

Graphical abstract

The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) appeared first in late December of 2019 in Wuhan, China.1 It has since spread globally, resulting in more than 110 million cases and 2.4 million deaths, with the United States, India and Brazil among the hardest hit nations to date (February 2021).2 SARS-CoV-2 is now the third highly pathogenic coronavirus to infect humans since the turn of the century, being preceded by SARS-CoV and MERS-CoV. Multiple vaccine trials3 have been conducted and several vaccines are now approved, but the need for a small molecule therapeutic remains critical, both to treat those who are already infected and for immunocompromised individuals who cannot receive a vaccine. Moreover, antiviral drugs targeting relatively constant viral proteins will be needed as part of the preparation against future coronavirus outbreaks, as vaccines developed against SARS-CoV-2 are likely to be ineffective against future strains. Early in 2020, the X-ray crystal structure of the SARS-CoV-2 main protease (Mpro, also known as 3CLpro) and its complex with an α-ketoamide inhibitor was reported.4 Mpro is known to be a critical component of viral replication, making it an extremely attractive target for antivirals.5 Unsurprisingly, several computational studies have been conducted to identify inhibitors of Mpro focusing primarily on existing drugs and/or natural products.6 In contrast, we decided to conduct a virtual screen of all commercially available drug-like molecules, giving a ligand pool with millions of candidates. By restricting our virtual screen to compounds which are already available, any identified hits would be readily available without the need for synthesizing them in-house.
We began our virtual screen by selecting the crystal structure of Mpro bound to an α-ketoamide inhibitor3 (PDB 6y2f) as our protein target (Fig. 1 ). We selected this protein structure as it was the only structure of the SARS-CoV-2 Mpro with a bound drug-like inhibitor at the time (early 2020).
Fig. 1.
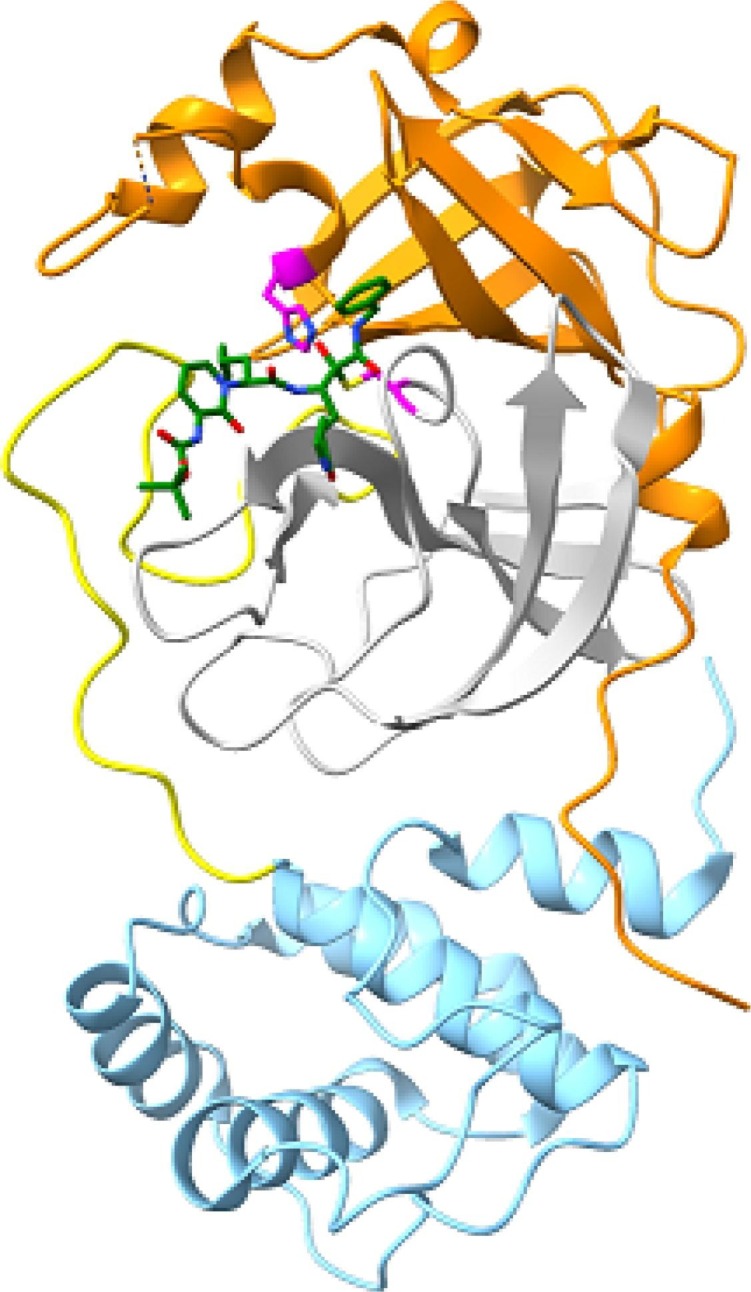
Crystal structure of Mpro (PDB 6y2f). Domain I is shown in orange, Domain II in grey, the connecting loop in yellow, and Domain III in teal. The inhibitor is shown with a green backbone, and the catalytic dyad residues shown with a magenta backbone.
