

Abstract
Background
Sarcoidosis is a multisystem granulomatous disease of unknown origin with a variable and often unpredictable course and pattern of organ involvement. In this study we sought to identify specific bronchoalveolar lavage (BAL) cell gene expression patterns indicative of distinct disease phenotypic traits.
Methods
RNA sequencing by Ion Torrent Proton was performed on BAL cells obtained from 215 well-characterised patients with pulmonary sarcoidosis enrolled in the multicentre Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study. Weighted gene co-expression network analysis and nonparametric statistics were used to analyse genome-wide BAL transcriptome. Validation of results was performed using a microarray expression dataset of an independent sarcoidosis cohort (Freiburg, Germany; n=50).
Results
Our supervised analysis found associations between distinct transcriptional programmes and major pulmonary phenotypic manifestations of sarcoidosis including T-helper type 1 (Th1) and Th17 pathways associated with hilar lymphadenopathy, transforming growth factor-β1 (TGFB1) and mechanistic target of rapamycin (MTOR) signalling with parenchymal involvement, and interleukin (IL)-7 and IL-2 with airway involvement. Our unsupervised analysis revealed gene modules that uncovered four potential sarcoidosis endotypes including hilar lymphadenopathy with increased acute T-cell immune response; extraocular organ involvement with PI3K activation pathways; chronic and multiorgan disease with increased immune response pathways; and multiorgan involvement, with increased IL-1 and IL-18 immune and inflammatory responses. We validated the occurrence of these endotypes using gene expression, pulmonary function tests and cell differentials from Freiburg.
Conclusion
Taken together, our results identify BAL gene expression programmes that characterise major pulmonary sarcoidosis phenotypes and suggest the presence of distinct disease molecular endotypes.
Shareable abstract (@ERSpublications)
Genome-wide BAL transcriptomics identified novel gene expression profiles associated with distinct phenotypic traits in sarcoidosis and is suggestive of the presence of novel molecular and clinical sarcoidosis endotypes https://bit.ly/3vf7VfT
Introduction
Sarcoidosis is a granulomatous disease of unknown aetiology which can affect almost every organ, but affects the lungs in majority of the cases (>90%). The patterns of organ involvement and disease course are often unpredictable, but a substantial number of patients suffer either a relapsing or progressive course with mortality estimated at 12% in advanced cases [1–4]. Despite significant advances in understanding the contribution of genetic predisposition, immune aberrations and the presence of microbial antigens in patients with sarcoidosis, little is known about the genetic networks and environmental factors that determine the phenotype in sarcoidosis. Similarly, treatment is still based on immunosuppression, with corticosteroids serving as first-line, and then use of “steroid-sparing” agents with limited evidence of long-term benefit [5, 6]. Genetics and genomics studies on sarcoidosis have focused on identifying DNA variants or gene signatures associated with sarcoidosis [7–13]. Genome-wide association studies have found associations between the major histocompatibility complex region and HLA-DRB1 variants and disease severity and sarcoidosis risk. Most previous whole-transcriptome studies focused on identifying gene signatures that distinguish sarcoidosis from control; progressive from nonprogressive disease; or from other granulomatous diseases such as tuberculosis [7–14]. While highly informative, they were mostly focused on the peripheral blood and limited in size, heterogeneity of disease manifestations and depth of phenotyping.
In this study, we performed a genome-wide transcriptome analysis of bronchoalveolar lavage (BAL) samples collected from a large cohort of well-characterised sarcoidosis patients recruited by the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study [15]. We conducted both supervised and unsupervised analysis on the measured gene expression profiles of these BAL samples to identify gene signatures that are associated with the heterogeneity in the clinical and phenotypic manifestations of sarcoidosis (figure 1). The results suggest the presence of novel molecular and clinical endotypes of sarcoidosis.
FIGURE 1.
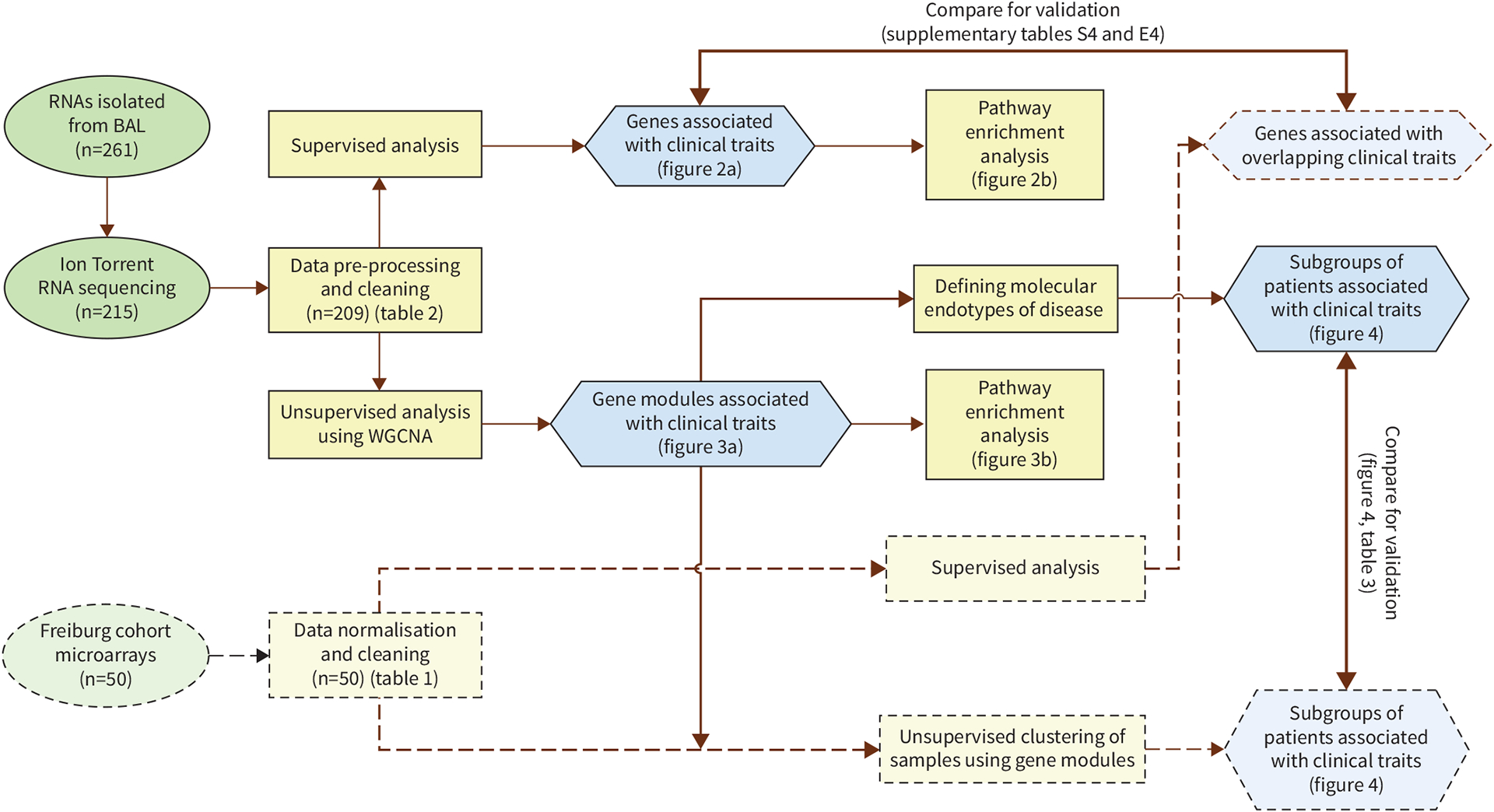
Study workflow. Summary of all steps in the supervised and unsupervised analyses of bronchoalveolar lavage (BAL) from the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) cohort (n=209) together with validation using BAL from the Freiburg cohort (n=50) is presented. WGCNA: weighted gene co-expression network analysis.
Methods
GRADS patient population
BAL samples were available from subjects enrolled in the GRADS study as described previously [15]. After informed consent and recruitment screening (supplementary figure S1), patients were grouped into pre-defined phenotypic groups based on a modified organ assessment instrument developed by the ACCESS study [16] and described by us in detail [15]. The collected phenotypic information according to the GRADS study protocol include physiological parameters, high-resolution computed tomography (CT), BAL and detailed questionnaires assessing dyspnoea, fatigue and quality of life, current and past medical information, occupational and environmental exposures, among others. All recruitment and consenting procedures were compliant with current United States Health Insurance Portability and Accountability Act regulations and approved by the local institutional review board (IRB) [15].
Freiburg validation cohort
BAL gene expression data from Affymetrix Human Gene 1.0 ST arrays were available from 50 individuals with sarcoidosis recruited independently in Freiburg, Germany (table 1, supplementary material). The consents were collected following institutional IRB protocols.
