

Abstract
Synthetic triple helix-forming oligodeoxyribonucleotides (TFOs) have been used to alter gene expression and to induce targeted genome modification in cells and animals. However, the efficacy of such oligodeoxyribonucleotides (ODNs) depends on efficient intracellular delivery. A novel vector system was tested for the production of single-stranded DNA (ssDNA) to serve as a TFO in mouse cells. Mouse cells carrying a substrate that can report triplex-stimulated intrachromosomal recombination were transfected with a series of ssDNA vectors, and induced recombination was assayed. Transfection with a vector set designed to generate a 34 nt G-rich ssDNA capable of triplex formation at a 30 bp polypurine target site within the reporter substrate yielded recombinants at a frequency of 196 × 10–6, versus a background frequency of 45 × 10–6 in mock transfected cells. No induction was seen when a vector set lacking the TFO sequence insert was tested or when the component vectors were transfected individually. Vectors engineered to express a C-rich 34 nt sequence (not expected to form triplex under physiological conditions) had no effect over background. Primer extension analyses on lysates from transfected cells confirmed the production of the intended ssDNAs. These results suggest that ssDNA molecules of a defined sequence can be generated intracellularly using a novel vector system and that such molecules are active in mediating triplex-dependent chromosomal events. The ability to produce active TFOs within cells may provide a new foundation for triplex-based gene targeting strategies.
INTRODUCTION
Oligodeoxyribonucleotides (ODNs) can bind to DNA in a sequence-specific manner to form triple helices (1,2). Triple helix formation has been shown to suppress gene expression (3,4) and to mediate targeted genome modification in mammalian cells via directed mutagenesis or induced recombination (5,6). The ability of triple helix-forming oligodeoxyribonucleotides (TFOs) to stimulate recombination has been shown to depend on XPA and Rad51 (7,8), factors involved in nucleotide excision repair and homologous recombination, respectively. These results are consistent with studies demonstrating that triplex structures provoke DNA repair (9).
In previous work, we showed that a G-rich 30mer TFO (AG30), when transfected into a mouse fibroblast cell line (FL-10), could induce recombination between direct repeat copies of the herpes simplex virus thymidine kinase (TK) gene in a chromosomal substrate in which the target site for triplex formation was situated between the genes (5). The AG30 TFO was 3′-end protected from degradation by modification with a propylamine group and transfected into cells either by co-mixture with cationic lipids or via direct microinjection. Although lipid-mediated transfection yielded specific and detectable induction of recombination, microinjection yielded a 300-fold higher frequency of recombinants. These results highlighted the need for improved delivery methods to achieve sufficient concentrations of TFOs within biologically active compartments in mammalian cells.
Recently, Chen et al. described a vector system designed to produce single-stranded DNA (ssDNA) in cells, and they demonstrated its use in generating either antisense or catalytic DNA to mediate degradation of a targeted mRNA (10). In this system, co-expression from one vector of a reverse transcriptase–MboII fusion protein along with a mRNA from a second vector containing an inverted repeat sequence downstream of the Moloney murine leukemia virus (MoMuLV) core promoter is designed to yield a cDNA molecule with a stem–loop structure. MboII cleavage at the base of the stem is intended to release the ssDNA sequence of interest contained in the loop.
In work reported here, we have tested the ability of this vector system to produce ssDNA in mouse cells capable of serving as a TFO for the purpose of inducing intrachromosomal recombination. Although the intended 34 nt ssDNA product would not be 3′-end protected like the synthetic TFO, AG30, and even though it would contain 4 extra nt at the 3′ end compared with AG30 (thereby reducing third-strand binding affinity), we found that the ssDNA expression system could produce detectable amounts of the desired TFO in cells, leading to levels of induced recombination 7-fold above that previously observed when synthetic AG30 was transfected into cells using cationic lipids (5). The results suggest that the ssDNA expression system may provide a useful means of generating active TFOs in vivo to mediate site-directed modification of genomic DNA.
MATERIALS AND METHODS
Vectors
The construction of the pssXA and pssXB vectors (Fig. 1A) has been previously described (10). The vectors contain 7869 and 5459 bp, respectively. To make vectors expressing the desired ssDNA containing the AG30 TFO sequence or the complement of it, synthetic oligonucleotides of the sequence: 5′-d(GGGCCGCAGGCTCCCCCTCCCCCACCACCCCCCCCTTCCTGC)-3′ and 5′-d(GGCCGCAGGAAGGGGGGGGTGGTGGGGGAGGGGGAGCCTGC)-3′ were annealed to produce a synthetic duplex with NotI cohesive ends and ligated into the NotI site of pssXB after removal of the stuffer fragment present in the original vector. Following transformation into Escherichia coli, colonies were identified by direct DNA sequencing that contained plasmids with the insert sequences in both possible orientations. The orientation designed to generate a 34 nt G-rich sequence incorporating the AG30 TFO is illustrated in Figure 1 and is designated pssXB(AG30). The reverse orientation vector is designated pssXB(rev).
