to the proper minimization and interaction between the native NF-κB p50 and analogs. SASA analysis was utilized to assess the measures of the interaction between the AGP/analog-native protein complex and the solvents. Herein, the average value of SASA for the simulation time period was computed to be 176.63, 178.51, 174.63, 176.64, 177.50, 174.37, and 179.46 nm2 for native protein, AGP complex, and Ana1-5 complex, respectively (Table S7). The complexes of AGP, Ana2, Ana3, and Ana5 showed better SASA values compared to the native protein suggesting good accessibility and more interaction for solvents. In addition, SASA values were observed to be relatively stable for all the complexes during the simulation period indicating highly interactive relationships of the analogs with binding pocket residues (Fig. 4d).
As the protein performs via combined atomic motions, PCA was employed to interpret the Cα atomic structural motions in all the AGP and hit analogs complexes with native protein. The PCA graph was constructed based on two eigenvectors of the first and the last eigenvalues to represent the essential collective motion of the NF-κB p50 protein-AGP/hit analog complexes. The eigenvectors of all protein–ligand complexes are plotted in Fig. 4f based on the diagonalization of the covariance matrix of the structural disturbance. The PCA results demonstrated that the binding of hit analogs considerably decreased the collective motion of the native NF-κB p50 protein in comparison to the unbound native protein. Further, we observed a fewer subspace of conformation for Ana2 and Ana5 than other analogs, indicating stable and less occupied conformational space.
Additionally, the MD simulation studies of mutated (Cys62 to Ala62) NF-κB p50 protein and its complex with AGP and Ana2 were also performed to observe the effect of mutation on the protein stability and binding ability after binding with the ligand. Ana2 showed the best binding affinity and pattern amongst all the studied compounds in docking studies with native as well as mutated protein and after simulation studies with native protein, Ana2 displayed the best results as compared to other studied analogs, therefore, in this comparative simulation study with the mutated protein, Ana2 was used as a reference analog out of screened AGP analogs. It can be noticed from Table S8 and Fig. 5a–f that after mutation the stability of the protein–ligand complex gets disturbed which signifies the importance of Cys62 in the interaction of the protein with the ligand. The MD simulation study suggested that Ana2 can be a choice of a natural AGP-based anti-inflammatory drug to inhibit NF-κB p50 transcription. Apart from this, it can also be concluded that Cys62 is an important residue in the interaction of AGP and analogs with NF-κB p50 protein. Additionally, we compared hit analogs and AGP interactions before and after MD simulations with native and mutant proteins. Figures 6 and S5 show that all the hit analogs with the exception of Ana4 and Ana5 interacted with the Cys62 residues at the protein binding site during the MD simulation and form a stable complex with the NF-κB p50 protein. In addition, the strong binding of the hit analogs to the active site of NF-κB p50 found during the simulations indicates that these compounds are potentially able to inhibit DNA binding to proteins, disrupt transcription, and induce anti-inflammatory effects. On the contrary, inconsistency was observed in the interaction of analogs with mutated protein which again proves the significance of the Cys62 residues in the molecular interaction analysis.
Fig. 5.

Graphical representation of a1–3 backbone RMSD, b1–3 RMSF, c1–3 Rg, d1–3 SASA, and e1–2 protein–ligand H-bond of AGP-complex (with both NF-κB p50 Cys62 and Cys62Ala), and Ana2-complex (with both NF-κB p50 Cys62 and Cys62Ala), respectively. f PCA of mutated protein, AGP-mutated protein complex, and Ana2-mutated protein complex
Fig. 6.

