

Abstract
Endogenous glucocorticoids are crucial to various physiological processes, including metabolism, development and inflammation. Since 1948, synthetic glucocorticoids have been used to treat various immune-related disorders. The mechanisms that underlie the immunosuppressive properties of these hormones have been intensely scrutinized, and it is widely appreciated that glucocorticoids have pleiotropic effects on the immune system. However, a clear picture of the cellular and molecular basis of glucocorticoid action has remained elusive. In this Review, we distil several decades of intense (and often conflicting) research that defines the interface between the endocrine stress response and the immune system.
In 1948 at Mayo Clinic (Minnesota, United States), a patient with rheumatoid arthritis began daily injections of ‘Compound E’, a synthetic version of a steroid hormone that was isolated from animal adrenal glands. Within 3 days, the patient was nearly asymptomatic. In 1950, the Nobel Prize in Physiology or Medicine was awarded to Philip S. Hench, Edward Kendall and Tadeus Reichstein for their discovery of ‘the adrenal cortical hormones’. Much excitement surrounded the miracle drug, Compound E, and it was referred to as a glucocorticoid or corticosteroid, although prolonged clinical use was discouraged upon the realization of adverse side effects (BOX 1). To this day, however, glucocorticoids remain the mainstays in the treatment of inflammatory and autoimmune pathologies, and they are used as immunosuppressants following organ transplantation and as lympholytics in chemotherapeutic regimens.
Box 1 |. Adverse effects of glucocorticoid excess: the CUSHINGOID mnemonic.
The term ‘cushingoid’ describes the collection of signs and symptoms that are associated with Cushing syndrome, which results from excess glucocorticoid activity (either endogenous or exogenous). The medical mnemonic below reveals the widespread roles of glucocorticoids in various physiological systems.
| Letter of mnemonic | Sign or symptom | Pathophysiology |
|---|---|---|
| C | Cataracts | Unknown but may involve perturbed migration of lens epithelial cells113 |
| U | Ulcers | Disputed, but may be due to the inhibition of gastric-protective prostaglandins, mucus production and/or bicarbonate secretion114 |
| S | Striae and skin thinning | Unclear but may involve decreased fibroblast proliferation and/or altered metabolism of the extracellular matrix115 |
| H | Hypertension and hirsutism (in women) | Increased plasma volume, cardiac output and peripheral vascular resistance occur through both mineralocorticoid and glucocorticoid effects116; hirsutism occurs due to dysregulated production of adrenal testosterone |
| I | Immunosuppression and infections | Discussed in the main text |
| N | Necrosis of femoral heads | Increased bone marrow fat, decreased bone perfusion and osteocyte apoptosis117 |
| G | Glucose elevation | Glucose intolerance and insulin insensitivity118 |
| O | Osteoporosis and obesity | Inhibition of osteoblast function and survival, decreased bone mass (osteoporosis)119 and redistribution of adipose tissue (obesity)118 |
| I | Impaired wound healing | Reduced proliferation of fibroblasts and epidermal cells, inhibition of collagen synthesis and reduced angiogenesis120 |
| D | Depression and mood changes | Psychological, cognitive and behavioural disturbances121 |
For decades, the clinical application of glucocorticoids outpaced our mechanistic understanding of their immunosuppressive properties, and research into glucocorticoid-mediated regulation of the immune system continues to be an intense field of investigation for several reasons. First, the modes of glucocorticoid action are manifold. Classically, glucocorticoid binding to glucocorticoid receptors activates or represses gene transcription, and glucocorticoids regulate up to 20% of the genome according to some studies1. However, studies in recent years have uncovered other mechanisms that underlie glucocorticoid activity, including the non-genomic effects of the liganded glucocorticoid receptors, ‘crosstalk’ of glucocorticoid receptors with other transcription factors, and possibly even receptor-independent effects. Second, glucocorticoid receptors are expressed by nearly all nucleated cells, but the functional effects of glucocorticoids differ by cell type. The physiological outcomes of glucocorticoid signalling therefore reflect a kaleidoscope of cell-specific effects. Cell-depletion studies and lineage-specific glucocorticoid receptor-knockout mice have allowed investigation of the cell-specific contributions to the in vivo effects of glucocorticoids (BOX 2). Finally, there is evidence that glucocorticoids promote immune responsiveness under certain conditions, which presents an often overlooked challenge to the clinical dogma of glucocorticoids acting solely as immunosuppressive agents.
A formidable amount of literature exists on the topic of glucocorticoid-mediated regulation of the immune system. Here, we provide an update on the effects of glucocorticoids on canonical immune processes — for example, on the production of inflammatory mediators, leukocyte migration, lymphocyte development and antigen receptor signalling — with the goal of providing a primer for readers who are interested in the interface between the neuroendocrine stress response and the immune system. We begin with a brief overview of steroidogenesis and the glucocorticoid receptor; the following sections are dedicated to discussing glucocorticoid-mediated regulation of innate immunity and inflammation, and adaptive immunity, and we finish with a working model that explains both the immune-potentiating and immunosuppressive actions of glucocorticoids.
Glucocorticoid production
Steroidogenesis.
Endogenous glucocorticoids (cortisol in humans and corticosterone in rodents) are essential for life, and they regulate myriad physiological and developmental processes. Like all steroid hormones, glucocorticoids are synthesized from cholesterol through an enzymatic process called steroidogenesis, which occurs in mitochondria2. Glucocorticoid bio-synthesis occurs in the adrenal cortex, although local glucocorticoid production has also been reported in the thymus, intestine and skin3. As shown in FIG. 1, adrenal glucocorticoid production is induced upon activation of the hypothalamic–pituitary–adrenal axis (HPA axis), which is a neural–endocrine ‘hub’ that coordinates physiological responses to external stimuli. Glucocorticoids released into the blood exert systemic effects by binding glucocorticoid receptors that are present in cells throughout the body, including cells in the hypothalamus and pituitary gland, which are part of a negative feedback loop that controls glucocorticoid production (FIG. 1). Notably, early clues about the effects of glucocorticoids on the immune system came from patients with Cushing syndrome (which is characterized by glucocorticoid excess; BOX 1) and patients with Addison disease (which is characterized by glucocorticoid deficiency).
Figure 1 |. Regulation of glucocorticoid production by the hypothalamic–pituitary–adrenal axis.
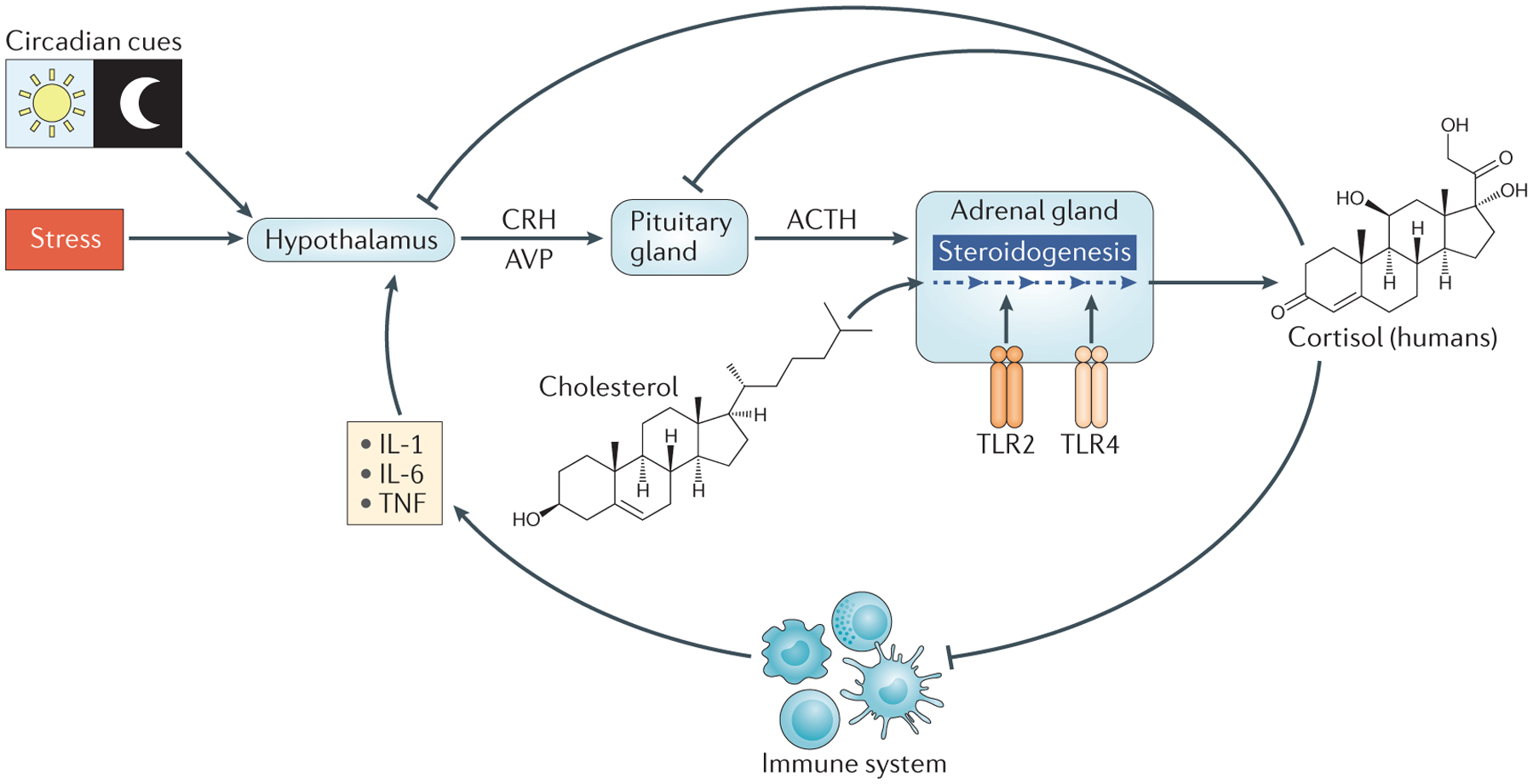
The hypothalamus responds to circadian cues, stress (real or perceived) and inflammatory cytokines by producing corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP). CRH and AVP act on the anterior pituitary gland to induce the synthesis and secretion of adrenocorticotropin hormone (ACTH). ACTH enters the circulation and binds receptors on adrenocortical cells to stimulate steroidogenesis (indicated by the dark blue dashed arrows). Activation of Toll-like receptor 2 (TLR2) and TLR4 on adrenocortical cells also triggers steroidogenesis. During steroidogenesis, cholesterol undergoes a series of enzymatic changes that result in the production of glucocorticoids. Cortisol and corticosterone are the primary glucocorticoids in humans and rodents, respectively. Glucocorticoids enter the circulation for distribution throughout the body. Glucocorticoids negatively regulate the hypothalamic–pituitary–adrenal (HPA) axis by feeding back on the hypothalamus and pituitary gland, and by decreasing the expression of pro-inflammatory cytokines. IL, interleukin; TNF, tumour necrosis factor.
Psychological distress, physical strain and tissue trauma activate the HPA axis, which in turn triggers rapid and substantial increases in glucocorticoid production. The hypothalamus is also stimulated by interleukin-1 (IL-1), tumour necrosis factor (TNF) and IL-6 (REF. 4) (FIG. 1). Cytokine-induced HPA activation promotes glucocorticoid secretion, which, as detailed below, potently suppresses pro-inflammatory cytokine expression; thus, a second feedback loop links the inflammatory response to the HPA axis (FIG. 1). Recent studies have demonstrated that direct ligation of Toll-like receptor 2 (TLR2) and TLR4 in adrenocortical cells induces glucocorticoid production, and this is indicative of an inflammatory pathway of glucocorticoid production that feeds into the HPA axis5 (FIG. 1).
The HPA axis also couples glucocorticoid production to circadian and ultradian rhythms6 (FIG. 1). Although the amplitudes of circadian and ultradian fluctuations in steroidogenesis are not as pronounced as those associated with a stress response, these small changes in glucocorticoid concentrations still modulate immune responsiveness and leukocyte trafficking7. Fluctuations in blood lymphocyte counts in humans, for example, inversely correlate with diurnal patterns of glucocorticoid secretion, such that there is a 40% decline in the number of circulating T cells from night to morning as a result of altered tissue homing8.
Local regulation of glucocorticoid activity.