Figure 1.

Design of the vector system to produce ssDNA in mammalian cells. (A) The vector pssXA expresses an RT–MboII fusion protein driven by a Rous sarcoma virus (RSV) promoter. It also carries a neomycin-resistance gene as a selectable marker. pssXB(AG30) is engineered to express from a cytomegalovirus (CMV) promoter a transcript containing a desired sequence insert situated within NotI sites. The transcript is designed to also incorporate a core promoter site for MoMuLV RT, thereby allowing generation of a cDNA containing one strand of the insert sequence. (B) Partial DNA sequence of the pssXB(AG30) vector. The sequence of the fragment inserted into the NotI sites of pssXB to produce pssXB(AG30) is shown in upper case letters, with the AG30 TFO sequence in bold. The MboII and NotI sites are indicated by underlining and italics, respectively. (C) Stem–loop structure predicted to be formed by the cDNA produced upon reverse transcription of the pssXB(AG30)-derived transcript. The MboII recognition site predicted to form in the stem and the expected sites of MboII cleavage are indicated. (D) Predicted 34 nt ssDNA product (AG34) to be produced from the combined transfection of pssXA and pssXB(AG30), based on the proposed pattern of MboII cleavage in (C). The 30 nt AG30 TFO sequence contained within AG34 is underlined, distinguishing this portion of AG34 from the extra 4 nt at the 3′ end.
Oligodeoxyribonucleotides
ODNs were synthesized by the Midland Certified Reagent Co. (Midland, TX) and purified by either gel electrophoresis or high-pressure liquid chromatography (HPLC), followed by Centricon-3 filtration in distilled water (Amicon, Beverly, MA). The ODNs consisted of phosphodiester linkages and in some cases (as indicated) were synthesized to contain a 3′ propylamine group (11).
Triplex binding assays
Electrophoretic mobility shift assays were performed to determine apparent equilibrium dissociation constants (Kds). ODNs (57 bp) containing the 30 bp polypurine TFO binding site were annealed to form a synthetic target duplex (12). Duplexes were radiolabeled on the 5′ end using T4 polynucleotide kinase and [γ-32P]ATP, gel purified, and incubated for 18 h at 37°C with increasing concentrations of the selected TFOs in a buffer containing 10 mM Tris–HCl (pH 7.6), 1 mM spermine and 10% glycerol. Samples were subjected to polyacrylamide gel electrophoresis in 12% native gels containing 89 mM Tris, 89 mM boric acid, pH 8.0, and 10 mM MgCl2 for 4 h at 60 V, followed by autoradiography.
Cells
The construction and characterization of mouse FL-10 cells were previously described (5). The cells were derived from LTK– cells and were determined to contain a single copy of the pTK2supF construct as a target substrate for triplex-induced intrachromosomal recombination. The FL-10 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Vector transfection and recombination assay
FL-10 cells at a density of 1.67 × 104 per cm2 (1 × 106 in 100-mm dishes) were transfected with 3 µg each of selected vector DNAs that were pre-mixed with 66 µl of GenePorter transfection reagent and diluted into a total of 2 ml serum-free media, as directed by the manufacturer (Gene Therapy Systems, San Diego, CA). After 5 h, the medium was replaced with standard growth medium. The cells were incubated for an additional 24 h in non-selective medium to allow recombination and expression of reconstituted TK genes to occur, after which the medium was changed to DMEM supplemented with 1 × 10–4 M hypoxanthine, 2 × 10–6 M aminopterin and 1.6 × 10–5 M thymidine (HAT) to select for potential recombinants expressing wild-type TK. Cells were maintained in HAT-containing medium for 10 days, at which point surviving colonies were counted.
ODN transfection
Cells at a density of 1.67 × 104 per cm2 (1 × 106 in 100-mm dishes) were transfected with 10 µg ODN DNA per dish mixed with 66 µl of GenePorter and diluted into a total of 2 ml serum-free media, as directed by the manufacturer (Gene Therapy Systems, San Diego, CA). As above, 5 h after transfection the cells were placed in full growth medium supplemented with 10% FBS. Medium was changed to HAT selection 24 h later.