TABLE 1.
Patient characteristics of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) and Freiburg cohorts for the 12 overlapping clinical features
| GRADS | Freiburg | p-value | |
|---|---|---|---|
| Total | 209 | 50 | |
| Scadding stage | |||
| 0 | 21 (10.1) | 0 (0.0) | 0.02 |
| 1 | 54 (25.8) | 5 (10.0) | |
| 2 | 73 (34.9) | 28 (56.0) | |
| 3 | 27 (12.9) | 11 (22.0) | |
| 4 | 33 (15.8) | 6 (12.0) | |
| NA | 1 (0.5) | 0 (0.0) | |
| Female | 112 (53.6) | 18 (36.0) | 0.04 |
| Age years | 51.6±10.2 26.0–74.9 |
46.3±13.2 21.0–81.0 |
2.9×10−3 |
| FEV1 L | 2.7±1.0 | 2.6±0.9 | 0.497 |
| FEV1 % pred | 85.1±25.3 | 72.9±20.9 | 2.0×10−4 |
| FVC L | 3.6±1.2 | 3.5±1.1 | 0.45 |
| FVC % pred | 86.5±23.0 | 81.4±20.3 | 0.05 |
| FEV 1 /FVC ratio | 0.8±0.1 | 0.8±0.1 | 0.92 |
| Macrophages % | 73.2±32.0 | 54.2±21.2 | 2.6×10−9 |
| Lymphocytes % | 10.5±11.5 | 41.5±21.6 | 1.4×10−19 |
| Neutrophils % | 1.2±2.5 | 2.8±4.4 | 9.0×10−5 |
| Eosinophils % | 0.21±0.95 | 1.08±1.29 | 6.1×10−13 |
Data are presented as n, n (%), mean±SD or range, unless otherwise stated. NA: not available; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity.
Sample preparation and transcriptomic analysis
Total RNA was extracted as described previously [17] (supplementary material). For GRADS samples, cDNA libraries were generated, amplified and sequenced on the Ion Proton System for next-generation sequencing to obtain sequencing depth of ~30 million single-end reads per sample with an average read length of 150 bps (supplementary table S1). Gene expression in Freiburg samples was quantified using the Affymetrix Human Gene 1.0 ST arrays (supplementary material).
Statistical analysis
Our analysis plan included a supervised analysis aiming to identify known associations between pre-defined parameters and unsupervised analysis aimed to identify novel relationships. The supervised analysis identified gene signatures associated with pre-defined clinically relevant sarcoidosis phenotypes including Scadding staging, pulmonary function tests (PFTs), CT scan parameters, age, gender, BAL cell counts and the eight phenotypic groups defined by the GRADS study [15] using nonparametric tests (supplementary material). Unless otherwise stated in the text we used the false discovery rate (FDR) to control for multiple testing error. The unsupervised analysis was performed using the weighted gene co-expression network analysis (WGCNA) [18] as described previously by us [19]. Genes from chosen WGCNA modules (p<0.05) were used to cluster the patients into subgroups using K-means clustering (supplementary material). Chi-squared test and Wilcoxon rank sum test were used to assess the difference for categorical and continuous patient characteristics (p<0.05), respectively, among the clusters for chosen gene modules (supplementary material). GeneGO MetaCore was applied to identify significant enriched pathways (FDR<0.05) for each gene module identified by the unsupervised analysis. The data are publicly available on the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/) under the accession number GSE109516. All analysis codes and results are available on github page (https://yale-p2med.github.io/SARC_BAL).
Results
Patient cohorts
For the GRADS cohort, based on RNA quality and quantity as well as the quality of RNA-Seq data of BAL samples, we included 209 BAL samples from eight pre-defined clinical phenotype groups [15]: n=25 stage I, n=34 stage II–III treated, n=42 stage II–III untreated, n=19 stage IV treated, n=12 stage IV untreated, n=14 acute sarcoidosis, n=40 remitting and n=23 subjects with multiorgan involvement (supplementary table S1). 53.6% were female and 23.4% were black (table 2). For the Freiburg cohort, 50 BAL samples were available; 36% were female and 100% were white. The clinical information collected included demographics and 12 clinical phenotypic traits overlapping with the GRADS protocol (table 1, supplementary table S3).
TABLE 2.
Demographic and clinical characteristics of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) cohort categorised by the phenotypic groups
| Multiorgan | Nonacute stage I untreated | Stage II-III treated | Stage II-III untreated | Stage IV treated | Stage IV untreated | Acute untreated | Remitting untreated | |
|---|---|---|---|---|---|---|---|---|
| Total | 23 | 25 | 34 | 42 | 19 | 12 | 14 | 40 |
| Scadding stage | ||||||||
| 0 | 4 (17.4) | 0 (0.0) | 1 (2.9) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (14.3) | 14 (35.0) |
| 1 | 7 (30.4) | 24 (96.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 9 (64.3) | 14 (35.0) |
| 2 | 6 (26.1) | 0 (0.0) | 23 (67.7) | 34 (81.0) | 0 (0.0) | 0 (0.0) | 3 (21.4) | 7 (17.5) |
| 3 | 4 (17.4) | 0 (0.0) | 10 (29.4) | 8 (19.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 5 (12.5) |
| 4 | 2 (8.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 19 (100) | 12 (100) | 0 (0.0) | 0 (0.0) |
| NA | 0 (0.0) | 1 (4.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Female | 12 (52.2) | 18 (72.0) | 18 (52.9) | 19 (45.2) | 9 (47.4) | 6 (50.0) | 9 (64.3) | 21 (52.5) |
| Age years | 54.6±10.8 30.9–69.6 |
46.2±10.9 27.3–64.9 |
54.3±9.9 34.8–74.9 |
50.2±9.3 26.0–68.2 |
52.9±8.0 39.3–69.5 |
49.8±10.4 32.7–63.6 |
51.4±12.2 30.8–66.9 |
52.2±9.8 26.3–71.5 |
| Race | ||||||||
| Caucasian/white | 15 (65.2) | 21 (84.0) | 22 (64.7) | 31 (73.8) | 13 (68.4) | 9 (75.0) | 9 (64.3) | 33 (82.5) |
| African American/black | 8 (34.8) | 4 (16.0) | 8 (23.5) | 10 (23.8) | 6 (31.6) | 3 (25.0) | 4 (28.6) | 6 (15.0) |
| Other | 0 (0.0) | 0 (0.0) | 4 (11.8) | 1 (2.4) | 0 (0.0) | 0 (0.0) | 1 (7.1) | 1 (2.5) |
| FEV1 L | 2.6±1.3 | 2.8±0.8 | 2.3±0.8 | 2.9±0.8 | 2.08±0.8 | 2.4±1.9 | 3.0±0.6 | 3.1±0.9 |
| FEV1 % pred | 90.0±13.4 | 84.5±30.9 | 78.6±20.2 | 84.24±26.2 | 69.5±21.3 | 84.2±25.9 | 86.1±39.4 | 96.2±21.4 |
| FVC L | 3.4±1.6 | 3.7±1.0 | 3.2±1.1 | 3.9±1.0 | 3.1±0.9 | 3.5±2.4 | 3.7±0.8 | 4.0±1.2 |
| FVC % pred | 89.2±11.4 | 84.8±29.4 | 82.5±18.8 | 85.5±24.4 | 78.2±16.2 | 91.3±23.9 | 83.1±38.0 | 93.9±20.4 |
| DLCO mL·min−1·mmHg−1 | 23.5±8.6 | 21.8±10.6 | 20.1±9.5 | 23.8±8.8 | 21.7±8.9 | 20.7±11.8 | 19.5±11.0 | 27.3±9.9 |
| DLCO % | 86.1±24.1 | 85.8±26.4 | 69.5±28.1 | 81.0±24.5 | 72.4±18.1 | 67.3±28.1 | 74.1±37.3 | 90.7±23.9 |
Data are presented as n, n (%), mean±SD or range. NA: not available; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
Gene expression patterns associated with hilar lymphadenopathy, parenchymal or airway involvement
Using supervised analysis, we assessed the association of gene expression of 209 BAL samples with each of 24 clinical traits that were pre-defined including Scadding staging, PFTs, CT scan parameters, age, gender, BAL cell counts and the eight phenotypic groups. This analysis revealed genes associated with each trait (FDR<0.05) as well as overlapping genes between traits (figure 2a, supplementary material).
FIGURE 2.
