

SUMMARY
Salivary adenoid cystic carcinoma (ACC) is a rare, biologically unique biphasic tumor that consists of malignant myoepithelial and luminal cells. MYB and Notch signaling have been implicated in ACC pathophysiology, but in vivo descriptions of these two programs in human tumors and investigation into their active coordination remain incomplete. We utilize single-cell RNA sequencing to profile human head and neck ACC, including a comparison of primary ACC with a matched local recurrence. We define expression heterogeneity in these rare tumors, uncovering diversity in myoepithelial and luminal cell expression. We find differential expression of Notch ligands DLL1, JAG1, and JAG2 in myoepithelial cells, suggesting a paracrine interaction that may support oncogenic Notch signaling. We validate this selective expression in three published cohorts of patients with ACC. Our data provide a potential explanation for the biphasic nature of low- and intermediate-grade ACC and may help direct new therapeutic strategies against these tumors.
In brief
Parikh et al. use single-cell RNA-seq to characterize the malignant and stromal cells in adenoid cystic carcinoma (ACC). Myoepithelial cells express Notch ligands, while luminal cells show Notch signaling, consistent with a paracrine mechanism and the biphasic nature of ACC. Recurrent ACC is more luminal, with increased Notch signaling.
Graphical Abstract
INTRODUCTION
Adenoid cystic carcinoma (ACC) of the head and neck is a rare tumor arising from the major and minor salivary glands. Histologically, low- and intermediate-grade ACC tumors are biphasic, with cribriform or tubular regions intermixed with solid components.1 At a cellular level, these tumors are characterized by luminal epithelial cells and abluminal myoepithelial cells.1
A hallmark of ACC is chromosomal translocations involving MYB (or MYBL1).2,3 A unifying feature of these rearrangements is that they juxtapose the MYB locus to super-enhancer regions that are bound by MYB protein, resulting in a positive feedback loop that drives MYB overexpression.4 MYB is subsequently able to target cell-cycle regulators CDK6 and GMNN that drive proliferation, as well as TP63, NOTCH1, Notch activators JAG1 and JAG2, and Notch repressor SPEN, which drive diverse expression programs across myoepithelial and luminal epithelial cells in these tumors.4 Recent genomic analyses have uncovered recurrent NOTCH1 mutations in ACC and suggested a critical role for oncogenic Notch signaling, particularly among high-grade and recurrent/metastatic tumors.5–7
Despite progress in identifying MYB and NOTCH1 as critical regulators in ACC, detailed descriptions of the expression programs within myoepithelial and luminal cells and mechanisms by which these two major cell types coordinate to drive oncogenesis are lacking. Furthermore, although these tumors have a low mutational burden, they have significant intra-tumoral genetic heterogeneity,5 suggesting that previously utilized bulk methods may be insufficient to adequately profile the cellular phenotypes present in these tumors.
Here, we utilize single-cell RNA sequencing (scRNA-seq) using the SMART-seq2 protocol8,9 to profile individual cells from treatment-naive human head and neck ACC tumors and systematically characterize the underlying landscape of expression heterogeneity.
RESULTS
A single-cell transcriptomic atlas for human salivary ACC
To systematically explore expression heterogeneity in ACC, we profiled individual cells from seven treatment-naive patients, as well as one matched local recurrence (Tables S1 and S2). The pathologic diagnosis of ACC was confirmed by a head and neck pathologist (W.C.F.) and a second pathologist with extensive experience with the histopathology of ACC (J.C.A.) based on the presence of classical cribriform morphology and an immunohistochemical staining profile consistent with ACC (Table S1). Freshly resected specimens were dissociated to single-cell suspensions using a protocol we optimized (see STAR Methods), flow sorted to deplete non-viable and CD45-positive cells, and profiled by SMART-seq2 (Figure 1A), which provided full-length transcripts for sequencing and analysis.8,9 After quality control, we retained transcriptomes for a total of 1,702 high-quality single cells.
Figure 1. Single-cell RNA sequencing (scRNA-seq) reveals expression heterogeneity in adenoid cystic carcinoma.
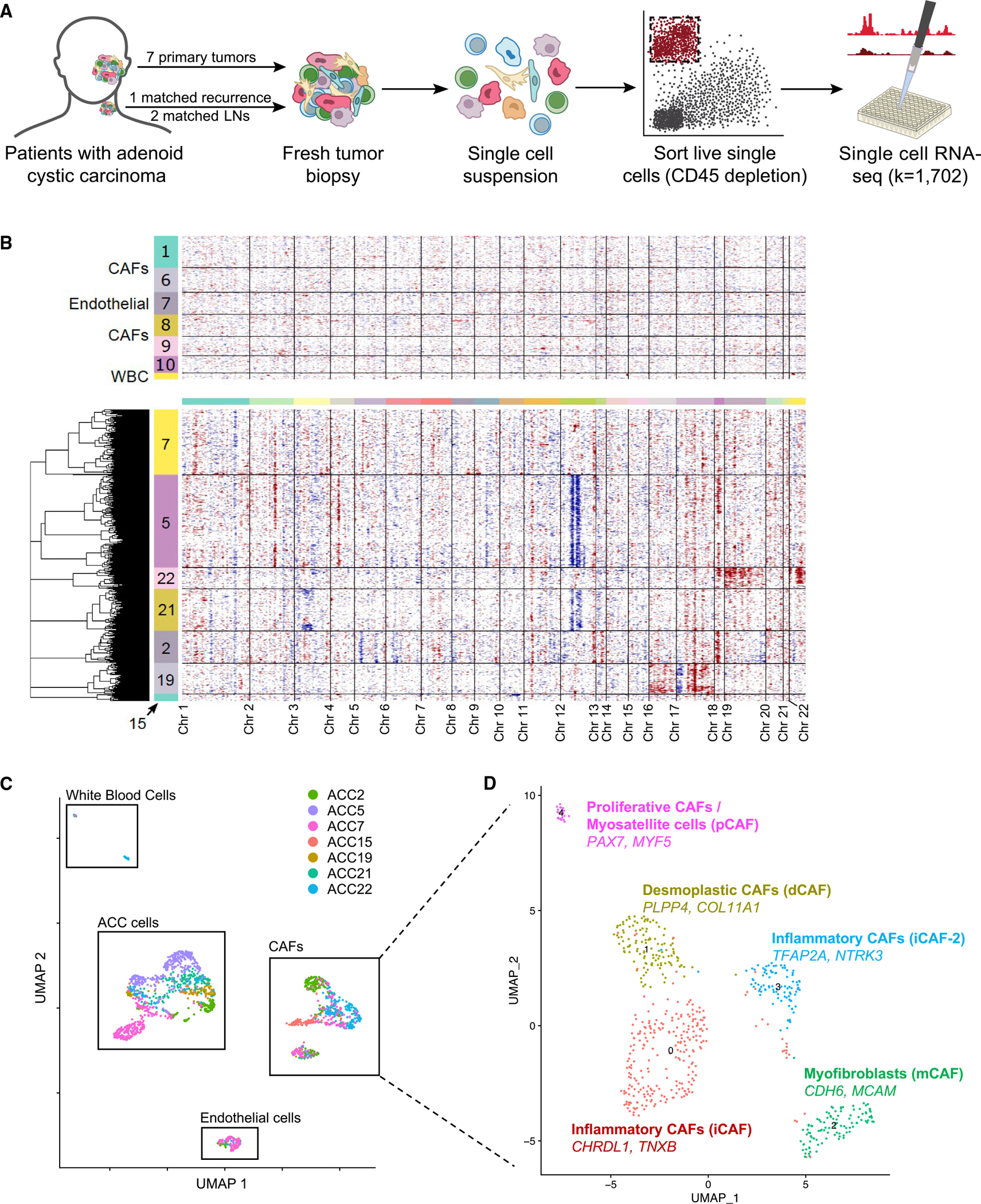
(A) Schematic shows scRNA-seq workflow. Freshly resected tumors were dissociated to single-cell suspensions and fluorescence-activated cell sorted (FACS) into 96-well plates after CD45 depletion. Library prep and sequencing were performed per the SMART-Seq2 protocol.
(B) Heatmap of inferred CNAs across all cells, as predicted by inferCNV, separates malignant cells from non-malignant cells. Each row represents a cell, and each column represents a genomic locus. Non-malignant cells are displayed in the top panel, with numbers in the color bar representing cluster number corresponding to Figure S1A. Malignant cells are shown in the bottom panel, with numbers in the color bar corresponding to the patient from whom the cells were obtained.