Detection of ssDNA in cells
Cells were transfected with the indicated vectors using cationic lipids, as above. The cells were harvested for analysis of the production of ssDNA 24 h later. The cell monolayers were washed three times with phosphate-buffered saline (PBS) and lysed by addition of 5 ml of Trizol reagent (Life Technologies, Gaithersburg, MD) per 60 mm dish. High molecular weight DNA in the lysate was sheared by repeated pipeting with a Pasteur pipette. The solution was transferred to a 50 ml tube, mixed with chloroform (0.2 ml per each 1 ml Trizol solution) and centrifuged for 30 min at 8000 r.p.m. using a Sorvall RC5C centrifuge. The aqueous supernatant was mixed with 0.5 ml isopropanol per ml of Trizol initially added and centrifuged again at 5500 r.p.m. for 30 min at 4°C. The resulting pellet was washed with 75% ethanol, air dried, and dissolved in water. RNaseA (20 µg/ml) was added and the solution was incubated at 37°C for 2 h. Following phenol/chloroform extraction, the DNA was precipitated with ethanol at –70°C and isolated by centrifugation at 14 000 r.p.m. for 30 min at 4°C. The pellet was washed with 75% ethanol, dissolved in water, and the sample was used in a primer extension reaction. For primer extension, 25 µl of each sample was mixed with the selected primer at 80 pmol in 34 µl (radiolabeled on the 5′ end using T4 polynucleotide kinase and [γ-32P]ATP), 2 µl each of dNTPs from 10 mM stock solutions, 2 µl vent polymerase (New England Biolabs, Beverly, MA) and 7 µl 10× Thermopol buffer (supplied with the vent polymerase). The reactions were heated to 95°C for 2 min in a thermocycler and then carried out for 30 cycles: step 1, 95°C 30 s; step 2, 48°C 1 min; step 3, 70°C 2 min, followed by 10 min at 70°C. The products were visualized by electrophoresis in a 15% denaturing polyacrylamide gel followed by autoradiography. The primers were 15mers designed to be complementary to the 3′ end of the expected products (either AG34 or rev34) and had the sequences 5′-d(CAGGCTCCCCCTCCC)-3′ and 5′-d(CAGGAAGGGGGGGGT)-3′, respectively.
Quantification of ssDNA in transfected cells
Twenty-four hours following transfection, the cells were washed three times with PBS and lysed for preparation of low molecular weight DNA (as above). At the same time, parallel samples were spiked with synthetic AG34 ODN [the predicted product of pssXA and pssXB(AG30)] at concentrations designed to mimic from 104 to 107 molecules per cell, assuming approximately 4 × 106 cells are present at the time of lysis. The rest of the analysis via primer extension was as above.
RESULTS
Design of the ssDNA vector system
The key features of the vector system designed to produce ssDNA in cells are shown in Figure 1A (10). The two-component system consists of one plasmid (pssXA) to express a reverse transcriptase (RT)–MboII fusion protein and a second plasmid (pssXB) to express an engineered RNA transcript from which the desired ssDNA can be generated. The pssXB construct contains an expression cassette incorporating the desired insert DNA sequence (in this case a duplex incorporating the AG30 TFO sequence) flanked by inverted repeats (Fig. 1B). The resulting transcript includes an MoMuLV core promoter and tRNA primer binding site at the 3′ end (10), from which RT can produce a cDNA that can form an internal stem–loop structure due to the inverted repeats (Fig. 1C). MboII cleavage of the stem is designed to release the loop as an ssDNA containing the insert sequence plus a few extraneous nucleotides. In the case of the insert incorporating the AG30 sequence, the expected 34 nt ssDNA is shown in Figure 1D. In the present work, two pssXB-derived vectors were used. One, pssXB(AG30), has the insert sequences oriented to produce the AG34 ssDNA shown in Figure 1D and the second has the NotI insert in the reverse orientation, thereby producing an ssDNA having a C-rich sequence with substantial but not complete complementarity to AG34 (see below).
Comparative binding of synthetic TFOs and potential ssDNA products
In the expected stem–loop structure within the cDNA product of the vector system, the cleavage by MboII occurs within the stem (Fig. 1C). Maintenance of the stem to preserve MboII cleavage site requires that the insert sequences incorporate complementary nucleotides at the 5′ and 3′ ends. In the case of the AG30 sequence this meant that, in the ssDNA ultimately produced, 4 extra nt would have to be included at either the 5′ or 3′ end (Fig. 1D). Although previous work had shown that the AG30 TFO binds with high affinity to the polypurine target site in the FL-10 cell recombination substrate, we were concerned that extra nucleotides at either the 5′ or 3′ end of the TFO might substantially diminish binding. To determine the effect of either a 5′ or 3′ tail on AG30 third-strand binding, gel mobility shift assays were carried out using a synthetic 57 bp duplex as a binding target (Fig. 2). For comparison, we also tested binding by the AG30 TFO with a 3′ propylamine [as used in our previous targeting experiments in mouse cells and mice (5,6)] and by the AG30 TFO with a 3′ OH, since the ssDNA expected to be produced intracellularly by the vector system would have a 3′ OH. As shown, both AG30 with a 5′ tail and AG30 with a 3′ tail show reduced binding relative to AG30, with Kds in the range of 5 × 10–7 M compared with 5 × 10–8 M for AG30. Hence, the 4 nt tails reduce but do not eliminate third-strand binding affinity. In the pssXB(AG30) construct used in our experiments, the predicted ssDNA product will have a 3′ tail relative to the AG30 sequence (Fig. 1D).