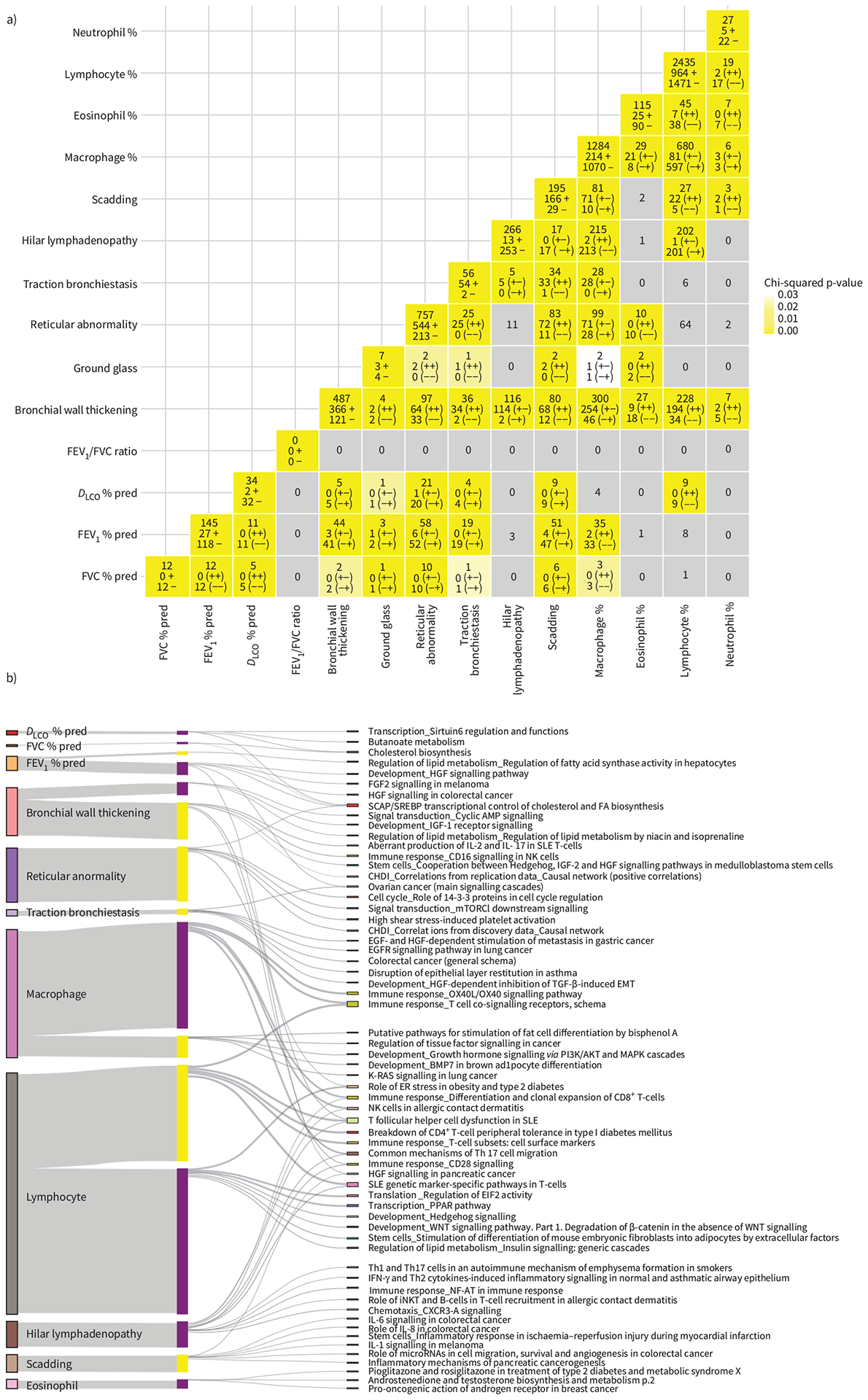
Association of gene expression with clinical traits and bronchoalveolar lavage (BAL) cell differentials identified using supervised analysis. Scadding staging, pulmonary function tests (PFTs), age, computed tomography (CT) scan features (with severity measurements) and BAL cell differentials were considered continuous variables and Spearman’s ρ test was used. Race, gender and CT scan features (without severity measurements) were considered as categorical and the Wilcoxon rank sum and Kruskal–Wallis tests were used. a) A matrix showing the number of genes associated with each clinical trait on diagonal in yellow and the overlap in assicated genes bewteen any two traits off the diagonal in grey to yellow. Each entry has three rows. For entries on the diagonal, the first row describes the total number of genes assocated with the trait. The second and third rows represent the number of positively and negatively associated genes marked by (+) and (−), respectively. For entries off the diagonal, the first row represents the number of overlapping genes associated with both traits. The second and third rows describe the number of genes positively and negatively associated with both traits marked by (++) and (−−), respectively. Entries coloured grey represent nonsignificant overlap between two traits (p>0.05), while white-to yellow-coloured entries represent a significant (p<0.05) overlap in genes associated with the two traits assessed with the significance assessed by Chi-squared test. b) The top five significant (false discovery rate (FDR) <0.05) enriched pathways for genes associated with each trait as well as overlap between mutliple traits are shown in a Sankey plot. Bars on the left represent genes significantly associated with each clinical trait, with their height representing the number of genes. In the middle of the plot, positively and negatively correlated genes for each clinical trait are represented by yellow and purple coloured bars, respectively, again with heights representing the number of genes. Each set of negatively or positively correlated genes was connected to its top five significant (FDR<0.05) enriched pathways (with at least three genes in the pathway), listed on the right, by grey lines.
Increased Scadding stage was significantly associated with increased expression in 166 genes and decreased expression in 29 genes (figure 2a). Of these genes, many were also significantly associated with CT traits such as hilar lymphadenopathy (17 genes) and reticular abnormalities (83 genes). Increased genes associated with both progressive Scadding staging and more reticular abnormalities include PLA2G7, ID1, LGMN and CCL2, and decreased genes include PDLIM1 and AOC3. SLC40A1 was increased with progressive Scadding staging, more reticular abnormalities and more hilar lymphadenopathy. These genes are known to be involved in fibrosis and chronic obstructive lung disease [20–22]. Genes increased with progressive Scadding staging were enriched for interleukin (IL)-1, IL-8 and IL-6 pathways (figure 2b), previously associated with inflammatory bowel disease [23] and lung fibrosis [20]. Interestingly, many of these genes are also increased with changes in clinical traits not included in the definition of Scadding stage, such as increased bronchial wall thickening (68 genes; e.g. TREM2, CHIT1, LGMN) and decreased forced expiratory volume in 1 s (FEV1) percentage and forced vital capacity (FVC) % predicted (47 and six genes, respectively, e.g. IDI1 and INSIG1), previously reported in sarcoidosis [24, 25].
Among the CT phenotypic traits, reticular abnormality, hilar lymphadenopathy and bronchial wall thickening were significantly associated with large number of genes (757, 266 and 487, respectively; figure 2a). Genes increased in the presence of hilar lymphadenopathy were enriched for T-helper type 1 (Th1) and Th17, interferon (IFN)-γ and nuclear factor of activated T-cells (NFAT) signalling (figure 2b), and included CD28, STAT1, CXCR3 and CCR4, previously reported in sarcoidosis immune response and implicated in pulmonary fibrosis [13, 26]. Genes increased with more severe bronchial wall thickening are enriched for aberrant IL-2 and IL-7 pathways (figure 2b) including MRC2, SLC40A1, F2R, IL7, PTPN7, ADORA2A, SPRY2, PLA2G7 and PTGS1. Reticular abnormalities on CT scan were positively correlated with many known fibrosis-associated genes such as TGFBR1, COL3A1, TLR3, ID1, TCF4, IGFBP6, PLA2G7, FADS1, ARGHAP12 and MMP10 [27–30]. Interestingly, ID1, HMGC1 and SEPP1 were also inversely correlated with diffusing capacity of the lung for carbon monoxide (DLCO) percentage and FVC % pred, potentially reflecting the shared biology underlying physiological restriction, loss of diffusion capacity and reticular abnormalities. Pathway analysis of genes positively correlated with reticular abnormality revealed an enrichment for mechanistic target of rapamycin (MTOR; cytochrome C, SC5D, HIF1A, PPAR-α) and cell cycle (CDC25C, CHK2, CDK1) signalling, which was also seen in genes increased with progressive Scadding staging (figure 2b).
Taken together, our results suggest that different transcriptional programmes affect the three major phenotypic manifestations of pulmonary involvement in sarcoidosis with Th1 and Th17 associated with hilar lymphadenopathy, transforming growth factor-β1 (TGFB1) and MTOR signalling for parenchymal involvement and IL-7 and IL-2 for airway involvement.