(C) Uniform manifold approximation and projection (UMAP) of all cells that passed quality control (QC) filtering shows distinct clusters of cancer cells, cancer-associated fibroblasts, endothelial cells, and white blood cells. Cells are colored by patient sample.
(D) UMAP of all cancer-associated fibroblasts (CAFs) that passed QC filtering, annotated by the two most differentially upregulated genes and by previously reported pan-cancer CAF subtypes,10 shows five putative subtypes of CAFs present across ACC tumors.
Clustering of all cells based on expression revealed 14 groups (Figure S1A). To identify malignant cell clusters, we inferred copy-number alterations (CNAs) from the transcriptomic data (see STAR Methods).11 Cells in clusters exhibiting high CNAs were classified as malignant (Figures 1B and S1B), and cells categorized as malignant also expressed a previously described ACC signature (Figure S1C),12 as well as MYB (Figure S1C), supporting their identity as ACC cells. The most common CNAs were loss of segments of chromosome 12 and gain of segments of chromosome 18 (Figure 1B). However, there were no coherent CNAs across tumors, consistent with prior analyses emphasizing a high degree of genetic diversity in ACC tumors.5,13 There were also no significant associations between specific CNAs and malignant cell clusters. The distinct clustering of malignant from non-malignant cells (Figure 1C), combined with the high concordance between these measures, provided confidence in our classification approach. In total, our dataset included 951 malignant and 751 non-malignant cells.
We then utilized major cell-type markers to classify non-malignant cells as cancer-associated fibroblasts (CAFs; Figure S1D), endothelial cells (Figure S1E), and white blood cells (Figure S1F), which did not exhibit CNAs (Figure 1B). Given the large number of CAFs sequenced, we further investigated CAF diversity by scoring for signatures of pan-cancer CAF subtypes (Figures 1D and S1G; Table S3).10 We identified proliferative CAFs expressing myosatellite cell markers PAX714 and MYF515 (Figure 1D, pink) that were present only in tumors originating in the oral cavity (ACC7, ACC21, and ACC22), consistent with the presence of submucosal muscle tissue in the oral cavity. We identified myofibroblasts (Figure 1D, green) expressing pericyte markers CDH6 and MCAM16 that were seen across tumors. Two other CAF clusters corresponded to inflammatory CAF subtypes. One expressed TFAP2A and NTRK3 (Figure 1D, blue), which have been associated with epithelial-to-mesenchymal transition (EMT) and poor prognosis in multiple cancer types,17,18 while the other expressed CHRDL1 and TNXB (Figure 1D, maroon), which have previously been associated with improved prognosis and decreased EMT, invasion, and migration in oral and breast cancers.19–21 The final cluster corresponded to desmoplastic CAFs expressing adipose stromal cell markers PLPP4 and COL11A1 (Figure 1D, yellow), which have previously been associated with poor prognosis.22 Taken together, our findings begin to characterize the ACC stromal microenvironment that may contribute to tumor biology and prognosis in previously unappreciated ways.
Defining in vivo myoepithelial and luminal programs in ACC
We next explored malignant cell expression heterogeneity. Globally, malignant cells clustered by patient (Figures 2A and 2B), consistent with other single-cell tumor studies,23–29 except for two clusters of cells from multiple patients (Figure 2B, clusters 3 and 4). When clusters were annotated based on known myoepithelial (Figure S2A) and luminal (Figure S2B) markers from prior studies, as well as global expression along a myoepithelial-luminal axis (Figure 2C), patient-specific clusters scored highly for myoepithelial markers, while those comprised of cells from multiple patients scored highly for luminal markers. These data suggest that myoepithelial cell expression may vary substantially between patients, while luminal cells are dominated by a shared expression program. Notably, both patient-specific and shared clusters have previously been described in breast cancer.29,30 We also demonstrated that myoepithelial and luminal markers were inversely related across cells (Figure S2C), supporting our use of the chosen markers for classification.
Figure 2. Heterogeneity of the malignant compartment defines in vivo myoepithelial and luminal programs in ACC.
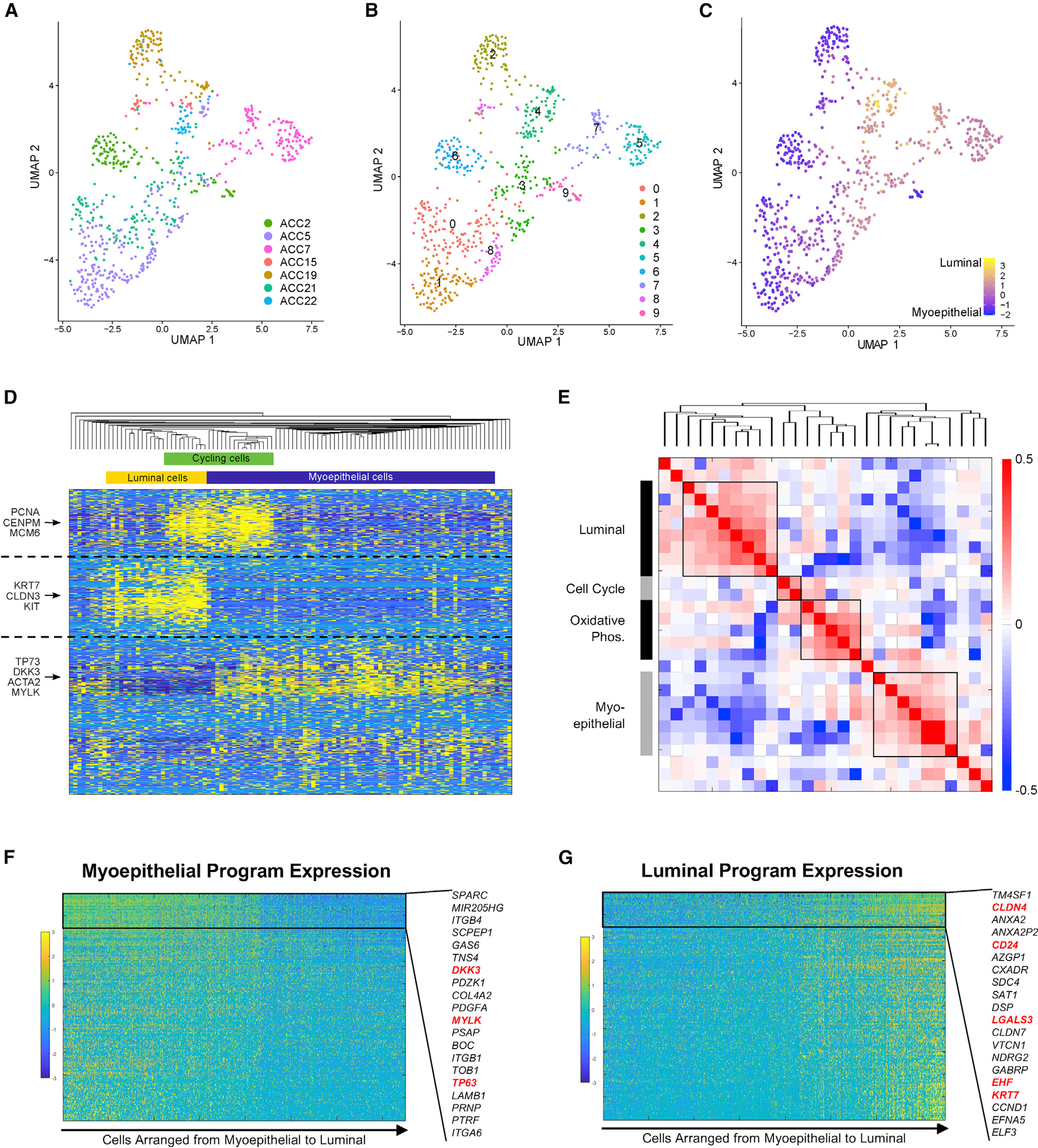
(A) UMAP shows malignant cells from primary tumors with cells colored by patient sample, demonstrating that inter-tumoral heterogeneity is prominent.
(B) UMAP shows 10 clusters identified by unsupervised Louvain clustering using the same UMAP coordinates as (A).
(C) UMAP shows all malignant cells from primary tumors colored on a gradient from myoepithelial (blue) to luminal (yellow), based on marker expression, using the same UMAP coordinates as (A) and (B). Luminal cells cluster together regardless of patient of origin.