Figure 2.
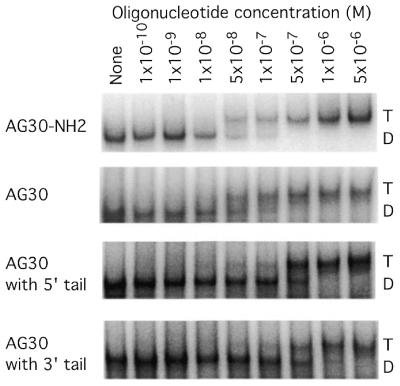
Gel mobility shift assays to determine the influence of extra nucleotides at the 5′ or 3′ end on the affinity of triplex formation by the TFO, AG30. The extra nucleotide ‘tails’ consisted of the sequence 5′-CCTG-3′. The oligomer with the 3′ tail matches the sequence of the expected product of the ssDNA vectors pssXA and pssXB(AG30) (as shown in Fig. 1). The target duplex was end-labeled and incubated with increasing concentrations of the listed oligonucleotides and subjected to native polyacrylamide gel electrophoresis. The lanes marked D or T indicate duplex or triplex, respectively.
Assay for induced intrachromosomal recombination in mouse cells
To test the ability of the vector system to produce ssDNA capable of acting as a TFO to bind to a chromosomal target site, we used an assay for triplex-induced intrachromosomal recombination. Previously we had established a subclone of mouse LTK– cells (FL-10) carrying a pair of mutant TK genes in a single locus as direct repeats (Fig. 3). In this construct, the region between the TK genes was engineered to contain the 30 bp G-rich polypurine sequence amenable to high-affinity third-strand binding in the anti-parallel triplex motif (13) by the AG30 TFO (12). The TK genes contain inactivating XhoI linker insertion mutations at different sites (positions 735 in TK 26 and 1220 in TK8). In the assay, recombination between the two TK genes has the potential to produce a functional gene. Since the parental LTK– cells lack the cellular TK, cells in which the mutant TK genes have recombined to generate a wild-type TK can be selected by growth in the presence of HAT medium. Induction of recombination by transfection with selected vectors or ODNs is quantified by enumerating the HAT-resistant colonies as a proportion of the total number of cells treated. In previous work, we found that the recombination substrate in the FL-10 cells is biased toward reporting gene conversion events rather than crossover recombination (5), and so the assay may actually underestimate the frequency of triplex-induced events.
Figure 3.

(A) Sequences of the ssDNA products, AG34 and rev34, expected to be produced by pssXA plus pssXB(AG30) and by pssXA plus pssXB(rev), respectively, in comparison with AG30. Note that AG34 has 4 extra nt at the 3′ end relative to AG30 (indicated by the underlining). (B) Target substrate designed to investigate induction of intrachromosomal recombination by TFOs. LTK– cells carrying, at a single chromosomal locus, two mutant copies of the TK gene as direct repeats flanking a polypurine third-strand binding site were used to test the ability of vector-generated ssDNAs to mediate triplex formation and recombination induction. The TK genes carry inactivating mutations consisting of XhoI linker insertions at the indicated positions. Potential recombinants are identified as TK+ clones growing in selective HAT-containing medium.
Induced recombination by ssDNA vectors
The FL-10 cells were transfected with a series of vectors, either individually or in pairs, and induction of HAT-resistant colonies was assayed in at least three separate experiments, with the combined results shown in Figure 4. The background frequency of recombination in this set of experiments was in the range of 45 × 10–6, and no induction above this level was seen when either pssXA, pssXB, or pssXA plus pssXB were transfected into the cells. However, when pssXA plus pssXB(AG30) were co-transfected, recombinants were produced at a frequency of 196 × 10–6. No effect was seen when pssXA was combined with pssXB(rev), containing the AG30 sequence insert in reverse orientation. This vector set is expected to express the C-rich rev34 nt ssDNA (Fig. 3), which does not form triplex at the polypurine target site under physiological pH conditions due to the need for cytosine protonation (data not shown).
Figure 4.

Intrachromosomal recombination induced in mouse FL-10 cells following transfection with the indicated vectors. The cells were transfected with the plasmid vectors by co-mixture with cationic lipids. One day later, the cells were placed in HAT-containing medium to select for TK+ recombinants, and surviving colonies were enumerated 10 days later. The results are presented as the frequency of HAT-resistant cells per 106 cells transfected. The mean values from at least three independent experiments are presented [except for A + B(rev), for which data from two experiments are shown]. The standard error is indicated by the error bars, and the compiled colony counts are listed above each bar. A, B, B(AG30) and B(rev) indicate the vectors pssXA, pssXB, pssXB(AG30) and pssXB(rev), respectively.