The protein structure was prepared using ChimeraX7 to remove the inhibitor and water molecules, leaving only the protein chain. The remaining structure was then adjusted to physiological pH (7.4) and converted to the PDBQT format using Open Babel.8
We next prepared a library of virtual compounds using the ZINC15 database, which contains more than 230 million compounds.9 We filtered this initial library by selecting only compounds that were commercially available and applied a series of Lipinski filters to select only drug-like molecules (molecular weight 200–500 g/mol, log P ≤ 5, number of rotatable bonds ≤ 7, total polar surface area ≤ 150 Å2, H-bond donors ≤ 5, and H-bond acceptors ≤ 10). We further filtered the library to remove PAINS scaffolds,10 then adjusted the pH of all compounds to 7.4 and compiled the library in PDBQT format. The final library consisted of 9,779,510 compounds.
Since several inhibitors of Mpro have been reported, we next decided to test which of the three docking programs available to us (AutoDock Vina11, iDock12 and Smina13) gave the best performance using these known inhibitors as a benchmark. We selected a test set of 7 inhibitors with IC50 values spanning 3 orders of magnitude13, 14 and then used the Spearman correlation coefficient (Rs) to rank the performance of each docking program (see supporting information, Table S1). We found that the iDock program performed the best (Rs = 0.745) and selected it for our virtual screen. We then selected the center of our search space as the center of mass of the bound inhibitor (x = 10.832, y = -0.291, z = 20.731) and defined the search area as a 22 Å x 22 Å x 22 Å cube to encompass the full binding surface. All other parameters of the iDock program were left at default settings, as a recent benchmarking study has found that increasing the settings to make the calculations more time intensive give little to no improvement.15 After concatenating the results, we selected the top 75 compounds as an initial arbitrary cutoff. These 75 compounds were then upload to Datawarrior16 where we applied additional drug-like filters (Druglikedness score > 0, DrugScore > 0.25) and removed any identified toxicophores, resulting in 45 hit compounds. We next applied a Tanimoto filter to these 45 compounds based on the FP2 path-based fingerprint17 using a threshold value of 0.4 to eliminate compounds with high similarity, resulting in a total of 28 unique hit compounds (see supporting information, Fig. S1). The ZINC IDs and several select properties of the top 28 hits (named CP-1, CP-2, etc.) as calculated by DataWarrior are shown in Table 1 .
Table 1.
Selected properties of the top hits from the virtual screen.