(D) Heatmap shows expression of the top 200 genes in each of the four NMF programs detected in an individual representative sample, ACC2. Key genes are annotated on the left, and inferred cell identity is shown on the top. Cells (columns) and genes (rows) are clustered by hierarchical clustering according to Pearson correlation. Myoepithelial, luminal, and cell-cycle programs are detected by unsupervised NMF analysis of a single tumor.
(E) Heatmap shows Spearman correlations between each NMF metagene from each patient, clustered by hierarchical clustering. This metaclustering analysis shows consistent luminal, cell-cycle, oxidative phosphorylation, and myoepithelial programs across tumors.
(F) Gene-expression heatmap shows expression of top 200 genes in the myoepithelial NMF program in malignant cells. Genes (rows) are sorted by variation across myoepithelial cells, and cells (columns) are sorted from myoepithelial to luminal according to myoepithelial and luminal marker scores. The 20 least variable genes are highlighted, and previously described myoepithelial markers are shown in red.
(G) Gene-expression heatmap shows expression of top 200 genes in the luminal NMF program in malignant cells. Genes (rows) are sorted by variation across luminal cells, and cells (columns) are sorted from myoepithelial to luminal. The 20 least-variable genes are highlighted, and previously described luminal markers are shown in red.
Given the prominent inter-tumoral heterogeneity of myoepithelial cells, we then defined expression heterogeneity in each tumor independently using unbiased non-negative matrix factorization (NMF) (Figure 2D, representative tumor, ACC2) and compared the resulting differential programs across patients in a metaclustering analysis (Figure 2E). This approach revealed four malignant cell expression programs present across multiple tumors in our cohort (Figure 2E; Table S3). Gene set enrichment analysis (Table S4) revealed that one program was enriched for genes (CLDN3, KRT19, KIT) with preferential expression in luminal-like cancer cells31 (p < 9 × 10−17) and developing epithelium (p < 10−5) (“luminal program”). A second program included myoepithelial markers (MYLK, ACTA2, TP63, CAV1) and was enriched for genes involved in EMT (p < 4 × 10−15) (“myoepithelial program”). A third program was enriched for genes (NDUFA1, COX5B, UQCR10) involved in oxidative phosphorylation (p < 10−24) (“OxPhos program”). The final program was enriched for genes expressed during the cell cycle (p < 4 × 10−28) (“cell-cycle program”).32 Classifying cells according to these shared programs (Figure S2D), there was significant overlap between the OxPhos and myoepithelial programs and a lesser degree of overlap between the OxPhos and luminal programs, suggesting that OxPhos cells may be myoepithelial or luminal cells undergoing a high degree of oxidative phosphorylation. We found similar overlaps between the cell-cycle program and myoepithelial and luminal programs, suggesting that these are cycling myoepithelial or luminal cells (Figures 2D and S2D).
To verify consistency between the myoepithelial and luminal meta-programs and our marker-based classification (Figure 2C), we plotted cells across a myoepithelial-luminal axis defined by known markers and examined the expression of the genes driving the myoepithelial (Figure 2F) and luminal (Figure 2G) meta-programs, which we identified de novo. There was clearly higher expression for the corresponding meta-program in the cells that scored highly for the known markers of each cell type, validating our classifications. We thus provide a detailed description of in vivo myoepithelial and luminal cell programs in ACC and demonstrate how they may vary across cells and tumors.29,30
MYB and Notch expression heterogeneity suggest a paracrine interaction that drives Notch signaling
We next assessed MYB and Notch expression heterogeneity given their established roles in ACC pathogenesis.4,33 We first scored malignant cells for expression of MYB (Figure 3A), as well as MYBL1 and MYBL2 (Figure S3A). MYB was expressed in a subset of cells in all tumors (Figure 3A), and immunohistochemistry validated diffuse MYB positivity (Figure 3B, brown).
Figure 3. MYB and Notch expression heterogeneity in the malignant compartment suggests paracrine interaction.
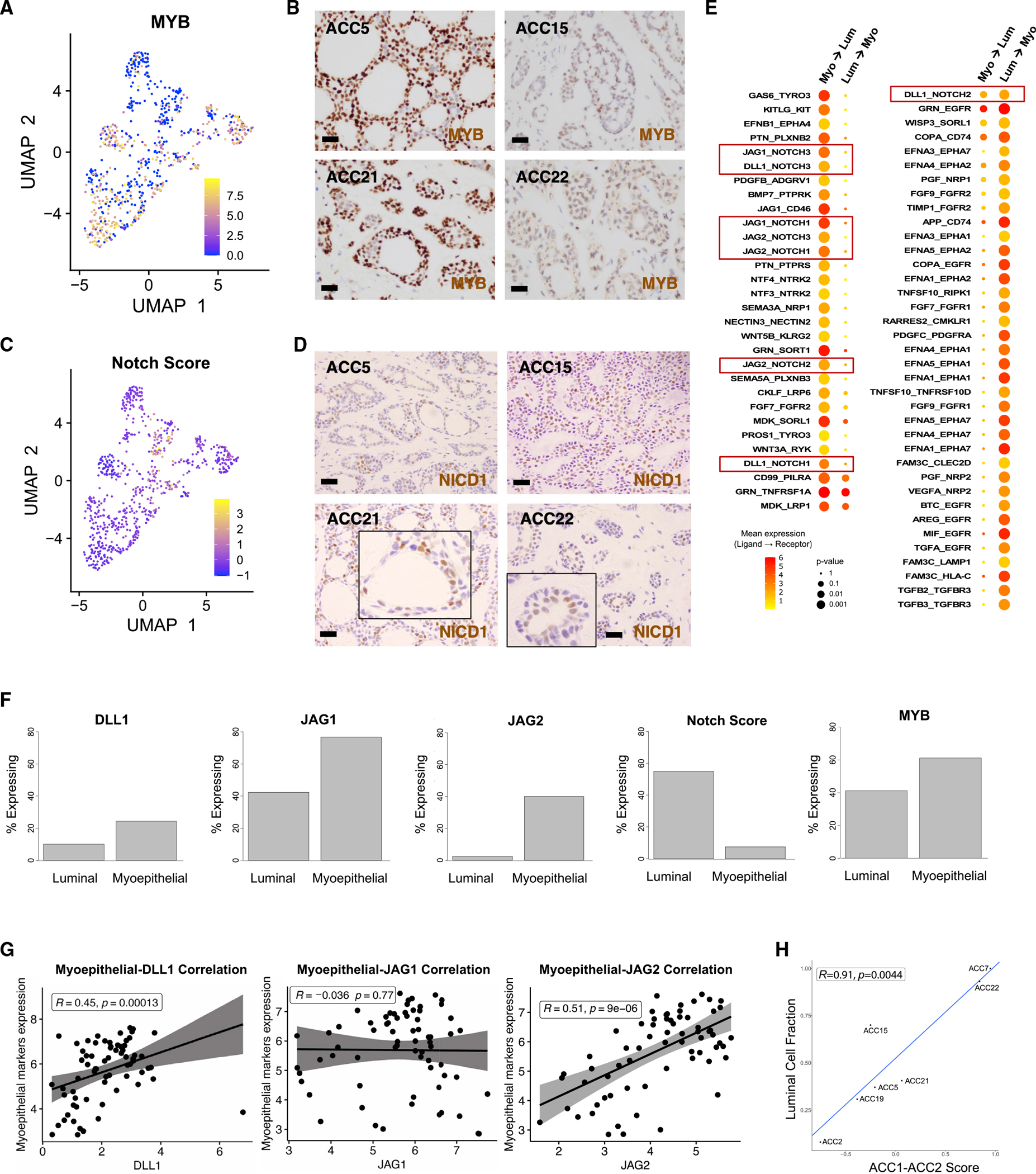
(A) UMAP shows malignant cells from primary tumors colored by MYB expression.
(B) 600× images of MYB immunohistochemistry in four representative tumors show strong and diffuse MYB staining. Scale bars represent 100 μm.
(C) UMAP shows malignant cells from primary tumors colored by Notch activation score.
(D) 600× images of NICD1 immunohistochemistry (brown) in four representative tumors shows localization of NICD1 to luminal cells. Scale bars represent 100 μm.
(E) Dot plot shows mean expression and significance of all putative ligand-receptor pairs identified in an unbiased fashion using CellPhoneDB. The color of the circles represents mean expression of the ligand (in myoepithelial cells, left column, or in luminal cells, right column) and the receptor (in luminal cells, left column, or in myoepithelial cells, right column), while the size of the circles represents significance. All ligand-receptor pairs that passed FDR <10% are shown. Red boxes highlight pairs involved in Notch signaling.
