

Abstract
Tregs are the immune system’s in-house combatants against pathological immune activation. Because they are vital to maintenance of peripheral tolerance, it is important to understand how they perform their functions. To this end, various mechanisms have been proposed for Treg-mediated immune inhibition. A major group of mechanisms picture Tregs as skilled thieves stealing a plethora of molecules that would otherwise promote immune effector functions. This suggests that several million years of evolution have endowed Tregs with efficient ways to deprive immune effectors of activating stimuli to prevent immunopathology for survival of the host. Although we are still long way from deciphering their complete set of tricks, this review will focus on the types of “crimes” committed by these master thieves in both secondary lymphoid organs and non-lymphoid tissue.
Introduction
Regulatory T cells play a nonredundant role in homeostasis via suppressing aberrant immune responses. The concept of immunotolerance evolved early in the metazoans coincidingly with immunity itself probably in the form of a simple discrimination of self from non-self using pattern recognition. In later metazoans however, compartmentalization of cells with similar functions into tissues and exposure of different barrier tissues to a uniquely diverse array of antigens rendered the simple binary system for self-discrimination insufficient. A more complex system required a set of changes in immune system by introducing mutations for randomly rearranging genes that encode antigen receptors1. Despite maximizing specificity of immune recognition, this random process came with a cost of self-reactivity that had to be counterbalanced via mechanisms such as clonal deletion in the primary lymphoid organs. Not surprisingly, central elimination of self-reactive cells does not completely prevent unwanted immune responses against innocuous foreign antigens such as those in the diet and or derived from commensals that are introduced only at the barrier sites after birth. Additionally, central deletion of autoreactive lymphocytes is not fully efficient, therefore some self-reactive lymphocytes inevitably escape to the periphery. These weaknesses were gradually counterbalanced by developing immunoregulatory pathways via natural selection. Foxp3 orthologs first appeared in jawed vertebrates and displayed ability to bind RORγt, supposedly to tolerize against commensal and dietary antigens2. However, fully functional Foxp3 with a proline rich region emerged in mammals possibly as a result of the need for protecting the fetus from the maternal immune system. Thus, regulatory T cells first emerged in placental mammals. However, it is worth noting that although a Foxp3 ortholog has not been identified in birds, CD4+ T cell subsets with high expression of IL-2Rα have in vitro suppressor activity2,3. One probable explanation for the evolution of Tregs as a distinct cellular component is the need for specialized immunoregulatory cells to detect and suppress aberrant immune responses against self-antigens, fetal antigens and innocuous foreign antigens deposited at barrier tissues. An alternative view is that Tregs, while favoring the recognition of self-antigens, can recognize pathogen-derived antigens and have evolved to protect the host against exuberant responses to pathogens. All placental mammals have Tregs and lack of Tregs result in devastating autoimmunity. Therefore, it is important to define and fully understand the immune inhibitory mechanisms used by Tregs.
Tregs constitute 5–10% of the peripheral CD4+ T cell pool with a unique transcriptional program controlled mainly by the transcription factor, Foxp3. Foxp3 is responsible for the stability and function of Tregs and congenital defects in Foxp3 expression cause a multiorgan autoimmunity syndrome called IPEX (immune dysregulation, polyendocrinopathy, enteropathy, and X-linked) in humans and a wasting disease in mice4–6. Similarly, strategies for chronic ablation of Foxp3 expressing cells in adult mice result in death due to severe immunopathology7. Tregs reside in and recirculate between lymphoid organs and non-lymphoid sites. Recently, they were found capable of adapting their transcriptional program to various different non-lymphoid niches such as skin, lamina propria of small and large intestine, liver, lung, adipose tissue, heart, skeletal muscle, and the tumor microenvironment8–13. In non-lymphoid sites, Tregs play tissue-specific immunoregulatory roles and help wound healing. As Tregs can be generated in both the thymus and peripheral lymphoid tissues, the heterogeneous make-up of the Treg population is a result both of their ability to be generated in the thymus (tTreg) and periphery (pTreg) as well as their capacity to adopt some of the transcriptional programs of T effector cells resident in non-lymphoid sites
Studies over the past two decades have resulted in the identification of more than a dozen Treg suppression mechanisms14,15. However, we still lack a clear explanation for the specific mechanisms used in in vivo partly because it has been difficult to translate from highly reductionist in vitro settings to the complex wiring of immune cells in vivo. Furthermore, one mechanism does not have to be mutually exclusive to another, and several mechanisms may work in unique combinations depending on the anatomic location and type of the immune response. Here, we will review the immunoinhibitory mechanisms used by Tregs and have classified these mechanisms into two categories (Fig. 1): “active” such as the production of suppressor cytokines and functioning as killer cells and “counter-active” including mechanisms where they have evolved to steal drivers of the immune response such as antigen, costimulatory molecules, cytokines, and inflammatory mediators.