Genes associated with race are also associated with hilar lymphadenopathy, parenchymal or airway involvement
308 genes were associated with race (188 increased in white subjects and 120 in black subjects; figure 2a). SLC22A16, NME4, PWP2, ASRGL1 and SCARB1 were the top increased genes in black subjects while CD300C, LAMA1, RNF135, ST14 were the top genes increased in white subjects (figure 2a). Expression of SLC22A16 and PWP2 were previously shown to be race dependent in cancer subjects [31]. Aberrant lipid trafficking, coenzyme A biosynthesis and arachidonic acid production were enriched in the genes higher in black subjects, while no enriched pathway was found for the genes higher in white subjects. Genes increased in black subjects had a significant overlap with genes increased with progressive Scadding stage (MYOZ1, SQRDL, FAM213A) and decreased PFTs (TCEA3, FAM213A, MYO1E, CYP51A1, SQLE), being consistent with findings from previous studies on the importance of race in disease severity[32]. Genes increased with reduced lung function (DLCO %, FVC %, FEV1 %), increased reticular abnormality and in black subjects were associated with SCAP/SREBP transcriptional control of cholesterol and fatty acid biosynthesis (figure 2b), potentially reflecting race-specific pathways of injury.
Genes increasing with higher lymphocyte fraction in BAL reveal shared transcriptional programmes related to hilar lymphadenopathy and bronchial wall thickening
Disease activity is known to be associated with changes in BAL cell composition [33]. While total BAL cell count was associated with only a small number of genes, lymphocyte and macrophage fractions in BAL were associated with the largest number of genes (2435 and 1284, respectively; figure 2a, supplementary material). Among the top genes (Spearman’s ρ>0.3, FDR<0.05) positively correlated with lymphocyte count were known markers of T-lymphocyte subpopulations such as CD2, CD3, CD6, CD5, CD96 and CD247 [34–37]. Among other positively correlated genes were markers of lymphocyte activation (ITK, LCK, CD28, CTLA4, IL2RB), granzymes (GZMA, GZMB and GZMH, ETS1), chemokines and their receptors (CCL5, CXCL9, CCR4, CXCR3,4,6), cytokines and their receptors (IFNG, IL6, IL26, IL32, IL2RB,G, IL12RB2, IL15RA, IL18RA, IL21RA) and inflammatory regulators (JAK3, NFATC, NKB2). Genes significantly correlated with lymphocyte count significantly overlapped with those genes associated with bronchial wall thickening, hilar lymphadenopathy and Scadding stage, but not with genes associated with reticular abnormality, PFT and demographics (figure 2a). Among the genes positively correlated with both lymphocyte count and bronchial thickening were CCR5, CCR6, CD84, CD28, IL12RB, IL18R, IL21R and IFNG, potentially reflecting activation of CD4+ T-lymphocytes, T:B lymphocyte interactions, Th1 inflammatory response and susceptibility to sarcoidosis [38–41]. Genes increasing with both increased lymphocyte count and increased hilar lymphadenopathy were LY9, GNAO1, IFNG, F2R, CCR 4/5/8, CD6, CD5 and KIF21B [42]. Genes increased with higher lymphocyte fractions were enriched for numerous immune T-cell responses (Th1/Th17, IFN-γ, OX40L/OX40), follicular Th-cell dysfunction, T-cell co-signalling receptors and systemic lupus erythematosus genetic marker genes (figure 2b, supplementary material).
WGCNA gene modules associate significantly with PFTs, CT imaging features and BAL cell differentials
To identify gene modules associated with important clinical features of sarcoidosis in an unbiased way, we ran a WGCNA analysis. This analysis identified 48 gene modules that correlated with PFTs, CT scan findings (mediastinal and hilar lymphadenopathy, traction bronchiectasis, micronodule, ground glass, reticular abnormality) and BAL cell differentials (supplementary figure S5). Among the 48 modules, five (modules 1, 4, 18, 33 and 47) had the largest number of genes significantly correlated (p<0.05) with the highest number or unique combination of clinical traits (figure 3a).
FIGURE 3.
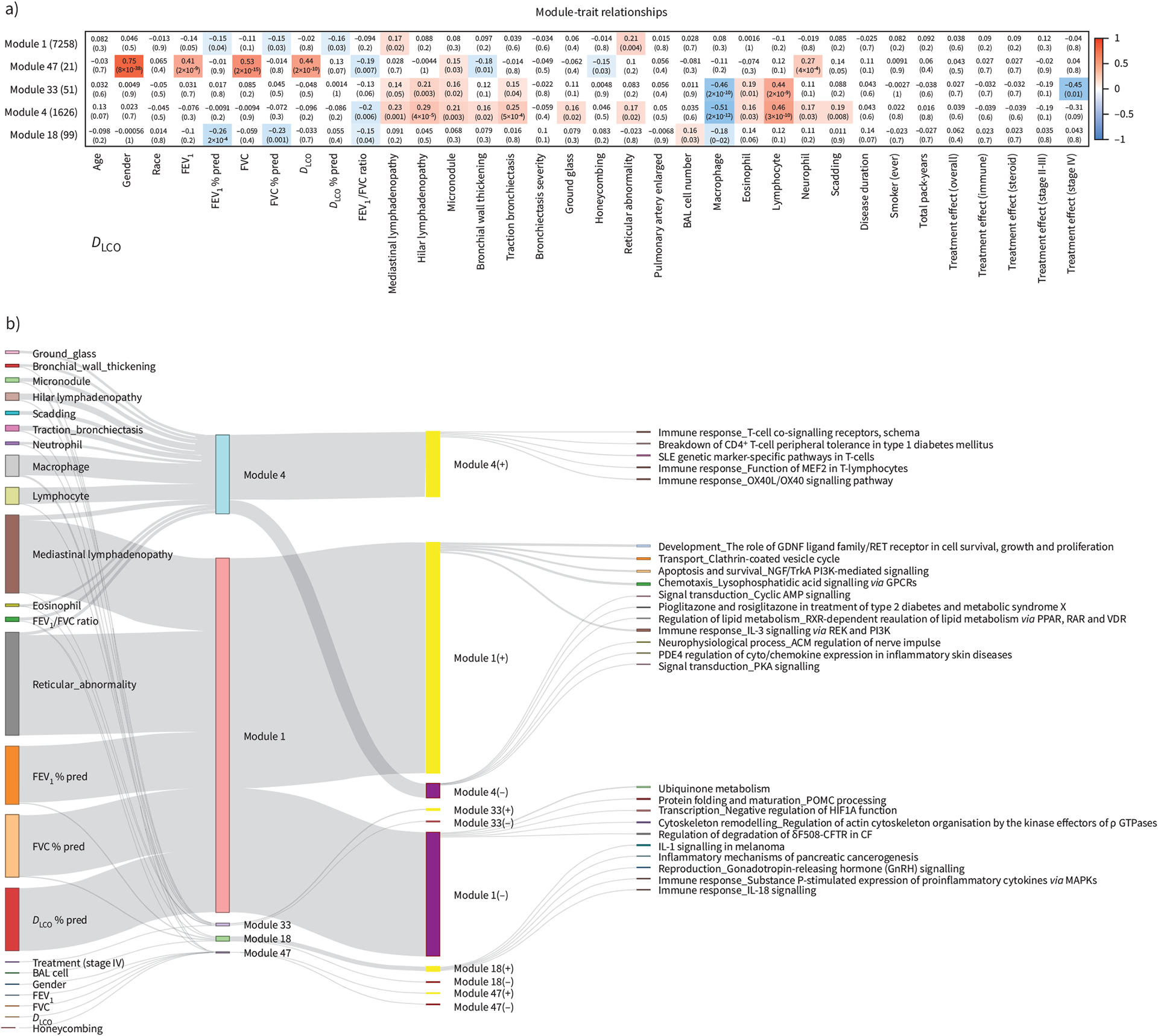
Idenification of gene modules that associate significantly with clinical traits and bronchoalveolar lavage (BAL) cell differentials using weighted gene co-expression network analysis. a) Heatmap showing the correlation (positive in red and negative in blue) between the Eigen genes of five chosen gene modules (1, 47, 4, 33, 18) and clinical traits (demographics, pulmonary function tests, computed tomography scan features, BAL cell differentials, Scadding stage and treatment within stage II–III and IV). b) Sankey plot visualising the significantly correlated clinical traits as well as the top five significantly enriched pathways (with at least three genes) for each gene module and their overlap. Bars on the left represent clinical traits and are connected to gene modules with which they are significantly correlated (p<0.05 in panel a). Bars in the middle represent the five chosen gene modules with bar heights describing module size. Each gene module was split into genes positively and negatively correlated with its eigen gene represented by yellow (+) and purple (−) bars, respectively. Finally, each gene set was connected to its top five significnatly enriched pathways represented by bars on the right, similarly with bar heights describing the number of module member genes from each pathway. Details of the gene modules can be found at https://yale-p2med.github.io/SARC_BAL. FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
Module 4 (1626 genes) was positively correlated with most CT imaging features, BAL cell differentials and Scadding staging, suggesting a plausible link between BAL gene expression, increased Scadding stage, increased lymphocyte and eosinophil differentials. This module was negatively correlated with macrophage differential and FEV1/FVC ratio (figure 3a). Module 1 had the largest number of genes (n=7258). It was negatively correlated with all PFT % pred values, and positively correlated with the presence of mediastinal lymphadenopathy and reticular abnormality. No correlation was found for BAL cell differentials or the total BAL cell counts, suggesting the existence of large number of gene signatures associated with lung function regardless of BAL cell differentials. In addition, module 18 (99 genes) was negatively correlated with FEV1 % pred, FVC % pred and FEV1/FVC ratio, as well as macrophage differential. Module 33 (51 genes) was positively correlated with the presence of mediastinal and hilar lymphadenopathy, micronodule, traction bronchiectasis and higher lymphocyte and eosinophil cell differentials (figure 3a). Module 47 contained 21 genes that correlated with sex and PFT but not % pred PFT values, reflecting the effect of sex on PFT. Disease duration, smoking and specific sarcoidosis treatment were not significantly associated with any gene module (figure 3a).