Subtracting the background of spontaneous recombination in the assay, transfection of the FL-10 cells with pssXA and pssXB(AG30) yielded, on average, an induced frequency of recombinants of 151 in 106. For comparison, in previous work, when AG30-NH2 (end-protected with a propylamine substitution for the 3′ OH) was transfected into the same cells by cationic lipids, the induction above background was 21 in 106 (5). For further comparison, in the current work we also tested the ability of a synthetic version of AG34 (designed to match the predicted ssDNA molecule and therefore made without propylamine protection) to induce recombination when transfected into the FL-10 cells using cationic lipids. No induced recombination was seen above background. Taken together, these results suggest that the use of the ssDNA system for intracellular generation of AG34 inside cells is substantially more effective in achieving chromosome targeting than is lipid-mediated transfection with the synthetic TFOs, either AG30-NH2 or AG34. In addition, the results demonstrate that, as expected, a synthetic version of AG34 is less active than AG30 when transfected into the FL-10 cells. This is consistent with the reduced binding affinity of AG34 compared with AG30 for the 30 bp polypurine target site in these cells, and also likely reflects the lack of end-protection on AG34 to block nuclease degradation.
To determine whether there is any toxicity associated with transfection with the vector system, we measured clonogenic survival following transfection (14). Relative to untreated cells, the clonogenic survival of cells transfected with cationic lipids alone (GenePorter reagent without any added DNA) was 93%, while that of cells transfected with the pssXA plus pssXB(AG30) DNAs via cationic lipids was 84% (data not shown). These results suggest that, as expected, lipofection itself is somewhat toxic to cells; however, the vectors may add to the toxicity, possibly due to expression of the RT–MboII fusion protein.
Detection of ssDNA products in cells
To confirm that the expected ssDNA molecules were generated by the transfected vectors, we isolated low molecular weight DNA from the cells 24 h following vector transfection and carried out a primer extension assay using primers radiolabeled at the 5′ end (Fig. 5). Lysates obtained from untransfected cells and from cells transfected with pssXA alone, pssXB alone, pssXA plus pssXB, or with pssXA plus pssXB(AG30) were assayed using a 15 nt primer designed to be complementary to the AG34 sequence (Figs 1D and 3). The lysate from cells transfected with pssXA and pssXB(rev) was assayed with a 15 nt primer designed to detect the rev34 sequence (Fig. 3). As shown, 34 nt ssDNA species were visualized only in lysates from cells transfected by pssXA plus pssXB(AG30) or pssXA plus pssXB(rev), using the AG34- and rev34-specific primers, respectively. But, as shown above (Fig. 4), only transfection with pssXA plus pssXB(AG30) yielded induced recombinants.
Figure 5.
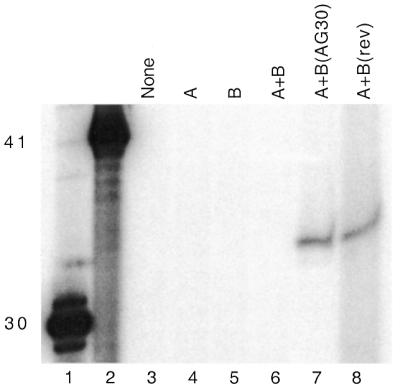
Direct detection of ssDNAs in lysates from vector-transfected cells. Mouse FL-10 cells were transfected with the indicated vectors, and 24 h later the cells were lysed for isolation of low molecular weight DNA. The samples were analyzed for the presence of the predicted ssDNA products, either AG34 or rev34, by primer extension with 15 nt primers 5′ end-labeled with 32P. Lanes 1 and 2 contain 5′ end-labeled synthetic oligonucleotides of length 30 and 41 as size standards to evaluate the sizes of the primer extension products. For lanes 3–7, the primer complementary to AG34 was used. For lane 8, the primer complementary to rev34 was used. Note that lanes 1 and 2 present oligonucleotide size standards only; these lanes are not intended to provide any information as to quantity or yield, and so the band intensities are not meaningful.
Quantification of ssDNA production in cells
To determine the approximate yield of ssDNA molecules produced per cell by the vector set, pssXA and pssXB(AG30), mouse FL-10 cells were transfected either with pssXA plus pssXB(AG30) (via co-mixture with cationic lipids) or, for comparison, with a synthetic ODN, AG34 (also by lipofection). The AG34 oligomer was 3′ end-protected with propylamine but was otherwise designed to exactly match the predicted product of the pssXA plus pssXB(AG30) vectors. As above, low molecular weight DNA was isolated from the cells 24 h following transfection, and a primer extension assay was used to visualize and quantify the ssDNA (Fig. 6). At the time of lysis, parallel samples were spiked with known quantities of AG34 at concentrations calculated to mimic 104–107 molecules per cell. The data were quantified by phosphorimager, and the standard curve was determined by linear regression (not shown). The quantity per cell of ssDNA molecules in the experimental samples was estimated by interpolation from the standard curve, yielding values of 6.2 × 105 molecules per cell for the pssXA plus pssXB(AG30) sample and 1.9 × 105 for the AG34 sample.
