

Abstract
Parkinson's disease (PD) is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). In this study, we generated a transgenic model by crossing germline Parkin–/– mice with PolgAD257A mice, an established model of premature aging and mitochondrial stress. We hypothesized that loss of Parkin–/– in PolgAD257A/D257A mice would exacerbate mitochondrial dysfunction, leading to loss of dopamine neurons and nigral-striatal specific neurobehavioral motor dysfunction. We found that aged Parkin–/–/PolgAD257A/D257A male and female mice exhibited severe behavioral deficits, nonspecific to the nigral-striatal pathway, with neither dopaminergic neurodegeneration nor reductions in striatal dopamine. We saw no difference in expression levels of nuclear-encoded subunits of mitochondrial markers and mitochondrial Complex I and IV activities, although we did observe substantial reductions in mitochondrial-encoded COX41I, indicating mitochondrial dysfunction as a result of PolgAD257A/D257A mtDNA mutations. Expression levels of mitophagy markers LC3I/LC3II remained unchanged between cohorts, suggesting no overt mitophagy defects. Expression levels of the parkin substrates, VDAC, NLRP3, and AIMP2 remained unchanged, suggesting no parkin dysfunction. In summary, we were unable to observe dopaminergic neurodegeneration with corresponding nigral-striatal neurobehavioral deficits, nor Parkin or mitochondrial dysfunction in Parkin–/–/PolgAD257A/D257A mice. These findings support a lack of synergism of Parkin loss on mitochondrial dysfunction in mouse models of mitochondrial deficits.
SIGNIFICANCE STATEMENT Producing a mouse model of Parkinson's disease (PD) that is etiologically relevant, recapitulates clinical hallmarks, and exhibits reproducible results is crucial to understanding the underlying pathology and in developing disease-modifying therapies. Here, we show that Parkin–/–/PolgAD257A/D257A mice, a previously reported PD mouse model, fails to reproduce a Parkinsonian phenotype. We show that these mice do not display dopaminergic neurodegeneration nor nigral-striatal-dependent motor deficits. Furthermore, we report that Parkin loss does not synergize with mitochondrial dysfunction. Our results demonstrate that Parkin–/–/PolgAD257A/D257A mice are not a reliable model for PD and adds to a growing body of work demonstrating that Parkin loss does not synergize with mitochondrial dysfunction in mouse models of mitochondrial deficits.
Keywords: mitochondria, mitophagy, parkin, Parkinson's disease, POLG
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disease characterized by the degeneration of dopamine neurons in the substantia nigra pars compacta (SNpc). Although a sporadic disease, mutations encoding the gene for the E3 ligase Parkin result in the second most common form of familial PD (Kitada et al., 1998; Bonifati, 2014). Data suggests that mutations in PRKN result in abnormal mitochondrial quality control, pointing to deficits in mitophagy, fission/fusion, transport, and/or biogenesis (Ge et al., 2020). The relative contributions of each arm of the mitochondrial quality control to dopamine cell death in PD is an area of active investigation (Grenier et al., 2013; Ge et al., 2020; Panicker et al., 2021).
We have previously reported that adult conditional Parkin–/– mice exhibit dopamine neurodegeneration, motor deficits, and abnormal mitochondria morphology and function. We further linked the loss of Parkin to the accumulation of PARIS, a Parkin substrate, which was found to suppress regulators of mitochondrial biogenesis (Shin et al., 2011; Pirooznia et al., 2020). Interestingly, germline Parkin knock-out (KO) mice (Parkin–/–) lack an obvious PD phenotype and corresponding dopaminergic loss (Fleming et al., 2005; Perez and Palmiter, 2005). In an effort to generate a mouse model of PD that does not stereotaxically or pharmacologically target dopaminergic neurons, thus a more etiologically relevant model, we generated a double transgenic line by crossing germline Parkin–/– mice with the mitochondrial DNA (mtDNA) PolgA D257A mice (PolgA+/D257A), a model of mitochondrial dysfunction and premature aging similar to a prior report (Pickrell et al., 2015). PolgAD257A/D257A mice display a progressive accumulation of mtDNA mutations, resulting in a reduction in oxidative phosphorylation (OXPHOS) function and a significantly shortened lifespan (Kujoth et al., 2005).
We initially generated the Parkin–/–/PolgAD257A/D257A double transgenic mouse model to better understand the role of mitochondrial quality control and the contributions of defects in mitophagy versus mitochondrial biogenesis in promoting dopaminergic neurodegeneration because it had been reported that the Parkin–/–/PolgAD257A/D257A mice exhibited loss of dopamine neurons and that the absence of Parkin in PolgAD257A/D257A mice lead to enhanced motor deficits and mitochondrial dysfunction (Pickrell et al., 2015). We hypothesized that deficits in mitochondrial biogenesis would predominate over deficits in mitophagy. To test our hypothesis we used behavioral, metabolic, and biochemical assessments in Parkin–/–/PolgAD257A/D257A; PolgAD257A/D257A, Parkin–/–, and wild-type (WT) littermate controls.
Materials and Methods
Animals
PolgAD257A/D257A mice were previously reported (Kujoth et al., 2005) and were obtained from The Jackson Laboratory: B6.129S7 (Cg)-Polgtm1TproI/J). Mice were genotyped with primer set: PolgA_F 5′-TCCACTGAGGGAGCTTCTGT-3′ and PolgA_R 5′-CTTCCCTAAAGACCGCAGGG-3′. Using these primers allowed for the amplification of the, knock-in allele, resulting in a positive band that was 171-bp higher than the wild-type band. These results were confirmed via sequencing. Parkin–/– mice were used as previously described (Von Coelln et al., 2004) in which exon 7 was deleted, generating a catalytically null mutant. All mice were backcrossed to C57Bl/6 mice (Charles River) for at least 10 generations. The four genotypes of mice used in this study were WT (Parkin+/+/PolgA+/+), Parkin–/–, PolgAD257A/D257A, and Parkin–/–/PolgAD257A/D257A. Animals were housed in a climate-controlled room with a 12/12 h light/dark cycle. Mice were given unlimited access to food and water. Both male and female mice were used.
Pole test
The pole test was performed as previously published (Ogawa et al., 1985; Matsuura et al., 1997) with slight modifications. Briefly, the mouse was placed face-up on top of a wrapped metal pole (diameter 8 mm; height 55 cm). To familiarize the mouse to its new environment, a tray with cage bedding was placed at the bottom of the pole. Each mouse was trained for 3 d. On the fourth day, the mouse was subjected to timed trials that were video recorded. Because of large variability trials, each mouse performed five trials, with a minimum of 1 min of rest in between each trial set. The time it took for the mouse to turn around and descend the pole was recorded. The mouse was given 120 s to turn and 120 s to descend the pole, giving each trial a maximum value of 240 s. If the mouse failed to turn in 120 s, the rater manually turned the mouse and started the clock, again recording the time of descent until a maximum of 120 s. If the mouse fell, it was given a maximum score of 120 s for descent. An average of all five trials was used with the total trial time (latency plus descent) used for analysis.
Openfield
The Photobeam Activity System (PAS)-Openfield suite from San Diego Instruments was used in the Johns Hopkins University School of Medicine Animal Behavior Core. Raters were trained in the use of the equipment, and equipment and animal handling were conducted per manufacturer and behavioral core protocol, respectively. Briefly, animals were brought into the suite for acclimation 1 h before testing. Each station was wiped down with 10% bleach before use. At the beginning of the testing period, each mouse was placed in the middle of the 16 × 16-inch photobeam box. The testing period concluded at the end of 15 min. Mice were placed back in their cages and each station was cleaned.
Grip strength
The Grip Strength Test from BIOSEB was used according to the manufacturer's protocol. Mice were trained 1 d before testing. In brief, mice were held by the tail and their forelimbs were allowed to grip the mesh strength plate. Mice were gently pulled backwards, away from the meter, in a horizontal plane. The force applied to the grid at peak tension (the point before the mouse lost its grip) was recorded in grams. The same protocol was used to determine hindlimb strength, although in this test the mouse was allowed to grip the grid with both its forelimbs and hindlimbs, giving a reading of total limb strength. Force readings were normalized to body weight for analysis.
Immunochemistry
Mice were perfused with ice cold 1× PBS (Quality Biologicals, pH 7.4), and the brains were dissected from the skulls. The left hemisphere was immediately placed in a 15-ml conical tube (Falcon) with cold 4% paraformaldehyde/PBS (Santa Cruz, pH 7.4). Conical tubes were placed on a rotator overnight at 4°C. After fixation, the 4% paraformaldehyde/PBS solution was replaced with cryoprotective 30% sucrose/PBS (pH 7.4) and continued to rotate at 4°C until the brains migrated to the bottom of the conical tubes. Brains were frozen at −80°C and then were cut in serial coronal sections (30-μm sections throughout the entire brain) using a HM440E microtome (Microm). Free floating sections were collected in a 12-well plate (Falcon), allowing for the collections of four complete z-stacks per brain. Sections were blocked with a 10% goat serum/PBS/0.2% Triton X-100 solution for 1 h and then incubated with a primary antibody specific to tyrosine hydroxylase (TH; rabbit polyclonal; Novus Biologicals, NB 300-109; 1:500). This was followed by incubations with a biotin-conjugated anti-rabbit antibody (Vector Laboratories; 1:1000), ABC reagents (Vector Laboratories), and SigmaFast DAB Peroxidase Substrate (Sigma). These sections were then mounted on glass slides (Fisher Scientific) and allowed to dry. Sections were counterstained with Nissl (0.09% thionin for 7 min, followed by formalin acetic acid for 1 min), and glass coverslips (Fisher Scientific) were mounted using DPX mountant (Sigma). Slides were allowed to dry for at least 24 h before stereological counting.
Stereological neuron counting
Neurons were quantified using an optical fractionator and a computer-based image analysis system. Slides were placed on a motorized stage (Ludl Electronics) attached to an Axiophot photomicroscope (Carl Zeiss Vision) containing a Hitachi HV C20 video camera. Quantification of neurons was conducted using Stereo investigator software (MicroBrightField) as previously described (Mandir et al., 1999). In brief, staining was done on every fourth section of the midbrain. Total number of TH-positive and Nissl-positive neurons were calculated via Stereo Investigator software for each genotype (n = 6–8). These quantifications were multiplied by a factor of two, as only the right hemisphere of each brain was used for immunohistochemistry.
Determination of striatal catecholamines
HPLC with electrochemical detection was performed to determine the concentration of striatal catecholamines. Snap-frozen striatal tissue from the left hemisphere of mice were weighed and sonicated in 0.2 ml of 0.1 m perchloric acid with 0.01% EDTA containing 25 g/ml 3,4-dihydroxybenzylamine (DHBA; Sigma) as an internal standard. After centrifugation (15,000 × g, 10 min, 4°C), 20 μl of the supernatant was injected onto a C-18 reverse phase Spheri 5, RP-18, 4.6 mm 25 cm catecholamine column (BASi). The mobile phase consisted of 0.15 m chloroacetic acid, 0.2 mm EDTA, and 0.86 mm sodium octyl sulfate, 4% acetonitrile and 2.5% tetrahydrofuran (pH 3.0). Flow rate was kept at 1.5 ml/min. Biogenic amines and their metabolites were detected by a Prostar ECD (Model 370) electrochemical detector (Varian), with the working electrode kept at 0.6 V. Data were collected and processed on a Star Chromatography Workstation 5.52 (Varian).
Western blot analysis
Mice were perfused with ice cold 1× PBS (Quality Biologicals, pH 7.4), and the brains were dissected from the skulls. The right hemisphere was regionally dissected on ice and immediately snap frozen. Tissue was homogenized in a 1× RIPA Buffer (Sigma) and protease/phosphatase inhibitor cocktail (CST). Once lysed, homogenates were freeze-thawed from dry ice to ice three times. Samples were vortexed for 10–20 s at the end of each thaw. Lysates were centrifuged at 14,000 rpm (Optima TLX micro-ultracentrifuges, TLA 100.3 rotor) at 4°C for 30 min and the fractions were collected. The protein concentration was measured via BCA assay (Pierce) and analyzed via Western blot. The Novex Bis-Tris system (including 4–12% Bis-Tris gel, MOPS buffer, LDS) was used according to manufacturer's protocol; 20 µg of protein was loaded per well. Protein was transferred to methanol-activated PVDF membranes. Membranes were stained with Ponceau to ensure even transfer. Membranes were blocked with 5% nonfat milk in 1× PBS-T for 1 h. Membranes were washed three times for 5 min each with PBS-T. Membranes were incubated with primary antibodies in 1× PBS-T at 4°C overnight. Antibodies used are as follows: NDUFA9 (Abcam; 1:1000), UQCRC2 (Proteintech; 1:1000), COXIV (Proteintech; 1:1000), PDHA (CST; 1:1000, SDHA (CST; 1:1000), VDAC (CST; 1:1000), LC3I/II (CST; 1:1000), Parkin (CST; 1:500), β-actin-HRP (Thermo; 1:10,000). Thermo luminol substrates were applied according to manufacturer's protocol and membranes were imaged on a GE AI600 Chemiluminescent Imager. Western blottings were quantified using ImageJ.