| Compound | ZINC ID | iDock Score (kcal/mol) | cLogP | HBA | HBD | tPSA (Å2) |
|---|---|---|---|---|---|---|
| CP-1 | ZINC000408813176 | −11.37 | 1.94 | 7 | 3 | 100 |
| CP-2 | ZINC000013724640 | −10.18 | 2.82 | 6 | 1 | 71 |
| CP-3 | ZINC000001787663 | −10.18 | 1.82 | 5 | 3 | 70 |
| CP-4 | ZINC000102176295 | −10.06 | 2.68 | 5 | 1 | 62 |
| CP-5 | ZINC000072140022 | −9.93 | 2.85 | 6 | 1 | 77 |
| CP-6 | ZINC000102277098 | −9.92 | 2.62 | 7 | 3 | 94 |
| CP-7 | ZINC000015016632 | −9.89 | 3.76 | 5 | 1 | 62 |
| CP-8 | ZINC000035399442 | −9.83 | 3.70 | 4 | 1 | 37 |
| CP-9 | ZINC000013644356 | −9.82 | 2.87 | 6 | 0 | 54 |
| CP-10 | ZINC000035373068 | −9.82 | 3.04 | 6 | 1 | 73 |
| CP-11 | ZINC000010156847 | −9.80 | 5.22 | 4 | 0 | 46 |
| CP-12 | ZINC000005421744 | −9.80 | 5.00 | 5 | 0 | 51 |
| CP-13 | ZINC000035429649 | −9.80 | 5.06 | 5 | 1 | 58 |
| CP-14 | ZINC000016955746 | −9.79 | 2.05 | 8 | 1 | 94 |
| CP-15 | ZINC000001568217 | −9.78 | 3.46 | 2 | 0 | 16 |
| CP-16 | ZINC000604418795 | −9.77 | 2.54 | 7 | 2 | 90 |
| CP-17 | ZINC000038880549 | −9.77 | 2.97 | 5 | 0 | 56 |
| CP-18 | ZINC000022085141 | −9.76 | 1.97 | 6 | 1 | 81 |
| CP-19 | ZINC000010336843 | −9.76 | 3.92 | 7 | 1 | 78 |
| CP-20 | ZINC000072143718 | −9.74 | 1.60 | 7 | 2 | 93 |
| CP-21 | ZINC000022606809 | −9.74 | 4.79 | 5 | 1 | 58 |
| CP-22 | ZINC000006475194 | −9.73 | 3.49 | 6 | 1 | 86 |
| CP-23 | ZINC000092038755 | −9.72 | 3.52 | 6 | 1 | 66 |
| CP-24 | ZINC000045995522 | −9.71 | 4.16 | 7 | 0 | 79 |
| CP-25 | ZINC000006757463 | −9.71 | 3.83 | 7 | 2 | 89 |
| CP-26 | ZINC000035478508 | −9.71 | 3.64 | 6 | 0 | 87.7 |
| CP-27 | ZINC000218427747 | −9.70 | 2.24 | 8 | 4 | 124 |
| CP-28 | ZINC000013777264 | −9.69 | 4.67 | 5 | 0 | 43 |
With our hit compounds identified, we began to order them from the associated vendors as indicated by the ZINC database. Due to the SARS-CoV-2 pandemic, however, some commercial vendors reported significant disruption in their transportation networks. As a result of these complications, we were unable to obtain eight of our hit compounds (CP-1, CP-4, CP-6, CP-15, CP-17, CP-18, CP-19, and CP-21). Rather than waiting for these compounds to become available, we decided to proceed to in vitro studies with the remaining 20 compounds.
With our compounds in hand, we next tested their inhibitory activity against Mpro. Briefly, a pH 7.3 buffer prepared with 20 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA and 1 mM DTT was utilized for the inhibitory assay of the hit compounds against SARS-CoV-2 Mpro in vitro. The substrate labeled with Dabcyl and Edans at the N and C-terminus, respectively, and comprising the cleavage site of Mpro ({Dabcyl}-KTSAVLQ↓SGFRKM-E{Edans}-NH2, cleavage site indicated by the arrow {↓}, GL Biochem, Shanghai) was employed in the FRET (fluorescence resonance energy transfer) based cleavage assay. Dequenching of the Edans fluorescence resulting from the cleavage of the substrate by the Mpro was monitored at the emission/excitation wavelength of 460 nm and 360 nm, respectively, using an Flx800 fluorescence spectrophotometer (BioTek). All compounds were dissolved in DMSO for preparation of the stock solutions. Initially, 2.5 μL of Mpro at a final concentration of 0.5 μM was pipetted into a well with 20 μL of buffer solution followed by the addition of 2.5 μL of the corresponding compound stock solution dissolved in DMSO, resulting in a final concentration of 50 μM in each well (2.5 μL of DMSO was used as the negative control). The protease and compound mixture was incubated at 37 °C for 10 min. Afterwards, 25 μL of FRET substrate dissolved in the reaction buffer at an overall concentration of 20 μM was added into the corresponding wells containing the protease and compound mixture to initiate the reaction. The relative fluorescence units per unit of time (ΔRFU/s) from the linear section of the curve were used for the calculation of the inhibition rate. The inhibition rate was determined by using the formula listed below where Vo represents the initial velocity of reaction of the negative control (DMSO only) and VI represents the initial velocity of reaction with inhibitor:
All experiments were performed in triplicate and the values are presented as the means ± the standard deviation (Table 2 ).