(F) Bar plots show percentage of cells expressing Notch ligands DLL1 (p < 5 × 10−4), JAG1 (p < 4 × 10−11), and JAG2 (p < 2 × 10−16), as well as overall Notch signaling (p < 2.0 × 10−14) and MYB expression (p < 2 × 10−4). While Notch signaling is higher in luminal cells, Notch ligands are preferentially expressed by myoepithelial cells.
(G) Scatterplots show Spearman correlation between myoepithelial markers and DLL1 (left panel), JAG1 (middle panel), or JAG2 (right panel) in Dou et al. cohort.34
(H) Scatterplot shows strong positive correlation between previously described ACC1-ACC2 score35 and fraction of luminal cells present across tumors in the cohort.
We next scored malignant cells for expression of NOTCH1 and NOTCH2 (Figure S3B), Notch target genes (Figure S3C), and a Notch expression signature (NRARP, NOTCH3, HES4, HEY1, and HEY2) (Figure 3C). Although NOTCH1 and NOTCH2 were variably expressed, the Notch target genes and expression signature, indicating active Notch signaling, were primarily expressed in luminal cells (Figures 3C and S3C). This finding was supported by higher Notch target expression in cells that were more luminal and less myoepithelial (Figure S3D), as well as Notch target expression in a higher fraction of luminal cells, compared with myoepithelial cells (Figure S3E). Staining for activated intracellular NOTCH1 (NICD1; Figure 3D, brown) confirmed localization to luminal cells, substantiating previous reports that Notch is only activated in luminal cells.36 For reference, classic ACC morphologies are shown in Figure S3F.
As Notch receptors are typically activated by ligands expressed on neighboring cells,37 we then performed an unbiased analysis to identify ligand-receptor interactions and frame Notch signaling within the context of overall cell-cell communication. We uncovered multiple putative ligand-receptor pairs (Figure 3E), suggesting bidirectional signaling between myoepithelial and luminal cells. Our analysis revealed eight potential Notch signaling interactions; in seven of these, the ligand was expressed by myoepithelial cells and the receptor by luminal cells (Figure 3E, red boxes). To further explore these interactions, we examined MYB and Notch ligand expression by cell type. Whereas Notch signaling was primarily detected in luminal cells (7.6% versus 55%, p < 3 × 10−16, Yates’ chi-squared test), we discovered a preferential expression of Notch ligands DLL1 (24% versus 10%, p < 6 × 10−4), JAG1 (77% versus 42%, p < 9 × 10−11), and JAG2 (40% versus 2.5%, p < 2.3 × 10−16) in myoepithelial cells (Figure 3F). We similarly found higher Notch ligand expression in cells that were more myoepithelial and less luminal (Figure S3G). This relative exclusivity between expression of Notch ligands and active Notch signaling supports a paracrine interaction between myoepithelial and luminal cells that drives Notch signaling in the latter. MYB was expressed in both cell types but in a higher fraction of myoepithelial cells (Figure 3F; 61% versus 41%, p < 2 × 10−4).
To independently validate selective expression of Notch ligands by myoepithelial cells, we examined the correlation between myoepithelial markers and Notch ligands in bulk RNA-seq data from three previously published cohorts12,35,34 with a total of 141 patients with ACC (Figures 3G and S3H). In the largest of these cohorts (n = 75),34 myoepithelial markers were strongly positively correlated with expression of DLL1 (r = 0.45, p < 2 × 10−4) and JAG2 (r = 0.51, p < 9 × 10−6) but not JAG1 (r = −0.036, p = 0.77) (Figure 3G); similar correlations were found in the other two cohorts (Figure S3H).12,35 Examining correlations between luminal markers and Notch ligands revealed complementary results (Figure S4A). We also assessed the correlations between Notch targets and myoepithelial (Figure S4B) and luminal (Figure S4C) markers in these same datasets. We found significant negative correlations of myoepithelial markers with HES4 in two datasets and HEY1 in one (Figure S4B); conversely, we found significant positive correlations of luminal markers with HEY1 in two datasets and HEY2 in one (Figure S4C). Together, these results support the selective activation of Notch in luminal cells and selective expression of Notch ligands in myoepithelial cells.
Finally, we leveraged our single-cell data to understand previously described ACC subtypes (ACC-1 and ACC-2) that notably differed by p63, MYC, and Notch expression, as well as activating Notch mutations.35 Given that p63 is a canonical myoepithelial marker,4 we sought to understand whether entire tumors correspond to a single subtype or each tumor is comprised of different proportions of cell populations, each corresponding to a given subtype. We found that all tumors contained both ACC-1 and ACC-2 cells. Luminal cells scored highly for the ACC-1 signature, while myoepithelial cells scored highly for the ACC-2 signature, regardless of whether the tumor was higher in ACC-1 or ACC-2, overall. Accordingly, the ACC-1/ACC-2 score captured the relative fractions of luminal and myoepithelial cells across tumors (Figure 3H). Moreover, in our small cohort, the two patients with the highest ACC-1/ACC-2 score both exhibited aggressive clinical disease, with distant metastasis and lower disease-free survival (Table S2), suggesting that previously demonstrated survival associations35 may, in fact, be related to luminal cell fraction.
Recurrent ACC demonstrates higher Notch signaling
Although ACC is a rare disease with delayed recurrences,33 our cohort did include one exceptional case (ACC5) of a patient with a both primary ACC and local recurrence profiled by scRNA-seq, offering the opportunity to perform a matched transcriptomic comparison. Globally, malignant cells from the recurrence clustered separately from cells from the primary tumor (Figures 4A and S5A), suggesting coherent differences in expression. We scored cells for luminal and myoepithelial signatures (Figure 4B), as well as for individual myoepithelial (Figure S5B) and luminal (Figure S5C) markers and found that these differences reflected a shift toward a more luminal expression program in the recurrence (Figure 4B). Accordingly, we also found significantly higher Notch signaling (p < 5 × 10−10, one-tailed Mann-Whitney test) and reduced MYB expression (p < 4 × 10−8, one-tailed Mann-Whitney test) in the recurrence (Figure 4C), consistent with the differential expression of Notch signaling in luminal cells.
Figure 4. Recurrent tumors may be more luminal and may demonstrate higher Notch signaling.
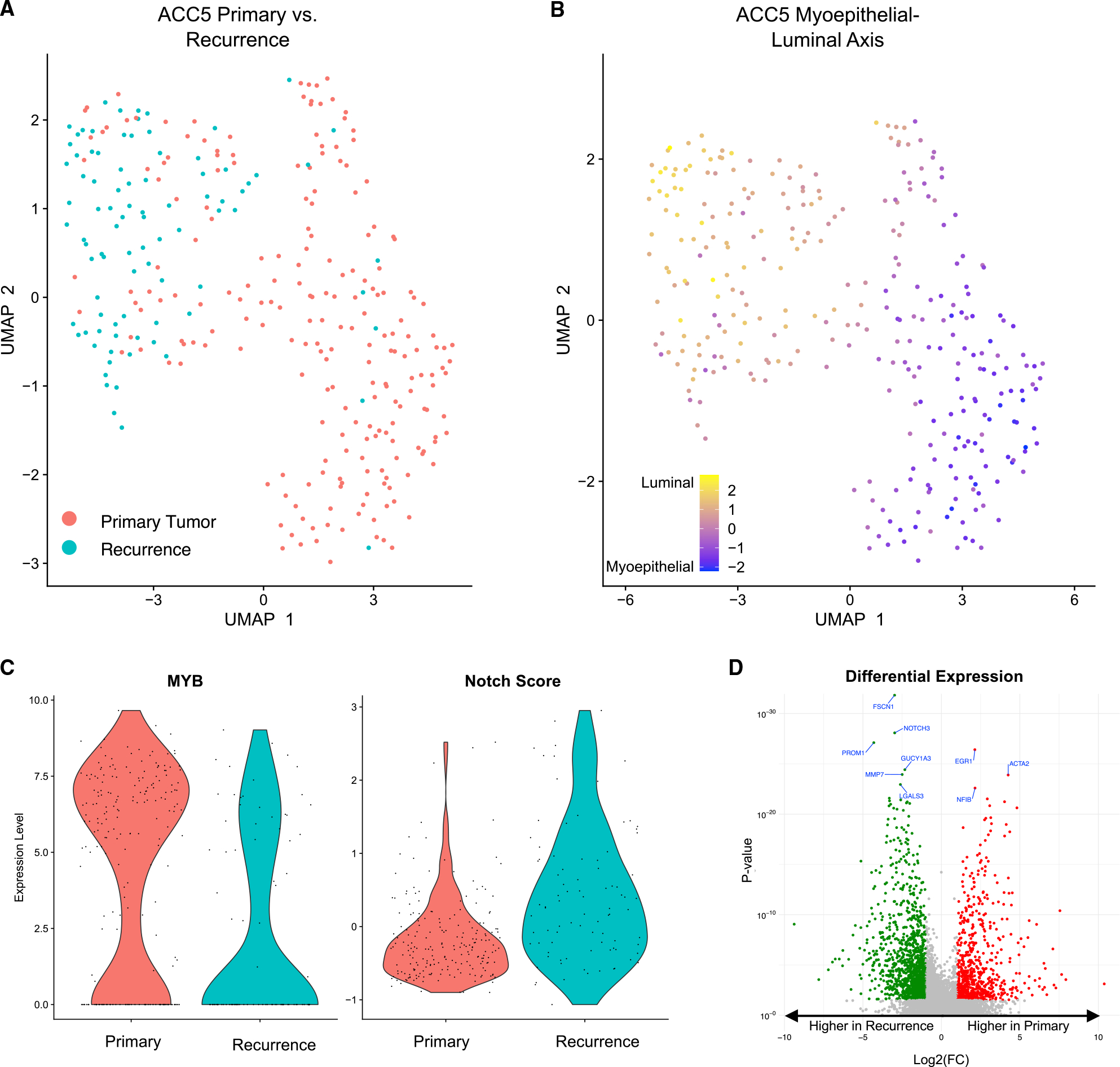
(A) UMAP shows malignant cells from ACC5. Cells from the primary tumor cluster separately from cells from the recurrence.