Adrenal steroidogenesis drives systemic changes in glucocorticoid concentrations, but extracellular binding proteins and intracellular enzymes regulate glucocorticoid activity locally. Corticosteroid-binding globulin (CBG) renders cortisol inactive and leaves only ~5% of circulating cortisol in a bioactive form. CBG is important for the systemic distribution of glucocorticoids via the circulation, but it may also have a tissue-specific role in glucocorticoid delivery. For example, neutrophil elastase cleaves CBG, thereby liberating bioactive glucocorticoids at inflammatory sites9. Within cells, 11β-hydroxysteroid dehydrogenase enzymes (11βHSD1 and 11βHSD2) regulate the interconversion of bioactive cortisol and inactive cortisone. Inflammatory signals, including TNF and IL-1β, modulate the expression of 11βHSD enzymes, thereby altering cellular sensitivity to endogenous glucocorticoids10.
Glucocorticoids in the clinic.
The earliest glucocorticoid used in the clinic was synthetic cortisone (Compound E, described above; note that inside cells, cortisone is converted into cortisol by 11βHSD1). Although this treatment was efficacious in treating rheumatoid arthritis, prolonged cortisone therapy causes mineral imbalance and fluid retention through the activation of mineralocorticoid receptors in the kidney. A decades-long pharmaceutical search for synthetic glucocorticoids that have minimal mineralocorticoid effects yielded several analogues that are routinely used in the clinic today, including prednisone, beclomethasone and fluticasone11. Each glucocorticoid analogue has distinct pharmacological properties, including fat solubility, half-life and mineralocorticoid activity. In general, synthetic glucocorticoids are more potent immunoregulators than is cortisol because they are not subject to endogenous inhibitors of cortisol activity, including CBG binding and 11βHSD inactivation. Moreover, synthetic glucocorticoids bind the glucocorticoid receptors with higher affinity and mineralocorticoid receptors with lower affinity than do endogenous glucocorticoids, thereby minimizing mineralocorticoid-based side effects.
Glucocorticoid signalling
Lipophilic glucocorticoids diffuse through cell membranes to bind cytosolic glucocorticoid receptors, which are ubiquitously expressed by nucleated cells. The human glucocorticoid receptor is encoded by the nine-exon gene NR3C1 (see Further information). Similar to other nuclear receptors, the glucocorticoid receptor comprises three domains: first, the amino-terminal domain, which interacts with co-regulators and the transcriptional machinery; second, the DNA-binding domain, which contains two zinc-finger motifs for genomic interactions; and third, the ligand-binding domain, which features a hydrophobic pocket for ligand binding12. A flexible hinge region — which promotes receptor dimerization, nuclear translocation and DNA binding — lies between the DNA-binding domain and the ligand-binding domain12. Glucocorticoid receptors contain multiple sites for post-translational modifications, including phosphorylation, acetylation, ubiquitylation and sumoylation sites, which have important roles in nuclear import and export, gene regulation and receptor degradation13.
FURTHER INFORMATION.
GeneCards entry for NR3C1: http://www.genecards.org/cgi-bin/carddisp.pl?gene=NR3C1
Nuclear Receptor Signaling Atlas entry for NR3C1: https://www.nursa.org/nursa/index.jsf
Online Mendelian Inheritance in Man entry for NR3C1: http://www.omim.org/entry/138040
The British Pharmacological Society and the International Union of Basic and Clinical Pharmacology (IUPHAR) Guide to PHARMACOLOGY entry for NR3C1: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=625
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
There is substantial heterogeneity in the proteins that are translated from the NR3C1 gene. The canonical receptor associated with transcriptional regulation by glucocorticoids is GRα. GRβ, by contrast, is a splice variant that does not bind a ligand but is thought to exert a dominant-negative effect on GRα, although some direct transcriptional activity has been reported13. Accumulating evidence indicates that inflammation affects the glucocorticoid receptor isoform expression profile, and enhanced GRβ expression has been associated with glucocorticoid resistance in several pathologies14. The repertoire of glucocorticoid receptor proteins is further expanded by the presence of eight alternative translation initiation sites; the resulting N-terminal glucocorticoid receptor isoforms exhibit distinct transcriptional activities, nuclear trafficking properties and tissue distribution patterns15. Differential expression of glucocorticoid receptor isoforms may, to some degree, explain cell-specific responses to glucocorticoids.
In the absence of ligand, GRα localizes to the cytoplasm in a multiprotein complex that contains heat-shock proteins, immunophilins and other chaperones, which enhance the affinity of the receptor for ligand and prevent receptor degradation (FIG. 2). Classically, ligand binding was thought to induce a conformational change in the glucocorticoid receptor that disassociates the chaperone complex, thus allowing nuclear translocation. Recent studies, however, provide evidence that glucocorticoid receptor chaperones are important for the nuclear import of the glucocorticoid receptor16. Within the nucleus, the glucocorticoid receptor interacts with DNA and with other proteins to exert biological effects (FIG. 2). The withdrawal of ligand results in calreticulin-mediated nuclear export of the glucocorticoid receptor to the cytoplasm, where the glucocorticoid receptor–chaperone complex reforms, poised for another round of ligand binding17.
Figure 2 |. Mechanisms of glucocorticoid activity.
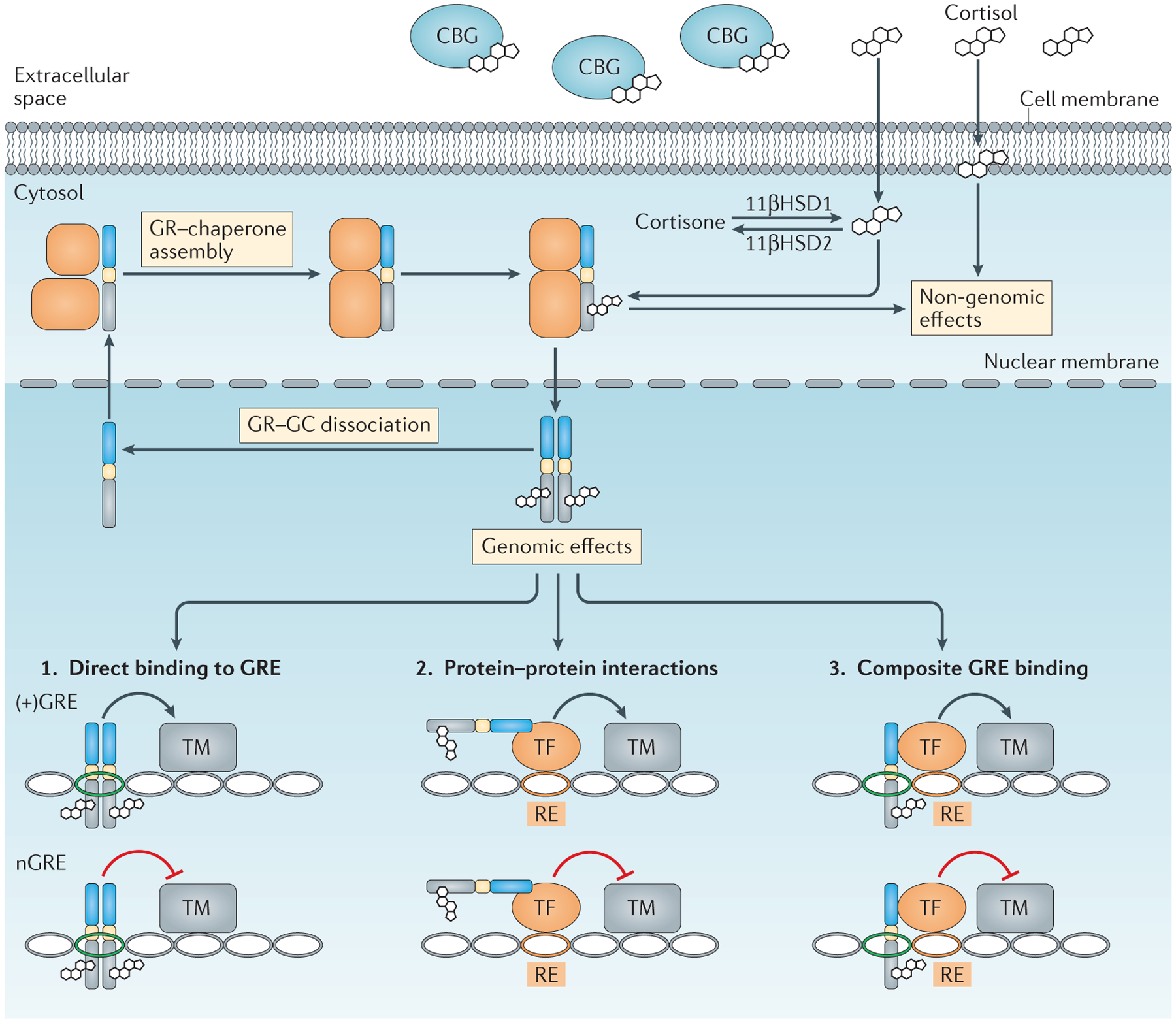
In the extracellular space, most endogenous glucocorticoids (GCs) are inactive owing to binding with corticosteroid-binding globulin (CBG). Unbound cortisol is lipid soluble and diffuses through cell membranes. Cortisol that enters the cytoplasm can be converted into inactive cortisone through enzymatic modification by 11β-hydroxysteroid dehydrogenase 2 (11βHSD2), whereas 11βHSD1 favours the reverse reaction. Cortisol intercalation into plasma membranes can exert non-genomic effects. Cytoplasmic cortisol binds the GC receptor (GR) as part of a chaperone complex. The ligand-bound GR translocates to the nucleus to exert genomic effects, or has non-genomic effects in the cytoplasm and mitochondria. Within the nucleus, the liganded GR alters gene expression through three basic mechanisms: direct binding to GC response elements (+GREs) or negative GREs (nGREs); protein–protein interactions with other transcription factors (TFs) that affect their transcriptional activity; and composite binding to DNA and protein substrates. Notably, positive and negative gene regulation is possible for each of these mechanisms. RE, response element; TM, transcriptional machinery.
As mentioned above, endogenous glucocorticoids also bind with high affinity to mineralocorticoid receptors (encoded by NR3C2), which regulate genes involved in sodium reabsorption. The expression pattern of mineralocorticoid receptors is more restricted than that of glucocorticoid receptors; high expression of mineralocorticoid receptors is found in the ducts and tubules of the kidney, in the colon, in the heart and in the hippo campus, but low expression is found in leukocytes. Blood concentrations of glucocorticoids are typically much higher than are concentrations of the endogenous mineralocorticoid aldosterone, but in classic mineralocorticoid-sensitive tissues, mineralocorticoid receptors specificity for aldosterone is enforced by 11βHSD2, which prevents glucocorticoid occupation of mineralocorticoid receptors through the inactivation of cortisol. There is little information about the roles of mineralocorticoid receptors in immunity, although it is notable that leukocytes do not express 11βHSD2, which raises the possibility that the leukocyte mineralocorticoid receptor is not ‘protected’ from occupation by endogenous glucocorticoids.
Gene regulation by glucocorticoid receptors.
Glucocorticoid-mediated immune modulation has classically been attributed to glucocorticoid receptor-induced alterations in gene expression. The genomic effects of glucocorticoids are categorized into three general mechanisms (FIG. 2). The first mode of regulation occurs through the function of the glucocorticoid receptor as a transcription factor and involves direct binding of the liganded glucocorticoid receptor to DNA. Glucocorticoid receptor homodimers bind glucocorticoid response elements (GREs; which have the consensus sequence GGAACAnnnTGTTCT, where ‘n’ is any base) to enhance gene expression18,19. The GRE-bound glucocorticoid receptor recruits co-regulators, including steroid receptor co-activator 1 (SRC1; also known as NCOA1), glucocorticoid receptor-interacting protein 1 (GRIP1; also known as NCOA2) and CREB-binding protein (CBP; also known as CREBBP), and then binds transcriptional activator BRG1 of the SWI/SNF chromatin-remodelling complex to form the pre-initiation complex and activate transcription. Glucocorticoid receptor monomers, by contrast, can bind negative GREs (nGREs; which have the consensus sequence CTCC(n)0–2GGAGA), and recruit the co-repressors nuclear receptor co-repressor 1 (NCOR1) and SMRT (also known as NCOR2) to inhibit gene transcription20. Classically, GREs and nGREs were thought to function as components of gene promoters, but recent genome-wide studies have revealed that most glucocorticoid receptor-binding sites lie in intragenic areas and regions distant from transcription start sites, which indicates that glucocorticoid receptors may exert regulatory effects over considerable genomic distances21. Whole-genome studies have also revealed that there is little overlap in glucocorticoid receptor-binding sites among tissues and cell types, which suggests that chromatin accessibility is a pre-requisite for the glucocorticoid receptor–DNA interaction22. GRE and nGRE accessibility probably contributes to the cell type-specific activities of glucocorticoids.