Mitochondrial enzyme activity
The activity of Complex I and IV activity in flash frozen ventral midbrain tissue was determined by using the Complex I Enzyme Activity Microplate Assay kit (ab109721, Abcam) and the Complex IV Rodent Enzyme Activity Microplate Assay kit (ab109911, Abcam) according to the manufacturer's instructions. Briefly, tissue of ventral midbrain was dissected and homogenized with PBS, centrifuged at 1000 × g for 10 min at 4°C, and the protein concentration of collected supernatant were measured using the Bradford Assay. Detergent was added to samples after adjusting sample concentration and incubated 30 min on ice. The supernatant was collected after centrifugation at 12,000 × g for 20 min at 4°C, 250-µg total protein per sample was resuspended in 200-µl assay buffer, added into plate and following 3-h incubation at room temperature. Based on the oxidation reaction, colorimetric changes were recorded at 450 nm (Complex I) or 550 nm (Complex IV). The complex activity was calculated from the change in absorbance using the extinction coefficients of the respective dyes.
mtDNA copy number
Total DNA was extracted from snap-frozen striatal tissue using Qiagen DNeasy kit. Aliquots of DNA were used for Real Time Quantitative PCR (Viaa7, Applied Biosystems ABI Prism 7700 Sequence Detection System, Applied Biosystems). PowerUP SYBR green reagent (Thermo) was used per manufacturer's instructions, with 10 ng of template DNA used for amplification. The primer sequences used are as follows: mtCOXI_6530F, 5′-AGGCTTCACCCTAGATGACACA-3′; mtCOXI_6647R, 5′-GTAGCGTCGTGGTATTCCTGAA-3′; mtCOXI_6620_F, 5′-AGGCTTCACCCTAGATGACACA-3′; mtCOX I_ 6647_R, 5′-GTAGCGTCGTGGTATTCCTGAA-3′; mtND4_F, 5′-AACGGATCCACAGCCGTA-3′; mtND4_R, 5′-AGTCCTCGGGCCATGATT-3′; mtdloop_F, 5′-TCCTCCGTGAAACCAACAA-3′; mtdloop_R, 5′-AGCGAGAAGAGGGGCATT-3′; mtCYTB_F, 5′-ATTCCTTCATGTCGGACGAG-3′; mtCYTB_R, 5′-TGAGCGTAGAATGGCGTATG-3′, GAPDH_6530_F, 5′-GCAGTGGCAAAGTGGAGATT-3′; GAPDH_6530_R, 5′-GAATTTGCCGTGAGTGGAGT-3′; GAPDH_6620_F, 5′-GCAGTGGCAAAGTGGAGATT-3′; GAPDH_6620_R, 5′-GAATTTGCCGTGAGTGGAGT-3′.
Sequences of primers used to measure mtDNA copy number; mtCOX1_6530F, mtCOX1_6647R, GAPDH_6530_F, and GAPDH_6530_R were previously reported in (Pickrell et al., 2015); mtCOX1_6620F, mtCOX1_6836R, mtCYTB_F, mtCYTB_R, GAPDH_6620_F, and GAPDH_6620_R were previously reported previously (Stevens et al., 2015).
Experimental design and statistical analysis
Power analysis was conducted using G*Power3.1.9.6. Raw data supplied by the first author from Figure 1B of the original Pickrell et al., 2015 article was used to determine the appropriate effect size (Pickrell et al., 2015). To be conservative, the maximum standard deviation between groups was used (2167.146). The effect size was determined to be 0.8557729. Using this effect size, an a priori power analysis was performed. Power was maintained at 80% and the resulting total sample size was determined to be 20 (n = 5/group).
Figure 1.
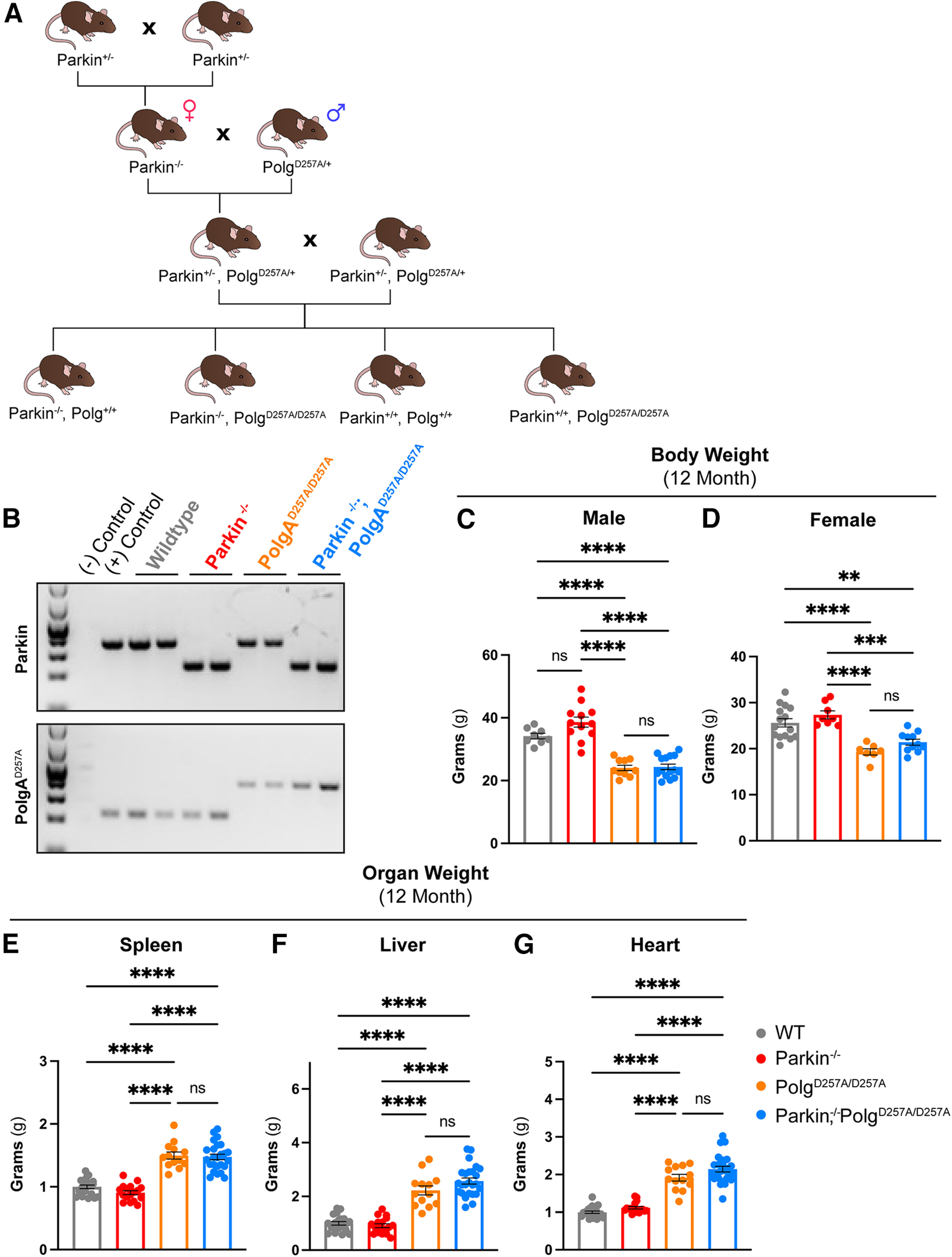
Physiologic data of 12-month-old mice. A, Schematic depicting breeding scheme to produce Parkin–/–/PolgAD257A/D257A mice. Female and male mice are depicted with the symbol for females and males respectively. B, Representative gel confirming genotypes of pups. C, D, Body weight of 12-month-old female and male mice. Results are the mean ± SEM, n = 7–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs did not significantly differ per Brown–Forsythe test of variance. Significance of means was analyzed via ordinary one-way ANOVA (C: F(3,41) = 40.94 ****p 0.0001; D: F(5,37) = 15.97 ****p 0.0001). Post hoc Tukey's test resulted in ****p 0.0001 for all PolgAD257A/D257A-expressing mice compared with wild-type and Parkin–/– for both male and female cohorts, ns, not significant. E, F, Mass of spleen and liver (organs with known defects in PolgAD257A/D257A mice) were recorded. Mass was normalized to body weight and then further normalized to the average wild-type organ mass/body weight ratio. Results are the mean ± SEM, n = 14–25 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. Brown–Forsythe test of variance found that SDs differed significantly; therefore, significance of means was determined via a Welch one-way ANOVA (E: W(3.0,42.16) = 65.89 ****p 0.0001; F: W(3.0,43.85) = 93.922 ****p 0.0001). Post hoc Dunnett's T3 test resulted in E: ****p 0.0001 for all PolgAD257A/D257A-expressing mice compared with wild-type and Parkin–/–; F: ****p 0.0001 for all PolgAD257A/D257A-expressing mice compared with wild-type and Parkin–/–, ns, not significant. G, Mass of hearts were recorded. Mass was normalized to body weight and then further normalized to the average wild-type organ mass/body weight ratio. Results are the mean ± SEM, n = 14–25 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs for heart mass did not significantly differ. Significance of means was analyzed via an ordinary one-way ANOVA (G: F(371) = 55.26 ****p 0.0001). Post hoc Tukey's test resulted ****p 0.0001 for all PolgAD257A/D257A-expressing mice compared with wild-type and Parkin–/–, ns, not significant. Analysis and graphs were produced using GraphPad Prism 8.4.3.
The remaining statistical analyses were conducted using GraphPad Prism version 8.4.3 and 9.4.1 software. ROUT analysis for outlier identification with a max false discovery rate at 0.1% was performed on each dataset. Next, the D'Agostino and Pearson test for normality was conducted. If data were distributed normally, the Brown–Forsythe test of variance was performed to determine whether SDs differed significantly between groups. If SDs did not differ, one-way or two-way ANOVA followed by Tukey's analyses was performed to determine the significance of differences among multiple experimental groups. If SDs significantly differed, significance of means was analyzed via Welch one-way ANOVA followed by post hoc Dunnett's T3 test of multiple comparisons. Data are expressed as the mean ± SEM, and values with p 0.05 considered statistically significant. If data were not normally distributed (50% of the groups were non-normally distributed), significance of medians was analyzed via Kruskal–Wallis test followed by post hoc Kruskal–Wallis test of multiple comparisons. Data in these analyses are expressed as the median ± SEM, and values with p 0.05 considered statistically significant.
Results
We generated Parkin–/–/PolgAD257A/D257A and appropriate littermate controls via the cross shown in Figure 1A. Because female mice are the primary contributor of mitochondria to pups and PolgAD257A/+ parents may still accumulate damaged mitochondria over time, we used PolgAD257A/+ male mice at every possible step in the breeding scheme, thus limiting the potential vertical transfer of PolgAD257A/+-dependent mtDNA mutations to newborn mice. Mice were born at expected Mendelian ratios and appeared normal. Genotypes were confirmed via PCR and gel electrophoresis (Fig. 1B). Both male and female mice harboring the PolgAD257A/D257A exhibited reduced body weight by 12 months (Fig. 1C,D). Previous reports stated that loss of Parkin partially rescued the splenomegaly phenotype observed in the PolgAD257A/D257A line (Pickrell et al., 2015). We did not observe worsening or amelioration of hepatosplenomegaly or cardiac hypertrophy in Parkin–/–/PolgAD257A/D257A mice compared with PolgAD257A/D257A mice (Fig. 1E–G).
Absence of nigro-striatal-dependent motor dysfunction in Parkin−/−/PolgAD257A/D257A mice
We first tested whether Parkin–/–/PolgAD257A/D257A mice displayed progressive motor dysfunction or behavioral deficits. Mice were subjected to Openfield and grip strength analysis every three months until 12 months of age and pole tests at 12 months of age, the time point at which broad respiratory chain dysfunction in PolgAD257A/D257A has been reported (Ross et al., 2013). The pole test is a widely used behavioral test for PD-related nigral-striatal-dependent motor dysfunction in PD mouse models (Ogawa et al., 1985). The pole test is defined by placing a mouse face up on top of a pole of a prespecified height and recording the time it takes the mouse to turn around and successfully run down the pole (Matsuura et al., 1997; Fig. 2A). We observed no significant difference in latency times for the pole test for 12-month-old Parkin–/–/PolgAD257A/D257A mice compared with wild-type (Fig. 2B).
Figure 2.