Representation of the binding site of NF-κB p50 protein (both native and mutated) and its interaction with AGP and Ana2 before and after the simulation. A Protein, B AGP, and C Ana2. Color code: light grey = protein, sky blue surface = binding site, yellow = Cys62 amino acid residue, green = Ala62 amino acid residue, red ball and stick model = AGP, and dark green ball and stick model = Ana2
MM/GBSA rescoring analysis
MM/GBSA technique is a popular method to evaluate the free energy of the interacting small chemical compounds to proteins (Genheden and Ryde 2015). Results revealed that Ana2 showed the best MM/GBSA score in terms of binding free energy (− 29.99 kcal/mol) while Ana4 displayed the least (− 15.99 kcal/mol) suggesting a strong interaction between Ana2 and NF-κB p50 protein. The order of binding energy for the analogs was found to be Ana2 > Ana3 > Ana5 > AGP > Ana1 > Ana4 (Fig. 7). Therefore, MM/GBSA rescoring analysis validated the molecular docking and simulation results, suggesting Ana2 could be a potential AGP analog that binds firmly to NF-κB p50 and inhibit DNA binding. Eventually, our studies show that Ana2 can efficiently inhibit DNA binding to NF-κB p50 and thereby obstruct its transcription activity.
Fig. 7.
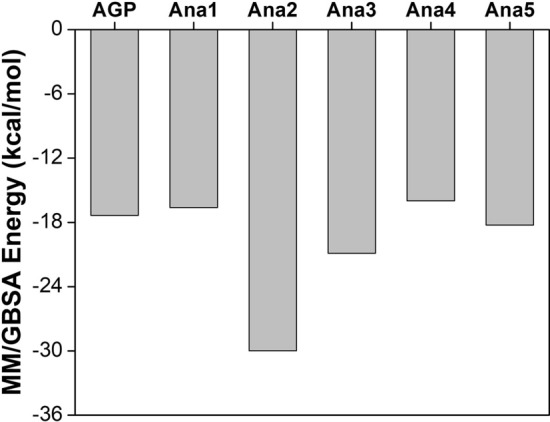
Illustration of MM/GBSA energy of AGP and selected analogs
Overall, our investigation reveals andrographolide analog Ana2 as the most potent and effective natural drug molecule against the NF-κB p50 protein. The efficacy of the Ana2 is found to be comparable or superior to other reported natural compounds. For example, the binding energy for Ana2 against the NF-κB p50 protein was found to be − 6.15 kcal/mol, which is comparable to curcumin (− 6.65 kcal/mol), ellagic acid (− 6.38 kcal/mol), and gallic acid (− 5.85 kcal/mol) (Kumar and Bora 2012). Advantageously, Ana2 interacts with the common residues (Arg57, Tyr60, Ala245, Pro246, Lys244, and Lys275) at the binding pocket like the DNA, which in contrast differs from the curcumin, ellagic acid, and gallic acid (Lys144, Asp206, Asp239, Leu207, and Lys241). This indicates better inhibition of the protein by the Ana2 compared to other natural compounds which were further validated with DFT analysis, MD simulation, and MM/GBSA in our study. To the extent of our knowledge, currently, no FDA-approved anti-inflammatory drug is available against NF-κB p50, our findings provide a plant-based natural small molecule that can be used as an anti-inflammatory agent in the early inflammation disease conditions like rheumatoid arthritis, asthma, and cancer. Although the computational study is a rapid, inexpensive, and reliable technique to identify the potential drug candidate from the large sample size at the pre-clinical drug discovery level, further experimental authentication is needed from the science community to expedite the drug development process.
Conclusion
In this study, a potent NF-κB p50 transcriptional inhibitor was identified by applying cheminformatics, and molecular and quantum mechanics. 237 andrographolide analogs were screened against NF-κB p50 through docking-based virtual screening. The AGP analogs having the BE < − 7.0 kcal/mol were further investigated sequentially by molecular docking study, DFT analysis, MD simulation, and MM/GBSA rescoring techniques to find out the potential inhibitor against DNA binding to NF-κB p50. The ADMET parameters of all the hit analogs were found to be in the acceptable range, establishing their drug-like candidacy. Interestingly, we found that hit analogs interact at the same binding pocket where DNA binds with NF-κB p50 protein and for the same, Cys62 residue plays an important role in the binding as evidenced by the site-directed mutagenesis analysis. The systematic investigation of the hit analogs showed that Ana2 (3Z,4S)-3-[2-[(4aR,6aS,7R,10aS,10bR)-3,3,6a,10b-tetramethyl-8-methylidene-1,4a,5,6,7,9,10,10a-octahydro-naphtho[2,1-d][1,3]dioxin-7-yl]ethylidene]-4-hydroxyoxolan-2-one) has the highest binding affinity and forms a stable complex with NF-κB p50 protein. Overall, the findings of this study strongly recommend that Ana2 would be a potential natural plant-based anti-inflammatory drug-like candidate to inhibit the DNA binding to NF-κB p50.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are thankful for the management of Vellore Institute of Technology, Vellore for providing the facilities to carry out this work.