Figure 6.

Quantification of ssDNAs in lysates from vector-transfected cells. Mouse FL-10 cells were transfected with either the pssXA plus pssXB(AG30) vector set or with the synthetic ODN, AG34 (both via lipofection), and 24 h later the cells were lysed for isolation of low molecular weight DNA. The samples were analyzed for the presence of the predicted ssDNA product or the presence of ODN by primer extension with a 15 nt primer 5′ end-labeled with 32P. Parallel samples were spiked with AG34 at concentrations calculated to mimic 104–107 molecules per cell. Lanes 1 and 2 contain 5′ end-labeled synthetic oligonucleotides of length 30 and 41 as size markers.
DISCUSSION
In the work presented here, we have found that a vector system designed to generate ssDNA in mammalian cells is able to produce a desired 34 nt TFO sequence in mouse cells, as documented by primer extension analyses performed on lysates from transfected cells. The ssDNA so generated appears to function as a TFO capable of stimulating intrachromosomal recombination in a mouse cell assay that was previously shown to report triplex-induced events (5). In particular, the combined vector set, pssXA and pssXB(AG30), specifically engineered to express the 34 nt G-rich ssDNA, was found to induce recombination between the mutant TK genes in the FL-10 cells at a frequency of 196 × 10–6, substantially above the background level of 45 × 10–6. In contrast, the component vectors used individually yielded no recombination above background. When pssXA plus pssXB(rev), the latter containing the exact same insert as in pssXB(AG30) but in the reverse orientation, were used, the expected C-rich ssDNA was detected in the cells by the primer extension assay, but no induced recombination was seen. The ineffectiveness of the C-rich ssDNA is in keeping with the inability of an ODN of this sequence to form a triple helix at the polypurine target site in FL-10 cells under physiological conditions.
Previously, we had not only established that transfected TFOs could induce recombination within the dual TK substrate in the FL-10 cells (5), but we also found that the assay could report induced recombination over a range of frequencies, from 10–6 to 10–2. Importantly, the prior work revealed that the level of TFO-induced recombination was very much dependent on the efficiency of intracellular delivery of the TFOs. When the AG30 TFO was transfected using cationic lipids, a frequency of induced recombination of 21 × 10–6 above background was seen (5). When the TFOs were introduced by microinjection, recombination frequencies in the range of 1% were detected.
In the work reported here, pssXA and pssXB(AG30) were co-transfected by mixture with cationic lipids, and the average induction above background was 151 × 10–6. This result can be correlated with quantification of intracellular ssDNA production based on a primer extension assay, which yielded an estimate of 6.2 × 105 molecules per cell generated by the pssXA plus pssXB(AG30) vector set. In contrast, transfection of the synthetic ODN, AG34, by lipofection yielded approximately 1.9 × 105 molecules per cell. The relative level of ssDNA production by the vector set is substantial, considering that in these experiments the co-transfection of the two vectors was not optimized; it is likely, therefore, that a sizable number of the cells were not transfected with both vectors. We expect that modifications to the vector system, such as consolidation of the key components into a single plasmid and possibly incorporation into a viral vector, will improve transfection efficiency and lead to increased activity.
In relative terms, the yield of recombinants generated by microinjection of the synthetic AG30 TFO into the FL-10 cells in our previous work (5) was 66-fold higher than that induced by pssXA plus pssXB(AG30) in the work here. On the other hand, the ssDNA vector system was found to produce recombinants above background at a frequency 7-fold higher than that stimulated by AG30 transfection using cationic lipids [data also from the previous study (5)]. The estimated numbers of ODNs or ssDNA molecules generated per cell by these methods are 7 × 104, 6 × 105 and 1.9 × 105, respectively. Clearly, microinjection results in the introduction of TFOs into a biologically active compartment in a manner superior to the other methods, since the activity of the microinjected TFOs is disproportionate to the estimated intracellular yield. However, the ability of the ssDNA vector system to generate biologically active ssDNA in cells to form triplexes at genomic sites is still impressive, considering some of the drawbacks inherent to the process.
In the first place, the ssDNA is generated biologically, and so no chemical modification is possible. Previous studies have suggested that ODNs modified by either backbone substitution (e.g. phosphoramidate instead of phosphodiester), sugar modification (2′-O-methyl or 2′-O-methoxyethyl), base substitution (7-deazaxanthine instead of adenine), or conjugation to intercalating agents (acridine or pyrene) can substantially enhance triplex binding affinity under physiological conditions and thereby boost intracellular activity (3,15,16).