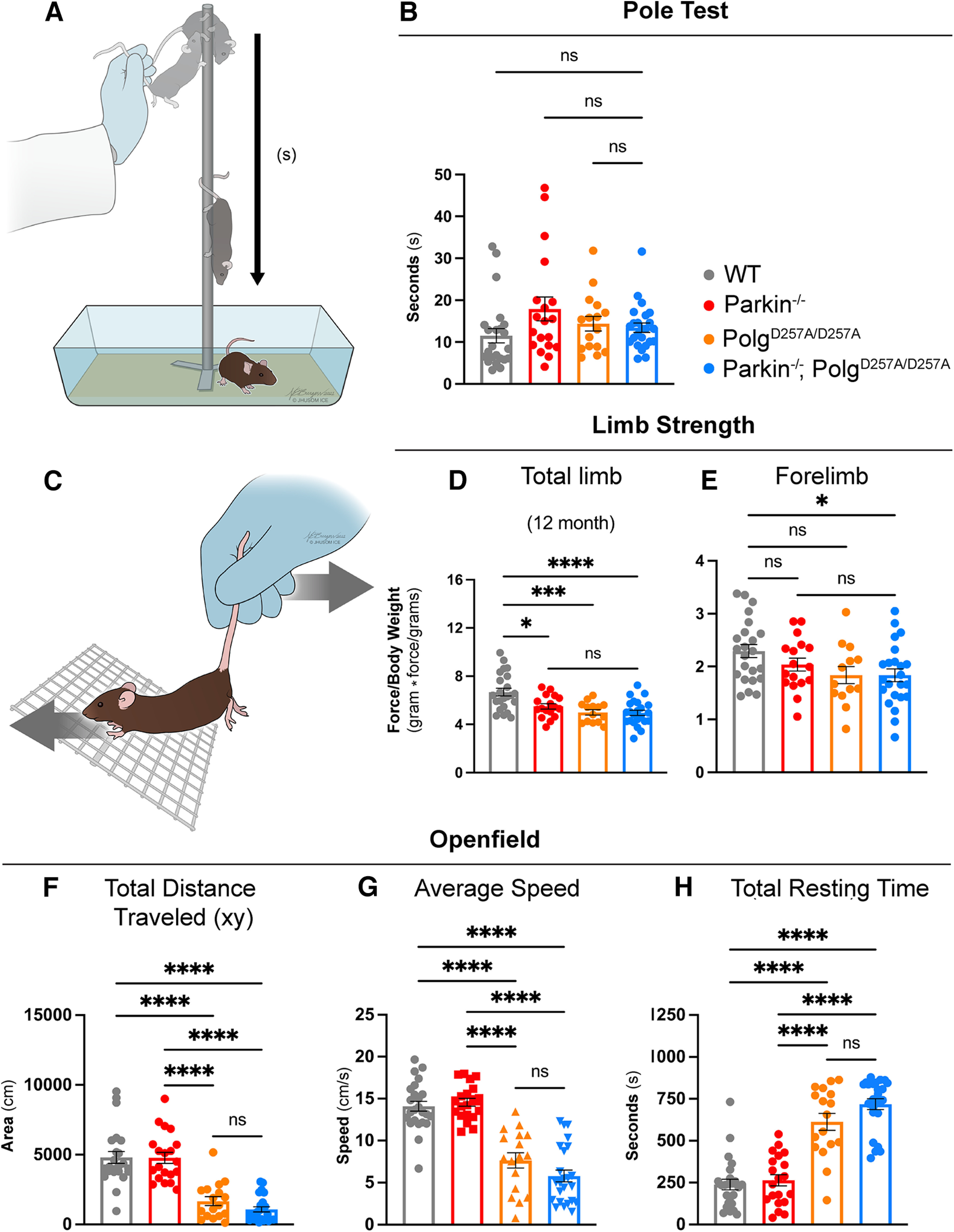
Behavioral and neuromuscular data of 12-month-old mice. A, Schematic of the pole test. B, Latency values of pole test of 12-month-old mice. Results are the median ± SEM, n = 17–25 per group with an average of five trials per mouse used for analysis. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be non-normally distributed via D'Agostino and Pearson test (with two out of the four groups being found to be non-normally distributed). Significance of medians was analyzed via Kruskal–Wallis nonparametric ANOVA (KW(4,82) = 7.271, p = 0.0637). Post hoc Kruskal–Wallis test for multiple comparisons resulted in no significant differences (ns) between groups. C, Schematic depicting directionality of force (mouse) versus tester in grip strength testing. D, E, Total limb and forelimb strength (in gram*force) normalized to body weight (grams). Results are the mean ± SEM, n = 13–23 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. Brown–Forsythe test of variance found no difference in SDs. Significance of means was analyzed via an ordinary one-way ANOVA (D: F(3,73) = 10.21 ****p 0.0001; E: F(3,72) = 3.012 *p = 0.0536). Post hoc Tukey's test was performed and resulted in D: *p = 0.0117 for wild-type versus Parkin–/–, ***p = 0.0003 for wild-type versus PolgAD257A/D257A, and ****p 0.0001 for wild-type versus Parkin–/–/PolgAD257A/D257A; E: *p = 0.0398 for wild-type versus Parkin–/–/PolgAD257A/D257A, ns, not significant. Forelimb and total limb strength at three, six, and nine months is provided in Extended Data Figure 2-1. F, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period. Results are the mean ± SEM, n = 17–24 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (W(5.0,51.85) = 36.71 ****p 0.0001). Post hoc Dunnett's T3 test resulted in ****p 0.0001 for wild-type versus all PolgAD257A/D257A-expressing mice and for Parkin–/– versus all PolgAD257A/D257A-expressing mice. G, Average speed of locomotion during 15-min test period. Results are the mean ± SEM, n = 17–24 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (F(5,121) = 52.80 ****p 0.0001). Post hoc Tukey's test was performed for and resulted in ****p 0.0001 for wild-type versus all PolgAD257A/D257A-expressing mice and for Parkin–/– versus all PolgAD257A/D257A-expressing mice, ns, not significant. H, Total resting time, defined as a period of four or more seconds with no photobeam breaks, in 15-min test period. Results are the mean ± SEM, n = 17–24 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (W(5.0,74.83) = 68.21 ****p 0.0001). Post hoc Dunnett's T3 test resulted in ****p 0.0001 for wild-type versus all PolgAD257A/D257A-expressing mice and for Parkin–/– versus all PolgAD257A/D257A-expressing mice, ns, not significant. Analysis and graphs were produced using GraphPad Prism 8.4.3. Distance traveled, average speed and total resting time at three, six, and nine months is provided in Extended Data Figure 2-2. Pole test, forelimb and total strength, distance traveled, average speed, and total resting time in male and female mice at 12 months is provided in Extended Data Figure 2-3.
Neuromuscular strength was analyzed via grip force readings of both forelimbs and total limbs (Fig. 2C). We observed significant reductions in total limb strength in all PolgAD257A/D257A -expressing lines compared with wild type at 12 months (Fig. 2D) as well as a reduction in forelimb strength between 12-month-old wild-type and Parkin–/–/PolgAD257A/D257A cohorts (Fig. 2E). Differences in forelimb strength were first seen in both PolgAD257A/D257A and Parkin–/–/PolgAD257A/D257A cohorts at six months (Extended Data Fig. 2-1A–C). However, these differences in forelimb strength were not present at nine months (Extended Data Fig. 2-2A–C). Furthermore, reductions in total limb strength were observed in Parkin–/–/PolgAD257A/D257A mice at six and nine months (Extended Data Fig. 2-1D–F). It should be noted that there were no differences between any PolgAD257A/D257A-expressing groups at 12 months, confirming that neuromuscular strength deficits at 12 months are ultimately a result of the PolgAD257A/D257A mutation (Fig. 2D,E). Unexpectedly, Parkin–/– mice displayed reductions in total limb strength at nearly all time points. Because Parkin–/– mice were observed to weigh slightly more than wild-type mice at nearly every time point, we believe that normalization to body weight contributed to some of the differences observed in the grip strengths. These findings are supported by analysis of non-normalized force readings, which demonstrated no differences between groups at early time points, with significant reductions in both forelimb and total limb strength in all PolgAD257A/D257A -expressing lines compared with wild-type and Parkin–/– at 12 months.
Locomotor activity was investigated using the Openfield test, a behavioral test commonly performed to assess locomotion, exploration, and emotionality in rodents. There was a progressive decline in the distance traveled in all cohorts with the PolgAD257A/D257A mutation when compared with wild-type and Parkin–/– mice at 12 months (Fig. 2F). This paralleled a previous report of reduced locomotor activity in aged PolgAD257A/D257A mice (Dai et al., 2013). All mice with the PolgAD257A/D257A background exhibited significant reductions in the average speed of locomotion at 12 months (Fig. 2G). Furthermore, they displayed an increase in resting time, quantified as a period of four or more seconds without movement, by 12 months (Fig. 2H). These changes were initially seen at six months, with both PolgAD257A/D257A and Parkin–/–/PolgAD257A/D257A mice demonstrating loss of speed and distance compared with controls (Extended Data Fig. 2-2B,E). These reductions progressed in significance and severity from six to nine months (Extended Data Fig. 2-2C,F). Similarly, an increase in resting time was observed in Parkin–/–/PolgAD257A/D257A at six months, and by nine months both PolgAD257A/D257A and Parkin–/–/PolgAD257A/D257A mice demonstrated significant increases in their time spent resting (Extended Data Fig. 2-2H,I). The loss of endogenous Parkin in the context of PolgAD257A/D257A did not significantly impact the neuromuscular behavioral defects observed. Furthermore, no sex effects were observed in behavior tests (Extended Data Fig. 2-3).
Dopaminergic neurons in the SNpc remain intact in Parkin−/−/PolgAD257A/D257A mice
Thus far, our data has suggested that behavioral changes were the result of the PolgAD257A/D257A genotype. To further explore any underlying neurochemical deficits in Parkin–/–/PolgAD257A/D257A, we performed immunohistochemistry for TH+ neurons in ventral midbrain tissue of 12-month-old mice (Fig. 3A). Unbiased stereologic cell counting revealed no observable changes in the amount of TH+ or Nissl+ neurons in the SNpc of any of the groups, including Parkin–/–/PolgAD257A/D257A mice (Fig. 3B) These data indicate that there was no dopaminergic neurodegeneration in the ventral midbrain.
Figure 3.
