

Abstract
Up to 40% of pheochromocytomas (PCCs) and paragangliomas (PGLs) are hereditary. Germline mutations/deletions in fumarate hydratase (FH) cause hereditary leiomyomatosis and renal cell carcinoma syndrome which manifests predominantly with FH-deficient uterine/cutaneous leiomyomas and renal cell carcinomas (RCCs)—tumors characterized by loss of immunohistochemical (IHC) expression of FH and/or positive staining for S-(2-succino)-cysteine. Occasional patients develop PCC/PGL. We investigated the incidence, morphologic, and clinical features of FH-deficient PCC/PGL. We identified 589 patients with PCC/PGLs that underwent IHC screening for FH and/or S-(2-succino)-cysteine. Eight (1.4%) PCC/PGLs were FH deficient (1.1% in an unselected population). The median age for FH-deficient cases was 55 (range: 30 to 77 y) with 50% arising in the adrenal. All 4 with biochemical data were noradrenergic. Two (25%) metastasized, 1 dying of disease after 174 months. Germline testing was performed on 7 patients, 6 of whom had FH missense mutations. None were known to have a significant family history before presentation or developed cutaneous leiomyomas, or FH-deficient RCC at extended follow-up. The patient wild-type for FH on germline testing was demonstrated to have somatic FH mutation and loss of heterozygosity corresponding to areas of subclonal FH deficiency in her tumor. One patient did not undergo germline testing, but FH mutation was demonstrated in his tumor. We conclude that FH-deficient PCC/PGL are underrecognized but can be identified by IHC. FH-deficient PCC/PGL are strongly associated with germline missense mutations but are infrequently associated with leiomyoma or RCC, suggesting there may be a genotype-phenotype correlation. FH-deficient PCC/PGL may have a higher metastatic risk.
Key Words: fumarate hydratase, FH, 2SC, pheochromocytoma, paraganglioma
Paragangliomas (PGLs) are tumors that arise from the neuroendocrine cells of paraganglia. PGLs that arise from the largest paraganglion in the body, the adrenal medulla, are known as pheochromocytomas (PCCs). As a group, PCC/PGL are rare, with a reported incidence of ∼2 to 8 cases per million per year1–3; although this is likely to be an underestimate, as incidental tumors are detected in up to 0.05% to 0.1% of autopsies.4 PCC/PGL carry one of the highest degrees of heritability among all human neoplasms, with up to 40% of cases being associated with germline mutations.3,5–8 In recent decades, advances in molecular genetics and genomics have led to remarkable improvements in our understanding of the genomic landscape of PCC/PGL.9 Currently, >20 susceptibility genes for PCC/PGL have been identified including RET, VHL, the SDH genes (SDHA, SDHB, SDHC, SDHD, and SDHAF2), NF1, TMEM127, MAX, KIF1B, FH, MDH2, EPAS1 (HIF2A), EGLN2 (PHD1), EGLN1 (PHD2), GOT2, SLC25A11, BAP1, MEN1, KMT2D (MLL2), DLST, IDH3B, and DNMT3A.3,8–12
The absence of a family history in no way excludes the possibility of syndromic disease, and in some series, the incidence of germline mutations in clinically apparently sporadic PCC/PGL has been as high as 35%.3,13–17 It has therefore been recommended that all patients with PCC/PGL be offered some degree of genetic counseling and testing, regardless of family history.3,12,15 This facilitates surveillance programs for other neoplasms which cluster with hereditary PCC/PGL in the index case and permits cascade genetic counseling and testing of family members with a view to early intervention.17–19 However, the degree of testing actually offered in clinical practice depends on a variety of factors including local resources/availability and the pretest probability of hereditary disease.
As a combined group, mutations in the succinate dehydrogenase complex (SDH) genes (SDHA, SDHB, SDHC, SDHD, and SDHAF2) are the most common cause of hereditary PCC/PGL.3,15 Immunohistochemistry (IHC) for SDHB is used routinely as a screening test for PCC/PGL associated with mutations in the SDH genes. Because SDHB IHC becomes negative whenever there is biallelic inactivation/mutation/deletion of any of the SDH genes, and somatic-only mutations in the SDH genes are very rare in the absence of germline mutation, loss of expression of SDHB in a PCC/PGL is strong prima facie evidence of a pathogenic germline SDH mutation.3,15,19 Furthermore, loss of expression of SDHB can also be used as functional evidence of pathogenicity when a variant of uncertain significance (VUS) is identified by sequencing one of the SDH genes.15,16,19,20 For this reason, reflex IHC for SDHB has been recommended for all PCC/PGL.12,15,19
The next most common germline mutations associated with PCC/PGL occur in RET (which causes MEN2 syndrome), VHL (von Hippel Lindau syndrome), and NF1 (neurofibromatosis type 1) which, together with mutations in the SDH genes, account for the great majority of hereditary PCC/PGL.3 Unfortunately, in our experience, no reliable surrogate IHC markers are identified for these mutations, but molecular testing for RET and VHL is widely available, and most patients with neurofibromatosis will have cutaneous manifestations by the time PCC/PGL are diagnosed.15,19 Therefore, most patients with these most common syndromes presenting with PCC/PGL are identified in most settings using current techniques. However, in routine clinical practice, the identification of patients with PCC/PGL who have any of the less common hereditary syndromes is more problematic. Many of these low-incidence genes are not included in molecular screening panels, and the cost of screening for all these genes, even if only performed in high-risk patients, may be prohibitive.
Pathogenic germline variants in fumarate hydratase (FH) cause the autosomal dominant hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome (OMIM reference 150800). HLRCC syndrome predominantly manifests with uterine and cutaneous leiomyomas and a distinctive subtype of renal carcinoma now termed FH-deficient renal cell carcinoma (RCC).21–24 IHC for FH and S-(2-succino)-cysteine (2SC) is widely used to screen leiomyomas and renal carcinomas with compatible morphology for biallelic FH inactivation/mutation—termed FH deficiency.21–24 Briefly, loss of expression of FH by IHC is highly specific for FH deficiency, while aberrant positive staining for 2SC is highly sensitive. In renal carcinoma, positive staining for 2SC lacks specificity; however, in leiomyomas, 2SC IHC appears to be both highly sensitive and specific.21–24 While the majority of patients with FH-deficient RCC will be shown to have a germline FH mutation (ie, HLRCC syndrome),21,22,24 the majority of patients with single FH-deficient uterine leiomyomas have somatic-only biallelic mutation/inactivation and do not have HLRCC syndrome.23
Occasionally, patients with HLRCC syndrome also develop PCC/PGL, and it is estimated that ∼1% of patients with PCC/PGL will have a germline FH mutation, often in the absence of a family history or other clinical manifestations.3,25–27 As the population prevalence of pathogenic germline FH variants is estimated to range from 0.0308% to 0.0390%,28 it is clear that germline FH mutation predisposes to PCC/PGL, but the penetrance of PCC/PGL in this syndrome is low. These patients with HLRCC syndrome presenting with PCC/PGL are difficult to identify as the diagnosis is often not considered, other clinical features are commonly absent, testing for FH mutation is not included on some molecular screening panels used to identify patients with hereditary PCC/PGL syndromes, and the resources required to offer genetic testing for rare causes of hereditary PCC/PGL are not available in all settings.