Four gene modules are suggestive of novel molecular endotypes of sarcoidosis
To define novel disease endotypes we performed K-means clustering analysis using genes from each of the WGCNA modules to identify clusters of sarcoidosis patients using 2289 clinical traits collected under the GRADS protocol, including 204 environmental factors relevant for pathogenesis of lung disease and outlined by GRADS investigators (figure 4).
FIGURE 4.
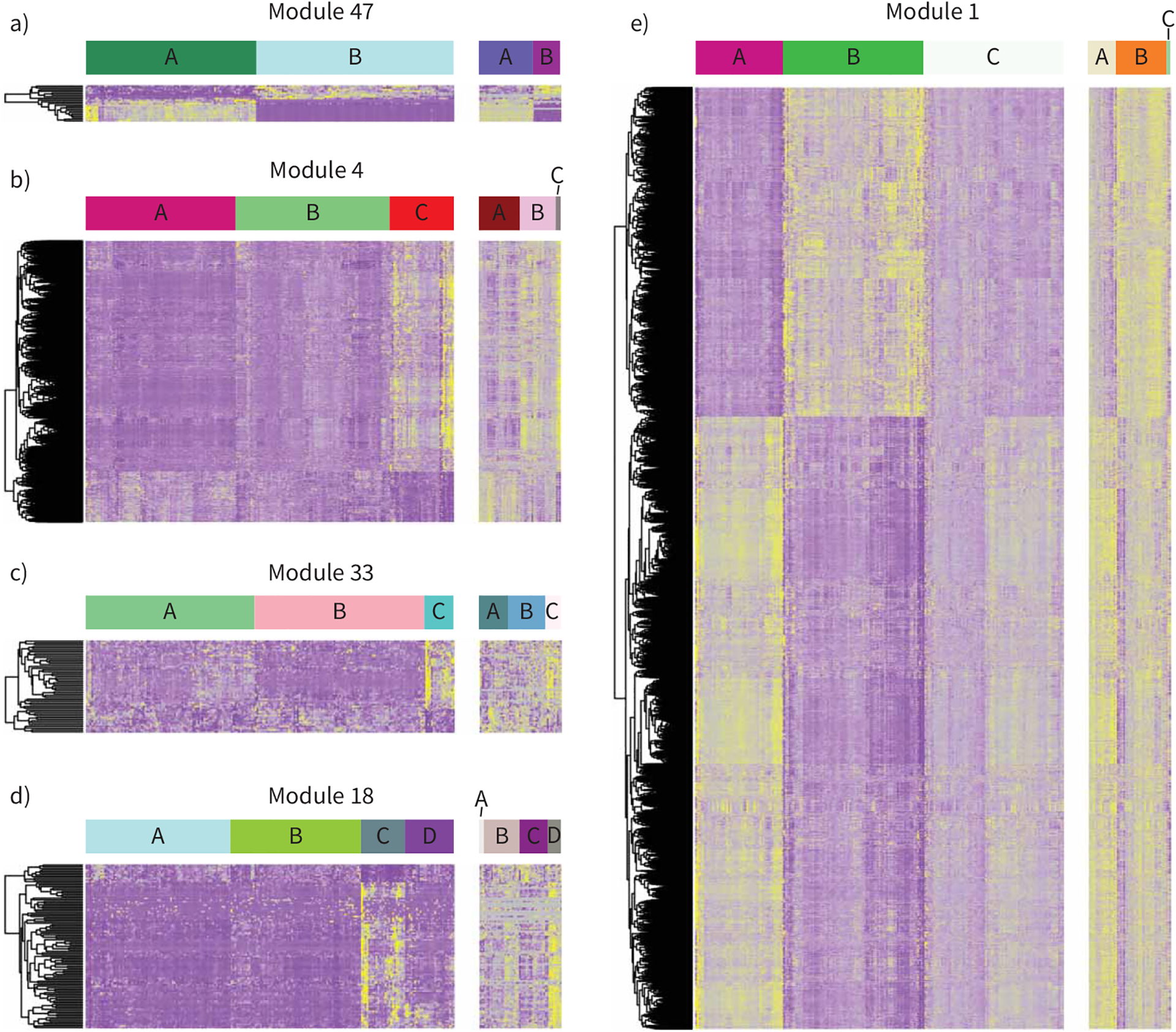
Identification of novel endotypes of sarcoidosis using weighted gene co-expression network analysis gene modules. Heatmaps show the gene expression pattern of five selected gene modules and their corresponding results of K-means clustering of patients in the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study as a discovery cohort (left panels) and Freiburg as a validation cohort (right panels). Each row represents a gene and each column represents a patient. Panels a) to e) are heatmaps of the modules 1, 47, 4, 33 and 18, respectively. The patient clusters (endotypes) identified by K-means clustering are labelled as clusters A, B, C and D at the top of the heatmaps.
Clustering based on module 47 revealed separation of sarcoidosis patients based on sex in two clusters (A: male, B: female) but no other phenotypic traits (figure 4a).
Clustering based on genes in module 4 identified clusters of patients who differed clinically with one group having significantly more hilar and mediastinal lymphadenopathy, larger lymph nodes, increased Scadding stage, less remitting phenotype and more lymphocytes in BAL (figure 4b, cluster C) than the others. These patients had a history of increased exposure to woodfire smoke and had more skin and kidney involvement. Increased T-cell immune response and decreased signal transduction via cAMP and protein kinase A was observed in this cluster of patients, suggesting that these patients have an acute lymphocytic inflammation (figure 4b, supplementary table E3).
Clustering patients based on genes in module 33 revealed clusters of patients who differed clinically with one group (cluster C; figure 4c), having significantly more lymphocytes in BAL and more lymph organ involvement, more exposure to sand, less fatigue and less work in the house than patients in clusterB. Patients in cluster C were more chronic and had higher multiorgan involvement (skin) than cluster A (figure 4c, supplementary table E3).
Clustering patients based on genes in module 18 identified clusters of patients who differed clinically and environmentally with one distinguished group (cluster C; figure 4d) having significantly more patients from US states Connecticut, New Jersey, New York, Pennsylvania and Tennessee; more lung, joint and kidney involvement; and increased urinary calcium; and were exposed to more wood coal before diagnosis. However, these patients have less mediastinal lymphadenopathy and micronodules than patients in cluster B (figure 4d, supplementary table E3). Increased IL-1 and IL-18 immune and inflammatory responses were observed in cluster C (figure 4d).
Clustering patients based on genes in module 1 revealed clusters of patients who differed clinically and environmentally with one group having significantly more mediastinal lymphadenopathy and reticular abnormality in the lung, less multiorgan phenotype (affected eyes), more environment effects (aluminum) and with more subjects living in US states Arizona and Tennessee. Upregulation of apoptotic, immune response and development pathways related to phosphoinositide 3-kinase (PI3K) activation was observed in the same cluster of patients (cluster A; figure 4e, supplementary table E3).