Table 2.
Inhibition of SARS-CoV-2 Mpro by hit compounds.
| Compound | Structure | Inhibition (%)b |
|---|---|---|
| CP-2 | 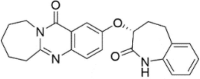 |
n.d. |
| CP-3 | 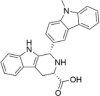 |
6.54 ± 3.60 |
| CP-5 | 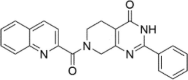 |
1.16 ± 6.12 |
| CP-7 | 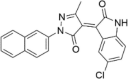 |
16.57 ± 3.12 |
| CP-8 | 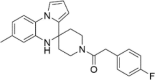 |
6.29 ± 3.32 |
| CP-9 | 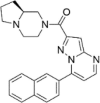 |
9.38 ± 6.75 |
| CP-10 | 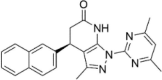 |
7.54 ± 1.30 |
| CP-11 | 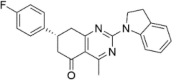 |
n.d.a |
| CP-12 | 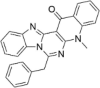 |
7.42 ± 2.39 |
| CP-13 | 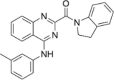 |
45.71 ± 8.98 |
| CP-14 | 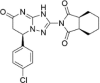 |
8.84 ± 1.45 |
| CP-16 | 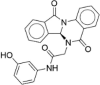 |
5.21 ± 1.07 |
| CP-20 | 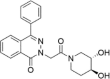 |
n.d. |
| CP-22 | 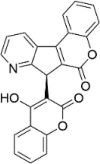 |
8.23 ± 3.77 |
| CP-23 | 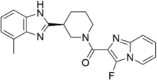 |
11.11 ± 0.90 |
| CP-24 | 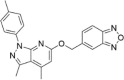 |
5.26 ± 4.51 |
| CP-25 | 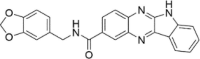 |
35.64 ± 10.24 |
| CP-26 | 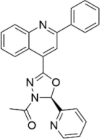 |
14.34 ± 9.23 |
| CP-27 | 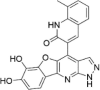 |
76.63 ± 55.40a |
| CP-28 |  |
6.87 ± 3.14 |
Autofluorescent compound.
Values are reported as the means ± SD of three measurements.
Since compounds CP-13 and CP-25 showed modest activity, we decided to subject them to molecular dynamics (MD) simulations carried out using AMBER18 to examine their ability to remain bound to the putative binding site under equilibrating conditions and also to provide a calculated binding energy at this site.18 We did not select CP-27 for MD given the large SD associated with its observed inhibition due to autofluorescence. Protein-ligand complexes of CP-13 and CP-25 with Mpro were subjected to 50 ns production runs at 300 K. Examination of the protein–ligand structure trajectories showed them to be stable throughout the production runs, with minimal reorganization of the ligands at the binding site and general stability of the protein structure. RMSD and potential energy data graphs are provided for CP-25 (Fig. S2) demonstrating that equilibration occurs within the first 35 ns, although a significant decrease in potential energy is not observed in this system. The RMSD-potential energy profile for CP-25 is representative of CP-13 with relative inhibitory activity, supporting the putative interactions of CP-13 and CP-25 with Mpro as shown in Fig. 2 and Fig. 3 . Additionally, the trajectories were analyzed by the MM/GBSA and MM/PBSA for computation of binding energies.19 ΔGbinding was estimated for CP-13 and CP-25 over a 100-ps window at the end of the 50 ns run, using the MM/GBSA method (-29.71 and −26.39 kcal/mol, respectively) and the MM/PBSA method (−23.97 and −18.97 kcal/mol, respectively). Notably the MM/GBSA and MM/PBSA methods both correctly ranked compounds CP-13 and CP-25 based on the experimental %inhibition data which indicates a greater level of inhibition achieved by CP-13.
Fig. 2.

Conformation of CP-13 after a 50 ns MD production run. Dashed green lines indicate non-polar interactions and catalytic dyad residues are marked in magenta.
Fig. 3.