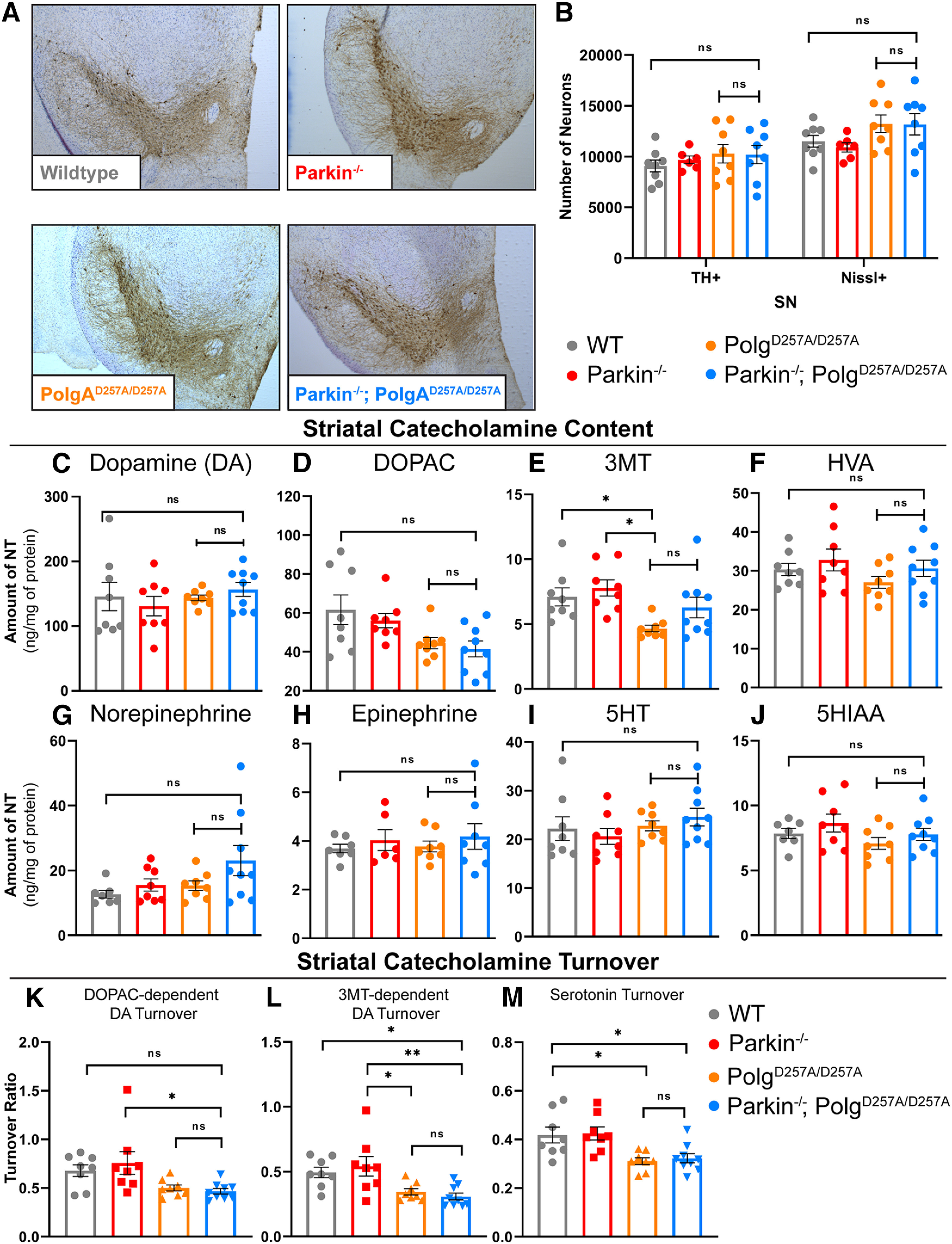
Neuropathological analysis of ventral midbrain tissue of 12-month-old mice. A, Representative images of immunohistochemical analysis of 12-month-old ventral midbrain sections. TH+ neurons are stained via, 3'-diaminobenzidine (D)AB (Brown) and Nissl+ (a pan-neuronal marker) are stained blue. B, Stereological quantification of TH+ and Nissl + neurons in the substantia nigra. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the mean ± SEM, n = 6–8 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. No outliers were removed. Data were found to be normally distributed via D'Agostino and Pearson test. Data analyzed using two-way ANOVA (Fc(3,52) = 2.143 p = 0.1060), ns, not significant. C, D, HPLC analysis of striatal dopamine and DOPAC content normalized to total protein concentration. Striatum of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the mean ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (D: W(3.0,13.86) = 0.6434 p = 0.5999; E: W(3.00,15.59) = 3.661 *p = 0.0357). Post hoc Dunnett's T3 test resulted in no significant differences between groups, ns, not significant. E, HPLC analysis of striatal 3MT content normalized to total protein concentration. Striatum of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the median ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be non-normally distributed per D'Agostino and Pearson test. Significance of medians was analyzed via Kruskal–Wallis nonparametric ANOVA (KW(4,33) = 13.17, *p = 0.0043). Post hoc Kruskal–Wallis test for multiple comparisons resulted in **p = 0.0024 for wild-type versus PolgAD257A/D257A and **p = 0.0042 for Parkin–/– versus PolgAD257A/D257A, ns, not significant. F–H, HPLC analysis of striatal HVA, norepinephrine, and epinephrine content normalized to total protein concentration. Striatum of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the median ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (G: F(3,29) = 1.2 p = 0.3050; H: F(3,28) = 2.4 p = 0.0868; H: F(2,25) = 0.3 p = 0.7653), ns, not significant. I, J, HPLC analysis of striatal 5HT and 5HIAA, a 5HT metabolite, content normalized to total protein concentration. Striatum of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the median ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (J: F(3,29) = 0.8779 p = 0.4642; K: F(3,28) = 1.529 p = 0.2286), ns, not significant. K, DOPAC-dependent dopamine turnover as assessed by (DOPAC+HVA)/dopamine. Significance of means analyzed via ordinary one-way ANOVA (F(2,29) = 4.367 *p = 0.0118). Post hoc Tukey's test resulted in *p = 0.0208 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. L, 3MT-dependent dopamine turnover as assessed by (3MT+HVA)/dopamine. Significance of means analyzed via ordinary one-way ANOVA (F(2,29) = 6.282 **p = 0.0020). Post hoc Tukey's test resulted in *p = 0.0310 for wild-type versus Parkin–/–/PolgAD257A/D257A, *p = 0.0254 for Parkin–/– versus PolgAD257A/D257A, and **p = 0.0049 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. M, Serotonin (5HT) turnover as assessed by 5HIAA/5HT. Significance of means analyzed via ordinary one-way ANOVA (F(2,29) = 6.449 **p = 0.0118). Post hoc Tukey's test resulted in *p = 0.0195 for wild-type versus PolgAD257A/D257A, *p = 0.0355 for wild-type versus Parkin–/–/PolgAD257A/D257A, *p = 0.0121 for Parkin–/– versus PolgAD257A/D257A, and **p = 0.0222 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. Analysis and graphs were produced using GraphPad Prism 8.4.3.
We next assessed striatal dopamine and dopamine degradation products in 12-month-old mice. In agreement with our stereological data, there were no significant changes in dopamine or its degradation products dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), norepinephrine, or epinephrine (Fig. 3C,D,F–H). Similar to prior reports (Pickrell et al., 2015), we did observe a reduction in 3-methoxytyramine (3-MT), a dopamine degradation product, in PolgAD257A/D257A mice compared with both wild-type and Parkin–/– mice; however, we saw no reduction in Parkin–/–/PolgAD257A/D257A mice (Fig. 3E).
Another method to assess striatal catecholamine dysregulation is to calculate turnover rates. Dopamine turnover can be determined by using the following equations: (1) DOPAC-dependent dopamine turnover [(DOPAC + HVA)/dopamine] or (2) 3MT dependent dopamine turnover [(3MT + HVA)/dopamine]. Using this approach, we found a significant reduction in DOPAC-dependent dopamine turnover in Parkin–/–/PolgAD257A/D257A mice compared with Parkin–/– mice, but not wild-type mice (Fig. 3K). Analysis of 3MT-dependent dopamine turnover showed substantial reductions in Parkin–/–/PolgAD257A/D257A mice and PolgAD257A/D257A mice compared with Parkin–/– mice (Fig. 3L). There was also a reduction in Parkin–/–/PolgAD257A/D257A mice versus wild-type mice (Fig. 3L). These results imply that both PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A mice exhibit some striatal dopamine dysregulation without dopaminergic neuronal loss. These results agree with previous findings, which report striatal dopamine dysregulation in PolgAD257A/D257A mice (Dai et al., 2013). No differences were observed between PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A mice in dopamine or in dopamine turnover, suggesting that dysregulation is a result of the PolgAD257A/D257A knock-in and is not significantly impacted by Parkin loss.
Serotonergic neurons from the raphe nuclei project, in part, to the striatum and are critical for a variety of neurologic functions including mood, memory processing, sleep, and cognition (Charnay and Léger, 2010). Analysis of striatal serotonin, also known as 5-hydroxytryptamine (5-HT) and its degradation product 5-hydroxyindoleacetic acid (5HIAA) demonstrated no differences between cohorts at 12 months of age (Fig. 3I,J). These data are contrary to previous work which reports a significant increase in serotonin levels in striatal tissue (Pickrell et al., 2015). Serotonin turnover can be calculated by normalizing 5HIAA to 5-HT. We observed a significant reduction in serotonin turnover of PolgAD257A/D257A mice compared with both wild-type and Parkin–/– mice (Fig. 3M), implying that aged PolgAD257A/D257A mice experience striatal catecholamine and indolamine dysregulation.
An early clinical sign that precedes motor dysfunction in PD patients is anosmia or the loss of one's sense of smell. A prior report indicated a significant increase in serotonin and norepinephrine levels in striatal and olfactory bulb tissue of Parkin–/–/PolgAD257A/D257A mice (Pickrell et al., 2015). They postulated that the upregulation of these neurotransmitters may be a compensatory mechanism by which neurons react to the loss of dopamine. Our analysis of these brain regions, however, displayed reductions in serotonin (5-HT) in Parkin–/–/PolgAD257A/D257A mice, as well as significant decreases in 5HIAA in all mice expressing the PolgAD257A/D257A mutation (Fig. 4E,F). Interestingly, there were no substantial differences in serotonin turnover, implying that serotonin dysregulation in the olfactory bulb occurs as a result of reduced serotonin levels, not in the ability to breakdown serotonin (Fig. 4H). Our analysis also revealed a significant decline in dopamine, DOPAC, HVA, and DOPAC-dependent dopamine turnover in all PolgAD257A/D257A-expressing mice (Fig. 4A,B,D). These differences are likely because of the effect of mtDNA mutations, as both PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A were affected and loss of Parkin did not lead to further decline in metabolite concentrations. Contrary to a prior report (Pickrell et al., 2015), we observed a reduction in olfactory bulb norepinephrine in Parkin–/–/PolgAD257A/D257A mice (Fig. 4C). Parkin–/– mice also displayed reductions in dopamine metabolites, suggesting that germline Parkin–/– mice themselves exhibit olfactory-bulb dopamine dysregulation at 12 months of age (Fig. 4B,G). This was expected as reductions in olfactory bulb dopamine were reported in this specific germline Parkin–/– line (Von Coelln et al., 2004).
Figure 4.
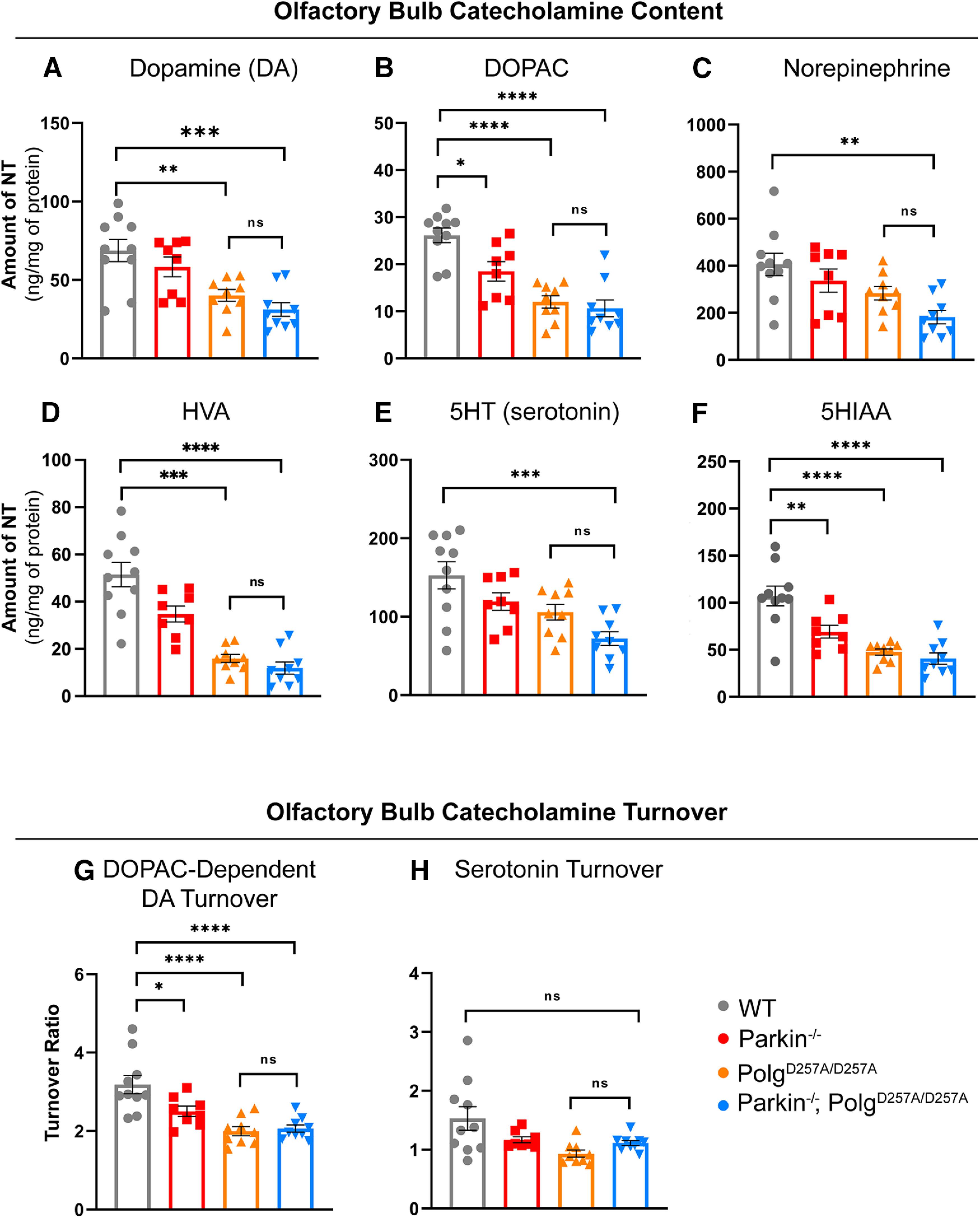
Neuropathological analysis of striatal tissue of 12-month-old mice. A–C, HPLC analysis of olfactory bulb dopamine, DOPAC, and norepinephrine content normalized to total protein concentration. Olfactory bulb of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the mean ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (A: F(3,32) = 9.4 ***p = 0.0001; B: F(3,32) = 2.4 ****p 0.00001; C: F(3,32) = 5.886 **p = 0.0026). Post hoc Tukey's test resulted in A: **p = 0.046 for wild-type versus PolgAD257A/D257A, ***p = 0.0002 for wild-type versus Parkin–/–/PolgAD257A/D257A, and *p = 0.0116 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; B: *p = 0.0154 for wild-type versus Parkin–/–, ****p 0.00001 for wild-type versus PolgAD257A/D257A and for wild-type versus Parkin–/–/PolgAD257A/D257A, and *p = 0.0153 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; C: **p = 0.0015 for wild-type versus Parkin–/–/PolgAD257A/D257A, ns, not significant. D, HPLC analysis of olfactory bulb HVA content normalized to total protein concentration. Olfactory bulb of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the mean ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (D: W(3.0,16.71) = 28.77 ****p 0.00001). Post hoc Dunnett's T3 test resulted in ***p = 0.046 for wild-type versus PolgAD257A/D257A, ****p 0.00001 for wild-type versus Parkin–/–/PolgAD257A/D257A, **p = 0.0031 for Parkin–/– versus PolgAD257A/D257A, and ***p = 0.0007 Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. E, F, HPLC analysis of olfactory bulb 5HT and 5HIAA content normalized to total protein concentration. Olfactory bulb of right hemisphere of 12-month-old mice were used. Quantifications multiplied by factor of two to extrapolate to whole-brain values. Results are the mean ± SEM, n = 8–9 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (E: F(3,32) = 7.249 ***p = 0.0008; F: F(3,32) = 17.31 ****p 0.00001). Post hoc Tukey's test resulted in E: ***p = 0.004 for wild-type versus Parkin–/–/PolgAD257A/D257A; F: **p = 0.0056 for wild-type versus Parkin–/–, ****p 0.00001 for wild-type versus PolgAD257A/D257A and for wild-type versus Parkin–/–/PolgAD257A/D257A, ns, not significant. G, DOPAC-dependent dopamine turnover as assessed by (DOPAC+HVA)/dopamine. Significance of means analyzed via ordinary one-way ANOVA (F(3,32) = 12.41 ****p 0.00001). Post hoc Tukey's test resulted in *p = 0.0260 for wild-type versus Parkin–/–, ****p 0.00001 for wild-type versus PolgAD257A/D257A and for wild-type versus Parkin–/–/PolgAD257A/D257A, ns, not significant. H, Serotonin (5HT) turnover as assessed by 5HIAA/5HT. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (D: W(3.0,17.32) = 4.339 *p = 0.0142). Post hoc Dunnett's T3 test resulted in no significant differences (ns) between groups. Analysis and graphs were produced using GraphPad Prism 8.4.3.