(B) UMAP shows malignant cells from ACC5 colored by myoepithelial-luminal axis. ACC5 recurrence is more luminal.
(C) Violin plots show MYB expression (left panel) and Notch score (right panel) in ACC5 malignant cells. ACC5 recurrence has lower MYB expression (p < 4 × 10−8, one-tailed Mann-Whitney test) and higher Notch signaling (p < 5 × 10−10, one-tailed Mann-Whitney test).
(D) Volcano plot shows differential gene expression across malignant cells in ACC5 primary tumor and recurrence. Genes colored in red had significantly higher expression (FDR <10%, >2-fold change) in the primary tumor, while genes colored in green had significantly higher expression (FDR <10%, >2-fold change) in the recurrence. Notch target NOTCH3 is a top upregulated gene in the recurrent setting.
We next compared overall gene expression in the recurrence and the primary (Figures 4D and S5D; Table S3). Notably, Notch target NOTCH3 (log2(fold change) = −2.98, p < 8.4 × 10−29; Table S3) was a top differentially expressed gene, consistent with upregulation of Notch signaling in the recurrent setting. Pathway analysis indicated that estrogen response may play a role in recurrence, consistent with previous studies describing a role for the estrogen receptor in ACC biology.38–40
To further explore differences across niches, we examined differential expression between the primary tumor and matched metastatic cervical lymph node (LN) for two patients (ACC7 and ACC22) for whom both samples were captured (Figure S5E; Table S3). There were several differentially expressed genes across niches in ACC7 (Wilcoxon test, false discovery rate [FDR] <5%; Table S3), with COL9A1, APP, and SERPINE2 being the top upregulated genes in the LN. In ACC22, only seven genes were differentially expressed (Table S3), and BST2 was the only upregulated gene in the LN. The only consistently differentially expressed gene was CRYAB, which was downregulated in the LN in both samples.
Finally, given the known prognostic importance of solid-type morphology in ACC,33 we explored expression differences between tumors in our cohort with solid regions (ACC7 and ACC19) and all others with cribriform morphology. We found significantly higher expression of luminal markers in solid-type tumors (Figure S5F; p < 2 × 10−9), consistent with previous descriptions.4 Given these differences, we separately analyzed differential expression in myoepithelial and luminal cells in solid and cribriform tumors. B2M and RPS4Y1 were more highly expressed in both myoepithelial (Table S3) and luminal (Table S3) cells in solid-type tumors compared with cribriform tumors. Among myoepithelial cells specifically, upregulated genes in solid-type tumors included KRT14 and KRT16P3 (Figure S5G; Table S3). Overall, there were more differentially expressed genes in myoepithelial than luminal cells, consistent with the greater variability in myoepithelial than luminal cells in our cohort (Figure 2C).
DISCUSSION
Here, we utilized scRNA-seq using full-length transcriptome profiling in fresh human head and neck ACC specimens to define cellular expression heterogeneity in these rare but deadly tumors. This analysis provides an important insight into the expression landscape of these human salivary gland tumors (Figure 1). Utilizing this approach, we defined in vivo myoepithelial and luminal cell programs (Figure 2; Table S3) and demonstrated distinct clustering of these two cell types (Figure 2), consistent with the known bicellular differentiation of ACC. While myoepithelial cells demonstrated considerable inter-tumoral heterogeneity, luminal cells from multiple patients clustered together, suggesting a more conserved luminal expression state across tumors (Figure 2). Interestingly, this finding mirrors previous results of single-cell profiling in triple-negative breast cancer, which also revealed shared luminal cell subpopulations.29
Our data also provide in vivo support for a paracrine interaction between myoepithelial and luminal cells that drives Notch signaling in the latter. We validated the localization of Notch signaling to luminal epithelial cells36 and demonstrated a selective expression of Notch ligands DLL1, JAG1, and JAG2 in myoepithelial cells (Figure 3F). We further validated this selective expression in bulk transcriptomic data from three published cohorts,12,35,34 demonstrating a strong correlation between myoepithelial markers and Notch ligands DLL1 and JAG2 (Figures 3G and S3H). Taken together with the previous identification of JAG1 and JAG2 as highly ranked putative MYB targets,4 our results suggest that MYB overexpression may drive oncogenesis in ACC, in part, via Notch ligand expression in myoepithelial cells.
Importantly, a paracrine myoepithelial-luminal interaction provides a direct biological explanation for the biphasic nature of ACC, a phenotype that is well described but poorly understood. By contrast, in high-grade solid-type ACC, which is strongly associated with gain-of-function mutations in NOTCH1 that lead to ligand-independent Notch activation,5 myoepithelial cells are largely or completely absent, consistent with the notion that paracrine signaling may no longer be required for Notch activation.36 Accordingly, our group previously demonstrated that BET inhibitors, which disrupt enhancer function, slowed tumor growth in low- and intermediate-grade ACC primagraft models but were ineffective against high-grade ACCs harboring activating Notch pathway mutations.4 Our data supporting a paracrine interaction also highlight the power of our approach using scRNA-seq as previous explorations into this possibility have been restricted by technical limitations in staining for Notch ligands.
Our cohort included a patient for whom we profiled both the primary tumor and a matched local recurrence. This matched transcriptomic comparison of a primary tumor and recurrence in ACC is notable given the rarity of surgically accessible recurrent disease. In our dataset, the recurrent sample demonstrated higher Notch signaling and reduced MYB expression compared with the primary tumor (Figure 4), consistent with previous reports of increased Notch signaling and escape mutants with wild-type MYB in recurrent ACC.5 Collectively, these findings suggest that MYB-independent Notch signaling may be increased in recurrent ACC. Furthermore, given that the primary tumor did not highly express Notch overall (Figure 4), our findings suggest that individual Notch-expressing cells within the primary tumor may have seeded a local recurrence. Larger matched cohorts are needed to support this model; however, understanding whether individual treatment-resistant “Notch-high” cells exist even in the absence of high overall Notch signaling is critical as it may support the use of Notch inhibitors in tumors without pervasive Notch activation—a strategy distinct from what is currently employed in most clinical trials.41,42
Our cohort also included two patients for whom we captured both the primary tumor and a matched metastatic LN at the time of surgical resection. Interestingly, there were few coherent expression differences across these two samples (Figure 4D), suggesting that LN metastasis may be more of a passage-drainage process than a biologically active process in these tumors, similar to our findings in oral cavity squamous cell carcinoma.23,24
Limitations of the study
Our findings are tempered by the small number of samples profiled. Larger cohorts, with primary tumors matched with recurrences or distant metastases, are critical to better understand the nuances of Notch signaling in these alternate settings. Computational approaches such as deconvolution of bulk data using single-cell data may be useful given the rarity of this disease and may now be possible with our detailed descriptions of in vivo myoepithelial and luminal programs. Additional functional and spatial characterization of this paracrine interaction will also be important in investigating its role in driving low- and intermediate-grade ACC. With the paucity of well-validated ACC cell lines, other in vitro approaches such as three-dimensional culture models may be useful in capturing the diverse cell populations present within these tumors.