Although there are isolated case reports of IHC for FH and 2SC being used to identify patients with HLRCC presenting with PCC/PGL,29,30 the sensitivity, specificity, and practicality of reflex screening IHC for FH and 2SC has not been explored in a large cohort. There have been suggestions that FH mutations are more likely to be associated with aggressive behavior of PCC/PGL, but this has not been validated.25 It is not clear if there are morphologic or anatomic clues to FH deficiency in PCC/PGL as there appears to be with SDH mutations.15,19,31 It is uncertain if FH mutations arising in PCC/PGL are more commonly somatic-only as they are in uterine leiomyoma or if they are usually associated with germline mutation as they are in RCC.21,23 Finally, it is unclear if there is a genotype-phenotype correlation, as there is with VHL syndrome,32 so that some classes of FH mutations may be more commonly associated with PCC/PGL, whereas other classes of mutation may be more commonly associated with the presence of the classic features of HLRCC syndrome-leiomyomas and RCCs.
For these reasons, we sought to share our experience of the incidence, clinical, morphologic, IHC, and molecular features of FH-deficient PCC/PGL.
MATERIALS AND METHODS
We searched the computerized database of the Department of Anatomical Pathology, Royal North Shore Hospital, for all cases of PCC/PGL diagnosed between January 1, 1991, and May 1, 2022. If IHC for FH and/or 2SC was performed at the time of primary diagnosis, these results were noted and included in the series. If this IHC was not performed at the time of diagnosis and formalin-fixed paraffin-embedded tissue blocks were available in the files, the IHC was performed on the archived tissue blocks. For consultation and external review cases, paraffin blocks were usually not available in the files, and therefore we had to rely on IHC performed at the time of primary reporting. For these external consultation cases, provided a paraffin block or sufficient unstained sections were available, we had performed IHC for FH routinely on all cases from 2016, and 2SC IHC from 2020.
For any cases recorded as actually or potentially demonstrating negative FH or positive 2SC IHC at the time of primary reporting, the material was retrieved from the archives or external referrer, and IHC was repeated for the purposes of this study. Available clinical data were collected for each FH-deficient case. This included demographic information, clinical presentation, personal and family history of neoplasia, and clinical follow-up. Pathologic data collected included gross appearance, tumor location, laterality, and size. At least 1 hematoxlin and eosin–stained section from each FH-deficient tumor was reviewed to permit morphologic correlation.
A PCC/PGL was defined as being FH-deficient if it demonstrated loss of expression of FH and/or positive staining for 2SC by IHC. Our IHC methods have previously been described in detail.21–23,33 Briefly, we used an automated staining platform—the Leica Bond III Autostainer (Leica Biosystems, Mount Waverley, Vic., Australia). For FH, a commercially available mouse monoclonal antibody was used at a dilution of 1 in 2000 (clone J-13, cat no sc-100743; Santa Cruz Biotechnology), with heat-induced epitope retrieval for 30 minutes at 97°C in the manufacturer’s alkaline retrieval solution ER2 (VBS part no: AR9640). For 2SC, a rabbit polyclonal antibody (crb2005017, Discovery Antibodies; Cambridge Research Biochemicals Cleveland, UK) was used at a 1 in 2000 dilution, with heat-induced epitope retrieval for 30 minutes at 97°C in the manufacturer’s acidic retrieval solution ER1 (VBS part no: AR9961).
The IHC was interpreted by a single observer (A.J.G.). Negative stating for FH was defined as absent staining in neoplastic cells in the presence of a positive internal control in non-neoplastic cells such as endothelial and stromal cells. If the tumor cells were negative but there was no internal positive control, staining was considered indeterminate and repeated. Focal expression was considered positive unless there was a clearcut expansile nodule of neoplastic cells with loss of expression of FH in the presence of excellent internal positive controls in the areas of negative staining, in which case it was interpreted as a subclonal loss. 2SC positivity was defined as strong and diffuse cytoplasmic or nuclear and cytoplasmic staining of all neoplastic cells. Again, if there was a clearcut expansile nodule of neoplastic cells with positive staining, this was considered subclonal positivity.
All patients with FH-deficient PCC/PGL were offered germline testing as part of their clinical care. Genetic testing was performed at various clinical laboratories in Australia using massively parallel sequencing (MPS) with Sanger confirmation. If paraffin blocks were available, FH mutation testing was also performed in our laboratory on DNA extracted from macrodissected neoplastic and non-neoplastic formalin-fixed paraffin-embedded tissue. This mutation testing was performed by both Sanger sequencing and MPS. For Sanger sequencing, previously described custom primer sets were used.23 For MPS, a MiSeq Platform and TruSeq Custom Amplicon Assay (Illumina, CA) was used. If a mutation was identified by MPS but not found on Sanger sequencing, repeat targeted Sanger sequencing of the exon of interest was performed before the mutation was considered confirmed. Loss of heterozygosity (LOH) studies were performed using a previously described set of 6 polymorphic short tandem repeat markers (D1S517, D1S2785, D1S180, AFM214xe11, D1S547, and D1S2842) surrounding the FH gene.23 Metabolomic profiling was performed on one of the tumors using high-performance liquid chromatography (Shimadzu, Australia) coupled to an API QTRAP 5500 spectrometer (SCIEX, Australia), according to previously described methods.33,34 This study was approved by the North Sydney Local Health District Medical Ethics Review Board.