The second mode of glucocorticoid receptor-mediated gene regulation occurs when the glucocorticoid receptor physically interacts with, or ‘tethers’, another transcription factor without contacting DNA23. This protein–protein interaction alters the capacity of the partnering transcription factor to bind DNA, or to recruit co-regulators and/or the transcriptional machinery. Tethering has received a considerable amount of attention as a mechanism for glucocorticoid-mediated inhibition of immune responses, as the glucocorticoid receptor interferes with key pro-inflammatory transcription factors, including nuclear factor-κB (NF-κB) and activator protein 1 (AP-1), as well as members of the signal transducer and activator of transcription (STAT), CCAAT/enhancer-binding protein (C/EBP) and nuclear factor of activated T cells (NFAT) families (BOX 3). Glucocorticoid receptor tethering is not a one-way relationship; numerous studies have revealed that tethering affects the ability of the glucocorticoid receptor to activate its target genes23.
Box 3 |. Transcription factors modulated by the glucocorticoid receptor.
Glucocorticoid receptors bind other transcription factors to modulate their effects on gene transcription. The table below lists various transcription factors that interact with the glucocorticoid receptor via protein–protein binding. For each partnering transcription factor, the type of protein–protein interaction is shown as either tethering (in which glucocorticoid receptor binds the indicated transcription factor but glucocorticoid receptor binding to DNA is unnecessary) or composite (in which glucocorticoid receptor binding to both the target transcription factor and DNA is required).
| Transcription factor | Type of interaction | Effects of interaction with glucocorticoid receptor | Refs |
|---|---|---|---|
| AP-1 | Tethering | AP-1 inhibited | 124 |
| C/EBPβ | Composite | C/EBPβ inhibited | 125 |
| CREB | Composite | Enhanced CREB2 activity | 126 |
| ETS2 | Composite | Enhanced ETS2 activity | 127 |
| GATA1 | Composite | GATA1 inhibited | 128 |
| NF-κB | Tethering | NF-κB inhibited | 129 |
| NR4A1 | Tethering | NR4A1 inhibited | 130 |
| POU2F1 | Tethering | POU2F1 inhibited | 131 |
| SMAD3 | Tethering | SMAD3 inhibited | 132 |
| STAT3 | Tethering | Enhanced STAT3 activity | 133 |
| STAT5 | Tethering | Enhanced STAT5 activity | 134 |
| STAT6 | Tethering | STAT6 inhibited | 135 |
| T-bet | Tethering | T-bet inhibited | 82 |
AP-1, activator protein 1; C/EBPβ, CCAAT/enhancer-binding protein-β; CREB, cAMP-responsive element-binding protein; GATA1, GATA-binding factor 1 (also known as erythroid transcription factor); NF-κB, nuclear factor-κB; NR4A1, nuclear receptor subfamily 4 group A member 1 (also known as NUR77); POU2F1, POU domain class 2 transcription factor 1 (also known as OCT1); SMAD3, mothers against decapentaplegic homologue 3; STAT, signal transducer and activator of transcription.
The third mode of action occurs when glucocorticoids regulate gene expression through glucocorticoid receptor binding to composite elements, which are DNA sequences that contain both a GRE and a response element for a distinct transcription factor24. Although it is common for the glucocorticoid receptor to inhibit a partner transcription factor at a composite element or in cases of tethering, it is important to note that glucocorticoid receptor binding can enhance the activity of the partnering transcription factor. For example, AP-1 is a common partner for the glucocorticoid receptor at composite elements throughout the genome25, but the effects of the glucocorticoid receptor depend on the AP-1 subunit composition; the glucocorticoid receptor robustly inhibits the activity of AP-1 heterodimers comprising JUN and FOS, whereas the function of AP-1 homodimers comprising JUN–JUN is less constrained by interaction with the glucocorticoid receptor and may even be enhanced26,27. Similarly, the glucocorticoid receptor differentially affects the activity of various STAT family members28 (BOX 3).
An attractive but controversial idea posits that glucocorticoids exert immunosuppressive effects primarily through glucocorticoid receptor tethering of transcription factors such as NF-κB and AP-1, which reduces the expression of pro-inflammatory genes (trans repression), whereas adverse side effects occur through gene activation via direct binding of the glucocorticoid receptor to GREs (transactivation). The notion that clinically beneficial and harmful properties of glucocorticoids are dissociable at the molecular level was suggested by observations of glucocorticoid receptor-dimerization mutants that fail to induce the expression of certain GRE-bearing genes yet still repress the activity of AP-1 and NF-κB29. These observations fuelled a pharmaceutical search for glucocorticoid receptor-binding compounds that selectively promote the transrepressive properties of the glucocorticoid receptor with minimal induction of transactivation30. It is generally appreciated now, however, that the transrepression-versus-transactivation concept is likely to be an oversimplification of the effects of glucocorticoids on immune processes, particularly in light of reports that glucocorticoid receptor-dimerization mutants retain some capacity to promote gene transcription31. Moreover, it is unclear whether monomers of glucocorticoid receptor-dimerization mutants retain the ability to bind nGREs and thus repress gene transcription. The degree of immunomodulation that occurs through glucocorticoid receptor-mediated gene regulation as opposed to that occurring through transcription factor inhibition is still unclear, but several genes that are directly targeted by glucocorticoids — as discussed below — exert robust regulatory effects on immune responses.
Non-genomic effects of glucocorticoids.
Whereas the genomic effects of glucocorticoids typically manifest over a period of hours, glucocorticoids also exert biological changes within seconds to minutes of exposure that do not arise from altered gene transcription. Glucocorticoid intercalation into membranes has been implicated as a glucocorticoid receptor-independent mechanism for altering cation transport through plasma membranes and promoting proton leak from mitochondria32. By contrast, glucocorticoid receptors are reported to interfere with cytoplasmic signalling complexes33. Moreover, ligand-dependent liberation of molecular components of the glucocorticoid receptor–chaperone complex has been implicated in attenuating signal transduction33. The ligand-bound glucocorticoid receptor has also been reported to translocate to mitochondria, resulting in apoptosis34.
Regulation of inflammation
Glucocorticoids are potent regulators of inflammation. The inflammatory response comprises a series of interconnected processes that can be generally divided into three sequential phases: first, the alarm phase, in which ‘danger’ signals trigger the release of inflammatory mediators; second, the mobilization phase, in which leukocytes infiltrate the injured site; and last, the resolution phase, in which the tissue is cleared of cellular debris. The successful progression and resolution of the inflammatory response are crucial to wound healing. As detailed below and in FIG. 3, glucocorticoids regulate processes in each of these phases.
Figure 3 |. Effects of glucocorticoids on inflammation.
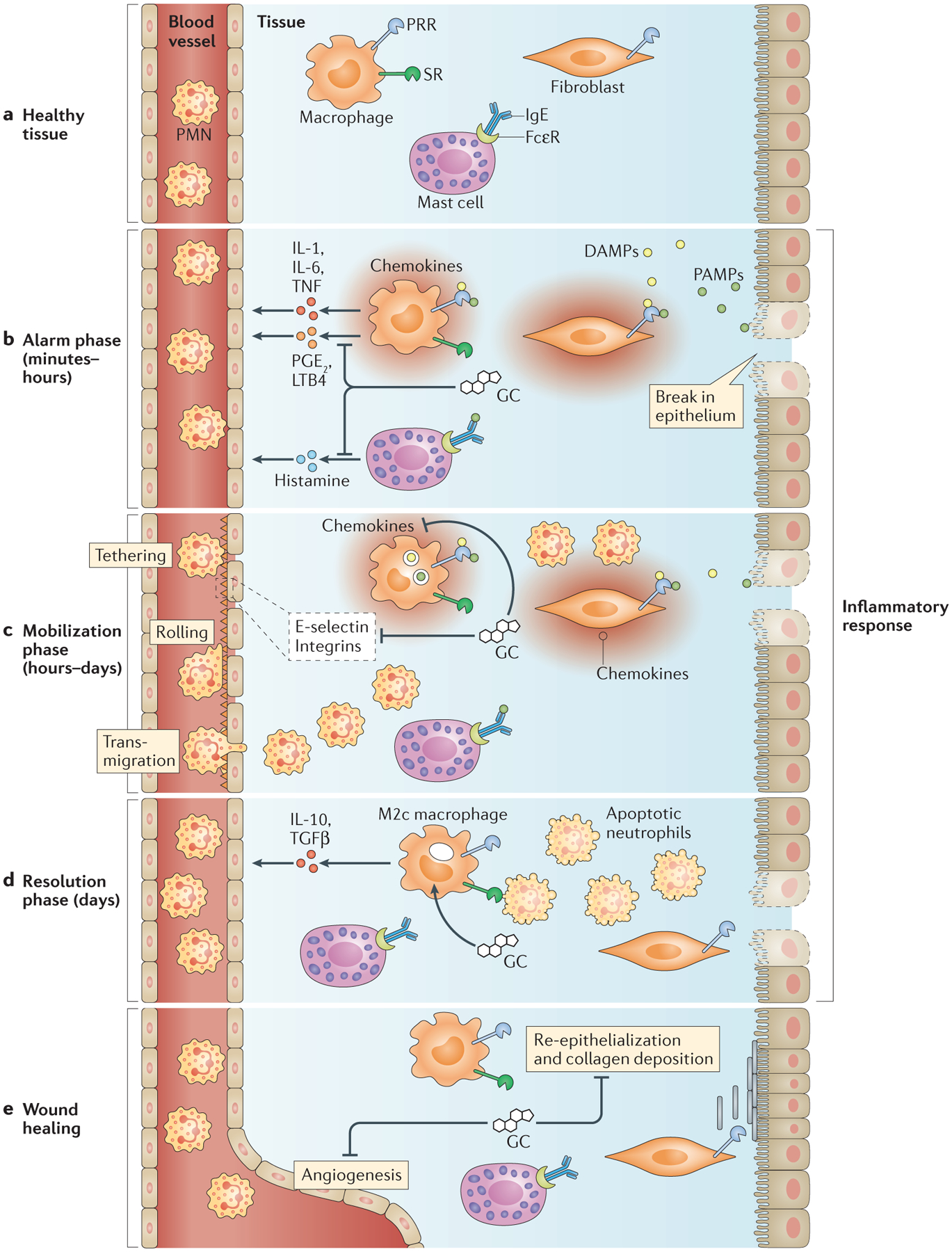
a | In healthy tissue, tissue-resident macrophages, fibroblasts and stromal cells express pattern recognition receptors (PRRs) and scavenger receptors (SRs). Mast cells bind soluble IgE via Fcε receptors (FcεRs). b | During the alarm phase of inflammation, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) trigger PRR signalling, which induces the production of inflammatory mediators, including cytokines, prostaglandin E2 (PGE2) and leukotriene B4 (LTB4). Antigens bind FcεR-bound IgE on mast cells to induce histamine release. Glucocorticoids (GCs) dampen signalling through PRRs, FcεRs and cytokine receptors. c | During the mobilization phase of inflammation, inflammatory mediators induce the display of adhesion molecules — including E-selectin, chemokines and integrins — on the vascular endothelium to recruit leukocytes, especially polymorphonuclear leukocytes (PMNs), into the tissue. Extravasating leukocytes follow chemokine gradients towards inflammatory sites. GCs inhibit the expression of E-selectin, chemokines and integrins to reduce leukocyte recruitment. d | During the resolution phase of inflammation, GCs promote the differentiation of alternatively activated ‘M2c’ macrophages, which clear apoptotic PMNs and secrete anti-inflammatory factors. e | The resolution of inflammation triggers wound healing, which is characterized by re-epithelialization, collagen deposition and angiogenesis. As GCs inhibit these processes, optimal wound healing probably depends on reduced glucocorticoid production. IL, interleukin; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor.
Effects of glucocorticoids on the alarm phase.