Validation using an independent cohort
Endotype validation
To validate the endotypes we discovered, we used the genome-wide BAL transcriptome data and clinical traits from an independent cohort of sarcoidosis patients (Freiburg cohort). 12 clinical traits (Scadding stage, age, gender, BAL cell differential and PFTs) were available for both the GRADS and the Freiburg cohorts (table 1). Patients in both cohorts were clustered using genes from each of the four novel modules and gender module independently and revealed a similar gene expression pattern visually (figure 4). The two extreme patient clusters defined by each module in both cohorts were compared for the 12 clinical common traits. Validated traits were considered if p-values from the two cohorts were either both significant (p<0.05) or both insignificant (p>0.05) (table 3 using original data, supplementary table S3 using data adjusted for BAL cell differentials). This analysis showed that the endotype of sex (module 47) was fully validated for its significant association with sex. Both modules 4 and 33 were mainly defined by CT scan features (figure 3a), which were not available in the Freiburg cohort for validation. However, for both modules, the significant association with macrophage, lymphocyte and neutrophil differential were validated and no association with age and PFTs (basal and predicted) was found in either cohort. Module 18, a chronic sarcoidosis endotype, was validated for its association with macrophage and lymphocyte differential. No association with any other traits was found in either cohort. Module 1, an extraocular organ involvement and PI3K activation, had a significant but weak association with PFT % pred values (figure 3a, table 3). This association was not validated possibly due to the weak association strength. No association with age, gender, Scadding stage, cell differentials, PFTs (basal) and FEV1/FVC ratio was found in either cohort. In general, validation for modules 47, 4, 33 and 18 were all significant with a less stringent significance level (hypergeometric test p<0.1) chosen due to the small number of overlapping features (table 3). Gene expression data adjustment for BAL cell differentials removed correlation with important clinical traits (supplementary table S3), suggesting that cell differentials are indeed indicative of sarcoidosis severity.
TABLE 3.
Association between chosen molecular endotypes and the 12 overlapping clinical traits in Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) and Freiburg cohorts for validation
| Module 47: gender (p<0.03) | Module 4: hilar lymphadenopathy and acute lymphocytic inflammation (p=0.08) | Module 33: multiorgan involvement with increased immune response (p<0.07) | Module 18: chronic sarcoidosis (p<0.01) | Module 1: extraocular organ involvement and PI3K activation (p=0.58) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GRADS (A versus B) p-value | Freiburg (A versus B) p-value | GRADS (A versus C) p-value | Freiburg (A versus C) p-value | GRADS (B versus C) p-value | Freiburg (B versus C) p-value | GRADS (B versus C) p-value | Freiburg (A versus D) p-value | GRADS (A versus B) p-value | Freiburg (B versus C) p-value | |
| Scadding stage | 0.02 | 0.11 | 0.01 | 0.34 | 0.53 | 0.06 | 0.24 | 0.15 | 0.44 | 0.79 |
| Age | 0.80 | 0.59 | 0.07 | 0.66 | 0.91 | 0.28 | 0.66 | 0.78 | 0.45 | 0.29 |
| Gender | <5×10 −4 | 2.1×10 −11 | 0.24 | 9×10 −3 | 0.43 | 0.29 | 0.64 | 0.08 | 0.86 | 1 |
| Macrophages | 0.75 | 0.05 | 7×10 −8 | 5×10 −3 | 2×10 −5 | 1×10 −3 | 0.04 | 0.01 | 0.53 | 0.33 |
| Lymphocytes | 0.53 | 0.05 | 9×10 −8 | 5×10 −3 | 2×10 −5 | 1×10 −3 | 0.03 | 0.01 | 0.35 | 0.44 |
| Neutrophils | 0.16 | 0.47 | 0.01 | 0.03 | 0.06 | 8×10 −3 | 0.16 | 0.05 | 0.46 | 0.004 |
| Eosinophils | 0.80 | 0.84 | 8×10 −5 | 0.39 | 0.03 | 0.38 | 0.85 | 0.14 | 0.11 | 0.40 |
| FVC | 9×10 −15 | 3×10 −3 | 0.23 | 0.33 | 0.23 | 0.30 | 0.82 | 0.5 | 0.11 | 0.19 |
| FEV 1 | 2×10 −9 | 8×10 −3 | 0.62 | 0.30 | 0.47 | 0.39 | 0.80 | 0.38 | 0.06 | 0.22 |
| FVC % pred | 0.98 | 0.14 | 0.79 | 0.64 | 0.97 | 0.08 | 0.82 | 0.33 | 0.01 | 0.19 |
| FEV1 % pred | 0.78 | 0.75 | 0.84 | 0.91 | 0.55 | 0.08 | 0.65 | 0.22 | 0.02 | 0.28 |
| FEV 1 /FVC ratio | 0.36 | 0.03 | 0.26 | 0.71 | 0.25 | 0.72 | 0.29 | 1.00 | 0.90 | 0.87 |
Each entry is a p-value for the difference of the given clinical trait between the two extreme clusters of patients identified using the corresponding gene module in the corresponding cohort. The p-values in the column title assess the significance of overlap/validation between the two cohorts calculated using a hypergeometic test. Bold type represents statistical significance. PI3K: phosphoinositide 3-kinase; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s.
Phenotypic trait validation using supervised analysis
We also examined the overlap in genes significantly associated with each of the 12 overlapping traits between GRADS and Freiburg cohorts for validation of individual phenotypic trait in supervised analysis. Due to the small sample size of Freiburg cohort, we considered genes with a p-value <0.05 as significant in each cohort for comparison. Significant overlap was observed in five out of the 10 phenotypic traits (Scadding stage, neutrophils %, lymphocytes %, FVC %, FEV1 %; supplementary table S4). 1682 genes overlapped for Scadding with SPRY2, PLA2G7, CHIT1, RFTN1 and MMP9 among the most positively correlated. For FVC % pred and FEV1 % pred in both cohorts (regardless of endotype), we found 16 and 39 genes, respectively (Chi-squared p-values 7.3×10−15 and 1.5×10−2, respectively; supplementary table E4). Among the top negatively correlated genes with FVC % pred in both cohorts were COL1A2, IGFBP6, MAT2A, AQP9, GJA1 and EPDR1. The 39 genes highly negatively correlated with FEV1 % pred included CYP51A1, FADS1, COL1A1, HSPA7, LDLR and NFKB1. SIGLEC6, GIMAP6, GEMIN7, RAPGEF5, A2M and GHRL were among the most positively correlated with lymphocyte percentage, suggestive of increased inflammatory and immune response.
The substantial replication of the results despite differences in cohorts support the validity of our results.
Discussion
Our study based on genome-wide transcriptome analysis of 209 BAL samples represents the largest effort to examine the BAL transcriptome in sarcoidosis with pulmonary involvement. Using both supervised and unsupervised analysis, our study revealed specific gene profiles correlated with activity and severity of disease including immune, inflammatory and profibrotic mediators. Most importantly, our study identified four new groups of sarcoidosis patients with specific molecular, clinical and environmental characteristics.
The unsupervised analysis identified four gene modules that were strongly correlated with CT imaging features of pulmonary involvement, Scadding stage and BAL cell differentials. These modules further identified groups of patients with 1) hilar lymphadenopathy and acute lymphocytic inflammation, 2) extraocular organ involvement and PI3K activation, 3) chronic sarcoidosis and 4) multiorgan involvement with increased immune response. Identification of BAL gene modules represents a robust and novel molecular approach to address clinical heterogeneity of sarcoidosis patients initially classified into eight clinical phenotypes in the GRADS study [15]. BAL gene modules had the strongest correlation with multiple clinical features suggestive of pulmonary involvement, which might be the top factor to consider for further patient phenotyping, as suggested previously [43]. The gender response gene module clearly separated female from male patients based on expression of 15 out of 21 genes, reflecting the accuracy and sensitivity of our method. The same gene modules were confirmed in the Freiburg cohort and were suggestive of the same endotypes regardless of clinical and demographic differences present between two cohorts. Previously, unsupervised clustering approaches used solely clinical patient characteristics to successfully subgroup sarcoidosis patients based on organ involvement (lung, abdominal, heart, muscle and extrapulmonary) or to modulate sarcoidosis based on personal and environmental factors [44, 45]. Our study is the first to combine multiple clinical and environmental factors with BAL transcriptome data, in an unsupervised approach, to phenotype sarcoidosis with pulmonary involvement.