RESULTS
Patient Characteristics and Clinical Data
We identified 589 different patients with PCC/PGL diagnosed between 1991 and 2022, where FH IHC was performed at the time of diagnosis or was able to be performed retrospectively for this study. Of these, results for 2SC were also available for 316 (54%) cases. Of the 589 patients with PCC/PGL, 414 (70.3%) were external consultation cases often sent to our institution primarily for SDHB IHC and therefore perhaps with a selection bias towards hereditary disease; whereas 175 (29.7%) patients had their primary treatment and surgery at our institution and therefore represent an unselected cohort. For the overall cohort, 311 patients (53%) were female (female:male=1.1:1) and the median age at diagnosis was 51 years (mean=50 y; range: 10 to 88 y).
A total of 8 PCC/PGL (1.4%) demonstrated negative staining for FH and/or positive staining for 2SC and were therefore classified as FH-deficient PCC/PGL. Six of these PCC/PGL patients were from consultation cases, whereas 2 of these patients were among the 175 patients from our institution, suggesting that the incidence in an unselected population is 1.1%. For the consultation cases, a variety of fixation and processing protocols would have been used, and some of the local cases had been in paraffin blocks for up to 30 years. Despite this, we had no difficulty in performing and interpreting IHC.
The clinical, pathologic, IHC, and molecular features of the FH-deficient PCC/PGL are summarized in Table 1. Of these cases, 7 (87.5%) showed both negative staining for FH and positive staining for 2SC, whereas 1 (12.5%) was positive for FH and positive for 2SC. The median age at the time of diagnosis of FH-deficient PCC/PGL was 55 years (mean=51 y; range: 30 to 77 y). There was no statistically significant difference in mean age at diagnosis between the FH-deficient and FH-retained cases (50 vs. 51 y, P=0.891). Five patients (63%) were female. Tumor locations were as follows: adrenal (4 cases), retroperitoneum (3 cases), and carotid body (1 case). Four patients had comprehensive biochemical profiling before surgery, and all were shown to have noradrenergic tumors. One patient, patient 2, did not have biochemical testing before surgery as the diagnosis was not suspected. However, she had clear clinical evidence of life-threatening catecholamine crises in the lead-up to diagnosis including an intrauterine growth retardation pregnancy and urgent preterm delivery; and acute Takotsubo-like cardiomyopathy with pulmonary edema requiring inotropic support. Immediately postoperatively, she developed acute respiratory distress syndrome, labile blood pressure requiring vasopressors and delayed extubation.
TABLE 1.
Clinical, Pathologic, IHC, and Molecular Features of 8 FH-deficient PCC/PGL Identified by Screening a Cohort of 589 Consecutive Cases
Patients 1, 5, and 6 are all from the same extended kindred. Patient 5 had been unaware of patient 1’s diagnosis of HLRCC and only after genetic counseling and molecular testing was undertaken, was patient 6 found to be related to this kindred.
A synonymous germline FH c.330T>C (p.Tyr110Tyr) VUS was detected.
A synonymous FH c.920C>T (p.Val306Val) variant was also detected in the FH-deficient part of the tumor.
Patient 2 biochemistry was not tested before removal. However, there was strong clinical evidence of catecholamine production including takotsubo-like cardiomyopathy, postoperative acute respiratory distress syndrome, labile blood pressure requiring vasopressors, and delayed extubation.
ANED indicates alive no evidence of disease; AWD, alive with disease; DOD, dead of disease; F, female; LVI, lymphovascular invasion; M, male; U, unknown/not tested; WT, wild-type.
None of the 8 patients were known to have a family history of HLRCC at the time of diagnosis. The mother of patient 1 had a large uterine leiomyoma removed in her 30s and was subsequently demonstrated to carry the family mutation after her daughter was diagnosed. This leiomyoma was not available for review or IHC. None of the other patients were known to have a personal or family history of other FH-deficient neoplasms such as cutaneous leiomyomas, clinically significant uterine leiomyomas or FH-deficient RCC even at long-term follow-up. Interestingly, it was only after patient 5 was diagnosed with FH-deficient PGL that she was made aware that she was a distant relative of patient 1; and only during the preparation of this paper was it found that patient 6, who shared the same germline mutation [c.1142C>T (p.Thr381Ile)], was also distantly related to these 2 patients. That is, although 3 of the patients were from the same large extended kindred, all 8 patients with FH-deficient PCC/PGL presented as clinically sporadic disease.
Three patients had apparently unrelated synchronous FH-retained neoplasms at the time of resection of their FH-deficient PCC/PGL. Patient 4 had non-Hodgkin lymphoma (diffuse large B-cell type) and patient 5 had ovarian high-grade serous carcinoma. The blocks from these other tumors were retrieved, the diagnoses confirmed, and the tumors demonstrated to be FH-sufficient (FH positive and 2SC negative). Patient 6 deserves special discussion as she was diagnosed with conventional clear cell RCC at the time of presentation at age 32. Suspecting that this may represent misdiagnosed FH-deficient RCC, we retrieved all the slides and paraffin blocks. The tumor demonstrated typical morphology for clear cell RCC and was FH-sufficient (positive for FH and negative for 2SC). Furthermore, on sequencing of the RCC, although the trace germline FH mutation (c.1142C>T) was detected, no somatic second hit or LOH was identified in the RCC. We therefore concluded that this RCC was indeed an incidental conventional clear cell RCC and not a FH-deficient RCC.
The median duration of follow-up was 54 months (mean=96 mo, range: 3 to 370 mo). Of the 8 patients, 2 (25%) had metastatic disease. Patient 8, who had lymph node metastases resected at presentation, was alive with disease (other lymph node metastases) at 6 months. Patient 7, who had a carotid body PGL, was diagnosed with widespread bone and liver metastases 10 years after his initial presentation and died of the disease at 174 months. The remaining 6 patients were alive with no evidence of disease at last follow-up (Table 1). One patient developed a metachronous PCC 22 years after her first tumor was diagnosed. None of the patients developed cutaneous leiomyomas, FH-deficient RCCs or clinically significant uterine leiomyomas during follow-up.
Morphologic Characteristics
The pathologic characteristics of the 8 FH-deficient PCC/PGLs were reviewed. The median tumor size was 65 mm (mean=62 mm, range: 28 to 100 mm). All FH-deficient tumors demonstrated retained expression of SDHB and SDHA. Of the 589 patients in our cohort, 103 (ie, 17.5%) showed loss of expression of SDHB by IHC (ie, they were SDH deficient) and of these patients, 7 were also negative for SDHA by IHC. As all these SDH deficient tumors demonstrated retained FH expression, this supports the suggestion that SDH deficiency and FH deficiency are mutually exclusive.