The alarm phase of the inflammatory response occurs upon detection of pathogens and/or tissue trauma. Tissue-resident cells express transmembrane and cytoplasmic proteins that function as ‘danger’ sensors; these sensors include pattern recognition receptors (PRRs), complement receptors and Fc receptors (FIG. 3a). PRRs bind pathogen-associated molecular patterns (PAMPs), which are evolutionarily conserved molecular motifs from microbial organisms, and damage-associated molecular patterns (DAMPs), which are endogenous cellular products that are released following tissue injury. Upon PRR activation, tissue macrophages, mast cells and stromal cells secrete inflammatory mediators, including lipid agents, vasoactive amines and cytokines (FIG. 3b). Glucocorticoids attenuate signalling pathways downstream of many danger sensors, thereby suppressing the production of inflammatory mediators. TLR signalling, for example, is subject to regulation by glucocorticoids at several points of signal transduction (FIG. 4). Liganded glucocorticoid receptors tether and inhibit transcription factors downstream of TLR signalling, including NF-κB, AP-1 and interferon-regulatory factor 3 (IRF3) (FIG. 4). Glucocorticoids also activate genes that encode inhibitors of TLR signalling, including dual-specificity protein phosphatase 1 (DUSP1), which attenuates the activity of mitogen-activated protein kinase 1 (MAPK1), and IL-1 receptor-associated kinase 3 (IRAK3), which is a negative regulator of TLR and IL-1 receptor signalling35 (FIG. 4). Another glucocorticoid gene target is NFKIA, which encodes IκBα, a potent cytoplasmic inhibitor of NF-κB36, although the physiological contribution of this gene to the effects of glucocorticoids is debated37. Glucocorticoids also induce the expression of glucocorticoid-induced leucine zipper protein (GILZ; also known as TSC22D3), which further inhibits NF-κB activity38. Notably, inflammatory signals can override some inhibitory effects of glucocorticoids; in plasma cytoid dendritic cells (pDCs), sustained signalling through TLR7 and TLR9 disrupts the capacity of the glucocorticoid receptor to inhibit NF-κB activity, which may contribute to autoimmune pathology39.
Figure 4 |. Glucocorticoids inhibit signalling through Toll-like receptors.
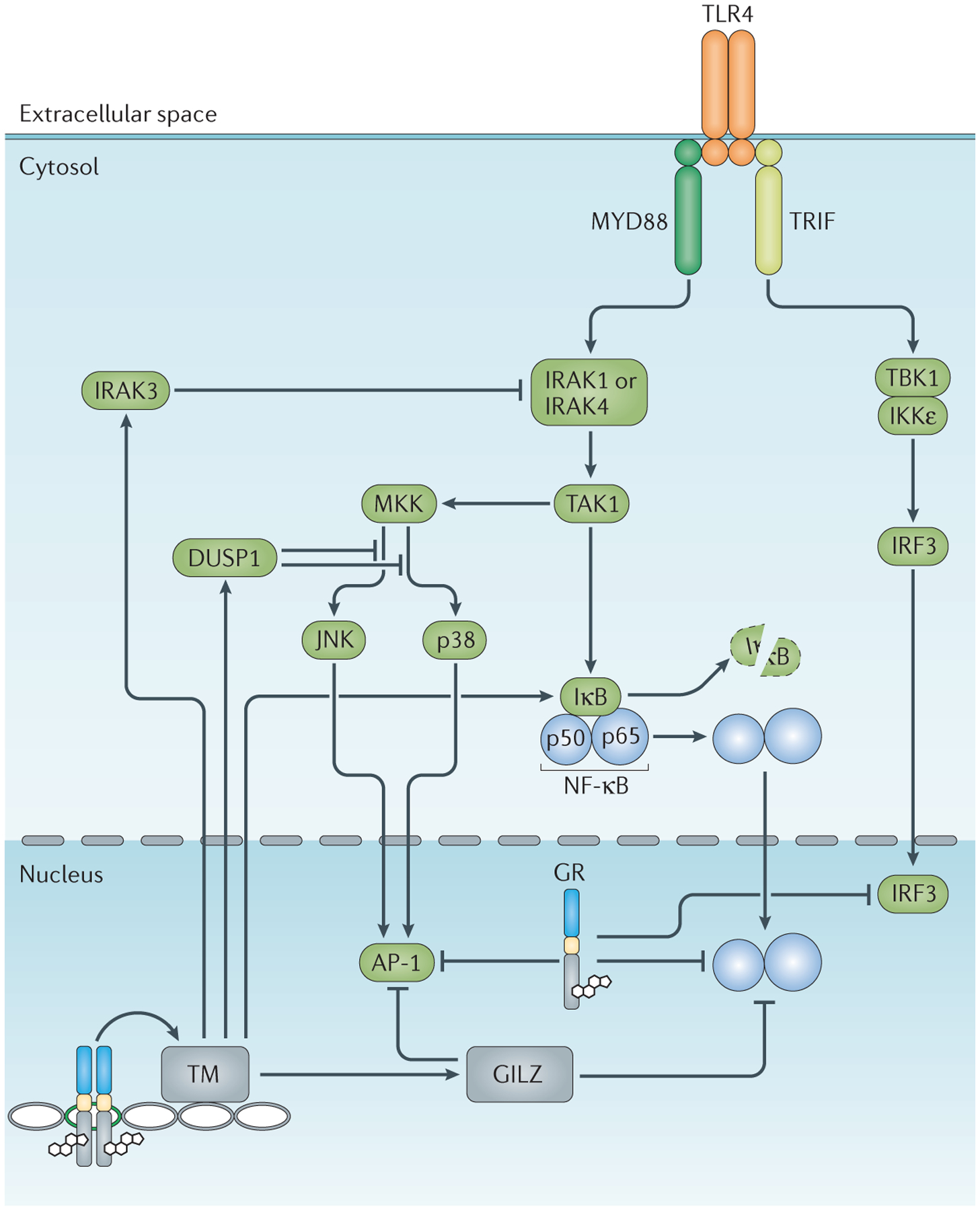
Glucocorticoid receptors (GRs) regulate various points in the Toll-like receptor (TLR) signalling cascade, and many of these steps are shared with other immune receptors. The activated GR enhances the transcription of genes encoding TLR signalling inhibitors, including interleukin1 receptor-associated kinase 3 (IRAK3), dual-specificity protein phosphatase 1 (DUSP1), inhibitor of nuclear factor-κB (IκB) and glucocorticoid-induced leucine zipper (GILZ). The liganded GR also represses the activity of pro-inflammatory transcription factors — including activator protein 1 (AP-1), nuclear factor-κB (NF-κB) and interferon-regulatory factor 3 (IRF3) — through protein–protein interactions. IKKε, IκB kinase-ε; JNK, JUN N-terminal kinase; MKK, mitogen-activated protein kinase kinase (also known as MAP2K); MYD88, myeloid differentiation primary response protein 88; TAK1, transforming growth factor-β-activated kinase 1 (also known as MAP3K7); TBK1, TANK-binding kinase 1; TM, transcriptional machinery; TRIF, TIR domain-containing adapter protein inducing IFNβ (also known as TICAM1).
Importantly, many of the TLR signalling components that are targeted by glucocorticoids, particularly NF-κB and AP-1, are shared by the signalling pathways of other danger sensors (as well as those of cytokine receptors). For example, glucocorticoids dampen signalling through Fcε receptors, thereby attenuating histamine release by mast cells40 (FIG. 3b). By inhibiting shared ‘modules’ of signal transduction, glucocorticoids suppress multiple signalling pathways that are involved in the detection of noxious agents and the propagation of inflammatory signals.
Inflammation is characterized by the dilation of local blood vessels, increased vascular permeability and the leakage of plasma into tissue. Glucocorticoids act on macrophages in inflamed tissue to inhibit the generation of eicosanoids, which are lipid mediators (such as prostaglandins and leukotrienes) that promote vascular dilation and permeability (FIG. 3b). The liganded glucocorticoid receptor activates the expression of annexin A1, which interferes with the phospholipase A2-catalysed release of arachidonic acid41, a fatty-acid intermediate of eicosanoid biosynthesis. Glucocorticoids further curb prostaglandin generation by inhibiting cyclooxygenase 2 (also known as PTGS2) through the NF-κB-suppressing properties of GILZ42. Glucocorticoids reduce blood flow to inflammatory sites by inducing the expression of angiotensin-converting enzyme and endothelin, by sensitizing endothelial cells to vasoconstrictors and by inhibiting the production of vasodilators, including bradykinin43. In an animal model of acute lung injury, Vettorazzi et al.44 found that glucocorticoids preserve the barrier function of the endothelium by upregulating macrophage expression of sphingosine kinase 1 (which is encoded by SPHK1), which inhibits vascular leakage44.
Danger signals detected by PRRs are propagated through cytokine induction. Cytokines bind to cognate receptors on neighbouring cells to stimulate biological effects, which often include the expression of more cytokines. Thus, cytokine networks amplify and shape the inflammatory response. Glucocorticoids inhibit the expression of many pro-inflammatory cytokines, including IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-11, IL-12, IL-13, IL-16, IL-17, interferon-γ (IFNγ), TNF and granulocyte–macrophage colony-stimulating factor (GM-CSF) (FIG. 3b). As described above, glucocorticoid-mediated attenuation of cytokine expression occurs downstream of danger sensor and cytokine receptor signalling45. In addition, nGREs have been identified in some cytokine genes, including IL1B (which encodes IL-1β), IL6 and TSLP (which encodes thymic stromal lymphopoietin)20. Glucocorticoids also regulate cytokine production at the post-transcriptional level; glucocorticoid receptor-mediated upregulation of tristetraprolin (also known as ZFP36) decreases the half-life of TNF mRNA46. In cytokine-responsive cells, glucocorticoids attenuate cytokine receptor signalling via protein–protein inter actions between the liganded glucocorticoid receptor and pro-inflammatory transcription factors (for example, STAT family members and NF-κB), as well as by induction of suppressor of cytokine signalling 1 (SOCS1), an inhibitor of Janus kinase (JAK)–STAT signalling47,48. The importance of glucocorticoid receptor signalling in dampening cytokine production is exemplified by mice with conditional glucocorticoid receptor ablation in macro phages or dendritic cells (DCs). These mice produce supranormal levels of IL-1β, IL-6, TNF and IL-12, and exhibit greater mortality during experimentally induced sepsis than do wild-type animals49–51.
Effects of glucocorticoids on the mobilization phase.
Inflammatory mediators produced in the alarm phase induce the expression of adhesion molecules and chemo attractants that recruit leukocytes from proximal blood vessels into inflammatory foci. The mobilization phase is crucial to the clearance of pathogens and cellular debris. Endothelial expression of E-selectin initiates the tethering of circulating neutrophils and monocytes to blood vessel walls, which facilitates the interaction between chemokines emanating from inflammatory sites and chemokine receptors on leukocytes (FIG. 3c). Chemokine ligand–receptor interactions promote leukocyte rolling on the endothelium and the binding of integrins to integrin ligands, which lead to firm adhesion and leukocyte transmigration through the blood vessel wall52 (FIG. 3c). Extravasated leukocytes then migrate through the tissue following chemokine gradients to inflammatory sites. Glucocorticoids attenuate leukocyte extravasation by inhibiting endothelial transcription of SELE (which encodes E-selectin), as well as the integrin ligands ICAM1 (intercellular adhesion molecule 1) and VCAM1 (vascular cell adhesion molecule 1)53,54. Glucocorticoids also downregulate the production of several chemokines and chemoattractants, including IL-8 (also known as CXCL8), IL-16, CC-chemokine ligand 2 (CCL2), CCL3, CCL5, CCL11, CCL24 and CCL26, thereby curbing leukocyte migration. Recent studies have also suggested that direct glucocorticoid receptor binding to chemokine-encoding mRNA transcripts, including CCL2 and CCL7, promotes their decay55. Glucocorticoids also reduce leukocyte expression of adhesion molecules, including CD44 and the integrins lymphocyte function-associated antigen 1 (LFA1) and very late antigen 4 (VLA4)56. Glucocorticoid-mediated induction of annexin A1 also impedes leukocyte recruitment57. In experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis, glucocorticoids inhibit T cell infiltration into the central nervous system56.
Roles of glucocorticoids in the resolution phase.