In addition, the intracellularly synthesized ssDNA also has no modification to resist nuclease degradation, such as the 3′ propylamine substitution (present on the microinjected AG30 TFO) that we and others have used to block 3′ exonucleases (6,11). Yet the primer extension analyses performed on the cell lysates obtained 24 h after vector transfection revealed persistence of full-length ssDNA product without obvious degradation. These results suggest that the ssDNA may be produced in a compartment at least partially protected from nuclease activity. In contrast, transfected ODNs are thought to be at high risk for degradation, especially from serum and lysosomal enzymes. Unprotected phosphodiester ODNs last only minutes in cell culture or in animals, and studies have shown that strategies to protect the oligomers, such as the 3′ end-capping or phosphorothioate backbone substitution, are needed (11).
Another drawback is that the design of the ssDNA system, with the requirement for MboII cleavage to release the ssDNA, results in the inclusion of a few extra nucleotides appended to the actual TFO sequence. In the case of the AG30 construct in pssXB(AG30), the predicted product would have 4 extra nt at the 3′ end and we did detect such a product in the primer extension analysis. However, the extra 4 nt create mismatches in the third-strand binding code for the anti-parallel triplex motif (2); they were found, as expected, to reduce the third-strand binding affinity. As shown in Figure 2, a synthetic 34mer matching the expected ssDNA product has a binding affinity ~10-fold lower than that of AG30 itself. That the ssDNA system was still effective in inducing recombination in spite of this decreased affinity further serves to demonstrate the power of the system and it suggests that substantial increases in TFO activity should be possible if the extra nucleotides can be eliminated in a revised design. In addition, this difference in affinity may partially explain why microinjection of the precise AG30 molecule is more effective than the ssDNA vector system (5).
Although we have measured induced recombination and have documented production of the predicted ssDNA species, we did not show directly that either RT or MboII activity was present in the transfected cells. However, Chen et al. (10), who first reported this approach for antisense DNA generation, did show evidence of RT activity in the vector-transfected cells. It is theoretically possible that either the RT activity or the MboII activity might have some deleterious consequences for the cells. In this regard, we found evidence that there might be some toxicity associated with the vector system. The clonogenic survival of the cells transfected with pssXA and pssXB(AG30) was reduced to 84% of untreated cells or to ∼90% of the survival of cells transfected with the lipofection reagent alone. In addition, several samples transfected with the pssXA vector showed reduced yields of HAT-resistant colonies compared with the untransfected control (Fig. 4), suggesting that some of the background recombinants might have been lost due to toxicity. [These samples were pssXA alone, pssXA plus pssXB and pssXA plus pssXB(rev). None of these would be expected to produce an ssDNA capable of forming a triplex at the target site in FL-10 cells, but all three could produce the RT–MboII fusion protein.] Note that in our analysis, the frequency of recombinants is calculated based on the number of transfected cells as a denominator, not on the number of surviving cells.
While, to our knowledge, this is the first use of a vector system to produce ssDNA in cells for chromosomal triplex formation, some previous studies have explored the use of intracellularly generated RNA transcripts for this purpose (17,18). However, the RNA-based approach has several disadvantages. Naturally occurring RNA is excluded from the anti-parallel purine motif for triplex formation that is otherwise favored at G:C bp-rich target sites, such as the one used in the experiments here (13,19,20). On the other hand, triplex formation in the parallel pyrimidine motif by naturally occurring RNA or DNA requires acidic pH because of the need for cytosine protonation (21,22). Strategies to overcome this require chemical modification of the C residues, which cannot be accomplished in the case of biologically generated molecules. In addition, no mechanism has yet been developed to post-transcriptionally modify the RNAs (in a manner analogous to the use of the MboII activity here) and so the transcripts typically carry a large number of extra nucleotides that reduce the third-strand binding affinity. The use of engineered RNA transcripts generated inside cells, on the other hand, has shown substantial promise for antisense applications (23).
In conclusion, the results presented here demonstrate that ssDNA can be effectively generated inside mammalian cells for the purpose of creating TFOs to target chromosomal sites. Although we studied only induced recombination, other applications of triplex technology, such as targeted gene knockout or transcription inhibition, should also be feasible using this approach. The ability to generate anti-gene TFOs with high efficiency in mammalian cells may offer an important new research tool and may provide the basis of a novel form of genetic therapy.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Macris, F. Rogers, L. Narayanan, A. F. Faruqi, R. Franklin, S. J. Baserga and L. Cabral for their help. This work was supported by the NIH (RO1 GM54731 and RO1CA64186) and by a Scholar Award to PMG from the Leukemia and Lymphoma Society. The ssDNA vector system was supplied by CytoGenix, Inc., Houston, TX.