Still, our data provide an important, highly plausible explanation for the biphasic nature of low- and intermediate-grade ACC given the frequent, highly conserved role of Notch signaling in regulating biphasic fate choices during normal development.43 The paracrine interaction we identify suggests there may be potential for therapeutic strategies directed at myoepithelial cells and their activated pathways, including MYB-driven overexpression of Notch ligands, particularly in low- and intermediate-grade tumors. Our work also highlights the importance of further studies of matched specimens to guide therapeutic strategies to prevent local recurrence and regional or distant metastasis, which remain significant challenges in ACC.
Conclusions
We utilize scRNA-seq in fresh human ACC specimens to define the landscape of cellular expression heterogeneity in these rare tumors, including descriptions of in vivo myoepthelial and luminal programs. Our data validate the known localization of Notch signaling to luminal cells and demonstrate localization of Notch ligands to myoepithelial cells, highlighting a potential oncogenic paracrine interaction between these two cell types. We also demonstrate a shift toward increased Notch signaling in recurrent ACC utilizing a comparison of matched primary and recurrent specimens. Our data provide a potential explanation for the biphasic nature of low- and intermediate-grade ACC and indicate the need for further study of matched primary and recurrent specimens to better guide therapeutic strategies targeting Notch signaling in low- and intermediate-grade ACC.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sidharth Puram (sidpuram@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Processed scRNA-seq data were deposited at GEO; raw data will be deposited at dbGaP. Accession numbers are listed in the key resources table.
This paper does not report original code.
Immunohistochemistry data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Monoclonal mouse CD45-vioblue, clone 5B1 | Miltenyi Biotec | Cat#130-092-880; RRID:AB_1103220 |
| Monoclonal rabbit c-MYB, clone EP769Y | Abcam | Cat#ab45150; RRID:AB_778878 |
| Monoclonal rabbit cleaved Notch1, clone D3B8 | Cell Signaling Technology | Cat#4147; RRID:AB_2153348 |
| Calcein AM | ThermoFisher | Cat#C3100MP |
| TO-PRO-3 iodide | ThermoFisher | Cat#T3605 |
|
| ||
| Biological samples | ||
|
| ||
| Human head and neck adenoid cystic carcinoma patient samples (ACC2, ACC5, ACC7, ACC15, ACC19, ACC21, ACC22) | Massachusetts Eye and Ear | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Human Tumor Dissociation Kit | Miltenyi Biotec | Cat#130-095-929 |
| BOND Polymer Refine Detection Kit | Leica | Cat#DS9800 |
|
| ||
| Deposited data | ||
|
| ||
| Processed scRNA-seq data | GEO | GSE210171 |
| Raw scRNA-seq | dbGaP | phs003070.v1 |
|
| ||
| Software and algorithms | ||
|
| ||
| MatLab version 2019b | MathWorks | N/A |
| STAR version 2.5.2 | https://github.com/alexdobin/STAR | N/A |
| Seurat version 4.0.1 | https://github.com/satijalab/seurat | N/A |
| inferCNV version 1.4.0 | https://bioconductor.org/packages/release/bioc/html/infercnv.html | N/A |
| cellphoneDB version 4 | https://github.com/ventolab/CellphoneDB | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Institutional review board approval was obtained. Patients undergoing surgical resection of primary head and neck salivary adenoid cystic carcinoma (ACC) at the Massachusetts Eye and Ear (MEE) were consented preoperatively to take part in the study. Demographic characteristics of human subjects providing samples are summarized in Table S1. Our cohort included seven patients of the following age/gender: 64/F (ACC2), 81/F (ACC5), 91/M (ACC7), 81/M (ACC15), 52/M (ACC19), 57/M (ACC21), and 50/F (ACC22). Fresh biopsies of head and neck salivary ACC were collected at the time of surgical resection. Post-operatively, H&E stained sections were reviewed by a dedicated head and neck pathologist (W.C.F.) and assessed for growth pattern and immunohistochemical staining profile to confirm the pathologic diagnosis of ACC, based upon histomorphologic criteria outlined in the WHO Classification of Head and Neck Tumours.44 Only cases unequivocally classified as ACC were included in the final dataset.
METHOD DETAILS
Tumor dissociation and cell sorting
Samples were minced and dissociated with a Human Tumor Dissociation Kit (Miltenyi Biotec) according to manufacturer guidelines. Single cell suspensions were filtered, and cells were resuspended in PBS with 1% bovine serum albumin (BSA; Sigma-Aldrich). Trypan blue (ThermoFisher Scientific) was used to confirm cell viability of >90% in all samples. Cells were stained with CD45-vioblue (Miltenyi Biotec), as well as 1 μM calcein AM (ThermoFisher Scientific), and 0.22 μM TO-PRO-3 iodide (ThermoFisher Scientific) for viability prior to sorting. Fluorescence-activated cell sorting (FACS) was performed on FACSAria Fusion Special Order System (BD Biosciences) using 488 nm (calcein AM, 530/30 filter), 640 nm (TO-PRO-3, 670/14 filter), and 405 nm (Vioblue, 450/50 filter) lasers. We captured singlet viable cells using standard forward scatter height versus area criteria, as well as calceinhigh and TO-PROlow gates. Subsquently, CD45- cells were captured to enrich for cancer cells and deplete immune cells. Individual cells were sorted into TCL- buffer (QIAGEN) with 1% β-mercaptoethanol in 96-well plates. Plates were snap frozen and stored at −80°C before cDNA synthesis and library construction.
Library construction and sequencing
Full-length single cell RNA-seq libraries were prepared using the SMART-seq2 protocol,8,9 with the following modifications23: Agencourt RNAClean XP beads (Beckman Coulter) were used to purify RNA prior to reverse transcription with Superscript II (ThermoFisher Scientific) or Maxima (ThermoFisher Scientific) reverse transcriptase. Whole transcriptome amplification was performed using the KAPA HiFi HotStart ReadyMix (KAPA Biosystems). cDNA libraries were tagmented using the Nextera XT Library Prep Kit (Illumina) and sequenced as paired-end 38 base reads on a NextSeq 500 (Illumina).
Staining and imaging of tissue sections
Formalin fixed, paraffin-embedded (FFPE) ACC specimens were obtained from the Massachusetts General Hospital (MGH) tissue archives. 5 μm thick sections were cut by the MGH Histopathology Core. MYB staining was performed by the MGH Histopathology Core per standard protocols using rabbit monoclonal c-MYB primary antibody clone EP769Y (Abcam) at 1:100 for 30 minutes, visualized with HRP-linked secondary antibodies, followed by diaminobenzidine (DAB; Dako). NICD1 staining was performed per previously published protocols,36 using rabbit monoclonal antibody clone D3B8 (Cell Signaling Technologies) at 1:50 for 60 minutes. DAB staining was then developed using the Bond Polymer Refine Detection Kit (Leica), and slides were counterstained with hematoxylin. All images were captured with a 60X objective and reviewed by a dedicated head and neck pathologist (W.C.F. or J.C.A.).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell RNA-sequencing data processing
scRNA-seq data were aligned to GRCh38 with STAR version 2.5.245 and counted with featureCounts.46 Clustering of single cells, UMAP, violin plots, and marker gene analyses were conducted with Seurat 4.0.1.47 log2(TPM+1) values were used for Seurat analyses and were normalized by regressing out the number of detected features and the percent of mitochondrial reads. Cells with less than 2000 transcripts or more than 30% mitochondrial reads were removed from the analysis.
Classification of malignant and non-malignant cells
Copy number profiles were estimated using inferCNV version 1.4.0.11 The copy number aneuploidy score was defined as the square root of the average of deviation of diploidy squared. Cells were clustered using the Louvain algorithm as implemented in Seurat, taking into account the first 10 principal components. Cell types were inferred from identified clusters by their expression profiles, and identity of the ACC cluster was further validated by copy number alterations and Gao et al. “ACC signature”. For the latter, specific ACC markers were taken from Gao et al.12 (Table S1, all genes with FC > 2) and their average normalized expression was used to define the “ACC signature” (Figure S1C).