As pathogens and noxious agents are eliminated from inflammatory sites, PRR signalling subsides and the production of inflammatory mediators diminishes. However, inflammation is fully resolved through an active programme of immunoregulation, in which glucocorticoids — along with lipoxins, resolvins, collectins and protectins — have important roles58. Important processes in the resolution phase include the clearance of cellular debris and the production of anti-inflammatory factors. Glucocorticoids initiate gene programmes in monocytes and macrophages that promote the phagocytosis of apoptotic cells and debris59–61. Indeed, glucocorticoid-mediated programming of macrophages has received enough attention to warrant the classification of an ‘M2c’ subtype of alternatively activated macrophages; macrophages in this state are characterized by high expression of scavenger receptors (CD163 (also known as M130), CD206 (also known as MRC1) and tyrosine-protein kinase MER (MERTK)) and by the secretion of the anti-inflammatory cytokines transforming growth factor-β (TGFβ) and IL-10 (REF. 62) (FIG. 3d). Another function of glucocorticoids during the resolution phase is to sensitize cells to other pro-resolving factors through the upregulation of the lipoxin A4 receptor63. Resolution of the inflammatory response is followed by wound healing, which restores tissue integrity and function. Glucocorticoids inhibit many wound-healing processes, including collagen deposition, re-epithelialization and angiogenesis; attenuating glucocorticoid activity may be an important component of the transition from inflammation to wound healing.
Glucocorticoid regulation of cell death
Glucocorticoids exert potent regulatory effects on cellular immunity, and affect the development, activation and polarization of T cells. Discerning the specific role of glucocorticoids in thymocyte development has been perplexing, and there are conflicting reports about glucocorticoid receptor-mediated regulation of thymopoiesis. Thymocytes are exquisitely sensitive to glucocorticoid-induced cell death in vitro and in vivo; moreover, adrenalectomy results in thymic hyperplasia, which suggests that glucocorticoid insufficiency allows the supranormal expansion of thymocyte populations. These observations are central to a long-standing hypothesis of endogenous glucocorticoids as negative regulators of thymopoiesis. However, the thymomegaly phenotype of adrenalectomized mice is not recapitulated in glucocorticoid receptor-deficient fetal liver chimaeras64. Moreover, mice with a T cell-specific ablation of the glucocorticoid receptor still exhibit thymic hyperplasia following adrenalectomy, which indicates that glucocorticoid insufficiency promotes thymocyte expansion independently of glucocorticoid receptor signalling (or the lack thereof) in these cells65. The issue of the roles of glucocorticoids in thymopoiesis is further complicated by reports of glucocorticoids having a positive, rather than negative, role in thymocyte survival. According to the ‘mutual antagonism’ hypothesis, crosstalk between glucocorticoid receptor signalling and T cell receptor (TCR) signalling alters the thresholds for positive selection and negative selection during thymocyte development. Supporting this hypothesis, a recent study of a strain of mice with a T cell-specific deletion of the glucocorticoid receptor reported reduced thymic cellularity, an altered TCR repertoire and impaired T cell responsiveness66. Although stress and pharmacological levels of glucocorticoids have clear negative effects on thymocyte survival, a role for endogenous glucocorticoids in the steady-state regulation of thymopoiesis is still unclear.
The lympholytic properties of glucocorticoids are routinely exploited for the treatment of certain haemato-logical malignancies. Although glucocorticoids were among the earliest described inducers of cell death in lymphocytes, the molecular mechanisms that induce apoptosis downstream of glucocorticoid receptor activation are still unclear. Thus, there is some irony in the common use of glucocorticoids to benchmark cell death in studies of novel pro-apoptotic and anti-apoptotic factors. The mechanisms of glucocorticoid-induced apoptosis have been comprehensively reviewed elsewhere (REFS. 33, 67). Importantly, the apoptotic mechanisms activated by glucocorticoids differ by activation state, lymphocyte type and even the stage of differentiation68. There seems to be considerable redundancy in the apoptotic programmes induced by glucocorticoids, as lymphocytes from many single-gene-knockout mice (for example, knockouts of individual caspase family members) are still sensitive to glucocorticoid receptor-mediated death. It is generally thought that glucocorticoid receptor-mediated apoptosis requires gene activation, although recent studies have also implicated non-genomic mechanisms, particularly glucocorticoid receptor translocation to mitochondria33. N-Terminal isoforms of the glucocorticoid receptor exhibit different capacities to induce apoptosis, so the glucocorticoid receptor isoform profile of a cell probably affects sensitivity to glucocorticoid-induced death69. Lastly, it is worth noting that lymphocytes are not the only cells that are sensitive to glucocorticoid-induced death, as DCs70, eosinophils71 and osteocytes72 also undergo apoptosis in response to glucocorticoids.
Effects of glucocorticoids on T cell activation
A crucial component of cellular immunity is TCR-mediated activation of antigen-specific T cells. Antigen-presenting cells (APCs) display peptide antigens complexed with MHC class I or MHC class II molecules to activate CD8+ or CD4+ T cells, respectively. DCs — which are professional APCs that integrate antigens and signals from the innate immune response to stimulate T helper (TH) cell responses — are cellular targets of glucocorticoids. The effects of glucocorticoids have been investigated for various DC types, including tissue-resident DCs, migratory DCs and pDCs, with the general conclusions being that glucocorticoids attenuate DC activity by inhibiting DC maturation; by downregulating the expression of MHC class II, the lipid-presentation molecule CD1a, co-stimulatory molecules (for example, CD80 and CD86) and pro-inflammatory cytokines (such as IL-12 and TNF); and by promoting the expression of anti-inflammatory cytokines, such as IL-10 (REF. 73) (FIG. 5a). Interestingly, glucocorticoid exposure during monocyte-to-DC maturation enhances the capacity of DCs to take up antigens but inhibits their function as APCs, which has led to the hypothesis that glucocorticoids promote the differentiation of ‘tolerogenic’ DCs74. Moreover, in both rodents and humans, pharmacological doses of glucocorticoids decrease DC numbers, and this is probably a result of increased apoptosis and tissue redistribution75,76.
Figure 5 |. Glucocorticoids modulate T cell activity.
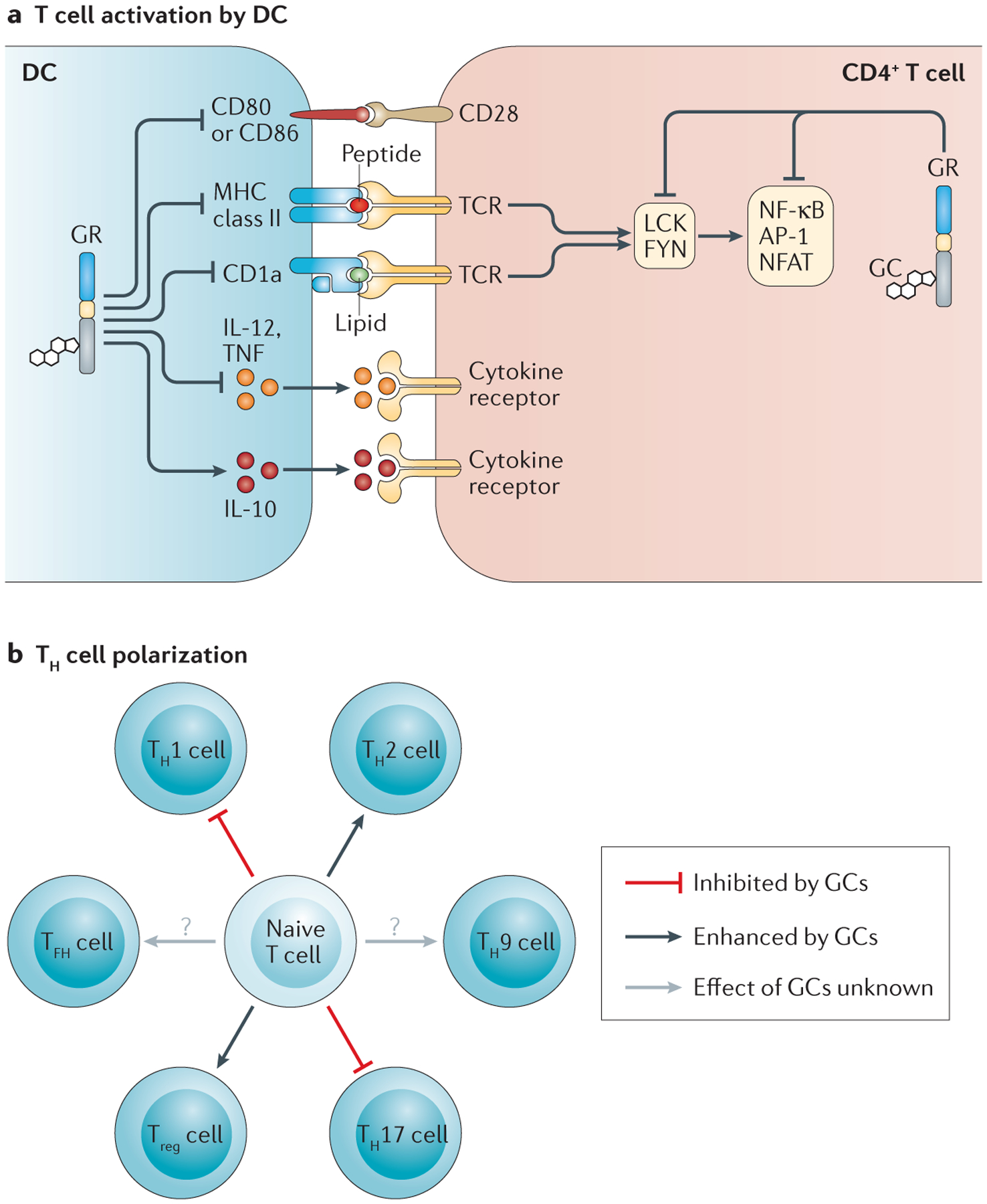
a | Glucocorticoids (GCs) suppress CD4+ T cell activation indirectly by modulating dendritic cell (DC) function (antigen presentation, co-stimulation and cytokine production) and directly by regulating T cell receptor (TCR) signalling. b | GCs influence the polarization of T helper (TH) cells, favouring the differentiation of TH2 cells and regulatory T (Treg) cells over that of TH1 cells and TH17 cells. The effects of GCs on the differentiation of TH9 cells and T follicular helper (TFH) cells require further investigation. AP-1, activator protein 1; GR, GC receptor; IL, interleukin; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-κB; TNF, tumour necrosis factor.
Glucocorticoids also dampen T cell activation by interfering with TCR signalling. Mechanisms implicated in glucocorticoid receptor-mediated attenuation of TCR signalling include the downregulation of FOS and interference with the activity of AP-1, NF-κB and NFAT77 (FIG. 5a). A recent study revealed that glucocorticoids also modulate the expression of several kinases that are involved in TCR signalling, including ITK, TXK and LCK78. Non-genomic actions of glucocorticoid receptors have been implicated in attenuating TCR signalling by reducing the activity of the kinases LCK and FYN79 (FIG. 5a). Glucocorticoid receptor-mediated tempering of TCR signalling results in reduced proliferative responses and diminished cytokine production, including reduced secretion of IL-2 (REF. 80).
Although the net effect of glucocorticoid action is diminished T cell activity, numerous studies suggest that glucocorticoids preferentially suppress the responses of TH1 cells and TH17 cells, while sparing, or possibly even promoting, the function of TH2 cells and regulatory T (Treg) cells (FIG. 5b). Mechanisms proposed for glucocorticoid-induced TH2 cell polarization include downregulation of the production of the TH1 cell-promoting cytokine IL-12 by APCs, attenuation of IL-12 receptor expression in T cells, inhibition of the TH1 cell master transcription factor T-bet, and enhanced production of the canonical TH2-type cytokines IL-4, IL-10 and IL-13 by TH2 cells81,82
Evidence for glucocorticoid-based regulation of TH17 cell polarization stems from the repressive effects of GILZ on the production of TH17 cell-promoting factors (that is, IL-1, IL-6 and IL-23) by DCs, and on the expression of genes involved in the differentiation and activity of TH17 cells (namely IL-17A, IL-23 receptor, RORγt, STAT3, BATF and IRF4)83,84. Studies in mice with a T cell-specific deletion of the glucocorticoid receptor revealed TH17 cells as crucial targets for glucocorticoid treatment in animal models of multiple sclerosis and rheumatoid arthritis56,85. These findings probably explain, at least in part, the capacity of glucocorticoids to ameliorate other auto immune and inflammatory pathologies, including psoriasis and spondyloarthropathy.