REFERENCES
- 1.Giovannangeli C. and Helene,C. (2000) Triplex-forming molecules for modulation of DNA information processing. Curr. Opin. Mol. Ther., 2, 288–296. [PubMed] [Google Scholar]
- 2.Chan P.P. and Glazer,P.M. (1997) Triplex DNA: fundamentals, advances, and potential applications for gene therapy. J. Mol. Med., 75, 267–282. [DOI] [PubMed] [Google Scholar]
- 3.Faria M., Wood,C.D., Perrouault,L., Nelson,J.S., Winter,A., White,M.R., Helene,C. and Giovannangeli,C. (2000) Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 97, 3862–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim H.G., Reddoch,J.F., Mayfield,C., Ebbinghaus,S., Vigneswaran,N., Thomas,S., Jones,D.E.,Jr and Miller,D.M. (1998) Inhibition of transcription of the human c-myc protooncogene by intermolecular triplex. Biochemistry, 37, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 5.Luo Z., Macris,M.A., Faruqi,A.F. and Glazer,P.M. (2000) High-frequency intrachromosomal gene conversion induced by triplex-forming oligonucleotides microinjected into mouse cells. Proc. Natl Acad. Sci. USA, 97, 9003–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasquez K.M., Narayanan,L. and Glazer,P.M. (2000) Specific mutations induced by triplex-forming oligonucleotides in mice. Science, 290, 530–533. [DOI] [PubMed] [Google Scholar]
- 7.Datta H.J., Chan,P.P., Vasquez,K.M., Gupta,R.C. and Glazer,P.M. (2001) Triplex-induced recombination in human cell-free extracts: Dependence on XPA and HsRad51. J. Biol. Chem., 27, 18018–18023. [DOI] [PubMed] [Google Scholar]
- 8.Faruqi A.F., Datta,H.J., Carroll,D., Seidman,M.M. and Glazer,P.M. (2000) Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol. Cell. Biol., 20, 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y., Ji,Y.J., Roxby,R. and Conrad,C. (2000) In vivo expression of single-stranded DNA in mammalian cells with DNA enzyme sequences targeted to C-raf. Antisense Nucleic Acid Drug Dev., 10, 415–422. [DOI] [PubMed] [Google Scholar]
- 11.Zendegui J.G., Vasquez,K.M., Tinsley,J.H., Kessler,D.J. and Hogan,M.E. (1992) In vivo stability and kinetics of absorption and disposition of 3′ phosphopropyl amine oligonucleotides. Nucleic Acids Res., 20, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beal P.A. and Dervan,P.B. (1991) Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science, 251, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 14.Fritzell J.A., Narayanan,L., Baker,S.M., Bronner,C.E., Andrew,S.E., Prolla,T.A., Bradley,A., Jirik,F.R., Liskay,R.M. and Glazer,P.M. (1997) Role of DNA mismatch repair in the cytotoxicity of ionizing radiation. Cancer Res., 57, 5143–5147. [PubMed] [Google Scholar]
- 15.Majumdar A., Khorlin,A., Dyatkina,N., Lin,F.-L.M., Powell,J., Liu,J., Fei,Z., Khripine,Y., Watanabe,K.A., George,J. et al. (1998) Targeted gene knockout mediated by triple helix forming oligonucleotides. Nature Genet., 20, 212–214. [DOI] [PubMed] [Google Scholar]
- 16.Faruqi A.F., Krawczyk,S.H., Matteucci,M.D. and Glazer,P.M. (1997) Potassium-resistant triple helix formation and improved intracellular gene targeting by oligodeoxyribonucleotides containing 7-deazaxanthine. Nucleic Acids Res., 25, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noonberg S.B., Scott,G.K., Garovoy,M.R., Benz,C.C. and Hunt,C.A. (1994) In vivo generation of highly abundant sequence-specific oligonucleotides for antisense and triplex gene regulation. Nucleic Acids Res., 22, 2830–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shevelev A., Burfeind,P., Schulze,E., Rininsland,F., Johnson,T.R., Trojan,J., Chernicky,C.L., Helene,C., Ilan,J. and Ilan,J. (1997) Potential triple helix-mediated inhibition of IGF-1 gene expression significantly reduces tumorigenicity of glioblastoma in an animal model. Cancer Gene Ther., 4, 105–112. [PubMed] [Google Scholar]
- 19.Roberts R.W. and Crothers,D.M. (1992) Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science, 258, 1463–1466. [DOI] [PubMed] [Google Scholar]
- 20.Semerad C.L. and Maher,L.J.,III (1994) Exclusion of RNA strands from a purine motif triple helix. Nucleic Acids Res., 22, 5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asensio J.L., Lane,A.N., Dhesi,J., Bergqvist,S. and Brown,T. (1998) The contribution of cytosine protonation to the stability of parallel DNA triple helices. J. Mol. Biol., 275, 811–822. [DOI] [PubMed] [Google Scholar]
- 22.Singleton S.F. and Dervan,P.B. (1992) Influence of pH on the equilibrium association constants for oligodeoxyribonucleotide-directed triple helix formation at single DNA sites. Biochemistry, 31, 10995–11003. [DOI] [PubMed] [Google Scholar]
- 23.Gorman L., Suter,D., Emerick,V., Schumperli,D. and Kole,R. (1998) Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc. Natl Acad. Sci. USA, 95, 4929–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]