Loss of parkin does not enhance mitochondrial dysfunction in PolgAD257A/D257A mice
To further investigate whether loss of endogenous Parkin affects mitochondrial dysfunction induced by the PolgAD257A/D257A mutation, we quantified the steady-state expression level of mitochondrial markers in the ventral midbrain of 12-month-old mice. Western blots were performed with a panel of commercially available antibodies. We found no differences in expression levels of most mitochondrial markers tested, including NDUFA9 (subunit of Complex I) and UQCRC2 (subunit of Complex III; Fig. 5B–F; Extended Data Fig. 5-1A,B). There were, however, significant reductions in the levels of Complex IV (Fig. 5G; Extended Data Fig. 5-1A,B). This is understandable as NDUFA9, UQCRC2, succinate dehydrogenase (SDHA) complex flavoprotein subunit A (a subunit of Complex II), pyruvate dehydrogenase E1 subunit α 1, and voltage-dependent anion-selective channel 1 (VDACI) are nuclear-encoded mitochondrially-targeted proteins, whereas COX4I1 (a subunit of Complex IV) is a mitochondrially-encoded subunit and would be directly affected by mtDNA mutations.
Figure 5.
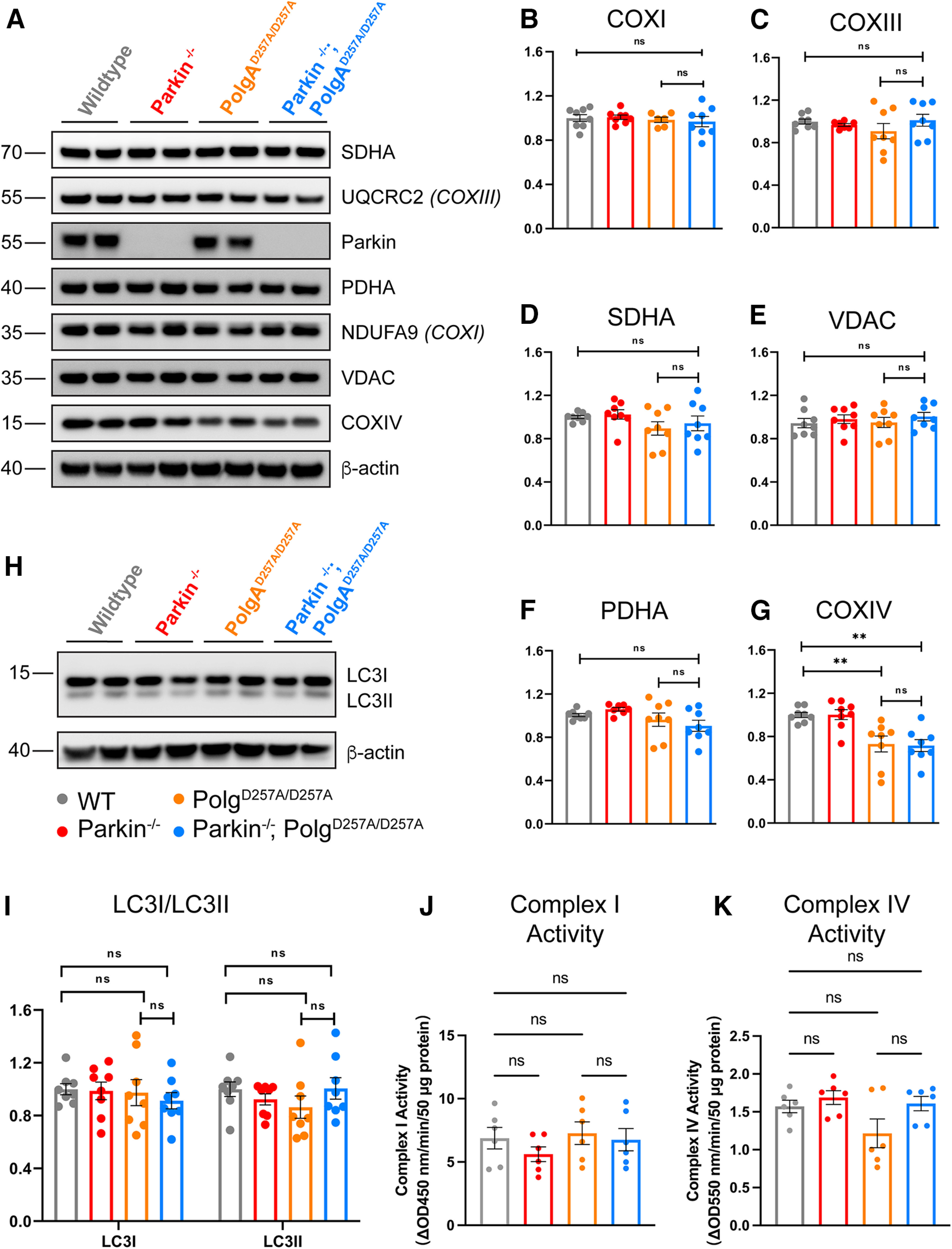
Steady-state expression of mitochondrial and mitophagy markers. A, Representative Western blot illustrating mitochondrial marker expression in ventral midbrain tissue of 12-month-old mice. B–E, Quantification of Western blot mitochondrial marker expression analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 8 mice per group. Experiments were run in a minimum of experimental triplicate with the average of results used for analysis. ImageJ was used for optical density analysis. Quantifications were normalized to β-actin to account for loading differences and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test, likely because of batch effect. Therefore, significance of means was analyzed via a Welch one-way ANOVA (B: W(3.0,13.89) = 0.2700 p = 0.8459; C: W(3,13.58) = 0.7985 p = 0.5156; D: W(3,13.98) = 3.607 *p = 0.0406; E: W(3,13.0) = 1.145 p = 0.3068). Post hoc Dunnett's T3 multiple comparisons test was conducted on D data and resulted in no significant (ns) differences between groups. F, G, Quantification of Western blot mitochondrial marker expression analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 8 per group. Experiments were run in a minimum of experimental triplicate with the average of results used for analysis. ImageJ was used for optical density analysis. Quantifications were normalized to β-actin to account for loading differences and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly per Brown–Forsythe test. Significance of means was analyzed via ordinary one-way ANOVA (F: F(3,28) = 9.173 ***p = 0.0002; G: F(3,28) = 0.4631 p = 0.9739). Post hoc Tukey's test for data in F resulted in **p = 0.0064 for wild-type versus PolgAD257A/D257A, **p = 0.0039 for wild-type versus Parkin–/–/PolgAD257A/D257A, **p = 0.0059 for Parkin–/– versus PolgAD257A/D257A, and **p = 0.0036 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. H, Representative Western blot illustrating blot mitophagy marker expression analysis in ventral midbrain tissue of 12-month-old mice. I, Quantification of Western blot mitophagy marker expression analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 8 per group. Experiments were run in a minimum of experimental triplicate with the average of results used for analysis. ImageJ was used for optical density analysis. Quantifications were normalized to β-actin to account for loading differences and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. Data were analyzed using two-way ANOVA (Fc(3,28) = 0.2892 p = 0.8328). Uncropped Western blottings for this figure are provided in Extended Data Figure 5-1. J, Mitochondrial Complex I activity of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 per group. Experiments were run in experimental duplicate on one plate with the average of results used for analysis. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via Shapiro–Wilk test. Data were analyzed using two-way ANOVA with repeated measures F(15,18) = 32.18 p < 0.0001 and a Geisser–Greenhouse's ε correction of 0.6569. Post hoc Tukey's multiple comparisons test was performed and demonstrated no significant (ns) differences between group. The results are as follows: p = 0.2569 for wild-type versus Parkin–/–/; p = 0.9281 for wild-type versus PolgAD257A/D257A, p = 0.995 for wild-type versus Parkin–/– versus PolgAD257A/D257A, and p = 0.9522 for PolgAD257A/D257A versus Parkin–/–/PolgAD257A/D257A. K, Mitochondrial Complex IV activity of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 per group. Experiments were run in experimental duplicate on one plate with the average of results used for analysis. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via Shapiro–Wilk test. Data were analyzed using two-way ANOVA with repeated measures F(15,18) = 1.758 p = 0.1269 and a Geisser–Greenhouse's ε correction of 0.6621. Post hoc Tukey's multiple comparisons test was performed and demonstrated no significant (ns) differences between group. The results are as follows: p = 0.6015 for wild-type versus Parkin–/–/; p = 0.2631 for wild-type versus PolgAD257A/D257A, p = 0.9822 for wild-type versus Parkin–/– versus PolgAD257A/D257A, and p = 0.3271 for PolgAD257A/D257A versus Parkin–/–/PolgAD257A/D257A. Analysis and graphs were produced using GraphPad Prism 9.4.1. The levels of the parkin substrates, NLRP3, AIMP2 normalized to β-actin are presented in Extended Data Figure 5-2.
We further assessed whether loss of endogenous Parkin impacted mitochondrial complex activity in flash-frozen ventral midbrain tissue of 12-month-old mice. We observed no differences in Complex I or Complex IV activities comparing wild-type to PolgD257A/D257A and Parkin–/–; PolgD257A/D257A (Fig. 5J,K). In addition, there were no changes in the levels of the parkin substrates AIMP2 or NLRP3 implicated in the death of DA neurons (Y. Lee et al., 2013; Panicker et al., 2022) in adult Parkin–/– mice (Extended Data Fig. 5-2A–D).
Because Parkin is known to be a key player in mitophagy, we investigated whether loss of Parkin effected expression levels of mitophagy markers. The conversion of soluble LC3I to activated, lipid-conjugated LC3II is considered to be necessary for autophagosome formation, a required step for mitophagy (Stevens et al., 2015). Therefore, we analyzed the steady-state levels of LC3I and LC3II as a readout for active mitophagy (Tanida et al., 2008; Stevens et al., 2015). We observed no significant differences between cohorts in LC3I or LC3II, suggesting that Parkin–/–/PolgAD257A/D257A mice maintain the ability to form autophagosomes (Fig. 5H,I).