Figure 1.
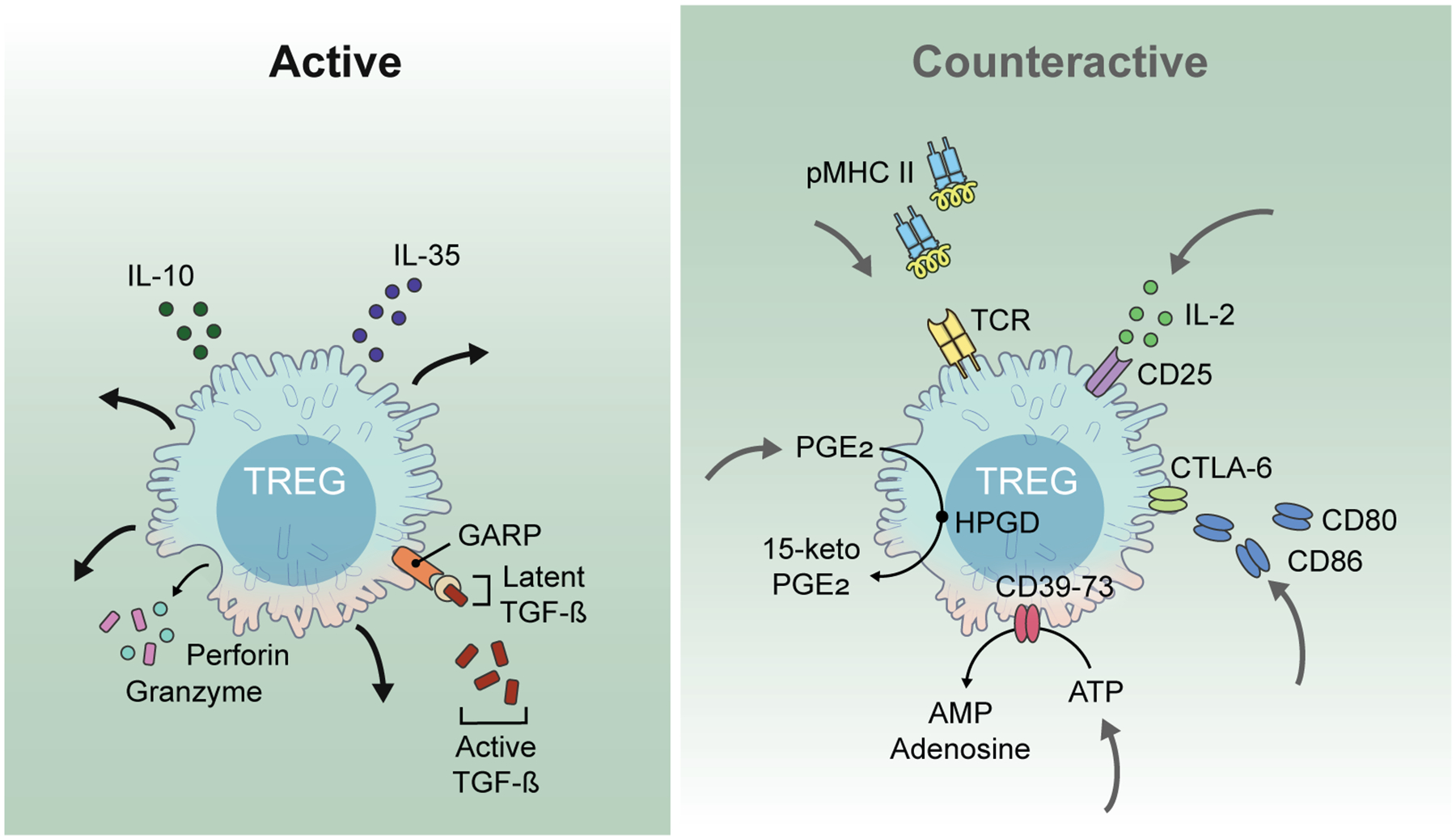
Two fundamentally different suppressor modes characterize Treg suppressor mechanisms. In the “active” mode, Treg secrete immunoinhibitory molecules that exert their effects on other cell types. In contrast, in the “counteractive” mode Treg are actively engaged in removing vital components from other cells types including antigen, costimulatory molecules, cytokines, and inflammatory signals thereby decreasing the activation of T effector cells.