Conformation of CP-25 after a 50 ns MD production run. Dashed green lines indicate non-polar interactions, dashed teal lines indicate polar interactions and catalytic dyad residues are marked in magenta.
In conclusion, our virtual screen of nearly 10 million commercially available compounds targeting SARS-CoV-2 Mpro and subsequent viral protease inhibition assay identified two compounds with modest inhibitory activity. The hit compounds, CP-13 and CP-25, are non-covalent inhibitors that bind with minimal reorganization and interact with protein residues primarily through non-polar interactions as indicated by MD simulation. While these compounds are not currently potent enough to advance to the clinic, they provide two additional candidates for development as antivirals to combat COVID-19. These compounds could also serve as a template to synthetically attach electrophilic “warheads” which have led to the development of powerful covalent inhibitors of Mpro.4 While additional improvement in potency is needed, we believe these findings help address the critical need for new treatment options to combat the raging pandemic as our identification of commercially available compounds with inhibitory activity against Mpro is a useful starting point for further optimization, either through purchasing or synthesizing additional analogues.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Bill and Linda Frost Fund at California Polytechnic State University (no associated grant number) and the European Commission through the SCORE program (grant agreement No. 101003627). The authors would like to thank Mr. Thomas W. Featherstone for his work in organizing the shipping and procurement of all tested samples.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2021.127990.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.(a) Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel Coronavirus Outbreak of Global Health Concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu F., Zhao S., Yu B., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu N., Zhang D., Wang W., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zhang, Y.; Zeng, G.; Pan, H.; Li, C.; Hu, Y.; Chu, K.; Han, W.; Chen, Z.; Tang, R.; Yin, W.; Chen, X.; Hu, Y.; Liu, X.; Jiang, C.; Li, J.; Yang, M.; Song, Y.; Wang, X.; Gao, Q.; Zhu, F. Safety, Tolerability, and Immunogenicity of an inactivated SARS-CoV-2 Vaccine in Healthy Adults Aged 18-59 Years: A Randomised, Double-Blind, Placebo-Controlled, Phase ½ Clinical Trial. Lancet Infect. Dis. 2020. DOI: 10.1016/ S1473-3099(20)30843-4. (b) Ramasay, M. H.; Minassain, A. M.; Ewer, K. J.; Flaxman, A. L.; Folegatti, P. M.; Owens, D. R.; Voysey, M.; Aley, P. K.; Angus, B.; Babbage, G.; Belij-Rammerstorfer, S.; Berry, L.; Bibi, S.; Bittaye, M.; Cathie, K.; Chappell, H.; Charlton, S.; Cicconi, P.; Clutterbuck, E. A.; Colin-Jones, R.; Dold, C.; Emary, K. R. W.; Fedosyuk, S.; Fuskova, M.; Gbesemete, D.; Green, C.; Hallis, B.; Hou, M. M.; Jenkin, D.; Joe, C. C. D.; Kelly, E. J.; Kerridge, S.; Lawrie, A. M.; Lelliott, A.; Lwin, M. N.; Makinson, R.; Marchevsky, N. G.; Mujadidi, Y.; Munro, A. P. S.; Pacurar, M.; Plested, E.; Rand, J.; Rawlinson, T.; Rhead, S.; Robinson, H.; Ritchie, A. J.; Ross-Russell, A. L.; Saich, S.; Singh, N.; Smilth, C. C.; Snape, M. D.; Song, R.; Tarrant, R.; Themistocleous, Y.; Thomas, K. M.; Villafana, T. L.; Warren, S. C.; Watson, M. E. E.; Douglas, A. D.; Hill, A. V. S.; Lambe, T.; Gilbert, S. C.; Faust, S. N.; Pollard, A. J. Safety and Immunogenicity of ChAd0x1 nCoV-19 Vaccine Administered in a Prime-Boost Regimen in Young and Old Adults (COV002): A Single-Blind, Randomised, Controlled, Phase 2/3 Trial. Lancet, 2020. DOI: 10.1016/ S0140-6736(20)32466-1. (c) Jackson, L. A.; Anderson, E. J.; Rouphael, N. G.; Roberts, P. C.; Makhene, M.; Coler, R. N.; McCullough, M. P.; Chappell, J. D.; Denison, M. R.; Stevens, L. J.; Pruijssers, A. J.; McDermott, A.; Flach, B.; Doria-Rosa, N. A.; Corbett, K. S.; Morabito, K. M.; O’Dell, S.; Schmidt, S. D.; Swanson II, P. A.; Padilla, M.; Mascola, J. R.; Neuzil, K. M.; Bennett, H.; Sun, W.; Peters, E.; Makowski, M.; Albert, J.; Cross, K.; Buchanan, W.; Pikaart-Tautges, R.; Ledgerwood, J. E.; Graham, B. S.; Beigel, J. H. An mRNA Vaccine against SARS-CoV-2 – Preliminary Report. N. Engl. J. Med. 2020, 383, 1920-1931.