Assessing mtDNA copy number is an indirect readout of the number of mitochondria per cell in a given amount of tissue (Malik and Czajka, 2013). It can also be used to investigate deficits in mitophagy, which would lead to an accumulation of damaged mitochondria and mtDNA, or mitochondrial biogenesis, which would result in a reduction of the mitochondrial pool and mtDNA. Because the depletion of mtDNA in whole-brain lysates of PolgAD257A/D257A mice was previously reported (Pickrell et al., 2015), suggesting defunct mitochondrial biogenesis, we next determined the mtDNA copy number in striatal tissue of 12-month-old mice (Stevens et al., 2015). This technique is done by isolating total DNA from tissue and performing qRT-PCR using mtDNA-specific primers (Fig. 6A; Materials and Methods). These results are then normalized to a nuclear encoded housekeeping gene, such as GAPDH, to to normalize the variance between samples, and then again to wild-type values to normalize against batch effect. We ran a panel of mtDNA primers probing several different loci, including ND4, COXI, CYTB, and the Dloop region (Pickrell et al., 2015; Stevens et al., 2015; Fig. 6G). During our initial analysis we used a mixed effects model with matching across rows, which assumes that all loci analyzed depend on the expression level of each other, appropriate in the case of mtDNA as the mitochondrial genome is circular dsDNA (Fig. 6C). This mixed-effects model also accounts for nonindependence between samples, meaning it accounts for the same biological samples being tested at each locus. When employing this statistical method, we observed no significant differences in the copy number of most mtDNA loci, indicating that mtDNA copy number remains unchanged in ventral midbrain tissue of PolgAD257A/D257A mice, regardless of the absence or presence of Parkin (Fig. 6G). Interestingly, the mixed-effects model found a consistent and substantial increase in the copy number of the Dloop region, the highly conserved origin of replication of mtDNA, in PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A mice.
Figure 6.
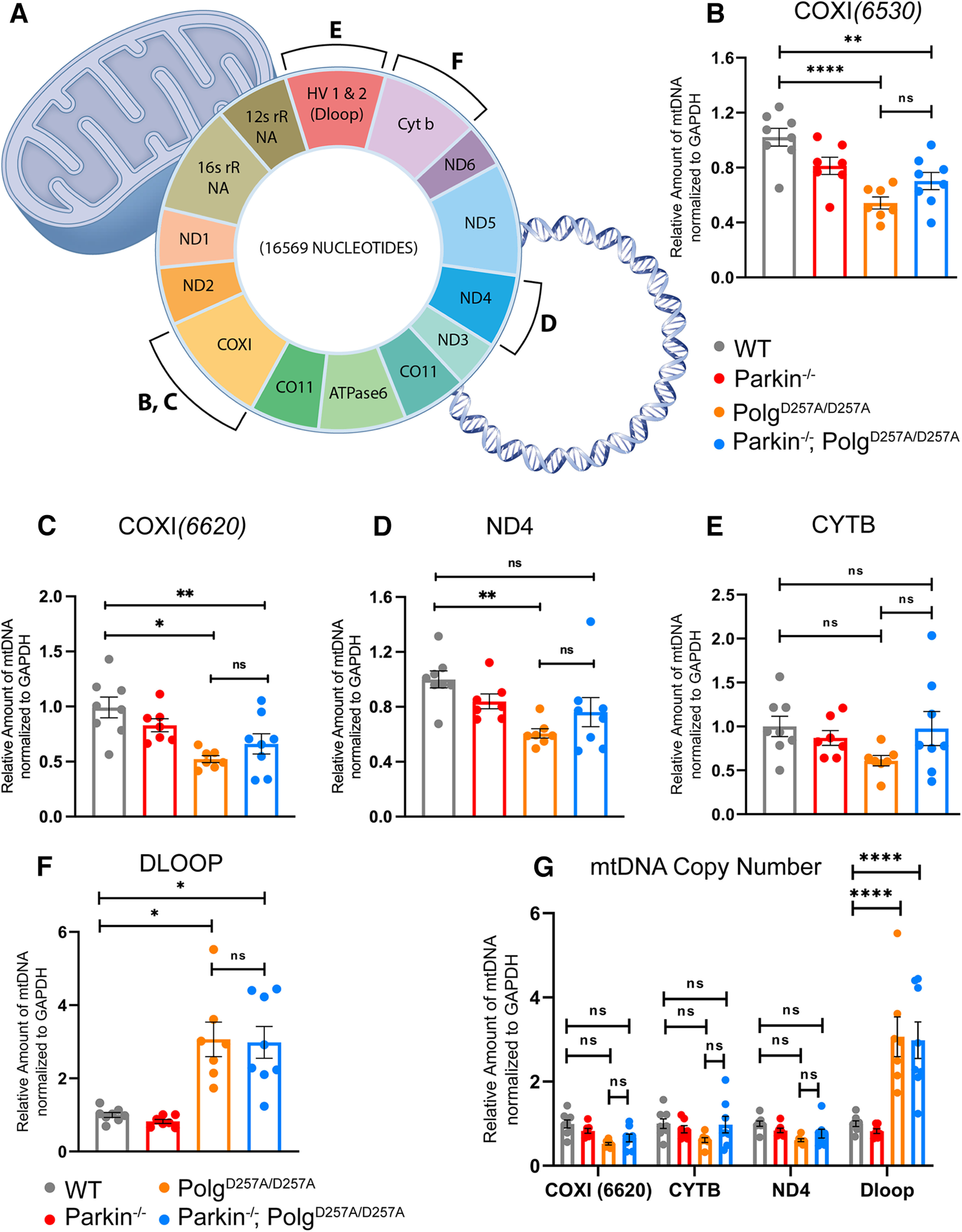
Analysis of mtDNA copy number in aged mice. A, Schematic denoting mtDNA genome and loci of selected primer sets. Sequences are listed in Materials and Methods. B–G, qRT-PCR analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 mice per group. Experiments run in experimental triplicate with the average of results used for analysis. Quantifications were normalized to GAPDH (COXI6520 to GAPDH_Neuron, while COXI6620, CYTB, ND4, and Dloop3' were normalized to GAPDH_PNAS) to normalize variance across groups and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. SDs did not significantly differ per Brown–Forsythe test. Significance of means analyzed via ordinary one-way ANOVA (B: F(3,26) = 11.33 ****p 0.00001; C: F(3,26) = 7.042 **p = 0.0013; D: F(3,26) = 5.130 **p = 0.0064; E: F(3,26) = 1.831 p = 0.1663). Post hoc Tukey's test resulted in B: ****p 0.00001 for wild-type versus PolgAD257A/D257A, **p = 0.0033 for wild-type versus Parkin–/–/PolgAD257A/D257A, and *p = 0.0236 for Parkin–/– versus PolgAD257A/D257A; C: **p = 0.0011 for wild-type versus PolgAD257A/D257A, and *p = 0.0205 for wild-type versus Parkin–/–/PolgAD257A/D257A; D: **p = 00.37 for wild-type versus PolgAD257A/D257A, ns, not significant. F, qRT-PCR analysis of Dloop of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 mice per group. Experiments run in experimental triplicate with the average of results used for analysis. Quantifications were normalized to GAPDH (GAPDH_PNAS primer set) to normalize variance across groups and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. SDs did significantly differ per Brown–Forsythe test. Significance of means were analyzed via a Welch one-way ANOVA W(3.0,13.01) = 14.24 ***p = 0.0002. Post hoc Dunnett's T3 multiple comparisons test resulted in *p = 0.0237 for wild-type versus PolgAD257A/D257A, *p = 0.0142 for wild-type versus Parkin–/–/PolgAD257A/D257A, *p = 0.0159 for Parkin–/– versus PolgAD257A/D257A, and **p = 0.0088 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. G, qRT-PCR analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 mice per group. Experiments run in experimental triplicate with the average of results used for analysis. Quantifications were normalized to GAPDH_PNAS to normalize variance across groups and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were analyzed using the mixed effects model (REML) with matching factors across rows Fc(3,80) = 2.660, Fr(3,80) = 27.20 ****p 0.00001, Fi(9,80) = 10.64 ****p 0.00001. Post hoc Tukey's test resulted in ****p 0.00001 for Dloop3' amplification for wild-type versus PolgAD257A/D257A, wild-type versus Parkin–/–/PolgAD257A/D257A, Parkin–/– versus PolgAD257A/D257A, and Parkin–/– versus Parkin–/–/PolgAD257A/D257A, ns, not significant. Analysis and graphs were produced using GraphPad Prism 8.4.3.
Because of the above results, we further analyzed each mtDNA loci individually via one-way ANOVAs as previously described (Pickrell et al., 2015). We found these results to be loci-dependent. Both primer sets to COXI exhibited significant reductions in mtDNA copy number in PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A mice, implying decreased mitochondrial mass in these cohorts (Fig. 6B,C). Although the level of statistical significance varied between COXI primer sets, these results are similar to mtDNA copy number analysis previously reported (Pickrell et al., 2015). Contrarily, there appeared to be no differences between mouse cohorts using primers specific to CYTB (Fig. 6D). There was a substantial decrease in the amplification of ND4 in PolgAD257A/D257A mice, but not Parkin–/–/PolgAD257A/D257A mice (Fig. 6E). Moreover, one-way ANOVA analysis further confirmed the increase in Dloop copy number in PolgAD257A/D257A mice and Parkin–/–/PolgAD257A/D257A mice (Fig. 6F). It should be noted that loss of Parkin did not lead to further reductions in mtDNA copy number, regardless of the gene loci tested. Taken together, these results suggest that quantifying mtDNA copy number via mtDNA amplification may not be a sufficient method to investigate mitochondrial mass in mouse lines that accumulate mtDNA mutations.
Discussion
In this study, we originally set out to expand on previous work which reported dopaminergic neurodegeneration and corresponding nigral-striatal dependent motor dysfunction in Parkin–/–/PolgAD257A/D257A mice (Pickrell et al., 2015). We planned to investigate PARIS (Parkin Interacting Substrate, ZNF746) expression and mitochondrial biogenesis dysfunction in this model, with the goal to understand the relative contributions of mitochondrial quality control pathways in PD pathology. Using a variety of behavioral tests, we observed substantial motor and muscular deficits in both PolgAD257A/D257A and Parkin–/–/PolgAD257A/D257A mice. These deficits were seen at six- and nine-month times with peak dysfunction found at 12 months. Behavioral testing concluded at 12 months as PolgAD257A/D257A mice die prematurely around 13 months of age (Dai et al., 2013; Perier et al., 2013). Locomotor deficits as assessed by openfield were progressive overtime, while deficits in grip strength appeared to fluctuate between timepoints. We speculate these changes in grip strength may be sensitive not only to loss of muscle mass seen in PolgAD257A/D257A-expressing mice, as previously reported (Trifunovic et al., 2004; Kujoth et al., 2005; Hiona et al., 2010; Safdar et al., 2011), but also to changes in body weight, as raw force readings were normalized to total body weight, and both PolgAD257A/D257A and Parkin–/–/PolgAD257A/D257A cohorts lost muscular mass and body weight overtime. Notably, these deficits were not worsened by the loss of Parkin. Finally, there were no deficits observed via the pole test in 12-month-old mice, demonstrating that behavioral deficits were nonspecific to the nigrostriatal pathway.
The major finding of this paper was that the absence of Parkin in PolgAD257A/D257A mice did not lead to the loss of DA neurons in the SNpc. Immunohistochemical analysis specific to TH+ neurons revealed no loss in the dopaminergic neuronal population of ventral midbrain tissue, and further HPLC assessment found no changes in striatal dopamine. We saw significant reductions in dopamine and serotonin turnover in all PolgAD257A/D257A mice -expressing lines. There was also a decrease in catecholamine concentrations in the olfactory bulb of all mice with the PolgAD257A/D257A mutation; however, loss of Parkin did not lead to further reductions in catecholamine content or turnover. Taken together, these data suggest that there is catecholamine and indolamine dysregulation as a result of the accumulation of mtDNA mutations in PolgAD257A/D257A mice and that loss of Parkin does not enhance this dysregulation.
We also failed to see any substantial changes in expression levels in several mitochondrial markers, including nuclear-encoded subunits of COXI-III. There was, however, a significant reduction in a mitochondrial-encoded subunit of COXIV, likely because of the accumulation of mtDNA indels. Notably, this reduction was not further enhanced by the absence of Parkin. We also failed to see statistically significant changes in Complex I and Complex IV activity between wild-type and PolgAD257A/D257A-expressing mice. This suggests that although there was a reduction in Complex IV expression in PolgAD257A/D257A-expressing mice, the activity of Complex IV did not decrease. In addition, the PolgA mutation did not seem to affect Parkin function, as there was no change in the steady state levels of ubiquitylation substrates of Parkin, including VDAC, AIMP2 and NLRP3 (Y. Lee et al., 2013; Ordureau et al., 2018; Panicker et al., 2022). Furthermore, we observed no changes in expression level of parkin substrates in Parkin–/– and Parkin–/–; PolgD257A/D257A, further providing evidence for the augmentation of developmental compensatory pathways in Parkin–/– mice.