Characterizing heterogeneity of CAFs
For the analysis of CAFs, the 7,500 most variable transcripts across all cells in the CAF clusters were normalized again among themselves (including regression by number of transcripts and percent of mitiochondrial reads). The cells were then reclustered and projected to UMAP coordinates using the same algorithms and parameters. Scoring of CAF clusters by signatures described in Galbo et al.10 was computed by the average expression of the genes in each signature, as described in Table S3 in their manuscript.
Characterizing heterogeneity of malignant cells
The identified cancer cells from each primary tumor and the 15,000 most variable transcripts across these cancer cells were normalized again among themselves (including regression by number of transcripts and percent of mitochondrial reads), and used for downstream analysis. These data were used to recluster cells and compute new UMAP coordinates, using the same algorithm and parameters. For nonnegative matrix factorization,48 we calculated z-scores of the log2(TPM+1) values of all cancer cells of each primary tumor, omitting genes with over 90% negative z-scores, and setting all negative values to 0. We used the MATLAB 2019b implementation of nnmf to identify four metagenes (expression programs) for each tumor and normalized the coefficients to sum to 1. We clustered all metagenes to identify shared programs using hierarchical clustering according to Spearman correlation and computed the average z-score of the metagenes in each of the four clusters. Gene set enrichment for the top 200 genes in each shared program (average metagene) was computed against MSigDB49 using fisher exact test as implemented by HOMER findGO.50
Defining malignant cell subtype scores
Average normalized expression of TP63, TP73, CAV1, CDH3, KRT5, KRT14, ACTA2, TAGLN, MYLK, and DKK3 was used as the myoepithelial score. Average normalized expression of KIT, EHF, ELF5, KRT7, CLDN3, CLDN4, CD24, LGALS3, LCN2, and SLPI was used as the luminal score. When the difference between luminal score and myoepithelial score was higher than 1, the cell was defined as luminal, and when it was less than −1 it was defined as myoepithelial. Average normalized expression of NRARP, NOTCH3, HES4, HEY1, and HEY2 was used as the Notch score, and a positive score was considered indicative of active notch signaling.
Cell-cell communication analysis
Cell-cell communication analysis was conducted with cellphoneDB v4.51 Only ligand-receptor interactions with FDR <10% were selected. Pairs were reordered to show ligand first and receptor second. The Wilcoxon rank sum test was used to detect significant differently expressed genes, considering all genes expressed in at least 10% of at least one of the compared groups of cells. False discovery rate (FDR) was controlled by the Benjamini-Hochberg procedure.
Analysis of bulk RNA-seq data
Bulk RNA-seq data of ACC were obtained from Gao et al.,12 Dou et al.34 and Ferrarotto et al.35 Outliers were identified by hierarchical clustering of samples by complete linkage according to Euclidean distance and filtering out the smallest first branch. This process was repeated twice for each dataset. Spearman correlation of myoepithelial score (as defined above) with Notch ligands was calculated in log2(TPM+1) values (for Dou et al.34 and Ferrarotto et al.35) or RMA values (for Gao et al.12). Scores of ACC-1 and ACC-2 subtypes were calculated as the average z-score of ACC-1 marker expression minus the average z-score of ACC-2 marker expression. ACC-1 markers (MYC, SOX6, SOX8, CTNND2, NOTCH3, BCL2) and ACC-2 markers (TP63, COL17A1, PDGFA, DKK3, EGFR, AXL, PDGFRA) were taken from Figure 3A in Ferrarotto et al.35
Supplementary Material
Highlights.
Single-cell RNA-seq reveals cellular heterogeneity in adenoid cystic carcinoma
Malignant luminal epithelial cells show active Notch signaling
Malignant myoepithelial cells express Notch ligands, suggesting paracrine signaling
Recurrent ACC is more luminal and shows increased Notch signaling
ACKNOWLEDGMENTS
This work was supported by the V Foundation (S.V.P.), Cancer Research Foundation (S.V.P.), Barnes-Jewish Hospital Foundation (S.V.P.), NCI K08CA237732 (S.V.P), NIDCR R21DE31072 (S.V.P.) NIDCR R21DE031366 (S.V.P.), the European Research Council Horizon 2020 grant 949029 (Y.D.), and the Adenoid Cystic Carcinoma Research Foundation (B.E.B., D.T.L., and J.C.A.). The funding sources had no involvement in the design, conduct, and reporting of the research.
Footnotes
DECLARATION OF INTERESTS
J.C.A. is a consultant to Ayala Pharmaceuticals, Remix Therapeutics, and Cellestia, Inc. B.E.B. declares outside interests in Fulcrum Therapeutics, Arsenal Biosciences, HiFiBio, Cell Signaling Technologies, and Chroma Medicine.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111743.
REFERENCES
- 1.Wysocki PT, Izumchenko E, Meir J, Ha PK, Sidransky D, and Brait M (2016). Adenoid cystic carcinoma: emerging role of translocations and gene fusions. Oncotarget 7, 66239–66254. 10.18632/oncotarget.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brayer KJ, Frerich CA, Kang H, and Ness SA (2016). Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov. 6, 176–187. 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persson M, Andrén Y, Mark J, Horlings HM, Persson F, and Stenman G (2009). Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 106, 18740–18744. 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJH, Kluk MJ, Carey CD, Rodig SJ, Sholl LM, Afrogheh AH, et al. (2016). An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 48, 265–272. 10.1038/ng.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AS, Ochoa A, Jayakumaran G, Ho AS, Valero Mayor C, Tepe J, Makarov V, Dalin MG, He J, Bailey M, et al. (2019). Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J. Clin. Invest. 129, 4276–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanna GJ, Bae JE, Lorch JH, Schoenfeld JD, Margalit DN, Tishler RB, Haddad RI, and Chau NG (2020). Long-term outcomes and clinico-genomic correlates in recurrent, metastatic adenoid cystic carcinoma. Oral Oncol. 106, 104690. 10.1016/j.oraloncology.2020.104690. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Yu Y, Fang Y, Huang H, Wu D, Fang H, Bai Y, Sun C, Yu A, Fan Q, et al. (2020). Whole-exome sequencing reveals genetic underpinnings of salivary adenoid cystic carcinoma in the Chinese population. J. Genet. Genomics 47, 397–401. 10.1016/j.jgg.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 9.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 10.Galbo PM, Zang X, and Zheng D (2021). Molecular features of cancer-associated fibroblast subtypes and their implication on cancer pathogenesis, prognosis, and immunotherapy resistance. Clin. Cancer Res. 27, 2636–2647. 10.1158/1078-0432.CCR-20-4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tickle T, Tirosh I, Georgescu C, Brown M, and Haas B (2019). inferCNV: Visualizing large-scale copy number variation in single-cell RNA-seq expression data. http://www.bioconductor.org/packages/devel/bioc/vignettes/infercnv/inst/doc/inferCNV.html.