The morphology of the tumors is illustrated in Figures 1–4. All cases demonstrated a somewhat nested architecture, although they lacked the strikingly well-developed, tight nesting pattern described in SDH-deficient PCC/PGL.31 Some tumors showed areas of trabecular architecture. There was no sheet-like growth. The neoplastic cells lacked the cytoplasmic clearing reported in SDH-deficient PGLs31 and all demonstrated distinctly granular eosinophilic to amphophilic cytoplasm. No cytoplasmic inclusions were seen. No spindle cell morphology was seen, and there was no significant pleomorphism. In contrast to FH-deficient leiomyoma and RCC, which are characterized by very prominent inclusion-like nucleoli,21–24 the nucleoli were inconspicuous. None of the tumors showed necrosis. Mitotic activity was low, with a maximum of 2 mitotic figures per 2 mm2. Two cases showed extension of tumor cells beyond the capsule and into the adjacent soft tissue. Capillary and lymphatic invasion was identified in 1 case. In summary, there were no clear morphologic clues to the presence of FH deficiency.
FIGURE 1.
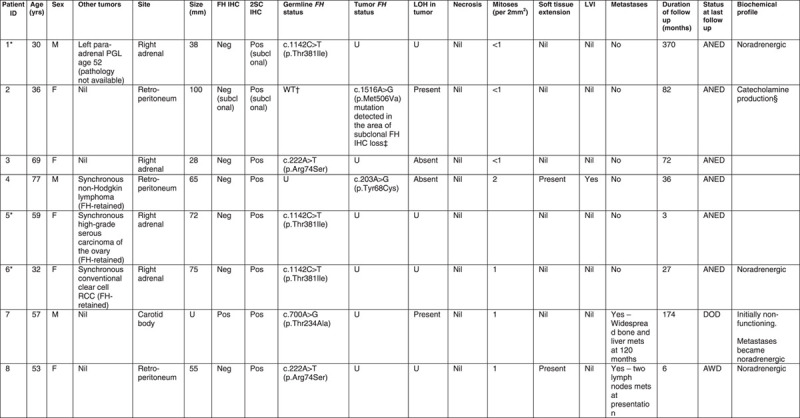
The variable morphology of FH-deficient PCC/PGL. A, Prominent vascular spaces. B. Typical nested and trabecular growth pattern with sustentacular cells. The cytoplasm is abundant and varies from amphophilic (C) to palely eosinophilic (D). E, Most cases were circumscribed and encapsulated, with a clear demarcation from the normal adrenal gland. F, Irregular extension of tumor cells into adjacent soft tissue was seen in a minority of cases.
FIGURE 4.
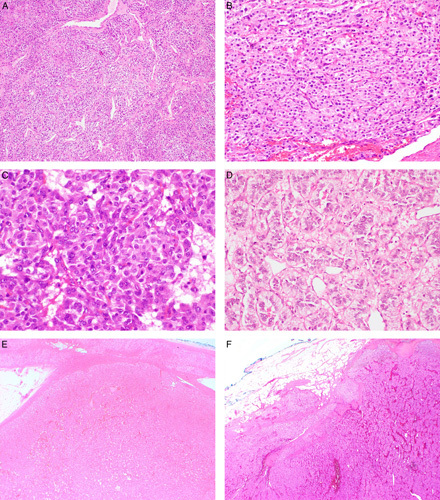
Serial sections stained with hematoxlin and eosin (A, C) and FH IHC (B, D) from patient 2. This tumor showed subclonal loss of FH expression with an area of preserved FH staining (asterisk). Of note, the area showing loss of FH staining demonstrates positive staining within the non-neoplastic endothelial/stromal cells (arrowheads) which serve as an internal control. In the FH-deficient areas, there is weak non-specific diffuse cytoplasmic expression which contrasts with the strong granular expression in the internal positive controls. This pattern of staining should be considered negative (FH deficient). This tumor was found to have a somatic FH mutation in this subclonal FH-deficient nodule but no germline mutation.
FIGURE 3.
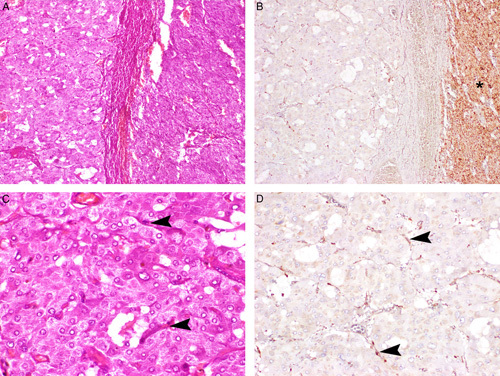
Serial hematoxlin and eosin–stained (A, C), 2SC IHC-stained (B), and FH IHC-stained (D) sections. The non-neoplastic adrenal gland (asterisk) is negative for 2SC IHC, while the tumor cells show diffuse cytoplasmic expression. The non-neoplastic endothelial cells within the tumor mass (arrowheads) demonstrate positive staining for FH, in contrast to the PCC cells which are completely negative.
The pattern of FH and 2SC staining is illustrated in Figures 2–4. All non-neoplastic tissues (including endothelial cells and adjacent non-neoplastic adrenal parenchyma) demonstrated positive staining for FH in a mitochondrial pattern (ie, granular and cytoplasmic), whereas the neoplastic cells of FH-deficient tumors were either completely negative for FH or exhibited only a weak cytoplasmic diffuse blush, which contrasted markedly to the distinctly granular expression seen in the non-neoplastic cells (Fig. 4). 2SC was diffusely strongly expressed in the cytoplasm of all neoplastic cells, occasionally also with nuclear expression. 2SC was not expressed in non-neoplastic tissue nor in any of the 308 FH-sufficient cases tested and not known to be associated with somatic or germline FH mutation.
FIGURE 2.