Using a mixed effects model, mtDNA copy number analysis revealed no significant differences in COXI, CYTB, or ND4 copy number between groups. However, there was a significant accumulation of Dloop, the highly conserved origin of replication of mtDNA, in mice with PolgAD257A/D257A knock-in. Individual one-way ANOVA analysis revealed inconsistent results between loci; however, the Dloop copy number remained significantly increased in all PolgAD257A/D257A-expressing mice. This suggests that the conserved Dloop region is significantly less prone to mutations compared with other the other genes tested. As such, the Dloop region amplifies appropriately, while other less conserved regions with mtDNA mutations do not. Alternatively, one could postulate that the PolgAD257A/D257A knock-in may promote the accumulation of advantageous mutations in the Dloop region, which in turn enhances primer annealing and consequential amplification. This may be beneficial to cells as they try to increase replication of mitochondria to combat severe mitochondria dysfunction. This finding is supported by prior work that suggested that accumulation of mitochondrial deletions SNpc dopaminergic neurons in PolgAD257A/D257A mice trigger a compensatory mechanism that include augmenting mtDNA copy number (Perier et al., 2013). Although these data further confirm mitochondrial dysfunction as a result of the accumulation mtDNA mutations because of the PolgAD257A knock-in, they imply that loss of endogenous Parkin in does not synergize mitochondrial dysfunction induced in PolgAD257A/D257A mice.
Studies using germline Parkin knock-out mice have failed to see neurodegeneration, implying that pathways in development likely compensate for its loss and impair the DA neuron degeneration (Fleming et al., 2005). Consistent with this notion are studies that show DA neuronal loss and neurobehavioral deficits when Parkin is conditionally knocked out in adult animals (Shin et al., 2011; Stevens et al., 2015; Y. Lee et al., 2017; Brahmachari et al., 2019; Jo et al., 2021; Panicker et al., 2022; Pirooznia et al., 2022). Because there has been a lack of an obvious Parkinsonian phenotype in germline Parkin null mice, there have been attempts to manipulate other genes in addition to Parkin to promote neurodegeneration. Because of the role of Parkin in mitochondrial quality control, there are a fair number of studies, listed below, in which Parkin–/– mice have been crossed to different mouse models of mitochondrial dysfunction. Like our results, the absence of parkin did not enhance the loss of DA neurons and progressive neurodegeneration of the MitoPark mice (Sterky et al., 2011). PD pathology was not worse in PD-mito-PstI mice, whose mtDNA undergoes double-strand breaks in dopaminergic neurons (Pinto et al., 2018). Crossing parkin KO mice to DJ-1, another autosomal recessive PD gene that affects mitochondrial function (Andres-Mateos et al., 2007) failed to cause a loss of DA neurons. The combined absence of parkin, PINK1 and DJ-1 did not lead to loss of DA neurons (Kitada et al., 2009). Studies in Drosophila in which mito-APOBEC1 flies, a model of mitochondrial dysfunction and motor deficits driven by an accumulation of mtDNA mutations in somatic tissues, failed to show an enhancement of mitochondrial dysfunction or worsening of motor deficits on loss of Parkin (J.J. Lee et al., 2020). A study analyzing the effect of DJ-1 found that DJ1−/−/PolgAD257A/D257A mice also failed to synergize with PolgAD257A as there was no dopaminergic neurodegeneration or nigral-striatal-dependent motor or enhanced mitochondrial deficits (Hauser et al., 2015).
What might account for the differences between the results presented here and other reports that indicate that mitochondrial dysfunction does not synergize with loss of parkin as reported by (Pickrell et al., 2015)? There are many factors to consider when evaluating biochemical and behavioral differences in Parkin–/–/PolgAD257A/D257A mice between studies. Animal housing may be a factor, as environment and diet could impact metabolism and gut microbiota, which in turn may affect brain function (Sudo, 2019). In this report we used a Parkin–/– line in which the first RING finger domain, encoded by exon 7, is deleted, leading to a catalytically null mutant line (Von Coelln et al., 2004). Because this line was previously reported to display noradrenergic neurodegeneration in the locus coeruleus, we hypothesized the additional mitochondrial dysfunction would surely synergize to induce dopaminergic neuronal loss in the SNpc. Pickrell et al. (2015) used a Parkin–/– line in which exon 3 is replaced with EGFP coding sequence, inducing a premature stop codon (Goldberg et al., 2003). This line has no reported dopaminergic neuronal degeneration. Regardless of this difference, we do not believe exon 7 deletion versus exon 3 skipping had a true effect on the study considering the other reports that mice with mitochondrial dysfunction do not synergize with the absence of parkin. Finally, it is important to consider the potential effects of an underpowered study. Our group was sure to sufficiently power our study to avoid a type 2 statistical error.
In conclusion, our results add to the bulk of work which reports germline Parkin knock-out mice do not display neurodegeneration of SNpc dopaminergic neurons. Furthermore, it contributes to findings that report a lack of synergism of Parkin loss on mitochondrial dysfunction in mouse models of mitochondrial deficits. A significant finding in this study is the use of a field accepted method to measure mitochondrial mass. This method relies on the quantification of mtDNA copy number at a DNA level. Here, we show conflicting results of mtDNA quantification. These results appear to be loci dependent, and we hypothesize that the accumulation of mtDNA mutations in PolgAD257A/D257A mice directly affect the ability of primers to anneal and amplify appropriately. Consequently, extreme care and consideration should be taken when analyzing mitochondrial mass in in vivo models of mtDNA mutations.
Parkin substrates. A, B, Representative Western blottings of parkin substrates. C, D, Quantification of Western blot parkin substrate expression analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 mice per group. Experiments were run in experimental triplicate with the average of results used for analysis. ImageJ was used for optical density analysis. Quantifications were normalized to beta-actin to account for loading differences and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via Shapiro–Wilk test. SDs were not found to differ significantly per Brown–Forsythe test. Significance of means were analyzed via an ordinary one-way ANOVA test (Extended Data Fig. 1-5C: F(3,20) = 1.350 p = 0.2867; Extended Data Fig. 1-5D: F(3,20) = 1.440 p = 0.2609). Post hoc Tukey's multiple comparisons test resulted in no significant differences between groups. Analysis and graphs were produced using GraphPad Prism 9.4.1. Download Figure 5-2, TIF file (3.4MB, tif) .
Representative uncropped Western blottings of mitochondrial markers. Nonspecific bands at 50 kDa (denoted in panel B by yellow arrow) were seen using Anti-NDUFA9. This banding pattern matched blots published on the manufacturer's website. To avoid interference/saturation issues from these nonspecific bands, blots probing for NDUFA9 were cut at 45–50 kDa. Download Figure 5-1, TIF file (11.4MB, tif) .
Evaluating for sex differences in 12-month behavioral studies. A, Latency values of pole test of 12-month-old mice. Results are the mean ± SEM, n = 7–15 per group with an average of five trials per mouse used for analysis. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via two-way ANOVA (Fi(43,74) = 0.7162, p = 0.5454). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within groups or between groups. B, C, Forelimb and total limb strength (in gram*force) normalized to body weight (grams). Results are the mean ± SEM, n = 5–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via a two-way ANOVA (Fi(3,69) = 2.327). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. D, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via a two-way ANOVA (Fi(3,76) = 2.448 p = 0.0701). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. E, Average speed of locomotion during the 15-min testing period 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means was analyzed via two-way ANOVA (Fi(3,77) = 1.036 p = 0.3814). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. F, Total resting time, defined as a period of four or more seconds with no photobeam breaks, in 15-min test period for 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means was analyzed via a two-way ANOVA (Fi(3,77) = 1.286 p = 0.2853). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. Analysis and graphs were produced using GraphPad Prism 9.4.1. Download Figure 2-3, TIF file (2.3MB, tif) .
Three-, six-, nine-month Openfield data. A, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for three-month-old mice. Results are the mean ± SEM, n = 15–20 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (W(3.0,45.27) = 2.338 **p = 0.0069). Post hoc Dunnett's T3 test resulted in no significant difference between groups. B, C, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for six- and nine-month-old mice. Results are the mean ± SEM, n = 19–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1B: F(3,87) = 7.665 ****p 0.0001; Extended Data Fig. 1-1C: F(3,87) = 13.94 ****p 0.0001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1B: *p = 0.0456 for wild type versus Parkin–/–/PolgAD257A/D257A, *p = 0.0148 for Parkin–/– versus PolgAD257A/D257A; ****p < 0.00001 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1C: ****p 0.0001 wild type versus PolgAD257A/D257A and wild type versus Parkin–/–/PolgAD257A/D257A, ***p = 0.003 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A and Parkin–/– versus PolgAD257A/D257A. D, F, Average speed of locomotion during 15-min test period for three-, six-, and nine-month-old mice. Results are the mean ± SEM, n = 15–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1D: F(3,68) = 3.818 *p = 0.0137; Extended Data Fig. 1-1E: F(3,88) = 4.632 **p = 0.0047; and Extended Data Fig. 1-1F: F(3,87) = 13.14 ****p 0.0001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1D: *p = 0.0189 for Parkin–/– versus PolgAD257A/D257A; Extended Data Figure 1-1E: *p = 0.0297 for Parkin–/– versus PolgAD257A/D257A, **p = 0.0033 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1F: ***p = 0.0001 for wild type versus PolgAD257A/D257A, ***p = 0.0007 for wild type versus Parkin–/–/PolgAD257A/D257A, ***p = 0.0002 for Parkin–/– versus PolgAD257A/D257A, and ****p 0.0001 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A. G–I, Total resting time, defined as a period of four or more seconds with no photobeam breaks, in 15-min test period for three-, six-, and nine-month-old mice. Results are the mean ± SEM, n = 15–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1G: F(3,67) = 8.461 p = 0.4735; Extended Data Fig. 1-1H: F(3,88) = 4.526 **p = 0.0053; Extended Data Fig. 1-1I: F(3,87) = 4.526 ****p < 0.00001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1H: *p = 0.0312 for wild type versus Parkin–/–/PolgAD257A/D257A, **p = 0.0054 Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1I: ***p = 0.0001 wild type versus -PolgAD257A/D257A, ***p = 0.0002 wild type versus Parkin–/–/PolgAD257A/D257A and Parkin–/– versus PolgAD257A/D257A, and ***p = 0.0003 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A. Analysis and graphs were produced using GraphPad Prism 8.4.3. Download Figure 2-2, TIF file (2.5MB, tif) .
Three-, six-, nine-month grip strength data. A, Forelimb strength (in gram*force) normalized to body weight (grams) for three-month-old mice. Results are the median ± SEM, n = 16–23 per group. Data were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data was found to be non-normally distributed per D'Agostino and Pearson test (2 out of 4 groups were non-normally distributed). Significance of medians was analyzed via Kruskal–Wallis nonparametric ANOVA (KW(4,77) = 8.878, *p = 0.0310). Post hoc Kruskal–Wallis test for multiple comparisons results in *p = 0.0226 for wild type versus Parkin–/–. B, C, Forelimb strength (in gram*force) normalized to body weight (grams) for six- and nine-month-old mice. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-2B: F(3,85) = 4.575 **p = 0.0051 and Extended Data Fig. 1-2C: F(3,85) = 4.575 **p = 0.0051). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-2B: *p = 0.0482 for wild type versus PolgAD257A/D257A and **p = 0.0028 for wild type versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-2C: *p = 0.0130 for wild type versus Parkin–/–. D–F, Total limb strength (in gram*force) normalized to body weight (grams). Results are the mean ± SEM, n = 11–24 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. Brown–Forsythe test of variance found no difference in SDs. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-2D: F(3,73) = 5.990 **p = 0.0010; Extended Data Fig. 1-2E: F(3,85) = 3.948 *p = 0.0109; Extended Data Fig. 1-2F: F(5,65) = 3.747 *p = 0.0151). Post hoc Tukey's test was performed for total limb strength and resulted in Extended Data Figure 1-1D: ***p = 0.0005 for wild type versus Parkin–/–; Extended Data Figure 1-1E: **p = 0.0096 for wild type versus Parkin–/–, *p = 0.0358 for wild type versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1F: *p = 0.0110 for wild type versus Parkin–/–/PolgAD257A/D257A. Analysis and graphs were produced using GraphPad Prism 8.4.3. Download Figure 2-1, TIF file (2MB, tif) .
Footnotes
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant P50 NS38377 and the JPB Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. V.L.D. and T.M.D. acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program M-2014, M-2016. This work was also supported by the National Science Foundation under Grant 2017238665 (to L.S.).
The authors declare no competing financial interests.