Studies in mice and humans have also implicated glucocorticoids in promoting Treg cell differentiation and activity. Glucocorticoid treatment is associated with increased frequencies of Treg cells in the circulation and/or inflamed tissue, and this effect is possibly mediated by glucocorticoid receptor-induced upregulation of the Treg cell master transcription factor forkhead box P3 (FOXP3)86, which is perhaps a result of the upregulation of GILZ87.
Glucocorticoids and humoral immunity
Our understanding of the effects of glucocorticoids on humoral immunity is limited. Nonetheless, glucocorticoids are effective in treating autoimmune diseases in which antibody contributes to pathology, such as rheumatoid arthritis. Moreover, recent studies have shown that B cells exhibit potent immunoregulatory functions, most notably through the expression of IL-10 (IL10 is a known glucocorticoid receptor target gene in macrophages), perhaps warranting a second look at the effects of glucocorticoids on B cells.
Effects on B cell development and survival.
In some ways, the actions of glucocorticoids on thymopoiesis are mirrored in B lymphopoiesis. Immature B cells are more sensitive to glucocorticoid-induced apoptosis than are mature B cells88,89, and adrenalectomy or treatment with the glucocorticoid receptor antagonist mifepristone results in the expansion of immature B cell populations in the bone marrow89. B cells express the glucocorticoid receptor throughout development90, and the glucocorticoid receptor affects several transcription factors downstream of B cell receptor (BCR) signalling (for example, AP-1, NF-κB and NFAT), which raises the possibility that glucocorticoids have effects on B cell selection. To our knowledge, however, mechanistic studies of crosstalk between glucocorticoid receptor signalling and BCR signalling are lacking.
Clues about the direct effects of glucocorticoids on B cell function may be gleaned from mice that lack the glucocorticoid target gene Gilz. In response to BCR stimulation, Gilz-deficient B cells exhibit an exaggerated proliferative response and, with age, Gilz-knockout mice develop a lupus-like syndrome91. Mice with a B cell-specific deficiency of Gilz do not recapitulate the autoimmune phenotype of global Gilz deficiency, which indicates a B cell-extrinsic role for GILZ in autoantibody production; however, mice that specifically lack GILZ expression in B cells exhibit systemic increases in B cell numbers, which suggests an intrinsic role for glucocorticoid receptor signalling in B cell survival and/or homeostasis92.
Effects on antibody production.
A consensus on the effects of glucocorticoids on humoral immunity has not been achieved, although, in general, pharmacological treatment with glucocorticoids is associated with reduced immunoglobulin concentrations. Only a few studies have addressed the effects of long-term glucocorticoid treatment on vaccination of humans, and little impact (if any) on antibody titres has been reported. Nonetheless, the regulation of IgE production by glucocorticoids has garnered some attention, as some clinical and in vitro studies suggest that glucocorticoids may, in certain situations, promote the production of this immunoglobulin isotype. In studies of patients with asthma who were undergoing glucocorticoid therapy, the serum concentrations of IgE rose, whereas the concentrations of other immunoglobulin isotypes were unaffected or decreased93,94. Proposed mechanisms for glucocorticoid-mediated enhancement of IgE production include direct effects on B cells, whereby glucocorticoids synergize with IL-4 to promote isotype class-switching95, as well as indirect effects on B cells via the actions of glucocorticoids on T cells and monocytes96–98. The efficacy of glucocorticoids in the treatment of asthma and allergies, however, suggests that glucocorticoid-mediated enhancement of IgE production (as well as TH2 cell activity) does not exacerbate pathology in these diseases. Indeed glucocorticoids exert potent effects on myeloid cells to suppress disease in animal studies of allergic dermatitis99.
In light of the clinical evidence that pharmacological treatment with glucocorticoids reduces blood concentrations of some non-IgE immunoglobulins, it is puzzling that endogenous glucocorticoids have also been implicated as positive regulators of antibody responses to immunization. For example, adrenalectomized rats mount poor IgM and IgG responses to the T cell-dependent antigen keyhole limpet haemocyanin100,101. The induction of T cell-dependent humoral responses involves multiple glucocorticoid receptor-expressing cell types, including DCs, CD4+ T cells and B cells. Determining the glucocorticoid-sensitive cells and processes that enhance antibody responses will provide important insights into the endocrine regulation of humoral immunity. In the next section, we address the enigmatic issue of glucocorticoids as potentiators of immune responses in more detail.
Immune-enhancing effects of glucocorticoids
This Review, like much of the literature on glucocorticoids that has been published in the last 60 years, has focused on the immunosuppressive properties of glucocorticoids. However, there is a considerable amount of work showing that glucocorticoids also enhance inflammation and immunity. In some studies, the potentiating — as opposed to suppressive — effects of glucocorticoids are linked to dose. For example, in macrophages activated by lipopolysaccharide (LPS) and IFNγ, high doses of corticosterone inhibit the transcription of inflammatory genes, whereas low doses of corticosterone enhance inflammatory gene expression102. The temporal relationship between a noxious stimulus and glucocorticoid exposure can also affect the directionality of the effects of glucocorticoids on inflammation. In rats, pro-inflammatory cytokine expression is enhanced if glucocorticoids are administered before LPS challenge but are dampened if glucocorticoids are administered following LPS treatment103. Similarly, acute low-dose corticosterone treatment before challenge enhances inflammation in a rat model of delayed-type hyper sensitivity, whereas high-dose or prolonged glucocorticoid treatment mitigates the delayed-type hypersensitivity response104.
Genome-wide expression studies of glucocorticoid-treated cells have yielded surprising results, and suggest that glucocorticoids largely spare or enhance gene pathways that are associated with innate immunity, but selectively suppress pathways that are involved in adaptive immunity (FIG. 6a). For example, the treatment of blood mononuclear cells with glucocorticoids leads to the upregulation of several PRRs, scavenger receptors, cytokine and chemokine receptors, complement components and other factors that are involved in innate immunity, but suppresses the expression of genes that encode TCR components as well as genes involved in T cell activation, including those that encode MHC class II and co-stimulatory molecules1 (FIG. 6a). Selective enhancement of innate immune gene expression by glucocorticoids at the expense of adaptive immune gene expression has also been observed in human macrophages105.
Figure 6 |. Glucocorticoid-induced sensitization of innate immunity.
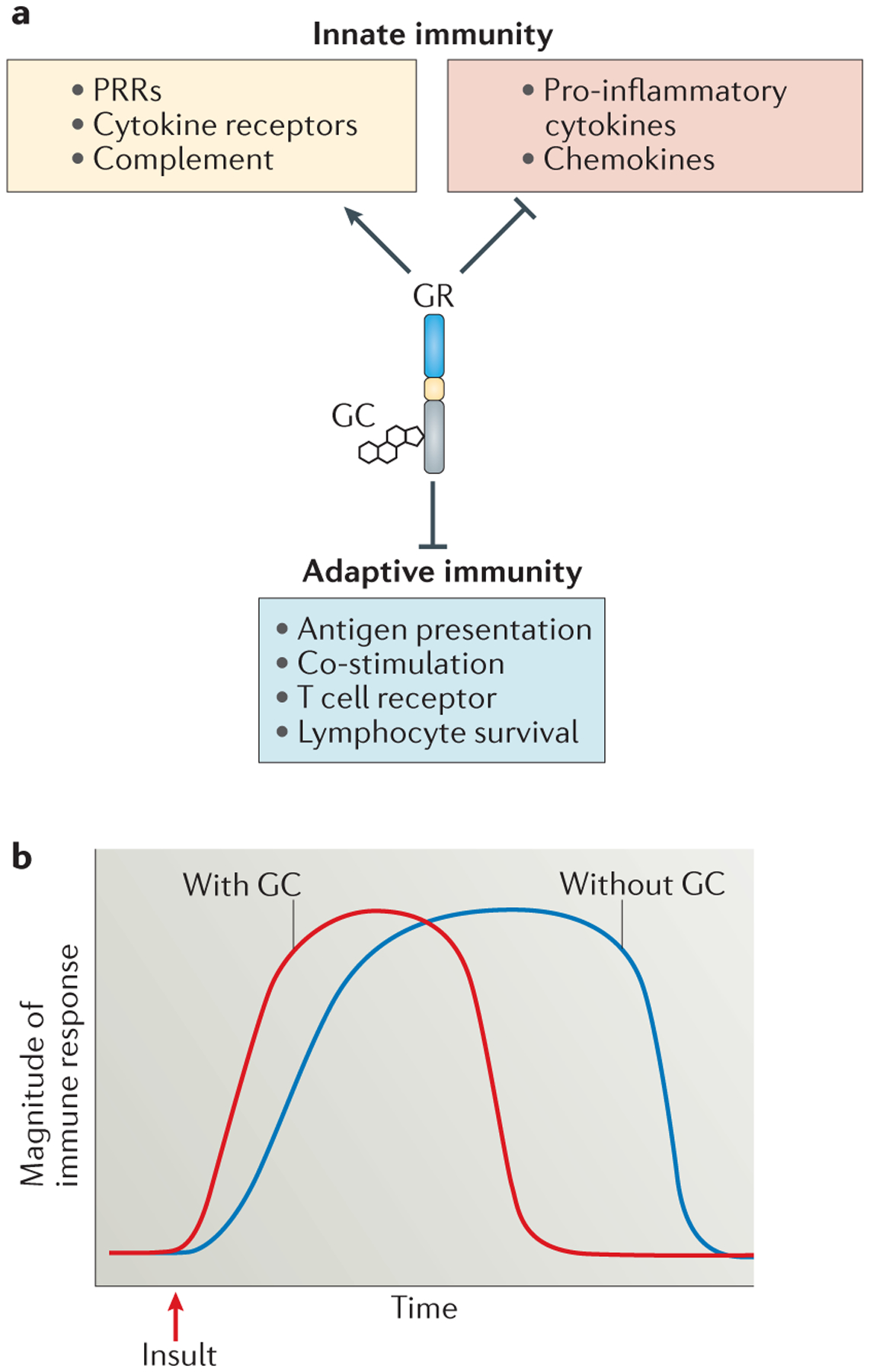
a | Genome-wide expression studies indicate that glucocorticoids (GCs) upregulate the expression of genes that are involved in the detection of pathogens and tissue trauma, including pattern recognition receptors (PRRs), cytokine receptors and complement factors, while inhibiting the expression of pro-inflammatory cytokines and chemokines. These studies have also revealed that genes involved in adaptive immunity are inhibited by GC exposure. b | A hypothetical timeline for immune responses occurring in the presence (red line) or absence (blue line) of GCs is shown. We propose that GCs regulate the immune system in a biphasic manner, such that low doses promote the expression of innate immune genes and rapid responses to insults, but stress and/or pharmacological concentrations suppress signalling through immune receptors. According to this hypothesis, GC-sufficient animals (red line) mount rapid immune responses to pathogens and tissue injury, but these responses are of controlled duration. In GC-insufficient animals (blue line), however, subnormal expression of PRRs and cytokine receptors results in a slower immune response. Furthermore, in the absence of GC-mediated suppression of immune receptor signalling, the duration of the immune response is prolonged. GR, GC receptor.
Clues about the molecular mechanisms that underlie the immunopotentiating effects of glucocorticoids might be found among glucocorticoid receptor target genes. Glucocorticoids promote the expression of TLR2, TLR4, the inflammasome component NOD-, LRR- and pyrin domain-containing 3 (NLRP3) and the purinergic receptor P2Y2R, which may sensitize cells to PAMPs and DAMPs106. Interestingly, although glucocorticoids are well-recognized to decrease cytokine secretion, they are reported to increase the expression of several cytokine receptors, including the receptors for TNF, IL-1, IL-6 and IFNγ107. Rapid increases in circulating glucocorticoid concentrations triggered by physiological stress may serve as a systemic warning system, and sensitize cells to DAMPs, PAMPs and inflammatory cytokines. Thus, glucocorticoids (along with catecholamines) are important contributors to altered immune function associated with chronic stress108,109.
A unified model
As discussed above, glucocorticoids have been shown to have both enhancing and suppressive effects on the immune system. To explain this apparent paradox of glucocorticoid action, Munck and Naray-Fejes-Toth posited that glucocorticoid physiology follows a biphasic dose–response curve, such that glucocorticoids have ‘permissive’ (that is, immunostimulatory) effects at low concentrations and suppressive effects at high concentrations110,111. Biphasic effects of glucocorticoids have also been observed in other physiological contexts, including neural plasticity112. Wiegers and Reul107 further hypothesized that the net cellular effect of concurrent glucocorticoid-mediated upregulation of cytokine receptors and downregulation of cognate cytokines is a bio logical response with a more rapid initiation but shorter duration.