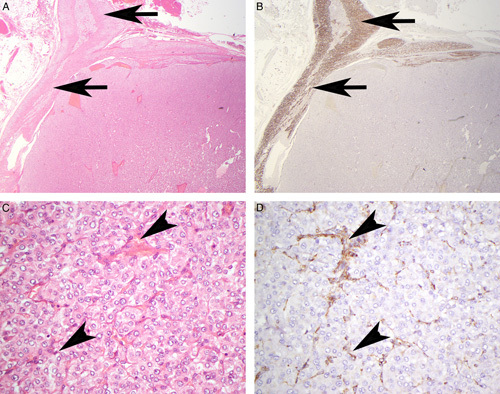
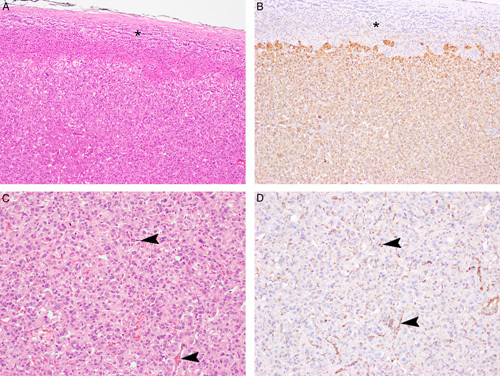
Serial sections stained with hematoxlin and eosin (A, C) and FH IHC (B, D). The neoplastic cells show completely negative staining for FH. In contrast, the non-neoplastic adrenal (arrows) and endothelial/stromal cells within the main tumor mass (arrowheads) demonstrate positive staining for FH in a granular cytoplasmic (mitochondrial) pattern and serve as an internal control.
One PGL (patient 2) demonstrated subclonal FH deficiency, defined as an expansile nodule of tumor cells which were FH negative and 2SC positive, arising from within a PGL with otherwise normal (FH positive and 2SC negative) IHC. The FH-deficient clone in this tumor accounted for ∼25% of the total tumor volume.
Molecular Analysis
All 8 patients had molecular testing performed and were found to have an FH mutation in their germline or tumor (summarized in Table 1). Seven patients had germline testing, of whom 6 were shown to have pathogenic variants in FH. Patient 2 with subclonal FH deficiency was wild-type in the germline on both sequencing and MLPA analysis. We macrodissected this tumor and were able to demonstrate a somatic FH c.1516A>G (p.Met506Val) mutation in the FH-deficient nodule, which was absent in the FH-sufficient regions and germline. In silico analyses predicted this variant to be damaging to protein function (PolyPhen-2, score 0.999; SIFT, score 0) and metabolomic profiling demonstrated an altered succinate:fumarate ratio. The subclonal area of FH deficiency demonstrated LOH. We therefore concluded that the addition of a somatic FH mutation with LOH in a PGL led to the subclonal FH deficiency.
Six patients had germline missense mutations within coding regions of the FH gene. The following mutations were detected: c.1142C>T (p.Thr381Ile) (3 patients, all subsequently found to be from one extended kindred), c.222A>T (p.Arg74Ser) (2 patients, unrelated to our knowledge), and c.700A>G (p.Thr234Ala) (1 patient). The c.222A>T (p.Arg74Ser) mutation has not previously been described in FH-deficient PCC/PGL and is currently classified as a VUS in ClinVar. However, given the IHC-confirmed loss of tumor FH protein expression and cosegregation with PCC/PGL in both patients 3 and 8 with this variant, we believe that there is sufficient evidence to classify it as likely pathogenic.
LOH testing on tumor tissue was performed on 4 tumors. As described above, patient 2 with somatic-only mutation demonstrated LOH. Patient 7 with germline FH c.700A>G (p.Thr234Ala) mutation was also shown to have LOH in his tumor. No LOH was found in patient 3 with a germline FH c.222A>T (p.Arg74Ser) mutation, however, we were unable to sequence the tumor and therefore are unable to exclude a somatic second hit mutation. Patient 4 did not have germline testing performed but was found to have an FH c.203A>G (p.Tyr68Cys) mutation in his tumor without LOH. Based on available data, we were unable to determine if this mutation was somatic-only or germline.
Role of FH and 2SC IHC to Support Pathogenicity in FH VUS
We reviewed the IHC findings of 3 patients identified with VUS in FH on germline sequencing. Two patients (a 60-y-old male and a 45-y-old female) harbored the FH germline variant c.1431_1433dupAAA (p.Lys477_Asn478insLys).35–37 This variant is known to be associated with the severe autosomal recessive metabolic disorder of FH deficiency and was initially assumed to also be pathogenic for HLRCC. However, both patients demonstrated normal IHC in their tumors (FH positive, 2SC negative) and lacked somatic second hits or LOH. Subsequently, convincing data have demonstrated that this FH variant is relatively common in the general population and is not associated with an increased risk of neoplasia.35–37 In fact, the 45-year-old female was eventually shown to have a germline pathogenic variant in exon 11 of RET c.1902C>G (p.Cys634Trp), subsequently developed medullary thyroid carcinoma, and was therefore diagnosed with MEN2A syndrome. Similar normal IHC staining was found in a 49-year-old female with PCC and an exon 10 variant c.1421C>T (p.Thr474Ile), which is currently considered a VUS but is likely benign.
That is, in all 3 patients, the normal staining (FH positive, 2SC negative) was clinically useful as it provided further support to the decision to consider these variants as likely non-pathogenic.
DISCUSSION
Our study has several direct implications for patient care. First, we provide proof that, like SDHB IHC, reflex FH and 2SC IHC on all PCC/PGL is a rational approach. If the presence of either germline or somatic mutation is considered the gold standard, FH/2SC IHC is 100% specific for FH deficiency. Our study was not designed nor intended to assess the sensitivity of FH/2SC IHC. However, we note that using screening IHC, we found that 1.4% of all PCC/PGLs were FH deficient (1.1% in an unselected population). Given that the combined results of other studies have estimated the incidence of FH germline mutation in PCC/PGL as being 1.1%,25,36 we conclude that FH/2SC IHC is also highly sensitive.
Whilst it could be argued that many of these patients will also undergo panel molecular testing and, in some sense, FH/2SC could be considered redundant, we note that molecular testing is not always completely sensitive and that identification of an FH-deficient PCC/PGL at diagnosis may permit a more targeted approach to genetic testing. Furthermore, as we have demonstrated, IHC has a clear role in assessing the potential pathogenicity of any VUS in the FH gene found by sequencing. Therefore, like SDHB IHC, we now perform FH and 2SC IHC on all PCC/PGL at the time of diagnosis. Although we only report 8 FH-deficient cases and therefore care should be taken before drawing firm conclusions, we note that no FH-deficient cases were also SDH deficient and that no FH loss was found in 103 SDH-deficient PCC/PGL. Furthermore, to date, co-mutation of SDH and FH have not been reported.25,26,34,36 Therefore, we conclude that SDH deficiency and FH deficiency are highly likely to be mutually exclusive and if cascade IHC testing is preferred, FH/2SC IHC may need only be performed on SDHB IHC-positive PCC/PGL.