The supervised analysis identified numerous divergent and convergent gene expression patterns associated with hilar lymphadenopathy, parenchymal or airway involvement in sarcoidosis. Our results suggest that different transcriptional programmes affect the three major phenotypic manifestations of pulmonary involvement in sarcoidosis with Th1 and Th17 associated with hilar lymphadenopathy, TGFB1 and MTOR signalling for parenchymal involvement and IL-7 and IL-2 for airway involvement. These responses were among identified pathways known to be involved in pathogenesis of sarcoidosis [7, 9, 46, 47]. An increase in reticular abnormality observed in chest radiology of sarcoidosis patients was associated with increased molecular signalling in BAL such as growth factor, apoptosis/survival, mTORC1 and immune CD28 signalling. The activation of these signalling pathways is suggestive of BAL cells involvement in granuloma formation and fibrosis in sarcoid lungs [48–50]. Genes associated with race (black) were also associated with hilar lymphadenopathy, aberrant lipids pathways, parenchymal or airway involvement in sarcoidosis when race was used as individual variable in supervised analysis, but not in combination with other clinical and environmental traits in unsupervised analysis. Thus, our data do not allow determination of whether gene expression differences are race specific or directly related to the presence of sarcoidosis, as described previously [51], or due to differences in disease severity and manifestations. Genes increasing with increased lymphocyte fraction in BAL reveal shared transcriptional programmes related to hilar lymphadenopathy and bronchial wall thickening. Genes increasing with increased macrophage fraction in BAL are associated with increased cell differentiation and development pathways such as PI3K/AKT, MAPK and BMP7 signalling, suggesting indirectly that these BAL macrophages were in contact with inflammatory sites and granuloma core in lungs [52, 53]. Genes positively correlated with decreased eosinophil fraction in BAL were associated with decreased airway thickness and could reflect the level of lung inflammation, as well as on severity and progression of sarcoidosis.
Our study has several limitations including sample size, heterogeneity of clinical and environmental features and confounding effect of BAL cell differentials. Although our study collected a relatively large sample size for transcriptomics analysis, the original design that aimed for equal recruitment of patients with a wide range of presentations of sarcoidosis [15] resulted in a substantial fragmentation and heterogeneity of the patient populations. Despite these limitations we have been able to identify statistically significant gene expression patterns that associated with clinical attributes in our supervised analysis, and four distinct endotypes in our unsupervised analysis. While the validation cohort was different from our cohort in size, racial and clinical diversity, and clinical information collected, we were able to replicate our major findings, supporting future more detailed prospective validations of our findings. Another key limitation of our study is the impact of the BAL cell differentials on gene expression data. In many of our findings it was impossible to distinguish between the effect of gene expression changes, and the effect of change in cell counts, as has happened for gene modules 4 (hilar lymphadenopathy and acute lymphocytic inflammation) and 33 (multiorgan involvement with increased immune response) but not for modules related to sex, chronic sarcoidosis (module 18) or extraocular organ involvement (module 1). While an important distinction, this does not detract from the value of the information, as clearly cell counts do not provide any information regarding molecular mechanisms and pathways, whereas our data provide the information about the genes differentially expressed and relevant to distinct clinical attributes regardless of the cells involved. Considering the statistically significant and biological plausible gene expression signals we uncovered for sarcoidosis clinical attributes and endotypes, our results support future studies utilising novel technologies such as single-cell RNA sequencing [54] to better understand the mechanistic role of our discoveries. Finally, our study was not designed to specifically study the effects of race, disease duration or specific therapy on gene expression and indeed we did not find significant gene expression changes associated with these attributes. Our results suggest that treatment [55] and race effects [56] should be studied specifically, prospectively in studies using repeated sampling and single cell technologies as previously mentioned.
In summary, our study identified gene profiles associated with major phenotypic manifestations of pulmonary involvement in sarcoidosis, as well as identified four novel endotypes that help to better stratify patients in the future. Our findings support the design of future studies to focused on specific attributes of sarcoidosis, and the use of novel single cell profiling technologies leading to novel therapeutic interventions and biomarkers.
Supplementary Material
Acknowledgements:
We thank all patients participating in the GRADS study for contributing samples.
Conflict of interest:
M. Vukmirovic has nothing to disclose. X. Yan has nothing to disclose. K.F. Gibson has nothing to disclose. M. Gulati reports grants from NIH, during the conduct of the study; personal fees for advisory board work and other (PI/publication committee) from Boehringer Ingelheim, other (lectures) from France Foundation, other (PI/centre director) from Pulmonary Fibrosis Foundation, grants from NIH and Sarcoidosis Research Foundation, outside the submitted work. J.C. Schupp has nothing to disclose. G. DeIuliis has nothing to disclose. T.S. Adams has nothing to disclose. B. Hu has nothing to disclose. A. Mihaljinec has nothing to disclose. T.N. Woolard has nothing to disclose. H. Lynn has nothing to disclose. N. Emeagwali has nothing to disclose.E.L Herzog reports grants from NIH, Sanofi, Bristol Myers and Promedior, personal fees for consultancy from Boehringer Ingelheim and Pfizer, outside the submitted work. E.S. Chen has nothing to disclose. A. Morris reports grants from NIH, during the conduct of the study. J.K. Leader has nothing to disclose. Y. Zhang has nothing to disclose. J.G.N. Garcia has nothing to disclose. L.A. Maier grants from NIH (1U01 HL112695-01, U01 HL112707-03) and NIH/NCRR (UL1TRR002535), during the conduct of the study; grants from National Institutes of Health (1R01 HL127461-01A1, R01HL136681-01A1, 1R01 HL140357-01A1, R01HL136681-01A1), FSR, University of Cinncinati under a Mallinckrodt foundation, MNK14344100, ATYR1923-C-002, outside the submitted work; and is a member of the FSR scientific advisory board, for which no compensation is received. R.G. Collman reports grants from National Institutes of Health, during the conduct of the study. W.P. Drake has nothing to disclose. M.J. Becich reports grants from NCATS, NCI, PCORI, NHLBI and CDC NIOSH, other (startup) from SpIntellx, during the conduct of the study; other (startup) from SpIntellx, outside the submitted work; and has patents SpIntellx (multiple) pending.H. Hochheiser has nothing to disclose. S.R. Wisniewski has nothing to disclose. P.V. Benos has nothing to disclose. D.R. Moller reports grants from NHLBI (1U01HL112708), during the conduct of the study; personal fees for consultancy from Merck, aTYR and Roivant, personal fees for advisory board work from SarcoMed, personal fees for consultancy/witness from Legal Expert, other (royalties) from Hodder Education and Taylor & Francis Group, outside the submitted work; has patents number 9,683,999 B2 issued, and number 9,977,029 B2 issued; is Chairman and Chief Technical Officer of Sarcoidosis Diagnostic Testing, LLC (a company whose goal is to develop a diagnostic blood test for sarcoidosis) and has received funding including past salary support under the NHLBI STTR programme, grant R41 HL129728 more than 3 years ago; and is a former member of the Scientific Advisory Board of the Foundation for Sarcoidosis Research. A. Prasse reports personal fees for lectures and consultancy and non-financial support for meeting attendance from Boehringer Ingelheim and Roche, personal fees for lectures from Novartis and AstraZeneca, personal fees for consultancy from Amgen, Pliant and Nitto Denko, outside the submitted work. L.L. Koth has nothing to disclose. N. Kaminski reports personal fees for consultancy and/or advisory board work from Biogen Idec, Boehringer Ingelheim, Third Rock, Samumed, NuMedii, Indaloo, Theravance, LifeMax, Three Lake Partners, RohBar and Pliant, non-financial support from Miragen, equity with Pliant, a grant from Veracyte; all outside the submitted work; and has a patent New Therapies in Pulmonary Fibrosis and on Peripheral Blood Gene Expression that have been licensed to Biotech.