We propose a unified model for glucocorticoid-mediated regulation of the immune response that incorporates the hypotheses of a biphasic dose–response proposed by Munck and Naray-Fejes-Toth110, and the kinetic effects of disparate cytokine–cytokine receptor regulation described by Wiegers and Reul107. We propose that in the absence of inflammation, basal levels of glucocorticoid receptor signalling — which are driven by circadian and ultradian rhythms of glucocorticoid production — sensitize cells to harmful stimuli by promoting the expression of PRRs, cytokine receptors and complement factors, which allow for the rapid detection of PAMPs and DAMPs, and promoting the induction of an inflammatory response upon tissue insult. During the inflammatory state, however, stress-induced (or pharmacological) concentrations of glucocorticoids restrain the immune response, primarily by blunting the propagation of PRR, Fc receptor and cytokine signals, thereby shortening the duration of the immune response (FIG. 6b). This model predicts that in the absence of glucocorticoids (via adrenalectomy or glucocorticoid antagonism), the immune response will be delayed in onset but prolonged in duration (FIG. 6b). Although this is likely to be an oversimplification of the actions of glucocorticoids, we offer this model as a framework for future research. Understanding the mechanisms by which and the conditions in which glucocorticoids shape an immune response, both positively and negatively, will probably provide important insights into optimized glucocorticoid therapies.
Conclusions and perspectives
The clinical efficacy of glucocorticoids in treating inflammatory and autoimmune disease is clear, yet the molecular mechanisms that underlie the immunoregulatory effects of glucocorticoids are still being elucidated. There are substantial gaps in our knowledge of glucocorticoid-mediated regulation of immunity, especially regarding cell lineage-specific functions and disease-specific roles, and possible sexually dimorphic effects. Moreover, the adverse effects of excess glucocorticoids are wide-ranging (BOX 1); a deeper understanding of the actions of glucocorticoids that are shared among physiological systems versus those that are unique to the immune system is necessary to achieve the ‘holy grail’ of safe glucocorticoid therapy.
Box 2 |. Cellular targets of glucocorticoid action in animal models of disease.
Mouse studies have revealed the crucial cellular targets for glucocorticoid treatment of various inflammatory and autoimmune models of human disease. In the studies cited in the table below, glucocorticoid efficacy in the indicated disease model was reduced by genetic ablation of glucocorticoid receptor expression in target cells or by in vivo depletion of specific cell types.
| Animal model | Human disease modelled | Crucial cellular target of glucocorticoids |
|---|---|---|
| Contact hypersensitivity | Allergic contact dermatitis | Myeloid cells99 |
| Experimental autoimmune encephalomyelitis | Multiple sclerosis | TH17 cells56 |
| Antigen-induced arthritis | Rheumatoid arthritis | TH17 cells85 |
| LPS-induced endotoxaemia | Sepsis | Myeloid cells50 and dendritic cells51 |
| Experimentally induced thrombocytopenia | Immune thrombocytopenia | CD8+ T cells122 |
| Doxorubicin-induced renal injury | Focal segmental glomerulosclerosis | Myeloid-derived suppressor cells123 |
LPS, lipopolysaccharide; TH17, T helper 17.
Acknowledgements
The authors’ research of the topic of this Review was supported, in part, by the Intramural Research Program of the US National Institutes of Health, National Institute of Environmental Health Sciences.
Glossary
- Lympholytics
Agents that cause the death of lymphocytes.
- Steroidogenesis
The enzymatic processing of cholesterol into steroid hormones.
- Hypothalamic–pituitary–adrenal axis
(HPA axis). The three-organ system that receives inputs from the endocrine, neural and immune systems, and controls physiological responses to stress.
- Sumoylation
A post-translational modification consisting of small ubiquitin-like modifier (SUMO) proteins.
- Immunophilins
Members of a family of highly conserved cytosolic isomerases, many of which have unknown cellular functions.
- Pattern recognition receptors
(PRRs). Transmembrane and cytosolic host receptors that recognize damage-associated molecular patterns and/or pathogen-associated molecular patterns.
- Scavenger receptors
Members of a subclass of pattern recognition receptors that are involved in the identification and removal of unwanted molecules and cellular debris.
- Positive selection
The process by which thymocytes expressing T cell receptors that bind self-peptide–MHC complexes are provided with survival signals during T cell development.
- Negative selection
The process through which thymocytes expressing highly self-reactive T cell receptors are induced to undergo cell death.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Galon J et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 16, 61–71 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Miller WL & Auchus RJ The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev 32, 81–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talaber G, Jondal M & Okret S Extra-adrenal glucocorticoid synthesis: immune regulation and aspects on local organ homeostasis. Mol. Cell. Endocrinol 380, 89–98 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Dunn AJ Cytokine activation of the HPA axis. Ann. NY Acad. Sci 917, 608–617 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Bornstein SR et al. The role of Toll-like receptors in the immune–adrenal crosstalk. Ann. NY Acad. Sci 1088, 307–318 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Spiga F, Walker JJ, Terry JR & Lightman SL HPA axis-rhythms. Compr. Physiol 4, 1273–1298 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Scheiermann C, Kunisaki Y & Frenette PS Circadian control of the immune system. Nat. Rev. Immunol 13, 190–198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimitrov S et al. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 113, 5134–5143 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pemberton PA, Stein PE, Pepys MB, Potter JM & Carrell RW Hormone binding globulins undergo serpin conformational change in inflammation. Nature 336, 257–258 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Woodward MJ et al. Tnfaip8 is an essential gene for the regulation of glucocorticoid-mediated apoptosis of thymocytes. Cell Death Differ. 17, 316–323 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Patrick G History of cortisone and related compounds. eLS 10.1002/9780470015902.a0003627.pub2 (2013). [DOI] [Google Scholar]
- 12.Kumar R & Thompson EB Gene regulation by the glucocorticoid receptor: structure:function relationship. J. Steroid Biochem. Mol. Biol 94, 383–394 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Oakley RH & Cidlowski JA Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J. Biol. Chem 286, 3177–3184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webster JC, Oakley RH, Jewell CM & Cidlowski JA Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative β isoform: a mechanism for the generation of glucocorticoid resistance. Proc. Natl Acad. Sci. USA 98, 6865–6870 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu NZ & Cidlowski JA Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 16, 301–307 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Vandevyver S, Dejager L & Libert C On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic 13, 364–374 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Holaska JM et al. Calreticulin Is a receptor for nuclear export. J. Cell Biol 152, 127–140 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strahle U, Klock G & Schutz GA DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc. Natl Acad. Sci. USA 84, 7871–7875 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luisi BF et al. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352, 497–505 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Surjit M et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145, 224–241 (2011). [DOI] [PubMed] [Google Scholar]; This whole-genome study of glucocorticoid receptor binding to chromatin reveals the identity of the nGRE and its surprising prevalence in the genome.
- 21.Biddie SC, John S & Hager GL Genome-wide mechanisms of nuclear receptor action. Trends Endocrinol. Metab 21, 3–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John S et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet 43, 264–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that chromatin accessibility is a key contributor to the tissue-selective effects of glucocorticoids.
- 23.Ratman D et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell. Endocrinol 380, 41–54 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Diamond MI, Miner JN, Yoshinaga SK & Yamamoto KR Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249, 1266–1272 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Biddie SC et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerppola TK, Luk D & Curran T Fos is a preferential target of glucocorticoid receptor inhibition of AP-1 activity in vitro. Mol. Cell. Biol 13, 3782–3791 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teurich S & Angel P The glucocorticoid receptor synergizes with Jun homodimers to activate AP-1-regulated promoters lacking GR binding sites. Chem. Senses 20, 251–255 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Lechner J, Welte T & Doppler W Mechanism of interaction between the glucocorticoid receptor and Stat5: role of DNA-binding. Immunobiology 198, 112–123 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Tuckermann JP et al. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J. Cell Biol 147, 1365–1370 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sundahl N, Bridelance J, Libert C, De Bosscher K & Beck IM Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol. Ther 152, 28–41 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Coutinho AE & Chapman KE The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol 335, 2–13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buttgereit F & Scheffold A Rapid glucocorticoid effects on immune cells. Steroids 67, 529–534 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Boldizsar F et al. Emerging pathways of non-genomic glucocorticoid (GC) signalling in T cells. Immunobiology 215, 521–526 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Sionov RV, Cohen O, Kfir S, Zilberman Y & Yefenof E Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J. Exp. Med 203, 189–201 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyata M et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat. Commun 6, 6062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheinman RI, Cogswell PC, Lofquist AK & Baldwin AS Jr. Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science 270, 283–286 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Heck S et al. IκBα-independent downregulation of NF-κB activity by glucocorticoid receptor. EMBO J. 16, 4698–4707 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beaulieu E & Morand EF Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat. Rev. Rheumatol 7, 340–348 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Guiducci C et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that sustained TLR signalling in pDCs overrides the capacity of the glucocorticoid receptor to inhibit NF-κB, which may contribute to glucocorticoid resistance in patients with lupus.
- 40.Oppong E, Flink N & Cato AC Molecular mechanisms of glucocorticoid action in mast cells. Mol. Cell. Endocrinol 380, 119–126 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kim SW et al. Inhibition of cytosolic phospholipase A2 by annexin I. Specific interaction model and mapping of the interaction site. J. Biol. Chem 276, 15712–15719 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Yang N, Zhang W & Shi XM Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J. Cell. Biochem 103, 1760–1771 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Perretti M & Ahluwalia A The microcirculation and inflammation: site of action for glucocorticoids. Microcirculation 7, 147–161 (2000). [PubMed] [Google Scholar]
- 44.Vettorazzi S et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat. Commun 6, 7796 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottlicher M, Heck S & Herrlich P Transcriptional cross-talk, the second mode of steroid hormone receptor action. J. Mol. Med 76, 480–489 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Smoak K & Cidlowski JA Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol. Cell. Biol 26, 9126–9135 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogatsky I & Ivashkiv LB Glucocorticoid modulation of cytokine signaling. Tissue Antigens 68, 1–12 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharyya S, Zhao Y, Kay TW & Muglia LJ Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl Acad. Sci. USA 108, 9554–9559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleiman A et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 26, 722–729 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Bhattacharyya S, Brown DE, Brewer JA, Vogt SK & Muglia LJ Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109, 4313–4319 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, specific deletion of the glucocorticoid receptor in myeloid cells reveals that the p38 MAPK pathway in macrophages is a crucial target for glucocorticoid-mediated suppression in an animal model of endotoxemia.
- 51.Li CC, Munitic I, Mittelstadt PR, Castro E & Ashwell JD Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. PLoS Biol. 13, e1002269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolaczkowska E & Kubes P Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F & Weissmann G A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl Acad. Sci. USA 89, 9991–9995 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atsuta J, Plitt J, Bochner BS & Schleimer RP Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am. J. Respir. Cell Mol. Biol 20, 643–650 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Ishmael FT et al. The human glucocorticoid receptor as an RNA-binding protein: global analysis of glucocorticoid receptor-associated transcripts and identification of a target RNA motif. J. Immunol 186, 1189–1198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wust S et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol 180, 8434–8443 (2008). [DOI] [PubMed] [Google Scholar]; This study shows that genetic ablation of the glucocorticoid receptor in T cells, but not in myeloid cells, renders mice resistant to glucocorticoid therapy in an animal model of multiple sclerosis.
- 57.Perretti M & Flower RJ Annexin 1 and the biology of the neutrophil. J. Leukoc. Biol 76, 25–29 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Serhan CN & Savill J Resolution of inflammation: the beginning programs the end. Nat. Immunol 6, 1191–1197 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Ehrchen J et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 109, 1265–1274 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Giles KM et al. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J. Immunol 167, 976–986 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Liu Y et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J. Immunol 162, 3639–3646 (1999). [PubMed] [Google Scholar]; This study shows that glucocorticoids promote macrophage phagocytosis of apoptotic neutrophils.
- 62.Martinez FO, Sica A, Mantovani A & Locati M Macrophage activation and polarization. Front. Biosci 13, 453–461 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Perretti M et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med 8, 1296–1302 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purton JF et al. Glucocorticoid receptor deficient thymic and peripheral T cells develop normally in adult mice. Eur. J. Immunol 32, 3546–3555 (2002). [DOI] [PubMed] [Google Scholar]; This study of rare glucocorticoid receptor-deficient mice suggests that the glucocorticoid receptor is dispensable for the development and selection of thymocytes, which is in contrast to the findings reported in reference 66.