In RCC and uterine leiomyoma, it has been estimated that, compared with 2SC, IHC for FH alone has a sensitivity of 80% to 90%.21,23,24,38–40 The finding that 7 of 8 PCC/PGL in our series were negative for FH, whereas all 8 were positive for 2SC, suggests a similar sensitivity of 87.5% for FH IHC in PCC/PGL compared with 2SC. Although 2SC IHC is imperfectly specific in RCC,21,24 it appears to be 100% specific in PCC/PGL (similar to uterine leiomyoma), and there was no significant staining at all in any of the other PCC/PGL. Therefore, an argument could be made to perform only 2SC IHC rather than combined FH and 2SC.
Most FH-deficient RCCs are associated with germline FH mutation, probably of the order of 80% to 90%,21,24 whereas the majority of FH-deficient uterine leiomyomas, particularly if solitary and occurring at an older age, are not associated with FH mutation.23,28,34 Of the 7 patients in our series who underwent genetic testing, 6 (85.7%) were found to have an FH germline mutation. Therefore, we conclude that most FH-deficient PCC/PGL are associated with germline mutations and therefore the diagnosis of an FH-deficient PCC/PGL is an absolute indication for genetic testing.
Review of different germline FH mutations associated with PCC/PGL reveals an interesting insight (Table 2). Including the 8 patients from our study, 11 different germline FH mutations have now been reported in 18 patients with FH-deficient PCC/PGL, of which 17 are missense mutations.25,26,34,36 All 8 patients in our series had missense mutations and were notable for having no evidence of FH-deficient leiomyoma or RCC. In contrast, truncating variants account for more than half of the mutations associated with HLRCC as currently defined based on leiomyomas and RCC.35 This genotype-phenotype distinction is reminiscent of VHL syndrome, in which patients with VHL large-scale deletions or pathogenic truncating or missense variants that are predicted to grossly disrupt the folding of the VHL protein have a very low risk of developing PCC but remain at risk of other features including hemangioblastoma, RCC, pancreatic cysts, and neuroendocrine tumors.32 These patients are considered to have VHL type 1. In contrast, patients with so-called VHL type 2 generally have pathogenic missense variants in the VHL gene. All these patients are at high risk of PCC and can be further subdivided into type 2a (PCC and hemangioblastoma but low risk of RCC), type 2b (PCC, hemangioblastoma, and high risk of RCC), and type 2c (PCC only).
TABLE 2.
Clinical and Molecular Features of Reported FH-deficient PCC/PGL Cases
| References | Patient ID | Germline DNA Change | Protein Change | Predicted Consequence | Age at Diagnosis (y) | Sex | Clinical Phenotype |
|---|---|---|---|---|---|---|---|
| Letouzé et al26 and Castro-Vega et al25 | 1 | c.349G>C | p.Ala117Pro | Missense | 63 | F | Unilateral PCC Metastatic |
| 2 | c.268-2A>G | (splice defect) | Splicing defect | 20 | F | PCC and PGL | |
| 3 | c.1142C>T | p.Thr381Ile | Missense | 28 | M | PGL Metastatic | |
| 4 | c.580G>A | p.Ala194Thr | Missense | 54 | F | PCC and PGL Metastatic | |
| 5 | c.986A>G | p.Asn329Ser | Missense | 70 | M | HNPGL | |
| Clark et al37 | 1 | c.1301G>A | p.Cys434Tyr | Missense | 6 | M | Unilateral PCC |
| 2 | c.157G>A | p.Glu53Lys | Missense | 41 | M | Unilateral PCC | |
| Richter et al35 | 1 | c.700A>G | p.(Thr234Ala) | Missense | 36 | F | Unilateral PCC Aberrant tumor fumarate:malate ratio on metabolomics, FH deficient by IHC |
| 2 | c.908T>C | p.(Leu303Ser) | Missense | 53 | F | Unilateral PCC Aberrant tumor fumarate:malate ratio on metabolomics, FH deficient by IHC | |
| 3 | c.816_836del | p.(Ala273_Val279del) | In-frame deletion leading to loss of 6 amino acids | U | U | Unilateral PCC Aberrant tumor fumarate:malate ratio on metabolomics and LOH detected | |
| This study | 1 | c.1142C>T | p.Thr381Ile | Missense | 30 | M | Unilateral PCC Family history of HLRCC Metachronous PGL diagnosed 22 y later (pathology not available) |
| 2 | WT | NA | NA | 36 | F | PGL with somatic FH c.1516A>G (p.Met506Val) mutation | |
| 3 | c.222A>T | p.Arg74Ser | Missense | 69 | F | Unilateral PCC No LOH | |
| 4 | U | NA | NA | 77 | M | PGL with somatic FH c.203A>G (p.Tyr68Cys) mutation No LOH | |
| 5 | c.1142C>T | p.Thr381Ile | Missense | 59 | F | Unilateral PCC Synchronous high-grade serous carcinoma of the ovary | |
| 6 | c.1142C>T | p.Thr381Ile | Missense | 32 | F | Unilateral PCC Synchronous clear cell RCC | |
| 7 | c.700A>G | p.Thr234Ala | Missense | 57 | M | PGL with LOH Metastatic | |
| 8 | c.222A>T | p.Arg74Ser | Missense | 53 | F | PGL Metastatic to lymph node |
F indicates female; M, male; NA, not available; U, unknown; WT, wild-type.
If the striking genotype-phenotype correlation in FH is upheld in other studies, it may be reasonable to subdivide FH mutation into 2 types—for example, FH deficiency type 1 which would have a high risk of RCC and leiomyoma without PCC/PGL and is associated with deletions, truncations or variants that disrupt the folding of the protein; and FH deficiency type 2 which would be associated with (possibly selected) missense variants and predominantly causes PCC/PGL.
Although these concepts require further studies to develop, at the very least it is critical to note that the types of FH mutations that cause PCC/PGL may be different to the types of mutations that cause RCC and leiomyoma and may also be different to the types of mutations that cause autosomal recessive FH deficiency. There is certainly ample evidence that the absence of leiomyomas and RCC should not be used as an argument against pathogenicity of these FH variants in patients with PCC/PGL. Failure to recognize this may lead to misclassification of the pathogenicity of variants and suboptimal patient care.