- 4.Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as a drug target. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ton A., Gentile F., Hsing M., Ban F., Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inf. 2020;39:2000028. doi: 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Khan S.A., Zia K., Ashraf S., Uddin R., Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]; (c) Hakmi M., Bouricha E.M., Kandoussi I., El Harti J., Ibrahimi A. Repurposing of known anti-virals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. Bioinformation. 2020;16:301–306. doi: 10.6026/97320630016301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meyer-Almes F. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and drug design. Comp. Bio. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jimenez-Alberto A., Ribas-Aparicio R.M., Aparicio-Ozores G., Castelan-Vega J.A. Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comp. Bio. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li Z., Huang Y., Wu Y., et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. PNAS. 2020;117:27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gahlawat A., Kumar N., Kumar R., et al. Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J. Chem. Inf. Model. 2020;60:5781–5793. doi: 10.1021/acs.jcim.0c00546. [DOI] [PubMed] [Google Scholar]
- 7.Goddard T.D., Huang C.C., Meng E.C., et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchinson G.R. Open babel: an open chemical toolbox. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sterling T., Irwin J.J. ZINC 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baell J., Walters M.A. Chemistry: chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 11.Trott O., Olson A.J. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, H.; Leung, K.; Wong, M. iDock: A Multithreaded Virtual Screening Tool for Flexible Ligand Docking. 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA , 2012, 77-84, DOI: 10.1109/CIBCB.2012.6217214.
- 13.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L., Lin D., Kusov Y., et al. α-Ketoamides as broad-spectrum inhibitors of coronoavirus and enterovirus replicatoin: structure-based design, synthesis and activity assessment. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 15.Masters L., Eagon S., Heying M. Evaluation of consensus scoring methods for AutoDock Vina, Smina and iDock. J. Mol. Graph. Model. 2020;96 doi: 10.1016/j.jmgm.2020.107532. [DOI] [PubMed] [Google Scholar]
- 16.(a) Lopez-Lopez E., Naveja J.J., Medina-Franco J.L. DataWarrior: an evaluation of the open-source drug discovery tool. Expert. Opin. Drug. Discov. 2019;14:335–341. doi: 10.1080/17460441.2019.1581170. [DOI] [PubMed] [Google Scholar]; (b) Sander T., Freyss J., von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 17.Butina D. Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: a fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999;39:747–750. [Google Scholar]
- 18.Case, D. A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.,; Gohlke, H.; Goetz, A.W.; Greene, D.; Harris, R.; Homeyer, N.; Izadi, S.; Kovalenko, A.; Kurtman, T.; Lee, T.S.; LeGrand, S.; Li, P.; Lin, C.; Liu, J.; Luchko, T.; Luo, R.; Mermelstein, D.J.; Merz, K.M.; Miao, Y.; Monard, G.; Nguyen, C.; Nguyen, H.; Omelyan, I.; Onufriev, A.; Pan, F.; Qi, R.; Roe, D.R.; Roitberg, A.; Sagui, C.; Schott-Verdugo, S.; Shen, J.; Simmerling, C.L.; Smith, J.; Salomon-Ferrer, R.; Swails, J.; Walker, R.C.; Wang, J.; Wei, H.; Wolf, R.M.; Wu, X.; Xiao, L.; York, D.M.; Kollman, P.A. AMBER 2018, University of California, San Francisco.
- 19.(a) Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Dis. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hou T., Wang J., Li Y., Wang W. Accessing the performance of the MM/PBSA and MM/GBSA Methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hou T., Wang J., Li Y., Wang W. Assessing the performance of the molecular mechanics/Poisson Bolzmann surface area and molecular mechanics/generalized born surface area methods. 2. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011;32:866–877. doi: 10.1002/jcc.21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.