Author contributions
All authors contributed to the study conception and design. Computational analysis, and the original draft writing was done by Priyanka Jain. Review and editing of the manuscript was performed by Dr. C. Sudandira Doss. All authors read and approved the final manuscript.
Data availability
All data generated or analyzed during this study are included in this article and its Supplementary Information files. Additional raw data or datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- Abraham MJ, Murtola T, Schulz R, et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Amadei A, Linssen ABM, Berendsen HJC. Essential dynamics of proteins. Proteins Struct Funct Bioinforma. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;2013:27–34. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, Orchard S, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, et al. The protein Data Bank www.rcsb.org. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, et al. Density-functional thermochemistry. III. The role of exact exchange. Nucleic Acids Res. 2015;21:1–16. doi: 10.1007/s00249-011-0700-9. [DOI] [Google Scholar]
- Bhardwaj VK, Purohit R. Computer simulation to identify selective inhibitor for human phosphodiesterase10A. J Mol Liq. 2021;328:115419. doi: 10.1016/j.molliq.2021.115419. [DOI] [Google Scholar]
- Bhardwaj VK, Oakley A, Purohit R. Mechanistic behavior and subtle key events during DNA clamp opening and closing in T4 bacteriophage. Int J Biol Macromol. 2022;208:11–19. doi: 10.1016/j.ijbiomac.2022.03.021. [DOI] [PubMed] [Google Scholar]
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- Cernuda-Morollón E, Pineda-Molina E, Javier Cañada F, Pérez-Sala D. 15-Deoxy-Δ12,14-prostaglandin J2 inhibition of NF-κB-DNA binding through covalent modification of the p50 Subunit. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- Chaudhary MK, Srivastava A, Singh KK, et al. Computational evaluation on molecular stability, reactivity, and drug potential of frovatriptan from DFT and molecular docking approach. Comput Theor Chem. 2020;1191:113031. doi: 10.1016/j.comptc.2020.113031. [DOI] [Google Scholar]
- Chen M, Xie C, Liu L. Solubility of andrographolide in various solvents from (288.2 to 323.2) K. J Chem Eng Data. 2010;55:5297–5298. doi: 10.1021/je100344z. [DOI] [Google Scholar]
- Chen M, Qin Y, Ma H, et al. Downregulating NF-κB signaling pathway with triterpenoids for attenuating inflammation: in vitro and in vivo studies. Food Funct. 2019;10:5080–5090. doi: 10.1039/c9fo00561g. [DOI] [PubMed] [Google Scholar]
- Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- Cockman ME, Lancaster DE, Stolze IP, et al. Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc Natl Acad Sci U S A. 2006;103:14767–14772. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concetti J, Wilson CL. NFKB1 and cancer: friend or foe? Cells. 2018;7:1–16. doi: 10.3390/cells7090133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Demarchi F, Bertoli C, Sandy P, Schneider C. Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J Biol Chem. 2003;278:39583–39590. doi: 10.1074/jbc.M305676200. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chen L, Wu W, et al. Andrographolide inhibits influenza A virus-induced inflammation in a murine model through NF-κB and JAK-STAT signaling pathway. Microbes Infect. 2017;19:605–615. doi: 10.1016/j.micinf.2017.08.009. [DOI] [PubMed] [Google Scholar]