- 65.Talaber G, Tuckermann JP & Okret S ACTH controls thymocyte homeostasis independent of glucocorticoids. FASEB J. 29, 2526–2534 (2015). [DOI] [PubMed] [Google Scholar]; This study challenges the paradigm that glucocorticoids act on thymocytes to control thymic homeostasis and suggests that adrenocorticotropin hormone acts on thymic epithelial cells to control thymic output.
- 66.Mittelstadt PR, Monteiro JP & Ashwell JD Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J. Clin. Invest 122, 2384–2394 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using T cell-specific glucocorticoid receptor-knockout mice, the authors of this paper show that T cell responsiveness to antigen is reduced owing to low affinity for self-MHC complexes, which is consistent with the ‘mutual antagonism’ hypothesis.
- 67.Herold MJ, McPherson KG & Reichardt HM Glucocorticoids in T cell apoptosis and function. Cell. Mol. Life Sci 63, 60–72 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang D, Muller N, McPherson KG & Reichardt HM Glucocorticoids engage different signal transduction pathways to induce apoptosis in thymocytes and mature T cells. J. Immunol 176, 1695–1702 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Lu NZ, Collins JB, Grissom SF & Cidlowski JA Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol. Cell. Biol 27, 7143–7160 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim KD, Choe YK, Choe IS & Lim JS Inhibition of glucocorticoid-mediated caspase-independent dendritic cell death by CD40 activation. J. Leukoc. Biol 69, 426–434 (2001). [PubMed] [Google Scholar]
- 71.Druilhe A, Létuvé S & Pretolani M Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis 8, 481–495 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Weinstein RS, Jilka RL, Parfitt AM & Manolagas SC Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of the deleterious effects on bone. J. Clin. Invest 102, 274–282 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szatmari I & Nagy L Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J. 27, 2353–2362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chamorro S et al. TLR triggering on tolerogenic dendritic cells results in TLR2 up-regulation and a reduced proinflammatory immune program. J. Immunol 183, 2984–2994 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Shodell M, Shah K & Siegal FP Circulating human plasmacytoid dendritic cells are highly sensitive to corticosteroid administration. Lupus 12, 222–230 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Cao Y et al. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood 121, 1553–1562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsitoura DC & Rothman PB Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J. Clin. Invest 113, 619–627 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petrillo MG et al. Transcriptional regulation of kinases downstream of the T cell receptor: another immunomodulatory mechanism of glucocorticoids. BMC Pharmacol. Toxicol 15, 35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lowenberg M, Verhaar AP, van den Brink GR & Hommes DW Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med 13, 158–163 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Gillis S, Crabtree GR & Smith KA Glucocorticoid-induced inhibition of T cell growth factor production. I. The effect on mitogen-induced lymphocyte proliferation. J. Immunol 123, 1624–1631 (1979). [PubMed] [Google Scholar]
- 81.Elenkov IJ Glucocorticoids and the Th1/Th2 balance. Ann. NY Acad. Sci 1024, 138–146 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Liberman AC et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein–protein interaction. FASEB J. 21, 1177–1188 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Yosef N et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones SA et al. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J. Autoimmun 61, 73–80 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Baschant U et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc. Natl Acad. Sci. USA 108, 19317–19322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karagiannidis C et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J. Allergy Clin. Immunol 114, 1425–1433 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Bereshchenko O et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling. Cell Rep. 7, 464–475 (2014). [DOI] [PubMed] [Google Scholar]
- 88.Garvy BA, King LE, Telford WG, Morford LA & Fraker PJ Chronic elevation of plasma corticosterone causes reductions in the number of cycling cells of the B lineage in murine bone marrow and induces apoptosis. Immunology 80, 587–592 (1993). [PMC free article] [PubMed] [Google Scholar]
- 89.Igarashi H et al. Early lymphoid progenitors in mouse and man are highly sensitive to glucocorticoids. Int. Immunol 17, 501–511 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Gruver-Yates AL, Quinn MA & Cidlowski JA Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology 155, 463–474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones SA et al. Glucocorticoid-induced leucine zipper (GILZ) inhibits B cell activation in systemic lupus erythematosus. Ann. Rheum. Dis 75, 739–747 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Bruscoli S et al. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood 126, 1790–1801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Posey WC, Nelson HS, Branch B & Pearlman DS The effects of acute corticosteroid therapy for asthma on serum immunoglobulin levels. J. Allergy Clin. Immunol 62, 340–348 (1978). [DOI] [PubMed] [Google Scholar]
- 94.Zieg G, Lack G, Harbeck RJ, Gelfand EW & Leung DY In vivo effects of glucocorticoids on IgE production. J. Allergy Clin. Immunol 94, 222–230 (1994). [DOI] [PubMed] [Google Scholar]
- 95.Jabara HH, Ahern DJ, Vercelli D & Geha RS Hydrocortisone and IL-4 induce IgE isotype switching in human B cells. J. Immunol 147, 1557–1560 (1991). [PubMed] [Google Scholar]
- 96.Jabara HH, Brodeur SR & Geha RS Glucocorticoids upregulate CD40 ligand expression and induce CD40L-dependent immunoglobulin isotype switching. J. Clin. Invest 107, 371–378 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bohle B et al. Hydrocortisone enhances total IgE levels — but not the synthesis of allergen-specific IgE — in a monocyte-dependent manner. Clin. Exp. Immunol 101, 474–479 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Akdis CA et al. Glucocorticoids inhibit human antigen-specific and enhance total IgE and IgG4 production due to differential effects on T and B cells in vitro. Eur. J. Immunol 27, 2351–2357 (1997). [DOI] [PubMed] [Google Scholar]
- 99.Tuckermann JP et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Invest 117, 1381–1390 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Friedman EM & Irwin M Central CRH suppresses specific antibody responses: effects of β-adrenoceptor antagonism and adrenalectomy. Brain Behav. Immun 15, 65–77 (2001). [DOI] [PubMed] [Google Scholar]
- 101.Fleshner M, Deak T, Nguyen KT, Watkins LR & Maier SF Endogenous glucocorticoids play a positive regulatory role in the anti-keyhole limpet hemocyanin in vivo antibody response. J. Immunol 166, 3813–3819 (2001). [DOI] [PubMed] [Google Scholar]
- 102.Lim HY, Muller N, Herold MJ, van den Brandt J & Reichardt HM Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 122, 47–53 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frank MG, Miguel ZD, Watkins LR & Maier SF Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav. Immun 24, 19–30 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Dhabhar FS & McEwen BS Enhancing versus suppressive effects of stress hormones on skin immune function. Proc. Natl Acad. Sci. USA 96, 1059–1064 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that glucocorticoids exhibit biphasic dose–response effects in an animal model of delayed-type hypersensitivity.
- 105.van de Garde MD et al. Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J. Immunol 192, 1196–1208 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Busillo JM & Cidlowski JA The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab 24, 109–119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wiegers GJ & Reul JM Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol. Sci 19, 317–321 (1998). [DOI] [PubMed] [Google Scholar]
- 108.Dhabhar FS, Malarkey WB, Neri E & McEwen BS Stress-induced redistribution of immune cells — from barracks to boulevards to battlefields: a tale of three hormones — Curt Richter Award winner. Psychoneuroendocrinol. 37, 1345–1368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dhabhar FS Stress-induced augmentation of immune function — the role of stress hormones, leukocyte trafficking, and cytokines. Brain Behav. Immun 16, 785–798 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Munck A & Naray-Fejes-Toth A The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol. Cell. Endocrinol 90, C1–C4 (1992). [DOI] [PubMed] [Google Scholar]
- 111.Sapolsky RM, Romero LM & Munck AU How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev 21, 55–89 (2000). [DOI] [PubMed] [Google Scholar]
- 112.Du J et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl Acad. Sci. USA 106, 3543–3548 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.James ER The etiology of steroid cataract. J. Ocul. Pharmacol. Ther 23, 403–420 (2007). [DOI] [PubMed] [Google Scholar]
- 114.Guslandi M & Tittobello A Steroid ulcers: a myth revisited. BMJ 304, 655–656 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cordeiro RC, Zecchin KG & de Moraes AM Expression of estrogen, androgen, and glucocorticoid receptors in recent striae distensae. Int. J. Dermatol 49, 30–32 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Cicala MV & Mantero F Hypertension in Cushing’s syndrome: from pathogenesis to treatment. Neuroendocrinology 92 (Suppl. 1), 44–49 (2010). [DOI] [PubMed] [Google Scholar]
- 117.Weinstein RS Glucocorticoid-induced osteonecrosis. Endocrine 41, 183–190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Patel R, Williams-Dautovich J & Cummins CL Minireview: new molecular mediators of glucocorticoid receptor activity in metabolic tissues. Mol. Endocrinol 28, 999–1011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weinstein RS Clinical practice. Glucocorticoid-induced bone disease. N. Engl. J. Med 365, 62–70 (2011). [DOI] [PubMed] [Google Scholar]
- 120.Guo S & Dipietro LA Factors affecting wound healing. J. Dent. Res 89, 219–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Judd LL et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am. J. Psychiatry 171, 1045–1051 (2014). [DOI] [PubMed] [Google Scholar]
- 122.Ma L et al. CD8+ T cells are predominantly protective and required for effective steroid therapy in murine models of immune thrombocytopenia. Blood 126, 247–256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li L et al. Role of myeloid-derived suppressor cells in glucocorticoid-mediated amelioration of FSGS. J. Am. Soc. Nephrol 26, 2183–2197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jonat C et al. Antitumor promotion and antiinflammation — down-modulation of AP-1 (Fos Jun) activity by glucocorticoid hormone. Cell 62, 1189–1204 (1990). [DOI] [PubMed] [Google Scholar]
- 125.Ki SH, Cho IJ, Choi DW & Kim SG Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPβ TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol 25, 4150–4165 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Imai E, Miner JN, Mitchell JA, Yamamoto KR & Granner DK Glucocorticoid receptor–cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J. Biol. Chem 268, 5353–5356 (1993). [PubMed] [Google Scholar]
- 127.Mullick J et al. Physical interaction and functional synergy between glucocorticoid receptor and Ets2 proteins for transcription activation of the rat cytochrome P-450c27 promoter. J. Biol. Chem 276, 18007–18017 (2001). [DOI] [PubMed] [Google Scholar]
- 128.Chang TJ, Scher BM, Waxman S & Scher W Inhibition of mouse GATA-1 function by the glucocorticoid receptor: possible mechanism of steroid inhibition of erythroleukemia cell differentiation. Mol. Endocrinol 7, 528–542 (1993). [DOI] [PubMed] [Google Scholar]
- 129.Ray A & Prefontaine KE Physical association and functional antagonism between the p65 subunit of transcription factor NF-κB and the glucocorticoid receptor. Proc. Natl Acad. Sci. USA 91, 752–756 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martens C, Bilodeau S, Maira M, Gauthier Y & Drouin J Protein–protein interactions and transcriptional antagonism between the subfamily of NGFI-B/Nur77 orphan nuclear receptors and glucocorticoid receptor. Mol. Endocrinol 19, 885–897 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Kutoh E, Stromstedt PE & Poellinger L Functional interference between the ubiquitous and constitutive octamer transcription factor 1 (OTF-1) and the glucocorticoid receptor by direct protein–protein interaction involving the homeo subdomain of OTF-1. Mol. Cell. Biol 12, 4960–4969 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Song CZ, Tian X & Gelehrter TD Glucocorticoid receptor inhibits transforming growth factor-β signaling by directly targeting the transcriptional activation function of Smad3. Proc. Natl Acad. Sci. USA 96, 11776–11781 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang Z, Jones S, Hagood JS, Fuentes NL & Fuller GM STAT3 acts as a co-activator of glucocorticoid receptor signaling. J. Biol. Chem 272, 30607–30610 (1997). [DOI] [PubMed] [Google Scholar]
- 134.Stocklin E, Wissler M, Gouilleux F & Groner B Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383, 726–728 (1996). [DOI] [PubMed] [Google Scholar]
- 135.Biola A et al. The glucocorticoid receptor and STAT6 physically and functionally interact in T-lymphocytes. FEBS Lett. 487, 229–233 (2000). [DOI] [PubMed] [Google Scholar]