This is well illustrated in our study where we identified familial PCC associated with FH c.1142C>T (p.Thr381Ile) (patients 1, 5 and 6); and another 2 apparently unrelated patients with FH-deficient PCC/PGL and germline FH c.222A>T (p.Arg74Ser) variants. The latter missense variant has never previously been associated with the classic HLRCC syndrome phenotype of leiomyoma and RCC and is currently listed as a VUS in ClinVar. However, our 2 patients with the same variant and FH-deficient PCC/PGL provide strong evidence that this variant is linked to an isolated PCC/PGL phenotype, with a low risk of leiomyomas and RCC. That is, some of these variants may be much more penetrant for PCC/PGL than leiomyoma or RCC. In fact, if we were to apply Bayesian analysis (methods described in the study by Benn et al42), the estimated penetrance for FH variants associated with PCC/PGL in our study is 22.5% (95% confidence interval: 3.8%–68.3%)—similar to the penetrance of SDHB mutation, and with similar metastatic potential.42
FH is a tumor suppressor gene, and therefore requires mutations in both alleles for initiation of tumorigenesis. LOH is one of the most common mechanisms accounting for the second hit. However, LOH was only detected in 2 of 5 tumors tested in this study. These findings are similar to those reported in SDH-deficient disease, in which LOH is only identified in 80% of SDHB-related and 50% of SDHD-related PCC/PGL.43,44 Possible alternative mechanisms for inactivation of the wild-type allele in these instances include large deletion events or hypermethylation, both of which would require different techniques for detection. Given that these tumors are known to be highly methylated, it is possible that hypermethylation contributes to tumorigenesis in some of the cases in this study and was not detected using our techniques.
Metastatic disease was reported in 2 of 8 patients (25%) in this study, one of whom ultimately died of widespread disease 174 months after initial presentation. When combined with other cases reported in the literature, the estimated incidence of metastasis in FH-deficient PCC/PGL is 28%.25,34 Although caution is required because of the small number of cases reported, this is significantly higher than the reported 15% rate of metastatic disease for PCC/PGL overall.3,6,10 We, therefore, hypothesize that FH-deficient PCC/PGL, similar to FH-deficient RCC, may be associated with more aggressive biological behavior and requires close and long-term follow-up.
Although the majority (86%) of FH-deficient PCC/PGL were associated with germline mutations, purely somatic mutations in the FH gene may rarely give rise to FH-deficient PCC/PGL, as was seen in one of the cases in this study (patient 2). This patient’s tumor demonstrated subclonal loss of FH staining by IHC (Fig. 4). Molecular testing found an FH c.1516A>G (p.Met506Val) mutation in the FH negative part of the tumor, which was confirmed to be pathogenic by metabolomic profiling and in silico analyses. Interestingly, germline testing in this patient showed a synonymous FH c.330T>C (p.Tyr110Tyr) VUS. A somatic synonymous FH c.920C>T (p.Val306Val) variant was also detected in the FH-deficient part of the tumor. The significance of these synonymous variants is uncertain. Although this patient would not be expected to demonstrate other syndromic manifestations of HLRCC, the absence of a germline mutation may not necessarily change the potentially aggressive clinical behavior associated with FH-deficient neoplasia, as is described in patients with non-syndromic FH-deficient RCC.21,22 However, additional follow-up studies will be required to determine the true clinical significance, if any, of PCC/PGL with somatic-only FH mutations.
In conclusion, FH deficiency occurs in ∼1% of all PCC/PGL and is associated with germline FH mutation in the majority (80% to 90%) of cases. This association is usually not suspected clinically as many of these patients present at an older age and without other syndromic features. Although there are no reliable morphologic clues, these tumors can be readily identified quickly and cheaply in routine clinical practice by 2SC IHC which is highly sensitive and specific, with or without FH IHC which is highly specific but less sensitive. FH-deficient PCC/PGL are commonly noradrenergic, may have a higher risk of metastasis, and there appears to be a genotype-phenotype correlation so that missense mutations are particularly associated with PCC/PGL but not with leiomyoma or RCC. We, therefore, recommend that reflex IHC for 2SC and/or FH be performed on all PCC/PGL.
ACKNOWLEDGMENTS
The authors thank Ying Zhu for assistance with bioinformatics; Mark Smithers, and Veronica Boyle for provision of clinical data, and Cornelia Nellgard for assistance with somatic testing.
Footnotes
Conflicts of Interest and Source of Funding: R.J.C.B. has served on advisory boards for Amgen, Eisai, Kyowa Kirin, Ipsen and received speaking honoraria from Amgen, Eisai, and Kyowa Kirin unrelated to the current work. For the remaining authors none were declared.
Contributor Information
Talia L. Fuchs, Email: tfuchs@dhm.com.au.
Catherine Luxford, Email: catherine.luxford@sydney.edu.au.
Adele Clarkson, Email: adele.clarkson@health.nsw.gov.au.
Amy Sheen, Email: amy.sheen@health.nsw.gov.au.
Loretta Sioson, Email: loretta.sioson@health.nsw.gov.au.
Marianne Elston, Email: marianne.elston@waikatodhb.health.nz.
Michael S. Croxson, Email: michaelc@adhb.govt.nz.
Trisha Dwight, Email: trisha.dwight@sydney.edu.au.
Diana E. Benn, Email: diana.benn@sydney.edu.au.
Lyndal Tacon, Email: lyndal.tacon@sydney.edu.au.
Michael Field, Email: mike.field@health.nsw.gov.au.
Mahsa S. Ahadi, Email: mahsa.seyedahadi@health.nsw.gov.au.
Angela Chou, Email: angelashihyuan.chou@health.nsw.gov.au.
Roderick J. Clifton-Bligh, Email: roderick.cliftonbligh@sydney.edu.au.
Anthony J. Gill, Email: affgill@med.usyd.edu.au.