- Espírito-Santo RF, Meira CS, Dos Santos CR, et al. The anti-inflammatory and immunomodulatory potential of braylin: pharmacological properties and mechanisms by in silico, in vitro and in vivo approaches. PLoS ONE. 2017;12:1–20. doi: 10.1371/journal.pone.0179174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill PMW, Johnson BG, Pople JA, Frisch MJ. The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. Chem Phys Lett. 1992;197:499–505. doi: 10.1016/0009-2614(92)85807-M. [DOI] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Bures M, Praestgaard J, Fesik SW. Privileged molecules for protein binding identified from NMR-based screening. J Med Chem. 2000;43:3443–3447. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Krappmann D, Wulczyn FG, Scheidereit C. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. EMBO J. 1999;18:4766–4778. doi: 10.1093/emboj/18.17.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BL, Neumann CA. Redoxins as gatekeepers of the transcriptional oxidative stress response. Redox Biol. 2019;21:101104. doi: 10.1016/j.redox.2019.101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- Huxford T, Ghosh G. A structural guide to proteins of the NF-kappaB signaling module. Cold Spring Harb Perspect Biol. 2009;1:1–16. doi: 10.1101/cshperspect.a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MD, Harrison SC. Structure of an IκBα/NF-κB complex. Cell. 1998;95:749–758. doi: 10.1016/S0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bora U. In silico inhibition studies of NF-κB p50 subunit by curcumin and its natural derivatives. Med Chem Res. 2012;21:3281–3287. doi: 10.1007/s00044-011-9873-0. [DOI] [Google Scholar]
- Kumar S, Kumar Bhardwaj V, Singh R, Purohit R. Explicit-solvent molecular dynamics simulations revealed conformational regain and aggregation inhibition of I113T SOD1 by Himalayan bioactive molecules. J Mol Liq. 2021;339:116798. doi: 10.1016/j.molliq.2021.116798. [DOI] [Google Scholar]
- Kumar S, Bhardwaj VK, Singh R, et al. Evaluation of plant-derived semi-synthetic molecules against BRD3-BD2 protein: a computational strategy to combat breast cancer. Mol Syst Des Eng. 2022;7:381–391. doi: 10.1039/D1ME00183C. [DOI] [Google Scholar]
- Kumar Bhardwaj V, Purohit R. Taming the ringmaster of the genome (PCNA): phytomolecules for anticancer therapy against a potential non-oncogenic target. J Mol Liq. 2021;337:116437. doi: 10.1016/j.molliq.2021.116437. [DOI] [Google Scholar]
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. doi: 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Biol. 2001;5:302–307. doi: 10.1016/S1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- Müller CW, Rey FA, Sodeoka M, et al. Structure of the NF-κB p50 homodimer bound to DNA. Nature. 1995;373:311–317. doi: 10.1038/373311a0. [DOI] [PubMed] [Google Scholar]
- Mussbacher M, Salzmann M, Brostjan C, et al. Cell type specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:1–31. doi: 10.3389/fimmu.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VS, Loh XY, Wijaya H, et al. Specificity and inhibitory mechanism of andrographolide and its analogues as antiasthma agents on NF-κB p50. J Nat Prod. 2015;78:208–217. doi: 10.1021/np5007179. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V, Sousa S, Ramos M. Direct covalent modification as a strategy to inhibit Nuclear Factor-Kappa B. Curr Med Chem. 2009;16:4261–4273. doi: 10.2174/092986709789578222. [DOI] [PubMed] [Google Scholar]
- Panwar U, Singh SK. In silico virtual screening of potent inhibitor to hamper the interaction between HIV-1 integrase and LEDGF/p75 interaction using E-pharmacophore modeling, molecular docking, and dynamics simulations. Comput Biol Chem. 2021;93:107509. doi: 10.1016/j.compbiolchem.2021.107509. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Sabe VT, Ntombela T, Jhamba LA, et al. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: a review. Eur J Med Chem. 2021;224:113705. doi: 10.1016/j.ejmech.2021.113705. [DOI] [PubMed] [Google Scholar]