- 12.Gao R, Cao C, Zhang M, Lopez MC, Yan Y, Chen Z, Mitani Y, Zhang L, Zajac-Kaye M, Liu B, et al. (2014). A unifying gene signature for adenoid cystic cancer identifies parallel MYB-dependent and MYB-independent therapeutic targets. Oncotarget 5, 12528–12542. 10.18632/oncotarget.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho AS, Kannan K, Roy DM, Morris LGT, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al. (2013). The mutational landscape of adenoid cystic carcinoma. Nat. Genet. 45, 791–798. 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maesner CC, Almada AE, and Wagers AJ (2016). Established cell surface markers efficiently isolate highly overlapping populations of skeletal muscle satellite cells by fluorescence-activated cell sorting. Skelet. Muscle 6, 35. 10.1186/s13395-016-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beauchamp JR, Heslop L, Yu DSW, Tajbakhsh S, Kelly RG, Wernig A, Buckingham ME, Partridge TA, and Zammit PS (2000). Expression of CD34 and Myf5 Defines the Majority of Quiescent Adult Skeletal Muscle Satellite Cells. J. Cell Biol. 151, 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng YC, Porfírio-Sousa AL, Ribeiro GM, Arend MC, da Silva Meirelles L, Chen ES, Rosa DS, and Han SW (2021). Analyses of the pericyte transcriptome in ischemic skeletal muscles. Stem Cell Res. Ther. 12, 183. 10.1186/s13287-021-02247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong Y, Feng Y, Zhao J, Lei J, Qiao T, Zhou Y, Lu Q, Jiang T, Jia L, and Han Y (2021). TFAP2A potentiates lung adenocarcinoma metastasis by a novel miR-16 family/TFAP2A/PSG9/TGF-β signaling pathway. Cell Death Dis. 12, 352. 10.1038/s41419-021-03606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Yu Y, Zhang P, Ma G, Zhang M, Liang Y, Jiao W, and Niu H (2021). Identification of NTRK3 as a potential prognostic biomarker associated with tumor mutation burden and immune infiltration in bladder cancer. BMC Cancer 21, 458. 10.1186/s12885-021-08229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cyr-Depauw C, Northey JJ, Tabariè s S, Annis MG, Dong Z, Cory S, Hallett M, Rennhack JP, Andrechek ER, and Siegel PM. (2016). Chordin-like 1 suppresses bone morphogenetic protein 4-induced breast cancer cell migration and invasion. Mol. Cell Biol. 36, 1509–1525. 10.1128/mcb.00600-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Q, Zheng Z, Zhang J, Piao Z, Xin M, Xiang X, Wu A, Zhao T, Huang S, Qiao Y, et al. (2022). Chordin-like 1 regulates epithelial-to-mesenchymal transition and metastasis via the MAPK signaling pathway in oral squamous cell carcinoma. Front. Oncol. 12, 862751. 10.3389/fonc.2022.862751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, Allen KG, Danilo C, Sotgia F, Bonuccelli G, et al. (2008). Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: implications for the response to hormonal therapy. Cancer Biol. Ther. 7, 1212–1225. 10.4161/cbt.7.8.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu K, Cai L, Cui C, de Los Toyos JR, and Anastassiou D (2021). Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adiposederived stromal cells into COL11A1- expressing cancer-associated fibroblasts. PLoS Comput. Biol. 17, e1009228. 10.1371/journal.pcbi.1009228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puram S.v., Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puram S.v., Parikh AS, and Tirosh I (2018). Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol. Cell. Oncol. 5, e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, et al. (2017). Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718. 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, You W-H, Li X, Wang P, Sha B, Liang Y, Qiu J, Zhou J, Hu H, and Lu L (2021). Single-cell RNA-seq reveals transcriptional landscape and intratumor heterogenicity in gallbladder cancer liver metastasis microenvironment. Ann. Transl. Med. 9, 889. 10.21037/atm-21-2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, Specht MC, Bernstein BE, Michor F, and Ellisen LW (2018). Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 9, 3588. 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, et al. (2017). Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 8, 15081. 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, et al. (2006). Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 25, 2273–2284. 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 32.Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, Van De Rijn M, Botstein D, and Brown PO (2004). Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2, E7. 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ, and Thomas CY (2016). Adenoid cystic carcinoma: a review of recent advances, molecular targets, and clinical trials. Head Neck 38, 620–627. 10.1002/hed.23925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dou S, Li R, He N, Zhang M, Jiang W, Ye L, Yang Y, Zhao G, Yang Y, Li J, et al. (2021). The immune landscape of Chinese head and neck adenoid cystic carcinoma and clinical implication. Front. Immunol. 12, 618367. 10.3389/fimmu.2021.618367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrarotto R, Mitani Y, McGrail DJ, Li K, Karpinets T.v., Bell D, Frank SJ, Song X, Kupferman ME, Liu B, et al. (2021). Proteogenomic analysis of salivary adenoid cystic carcinomas defines molecular subtypes and identifies therapeutic targets. Clin. Cancer Res. 27, 852–864. 10.1158/1078-0432.CCR-20-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sajed DP, Faquin WC, Carey C, Severson EA, Afrogheh AH, Johnson C, Blacklow SC, Chau NG, Lin DT, Krane JF, et al. (2017). Diffuse staining for activated NOTCH1 correlates with NOTCH1 mutation status and is associated with worse outcome in adenoid cystic carcinoma. Am. J. Surg. Pathol. 41, 1473–1482. 10.1097/PAS.0000000000000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aster JC, Pear WS, and Blacklow SC (2017). The varied roles of notch. Annu. Rev. Pathol. 12, 245–275. 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques YMFS, Giudice FS, Freitas VM, Abreu e Lima M.d.C.C.,Hunter KD, Speight PM, Machado de Sousa SCO, and Machado de Sousa SCO (2012). Oestrogen receptor β in adenoid cystic carcinoma of salivary glands. Histopathology 60, 609–616. 10.1111/j.1365-2559.2011.04095.x. [DOI] [PubMed] [Google Scholar]
- 39.Sumida T, Ishikawa A, Kamata YU, Nakano H, Yamada T, and Mori Y (2016). Estrogen enhances malignant phenotypes in human salivary adenoid cystic carcinoma cells. Anticancer Res. 36, 2793–2798. [PubMed] [Google Scholar]
- 40.Barrera JE, Shroyer KR, Said S, Hoernig G, Melrose R, Freedman PD, Wright TA, and Greer RO (2008). Estrogen and progesterone receptor and p53 gene expression in adenoid cystic cancer. Head Neck Pathol. 2, 13–18. 10.1007/s12105-007-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrarotto R, Ho A, Wirth LJ, Muzaffar J, Rodriguez C, Dekel E, Walker RM, Nadri-Shay C, and Vergara-Silva A (2019). ACCURACY a phase (P) II trial of AL101, a pan-Notch inhibitor, in recurrent/metastatic (R/M) adenoid cystic carcinoma (ACC) patients (pts) with Notch activating mutations (Notch act mut): preliminary safety and efficacy data. Ann. Oncol. 30, v465–v466. 10.1093/annonc/mdz252.040. [DOI] [Google Scholar]
- 42.Ferrarotto R, and Heymach JV (2017). Taking it up a NOTCH: a novel subgroup of ACC is identified. Oncotarget 8, 81725–81726. 10.18632/oncotarget.20879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siebel C, and Lendahl U (2017). Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294. 10.1152/physrev.00005.2017.-Notch. [DOI] [PubMed] [Google Scholar]
- 44.Tumours of the salivary glands. In WHO Classification of Head and Neck Tumours, El-Naggar A, Chan J, Grandis J, Takata T, and PJ S, eds. (Lyon IARC press; ), pp. 159–202. [Google Scholar]
- 45.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 47.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. 10.1038/nbt.4096.Integrating. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berry MW, Browne M, Langville AN, Pauca VP, and Plemmons RJ (2007). Algorithms and applications for approximate nonnegative matrix factorization. Comput. Stat. Data Anal. 52, 155–173. 10.1016/j.csda.2006.11.006. [DOI] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.3969/j.issn.0372-2112.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Alonso L, Handfield LF, Roberts K, Nikolakopoulou K, Fernando RC, Gardner L, Woodhams B, Arutyunyan A, Polanski K, Hoo R, et al. (2021). Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 53, 1698–1711. 10.1038/s41588-021-00972-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed scRNA-seq data were deposited at GEO; raw data will be deposited at dbGaP. Accession numbers are listed in the key resources table.
This paper does not report original code.
Immunohistochemistry data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Monoclonal mouse CD45-vioblue, clone 5B1 | Miltenyi Biotec | Cat#130-092-880; RRID:AB_1103220 |
| Monoclonal rabbit c-MYB, clone EP769Y | Abcam | Cat#ab45150; RRID:AB_778878 |
| Monoclonal rabbit cleaved Notch1, clone D3B8 | Cell Signaling Technology | Cat#4147; RRID:AB_2153348 |
| Calcein AM | ThermoFisher | Cat#C3100MP |
| TO-PRO-3 iodide | ThermoFisher | Cat#T3605 |
|
| ||
| Biological samples | ||
|
| ||
| Human head and neck adenoid cystic carcinoma patient samples (ACC2, ACC5, ACC7, ACC15, ACC19, ACC21, ACC22) | Massachusetts Eye and Ear | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Human Tumor Dissociation Kit | Miltenyi Biotec | Cat#130-095-929 |
| BOND Polymer Refine Detection Kit | Leica | Cat#DS9800 |
|
| ||
| Deposited data | ||
|
| ||
| Processed scRNA-seq data | GEO | GSE210171 |
| Raw scRNA-seq | dbGaP | phs003070.v1 |
|
| ||
| Software and algorithms | ||
|
| ||
| MatLab version 2019b | MathWorks | N/A |
| STAR version 2.5.2 | https://github.com/alexdobin/STAR | N/A |
| Seurat version 4.0.1 | https://github.com/satijalab/seurat | N/A |
| inferCNV version 1.4.0 | https://bioconductor.org/packages/release/bioc/html/infercnv.html | N/A |
| cellphoneDB version 4 | https://github.com/ventolab/CellphoneDB | N/A |