Support statement:
This work is supported by NIH grants: U01 HL112707, U01 HL112694, U01 HL112695, U01 HL112696, U01 HL112702, U01 HL112708, U01 HL112711, U01 HL112712, UL1 RR029882, UL1 RR025780, R01 HL110883, R01 HL114587, R01 HL127349, U01 HL137159, CTSI U54 grant 9 UL1 TR000005, CDC NMVB 5U24 OH009077, CTSA UL1 TR002535, R21 LM012884, German Network for Lung Research (DZL/ BREATH) and Fraunhofer CIMD Forschungscluster. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999; 160: 736–755. [DOI] [PubMed] [Google Scholar]
- 2.Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357: 2153–2165. [DOI] [PubMed] [Google Scholar]
- 3.Swigris JJ, Olson AL, Huie TJ, et al. Sarcoidosis-related mortality in the United States from 1988 to 2007. Am J Respir Crit Care Med 2011; 183: 1524–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and detection of sarcoidosis. An Official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med 2020; 201: e26–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baughman RP, Costabel U, du Bois RM. Treatment of sarcoidosis. Clin Chest Med 2008; 29: 533–548. [DOI] [PubMed] [Google Scholar]
- 6.Morgenthau AS, Iannuzzi MC. Recent advances in sarcoidosis. Chest 2011; 139: 174–182. [DOI] [PubMed] [Google Scholar]
- 7.Rosenbaum JT, Pasadhika S, Crouser ED, et al. Hypothesis: sarcoidosis is a STAT1-mediated disease. Clin Immunol 2009; 132: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lockstone HE, Sanderson S, Kulakova N, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med 2010; 181: 1367–1375. [DOI] [PubMed] [Google Scholar]
- 9.Gharib SA, Malur A, Huizar I, et al. Sarcoidosis activates diverse transcriptional programs in bronchoalveolar lavage cells. Respir Res 2016; 17: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou T, Zhang W, Sweiss NJ, et al. Peripheral blood gene expression as a novel genomic biomarker in complicated sarcoidosis. PLoS One 2012; 7: e44818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maertzdorf J, Weiner J 3rd, Mollenkopf HJ, et al. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci USA 2012; 109: 7853–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koth LL, Solberg OD, Peng JC, et al. Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am J Respir Crit Care Med 2011; 184: 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su R, Li MM, Bhakta NR, et al. Longitudinal analysis of sarcoidosis blood transcriptomic signatures and disease outcomes. Eur Respir J 2014; 44: 985–993. [DOI] [PubMed] [Google Scholar]
- 14.Schupp JC, Vukmirovic M, Kaminski N, et al. Transcriptome profiles in sarcoidosis and their potential role in disease prediction. Curr Opin Pulm Med 2017; 23: 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moller DR, Koth LL, Maier LA, et al. Rationale and design of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study. Sarcoidosis protocol. Ann Am Thorac Soc 2015; 12: 1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med 2004; 170: 1324–1330. [DOI] [PubMed] [Google Scholar]
- 17.Kim S, Herazo-Maya JD, Kang DD, et al. Integrative phenotyping framework (iPF): integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genomics 2015; 16: 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu JH, Zang W, Vukmirovic M, et al. Gene coexpression networks reveal novel molecular endotypes in alpha-1 antitrypsin deficiency. Thorax 2021; 76: 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer Y, Tedrow J, de Bernard S, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015; 52: 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poliska S, Csanky E, Szanto A, et al. Chronic obstructive pulmonary disease-specific gene expression signatures of alveolar macrophages as well as peripheral blood monocytes overlap and correlate with lung function. Respiration 2011; 81: 499–510. [DOI] [PubMed] [Google Scholar]
- 22.Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 2010; 53: 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap LM, Lottaz D, Ahmad T, et al. Polymorphisms in the MEP1A gene: a role in IBD? Inflamm Bowel Dis 2006; 12: Suppl. 2, S19. [Google Scholar]
- 24.Bucova M, Suchankova M, Tibenska E, et al. Diagnostic value of TREM-1 and TREM-2 expression in bronchoalveolar lavage fluid in sarcoidosis and other lung diseases. Bratisl Lek Listy 2015; 116: 707–713. [DOI] [PubMed] [Google Scholar]
- 25.Bergantini L, Bianchi F, Cameli P, et al. Prognostic biomarkers of sarcoidosis: a comparative study of serum chitotriosidase, ACE, lysozyme, and KL-6. Dis Markers 2019; 2019: 8565423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pignatti P, Brunetti G, Moretto D, et al. Role of the chemokine receptors CXCR3 and CCR4 in human pulmonary fibrosis. Am J Respir Crit Care Med 2006; 173: 310–317. [DOI] [PubMed] [Google Scholar]
- 27.Burgstaller G, Oehrle B, Gerckens M, et al. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J 2017; 50: 1601805. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol 2012; 3: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Königshoff M, Balsara N, Pfaff E-M, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PloS One 2008; 3: e2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunaga H, Matsui H, Ueno M, et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat Commun 2013; 4: 2563. [DOI] [PubMed] [Google Scholar]
- 31.Lal S, Wong ZW, Jada SR, et al. Novel SLC22A16 polymorphisms and influence on doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenomics 2007; 8: 567–575. [DOI] [PubMed] [Google Scholar]
- 32.Burke RR, Stone CH, Havstad S, et al. Racial differences in sarcoidosis granuloma density. Lung 2009; 187: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer KC, Raghu G. Bronchoalveolar lavage for the evaluation of interstitial lung disease: is it clinically useful? Eur Respir J 2011; 38: 761–769. [DOI] [PubMed] [Google Scholar]
- 34.Gimferrer I, Farnós M, Calvo M, et al. The accessory molecules CD5 and CD6 associate on the membrane of lymphoid T cells. J Biol Chem 2003; 278: 8564–8571. [DOI] [PubMed] [Google Scholar]
- 35.Brown MH, Cantrell DA, Brattsand G, et al. The CD2 antigen associates with the T-cell antigen receptor CD3 antigen complex on the surface of human T lymphocytes. Nature 1989; 339: 551–553. [DOI] [PubMed] [Google Scholar]
- 36.Georgiev H, Ravens I, Papadogianni G, et al. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol 2018; 9: 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudemiller N, Lund H, Jacob HJ, et al. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension 2014; 63: 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuenca M, Sintes J, Lányi A, et al. CD84 cell surface signalling molecule: an emerging biomarker and target for cancer and autoimmune disorders. Clin Immunol 2019; 204: 43–49. [DOI] [PubMed] [Google Scholar]
- 39.Ebert LM, McColl SR. Up-regulation of CCR5 and CCR6 on distinct subpopulations of antigen-activated CD4+ T lymphocytes. J Immunol 2002; 168: 65–72. [DOI] [PubMed] [Google Scholar]
- 40.Facco M, Baesso I, Miorin M, et al. Expression and role of CCR6/CCL20 chemokine axis in pulmonary sarcoidosis. J Leukoc Biol 2007; 82: 946–955. [DOI] [PubMed] [Google Scholar]
- 41.Spagnolo P, Renzoni EA, Wells AU, et al. C-C chemokine receptor 5 gene variants in relation to lung disease in sarcoidosis. Am J Respir Crit Care Med 2005; 172: 721–728. [DOI] [PubMed] [Google Scholar]
- 42.Cuenca M, Puñet-Ortiz J, Ruart M, et al. Ly9 (SLAMF3) receptor differentially regulates iNKT cell development and activation in mice. Eur J Immunol 2018; 48: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culver DA, Baughman RP. It’s time to evolve from Scadding: phenotyping sarcoidosis. Eur Respir J 2018; 51: 1800050. [DOI] [PubMed] [Google Scholar]
- 44.Schupp JC, Freitag-Wolf S, Bargagli E, et al. Phenotypes of organ involvement in sarcoidosis. Eur Respir J 2018; 51: 1700991. [DOI] [PubMed] [Google Scholar]
- 45.Ramos-Casals M, Kostov B, Brito-Zerón P, et al. How the frequency and phenotype of sarcoidosis is driven by environmental determinants. Lung 2019; 197: 427–436. [DOI] [PubMed] [Google Scholar]
- 46.Broos CE, Hendriks RW, Kool M. T-cell immunology in sarcoidosis: disruption of a delicate balance between helper and regulatory T-cells. Curr Opin Pulm Med 2016; 22: 476–483. [DOI] [PubMed] [Google Scholar]
- 47.Chen ES. Innate immunity in sarcoidosis pathobiology. Curr Opin Pulm Med 2016; 22: 469–475. [DOI] [PubMed] [Google Scholar]
- 48.Schnerch J, Prasse A, Vlachakis D, et al. Functional toll-like receptor 9 expression and CXCR3 ligand release in pulmonary sarcoidosis. Am J Respir Cell Mol Biol 2016; 55: 749–757. [DOI] [PubMed] [Google Scholar]
- 49.Bonham CA, Strek ME, Patterson KC. From granuloma to fibrosis: sarcoidosis associated pulmonary fibrosis. Curr Opin Pulm Med 2016; 22: 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linke M, Pham HT, Katholnig K, et al. Chronic signalling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol 2017; 18: 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salazar A, Pintó X, Mañá J. Serum amyloid A and high-density lipoprotein cholesterol: serum markers of inflammation in sarcoidosis and other systemic disorders. Eur J Clin Invest 2001; 31: 1070–1077. [DOI] [PubMed] [Google Scholar]
- 52.Facco M, Cabrelle A, Teramo A, et al. Sarcoidosis is a Th1/Th17 multisystem disorder. Thorax 2011; 66: 144–150. [DOI] [PubMed] [Google Scholar]
- 53.Ostadkarampour M, Eklund A, Moller D, et al. Higher levels of interleukin IL-17 and antigen-specific IL-17 responses in pulmonary sarcoidosis patients with Löfgren’s syndrome. Clin Exp Immunol 2014; 178: 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams TS, Schupp JC, Poli S, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 2020; 6: eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baughman RP, Lower EE. Treatment of sarcoidosis. Clin Rev Allergy Immunol 2015; 49: 79–92. [DOI] [PubMed] [Google Scholar]
- 56.Rybicki BA, Sinha R, Iyengar S, et al. Genetic linkage analysis of sarcoidosis phenotypes: the sarcoidosis genetic analysis (SAGA) study. Genes Immun 2007; 8: 379–386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.