References
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL (2007) DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A 104:14807–14812. 10.1073/pnas.0703219104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V (2014) Genetics of Parkinson's disease–state of the art, 2013. Parkinsonism Relat Disord 20 [Suppl 1]:S23–S28. 10.1016/S1353-8020(13)70009-9 [DOI] [PubMed] [Google Scholar]
- Brahmachari S, et al. (2019) Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson's disease. Brain 142:2380–2401. 10.1093/brain/awz172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnay Y, Léger L (2010) Brain serotonergic circuitries. Dialogues Clin Neurosci 12:471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Kiselak T, Clark J, Clore E, Zheng K, Cheng A, Kujoth GC, Prolla TA, Maratos-Flier E, Simon DK (2013) Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion 13:282–291. 10.1016/j.mito.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Fernagut PO, Chesselet MF (2005) Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx 2:495–503. 10.1602/neurorx.2.3.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge P, Dawson VL, Dawson TM (2020) PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener 15:20. 10.1186/s13024-020-00367-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635. 10.1074/jbc.M308947200 [DOI] [PubMed] [Google Scholar]
- Grenier K, McLelland GL, Fon EA (2013) Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front Neurol 4:100. 10.3389/fneur.2013.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser DN, Primiani CT, Langston RG, Kumaran R, Cookson MR (2015) The Polg mutator phenotype does not cause dopaminergic neurodegeneration in DJ-1-deficient mice. eNeuro 2:ENEURO.0075-14.2015. 10.1523/ENEURO.0075-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S, Barger JL, Portero-Otín M, Tanokura M, Prolla TA, Leeuwenburgh C (2010) Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5:e11468. 10.1371/journal.pone.0011468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo A, et al. (2021) PARIS farnesylation prevents neurodegeneration in models of Parkinson's disease. Sci Transl Med 13:eaax8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- Kitada T, Tong Y, Gautier CA, Shen J (2009) Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem 111:696–702. 10.1111/j.1471-4159.2009.06350.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484. 10.1126/science.1112125 [DOI] [PubMed] [Google Scholar]
- Lee JJ, Andreazza S, Whitworth AJ (2020) The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci Rep 10:2693. 10.1038/s41598-020-59647-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D, Jiang H, Kang SU, Lee BD, Kang HC, Kim D, Tessarollo L, Dawson VL, Dawson TM (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci 16:1392–1400. 10.1038/nn.3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, et al. (2017) PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep 18:918–932. 10.1016/j.celrep.2016.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Czajka A (2013) Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 13:481–492. 10.1016/j.mito.2012.10.011 [DOI] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM (1999) Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A 96:5774–5779. 10.1073/pnas.96.10.5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Kabuto H, Makino H, Ogawa N (1997) Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods 73:45–48. 10.1016/s0165-0270(96)02211-x [DOI] [PubMed] [Google Scholar]
- Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y (1985) A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol 50:435–441. [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang W, Ahfeldt T, Zhang J, Cohn EF, Hou Z, Heo JM, Rubin LL, Sidhu SS, Gygi SP, Harper JW (2018) Dynamics of PARKIN-dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol Cell 70:211–227.e8. 10.1016/j.molcel.2018.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker N, Ge P, Dawson VL, Dawson TM (2021) The cell biology of Parkinson's disease. J Cell Biol 220:e202012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker N, Kam TI, Wang H, Neifert S, Chou SC, Kumar M, Brahmachari S, Jhaldiyal A, Hinkle JT, Akkentli F, Mao X, Xu E, Karuppagounder SS, Hsu ET, Kang SU, Pletnikova O, Troncoso J, Dawson VL, Dawson TM (2022) Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson's disease. Neuron 110:2422–2437.e9. 10.1016/j.neuron.2022.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD (2005) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A 102:2174–2179. 10.1073/pnas.0409598102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, Bender A, García-Arumí E, Melià MJ, Bové J, Laub C, Klopstock T, Elstner M, Mounsey RB, Teismann P, Prolla T, Andreu AL, Vila M (2013) Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 136:2369–2378. 10.1093/brain/awt196 [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87:371–381. 10.1016/j.neuron.2015.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto M, Nissanka N, Moraes CT (2018) Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson's disease. J Neurosci 38:1042–1053. 10.1523/JNEUROSCI.1384-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia SK, Yuan C, Khan MR, Karuppagounder SS, Wang L, Xiong Y, Kang SU, Lee Y, Dawson VL, Dawson TM (2020) PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener 15:17. 10.1186/s13024-020-00363-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia SK, Wang H, Panicker N, Kumar M, Neifert S, Dar MA, Lau E, Kang BG, Redding-Ochoa J, Troncoso JC, Dawson VL, Dawson TM (2022) Deubiquitinase CYLD acts as a negative regulator of dopamine neuron survival in Parkinson's disease. Sci Adv 8:eabh1824. 10.1126/sciadv.abh1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JM, Stewart JB, Hagström E, Brené S, Mourier A, Coppotelli G, Freyer C, Lagouge M, Hoffer BJ, Olson L, Larsson NG (2013) Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501:412–415. 10.1038/nature12474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA (2011) Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A 108:4135–4140. 10.1073/pnas.1019581108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144:689–702. 10.1016/j.cell.2011.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A 108:12937–12942. 10.1073/pnas.1103295108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM (2015) Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A 112:11696–11701. 10.1073/pnas.1500624112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo N (2019) Role of gut microbiota in brain function and stress-related pathology. Biosci Microbiota Food Health 38:75–80. 10.12938/bmfh.19-006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. Methods Mol Biol 445:77–88. 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423. 10.1038/nature02517 [DOI] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A 101:10744–10749. 10.1073/pnas.0401297101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Parkin substrates. A, B, Representative Western blottings of parkin substrates. C, D, Quantification of Western blot parkin substrate expression analysis of ventral midbrain tissue of 12-month-old mice. Results are the mean ± SEM, n = 6 mice per group. Experiments were run in experimental triplicate with the average of results used for analysis. ImageJ was used for optical density analysis. Quantifications were normalized to beta-actin to account for loading differences and then to wild-type values to normalize against batch effect. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via Shapiro–Wilk test. SDs were not found to differ significantly per Brown–Forsythe test. Significance of means were analyzed via an ordinary one-way ANOVA test (Extended Data Fig. 1-5C: F(3,20) = 1.350 p = 0.2867; Extended Data Fig. 1-5D: F(3,20) = 1.440 p = 0.2609). Post hoc Tukey's multiple comparisons test resulted in no significant differences between groups. Analysis and graphs were produced using GraphPad Prism 9.4.1. Download Figure 5-2, TIF file (3.4MB, tif) .
Representative uncropped Western blottings of mitochondrial markers. Nonspecific bands at 50 kDa (denoted in panel B by yellow arrow) were seen using Anti-NDUFA9. This banding pattern matched blots published on the manufacturer's website. To avoid interference/saturation issues from these nonspecific bands, blots probing for NDUFA9 were cut at 45–50 kDa. Download Figure 5-1, TIF file (11.4MB, tif) .
Evaluating for sex differences in 12-month behavioral studies. A, Latency values of pole test of 12-month-old mice. Results are the mean ± SEM, n = 7–15 per group with an average of five trials per mouse used for analysis. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via two-way ANOVA (Fi(43,74) = 0.7162, p = 0.5454). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within groups or between groups. B, C, Forelimb and total limb strength (in gram*force) normalized to body weight (grams). Results are the mean ± SEM, n = 5–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via a two-way ANOVA (Fi(3,69) = 2.327). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. D, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means were analyzed via a two-way ANOVA (Fi(3,76) = 2.448 p = 0.0701). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. E, Average speed of locomotion during the 15-min testing period 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means was analyzed via two-way ANOVA (Fi(3,77) = 1.036 p = 0.3814). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. F, Total resting time, defined as a period of four or more seconds with no photobeam breaks, in 15-min test period for 12-month-old mice. Results are the mean ± SEM, n = 8–15 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Significance of means was analyzed via a two-way ANOVA (Fi(3,77) = 1.286 p = 0.2853). Post hoc Tukey's multiple comparisons test resulted in no significant differences between sexes within each phenotype. Analysis and graphs were produced using GraphPad Prism 9.4.1. Download Figure 2-3, TIF file (2.3MB, tif) .
Three-, six-, nine-month Openfield data. A, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for three-month-old mice. Results are the mean ± SEM, n = 15–20 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were found to differ significantly per Brown–Forsythe test of variance. Therefore, significance of means was analyzed via a Welch one-way ANOVA (W(3.0,45.27) = 2.338 **p = 0.0069). Post hoc Dunnett's T3 test resulted in no significant difference between groups. B, C, Openfield data analyzing total distance traveled in the xy direction during the 15-min testing period for six- and nine-month-old mice. Results are the mean ± SEM, n = 19–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1B: F(3,87) = 7.665 ****p 0.0001; Extended Data Fig. 1-1C: F(3,87) = 13.94 ****p 0.0001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1B: *p = 0.0456 for wild type versus Parkin–/–/PolgAD257A/D257A, *p = 0.0148 for Parkin–/– versus PolgAD257A/D257A; ****p < 0.00001 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1C: ****p 0.0001 wild type versus PolgAD257A/D257A and wild type versus Parkin–/–/PolgAD257A/D257A, ***p = 0.003 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A and Parkin–/– versus PolgAD257A/D257A. D, F, Average speed of locomotion during 15-min test period for three-, six-, and nine-month-old mice. Results are the mean ± SEM, n = 15–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1D: F(3,68) = 3.818 *p = 0.0137; Extended Data Fig. 1-1E: F(3,88) = 4.632 **p = 0.0047; and Extended Data Fig. 1-1F: F(3,87) = 13.14 ****p 0.0001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1D: *p = 0.0189 for Parkin–/– versus PolgAD257A/D257A; Extended Data Figure 1-1E: *p = 0.0297 for Parkin–/– versus PolgAD257A/D257A, **p = 0.0033 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1F: ***p = 0.0001 for wild type versus PolgAD257A/D257A, ***p = 0.0007 for wild type versus Parkin–/–/PolgAD257A/D257A, ***p = 0.0002 for Parkin–/– versus PolgAD257A/D257A, and ****p 0.0001 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A. G–I, Total resting time, defined as a period of four or more seconds with no photobeam breaks, in 15-min test period for three-, six-, and nine-month-old mice. Results are the mean ± SEM, n = 15–26 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-1G: F(3,67) = 8.461 p = 0.4735; Extended Data Fig. 1-1H: F(3,88) = 4.526 **p = 0.0053; Extended Data Fig. 1-1I: F(3,87) = 4.526 ****p < 0.00001). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-1H: *p = 0.0312 for wild type versus Parkin–/–/PolgAD257A/D257A, **p = 0.0054 Parkin–/– versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1I: ***p = 0.0001 wild type versus -PolgAD257A/D257A, ***p = 0.0002 wild type versus Parkin–/–/PolgAD257A/D257A and Parkin–/– versus PolgAD257A/D257A, and ***p = 0.0003 for Parkin–/– versus Parkin–/–/PolgAD257A/D257A. Analysis and graphs were produced using GraphPad Prism 8.4.3. Download Figure 2-2, TIF file (2.5MB, tif) .
Three-, six-, nine-month grip strength data. A, Forelimb strength (in gram*force) normalized to body weight (grams) for three-month-old mice. Results are the median ± SEM, n = 16–23 per group. Data were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data was found to be non-normally distributed per D'Agostino and Pearson test (2 out of 4 groups were non-normally distributed). Significance of medians was analyzed via Kruskal–Wallis nonparametric ANOVA (KW(4,77) = 8.878, *p = 0.0310). Post hoc Kruskal–Wallis test for multiple comparisons results in *p = 0.0226 for wild type versus Parkin–/–. B, C, Forelimb strength (in gram*force) normalized to body weight (grams) for six- and nine-month-old mice. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. SDs were not found to differ significantly. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-2B: F(3,85) = 4.575 **p = 0.0051 and Extended Data Fig. 1-2C: F(3,85) = 4.575 **p = 0.0051). Post hoc Tukey's test was performed for and resulted in Extended Data Figure 1-2B: *p = 0.0482 for wild type versus PolgAD257A/D257A and **p = 0.0028 for wild type versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-2C: *p = 0.0130 for wild type versus Parkin–/–. D–F, Total limb strength (in gram*force) normalized to body weight (grams). Results are the mean ± SEM, n = 11–24 per group. Datasets were unbiasedly analyzed using ROUT outlier analysis with a maximum false discovery rate (q) of 0.1%. Data were found to be normally distributed via D'Agostino and Pearson test. Brown–Forsythe test of variance found no difference in SDs. Significance of means was analyzed via an ordinary one-way ANOVA (Extended Data Fig. 1-2D: F(3,73) = 5.990 **p = 0.0010; Extended Data Fig. 1-2E: F(3,85) = 3.948 *p = 0.0109; Extended Data Fig. 1-2F: F(5,65) = 3.747 *p = 0.0151). Post hoc Tukey's test was performed for total limb strength and resulted in Extended Data Figure 1-1D: ***p = 0.0005 for wild type versus Parkin–/–; Extended Data Figure 1-1E: **p = 0.0096 for wild type versus Parkin–/–, *p = 0.0358 for wild type versus Parkin–/–/PolgAD257A/D257A; Extended Data Figure 1-1F: *p = 0.0110 for wild type versus Parkin–/–/PolgAD257A/D257A. Analysis and graphs were produced using GraphPad Prism 8.4.3. Download Figure 2-1, TIF file (2MB, tif) .