- Sagendorf JM, Berman HM, Rohs R. DNAproDB: an interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2017;45:W89–W97. doi: 10.1093/nar/gkx272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Schmid N, Eichenberger AP, Choutko A, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf AW, Van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science (80-) 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Seo EJ, Fischer N, Efferth T. Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol Res. 2018;129:262–273. doi: 10.1016/j.phrs.2017.11.030. [DOI] [PubMed] [Google Scholar]
- Simossis VA, Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005;33:289–294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Singh R, Bhardwaj VK, Das P, Purohit R. Identification of 11β-HSD1 inhibitors through enhanced sampling methods. Chem Commun. 2022 doi: 10.1039/d1cc06894f. [DOI] [PubMed] [Google Scholar]
- Singh R, Bhardwaj VK, Purohit R. Computational targeting of allosteric site of MEK1 by quinoline-based molecules. Cell Biochem Funct. 2022;40:481–490. doi: 10.1002/cbf.3709. [DOI] [PubMed] [Google Scholar]
- Singh R, Kumar S, Bhardwaj VK, Purohit R. Screening and reckoning of potential therapeutic agents against DprE1 protein of Mycobacterium tuberculosis. J Mol Liq. 2022;358:119101. doi: 10.1016/j.molliq.2022.119101. [DOI] [Google Scholar]
- Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98:11623–11627. doi: 10.1021/j100096a001. [DOI] [Google Scholar]
- Sumbalova L, Stourac J, Martinek T, et al. HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 2018;46:W356–W362. doi: 10.1093/nar/gky417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Chen C, Lei X, et al. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran QTN, Wong WSF, Chai CLL. Labdane diterpenoids as potential anti-inflammatory agents. Pharmacol Res. 2017;124:43–63. doi: 10.1016/j.phrs.2017.07.019. [DOI] [PubMed] [Google Scholar]
- Tran QTN, Tan DWS, Wong WSF, Chai CLL. From irreversible to reversible covalent inhibitors: harnessing the andrographolide scaffold for anti-inflammatory action. Eur J Med Chem. 2020;204:112481. doi: 10.1016/j.ejmech.2020.112481. [DOI] [PubMed] [Google Scholar]
- Tuszynska I, Magnus M, Jonak K, et al. NPDock: a web server for protein-nucleic acid docking. Nucleic Acids Res. 2015;43:W425–W430. doi: 10.1093/nar/gkv493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tan XF, Nguyen VS, et al. A quantitative chemical proteomics approach to profile the specific cellular targets of andrographolide, a promising anticancer agent that suppresses tumor metastasis. Mol Cell Proteomics. 2014;13:876–886. doi: 10.1074/mcp.M113.029793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang X, Li Y, et al. FarPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics. 2019;35:1777–1779. doi: 10.1093/bioinformatics/bty879. [DOI] [PubMed] [Google Scholar]
- Xia Y-F, Ye B-Q, Li Y-D, et al. Andrographolide attenuates inflammation by inhibition of NF-κB activation through covalent modification of reduced cysteine 62 of p50. J Immunol. 2004;173:4207–4217. doi: 10.4049/jimmunol.173.6.4207. [DOI] [PubMed] [Google Scholar]
- Xiong G, Wu Z, Yi J, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–W14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Liu S, Zhou J, et al. Andrographolide attenuates microglia-mediated Aβ neurotoxicity partially through inhibiting NF-κB and JNK MAPK signaling pathway. Immunopharmacol Immunotoxicol. 2017;39:276–284. doi: 10.1080/08923973.2017.1344989. [DOI] [PubMed] [Google Scholar]
- Zhan CG, Nichols JA, Dixon DA. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J Phys Chem A. 2003;107:4184–4195. doi: 10.1021/jp0225774. [DOI] [Google Scholar]
- Zheng Y, Zheng M, Ling X, et al. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole-benzimidazole derivatives as potent Aurora A/B kinase inhibitors. Bioorganic Med Chem Lett. 2013;23:3523–3530. doi: 10.1016/j.bmcl.2013.04.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article and its Supplementary Information files. Additional raw data or datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.