REFERENCES
- 1.Beard CM, Sheps SG, Kurland LT, et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58:802–804. [PubMed] [Google Scholar]
- 2.Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turchini J, Cheung VKY, Tischler AS, et al. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72:97–105. [DOI] [PubMed] [Google Scholar]
- 4.Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56:354–360. [PubMed] [Google Scholar]
- 5.Papathomas TG, Giordano TJ, Maher ER, et al. Boffetta P. Adrenal glands tumors: pathology and genetics. Hainaut PBT-E of C (Third Edition). Oxford, UK: Academic Press; 2019:18–29. [Google Scholar]
- 6.Dahia PLM. Pheochromocytomas and paragangliomas, genetically diverse and minimalist, all at once!. Cancer Cell. 2017;31:159–161. [DOI] [PubMed] [Google Scholar]
- 7.Papathomas TG, Suurd DPD, Pacak K, et al. What have we learned from molecular biology of paragangliomas and pheochromocytomas? Endocr Pathol. 2021;32:134–153. [DOI] [PubMed] [Google Scholar]
- 8.Jhawar S, Arakawa Y, Kumar S, et al. New insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. Cancers. 2022;14:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Cancer. 2014;14:108–119. [DOI] [PubMed] [Google Scholar]
- 11.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11:101–111. [DOI] [PubMed] [Google Scholar]
- 12.WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Series, 5th Edition, Volume 8: Endocrine and Neuroendocrine tumours. Lyon, France: International Agency for Research on Cancer; 2022. [Google Scholar]
- 13.Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70–87. [DOI] [PubMed] [Google Scholar]
- 14.Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915–1942. [DOI] [PubMed] [Google Scholar]
- 15.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–116. [DOI] [PubMed] [Google Scholar]
- 16.Garrett A, Loveday C, King L, et al. Quantifying evidence toward pathogenicity for rare phenotypes: the case of succinate dehydrogenase genes, SDHB and SDHD. Genet Med. 2022;24:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King KS, Prodanov T, Kantorovich V, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29:4137–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shuch B, Ricketts CJ, Metwalli AR, et al. The genetic basis of pheochromocytoma and paraganglioma: implications for management. Urology. 2014;83:1225–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44:285–292. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs TL, Maclean F, Turchini J, et al. Expanding the clinicopathological spectrum of succinate dehydrogenase-deficient renal cell carcinoma with a focus on variant morphologies: a study of 62 new tumors in 59 patients. Mod Pathol. 2022;35:836–849. [DOI] [PubMed] [Google Scholar]
- 21.Trpkov K, Hes O, Agaimy A, et al. Fumarate hydratase deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. 2016;40:865–875. [DOI] [PubMed] [Google Scholar]
- 22.Lau HD, Chan E, Fan A, et al. A clinicopathologic and molecular analysis of fumarate hydratase-deficient renal cell carcinoma in 32 patients. Am J Surg Pathol. 2020;44:98–110. [DOI] [PubMed] [Google Scholar]
- 23.Harrison WJ, Andrici J, Maclean F, et al. Fumarate hydratase deficient uterine leiomyomas occur in both the syndromic and sporadic settings. Am J Surg Pathol. 2016;40:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trpkov K, Hes O, Williamson S, et al. New developments in existing WHO entities and evolving molecular concepts: the Genitourinary Pathology Society (GUPS) Update on Renal Neoplasia. Mod Pathol. 2021;34:1392–1424. [DOI] [PubMed] [Google Scholar]
- 25.Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. [DOI] [PubMed] [Google Scholar]
- 26.Letouzé E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. [DOI] [PubMed] [Google Scholar]
- 27.Muller M, Ferlicot S, Guillaud-Bataille M, et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. 2017;92:606–615. [DOI] [PubMed] [Google Scholar]
- 28.Popp B, Erber R, Kraus C, et al. Targeted sequencing of FH-deficient uterine leiomyomas reveals biallelic inactivating somatic fumarase variants and allows characterization of missense variants. Mod Pathol. 2020;33:2341–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung VKY, Gill AJ, Chou A. Old, new, and emerging immunohistochemical markers in pheochromocytoma and paraganglioma. Endocr Pathol. 2018;29:169–175. [DOI] [PubMed] [Google Scholar]
- 30.Udager AM, Magers MJ, Goerke DM, et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma/pheochromocytoma syndromes. Hum Pathol. 2018;71:47–54. [DOI] [PubMed] [Google Scholar]
- 31.Turchini J, Gill AJ. Morphological clues to succinate dehydrogenase (SDH) deficiency in pheochromocytomas and paragangliomas. Am J Surg Pathol. 2020;44:422–424. [DOI] [PubMed] [Google Scholar]
- 32.Chou A, Toon C, Pickett J, Gill AJ. von Hippel-Lindau syndrome. Front Horm Res. 2013;41:30–49. [DOI] [PubMed] [Google Scholar]
- 33.Kim E, Wright MJP, Sioson L, et al. Utility of the succinate:fumarate ratio for assessing SDH dysfunction in different tumor types. Mol Genet Metab Rep. 2017;10:45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter S, Gieldon L, Pang Y, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med. 2019;21:705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Walsh MF, Jairam S, et al. Fumarate hydratase FH c.1431_1433dupAAA (p.Lys477dup) variant is not associated with cancer including renal cell carcinoma. Hum Mutat. 2020;41:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark GR, Sciacovelli M, Gaude E, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab. 2014;99:E2046–E2050. [DOI] [PubMed] [Google Scholar]
- 37.Whitworth J, Smith PS, Martin JE, et al. Comprehensive Cancer-predisposition gene testing in an adult multiple primary tumor series shows a broad range of deleterious variants and atypical tumor phenotypes. Am J Hum Genet. 2018;103:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alsolami S, El-Bahrawy M, Kalloger SE, et al. Current morphologic criteria perform poorly in identifying hereditary leiomyomatosis and renal cell carcinoma syndrome-associated uterine leiomyomas. Int J Gynecol Pathol. 2014;33:560–567. [DOI] [PubMed] [Google Scholar]
- 39.Reyes C, Karamurzin Y, Frizzell N, et al. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: detailed morphologic analysis and correlation with S-(2-succino)-cysteine immunohistochemistry. Mod Pathol. 2014;27:1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinek P, Grossmann P, Hes O, et al. Genetic testing of leiomyoma tissue in women younger than 30 years old might provide an effective screening approach for the hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC). Virchows Arch. 2015;467:185–191. [DOI] [PubMed] [Google Scholar]
- 42.Benn DE, Zhu Y, Andrews KA, et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J Med Genet. 2018;55:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber A, Hoffmann MM, Neumann HPH, et al. Somatic mutation analysis of the SDHB, SDHC, SDHD, and RET genes in the clinical assessment of sporadic and hereditary pheochromocytoma. Horm Cancer. 2012;3:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacFarlane J, Cheah Seong K, Bisamber C, et al. A review of the tumour spectrum of germline succinate dehydrogenase gene mutations: beyond phaeochromocytoma and paraganglioma. Clin Endocrinol. 2020;93:528–538. [DOI] [PubMed] [Google Scholar]