Active Suppression Mechanisms Used by Tregs
Tregs have the ability to produce and utilize cytokines that have immunoregulatory functions. Most prominent immunoregulatory cytokines actively produced by Tregs are IL-10, TGF-β and IL-35. Tregs can also function as classical cytotoxic T cells by producing cytotoxic molecules such as granzyme and perforin resulting in target cell lysis in a contact-dependent manner in limited in vivo settings.
IL-10 is a pleiotropic cytokine produced by leukocytes, epithelial cells and keratinocytes. The heterotetrameric receptor for IL-10 is comprised of a ligand binding IL-10R1 homodimer and signaling IL-10R2 homodimer and is expressed on all the hematopoietic cells. While IL-10 can inhibit phagocyte function, antigen presentation, co-stimulatory molecule expression, T cell proliferation, IL-2 and IFNγ production, it can also promote NK cell activity, B cell activation and isotype switching16. The immunoregulatory role of IL-10 at mucosal tissues is well conserved among mammalian and non-mammalian vertebrates and defects in IL-10 signaling result in inflammatory bowel disease in mouse and man17. This suggests that IL-10 is employed to promote tolerance against microbiota and dietary antigens and to protect the integrity of epithelial barriers18. Mice with a Treg specific deletion of IL-10 display spontaneous colitis and develop augmented epithelial pathology during GI and lung immune challenges19. Thus, IL-10 produced by Tregs plays a nonredundant role in promoting tolerance in intestinal mucosa. However, mice with a Treg-specific deletion of IL-10 do not recapitulate the severe multi-organ autoimmunity associated with the germline defects of Foxp3 or systemic ablation of Tregs confirming the existence of multiple Treg suppressor mechanisms19.
The TGF-β superfamily emerged approximately 1.1 billion years ago as a common ancestor of arthropods and vertebrates, playing crucial roles in homeostasis in a multitude of species20. TGF-β, the prototypical member of the superfamily, can be produced by all immune cells and signals through the heterotetrameric TGFBR1-R2 complex to regulate functions of all innate and adaptive immune cells. TGF-β1 is the most common isoform associated with immunoregulatory functions and primarily signals through phosphorylation of Smad proteins in all immune cells21. Global deletion of TGF-β1 results in an early onset wasting disease with multiorgan immunopathology22,23. TGF-β1 provided by the innate immune system plays important roles in the Treg-independent restriction of autoreactive T cells and also in the development of thymic and peripheral Tregs24–26. However, the immunoregulatory effect of TGF-β1 produced by Tregs is controversial. While initial studies described a key role for TGF-β secreted by Tregs in inhibiting immune responses in vitro, subsequent studies failed to confirm these experiments27–29. Nevertheless, subsequent studies demonstrated that Treg and activated platelets express a cell surface molecule, Glycoprotein-A repetitions predominant protein (GARP), which is rapidly mobilized to Treg surface upon activation. GARP binds latent-TGF-β intracellulary and transports it to the cell surface. It is also capable of binding secreted latent-TGF-β30,31. Latent TGF-β is comprised of the TGF-β homodimer non-covalently linked to Latency Associated Protein (LAP). Release of active TGF-β requires mechanical dissociation or proteolytic cleavage of LAP by αV integrin heterodimers that bind to the integrin binding Arginine-Glycine-Aspartate motif on LAP32,33. While Tregs are the only lymphoid cell type that expresses the GARP-latent TGF-β complex, the function of this complex remains unclear. One role might be to provide active TGF-β locally to antigen-specific effector T cells in an integrin dependent manner to convert them into antigen-specific induced Tregs, or to Th17 cells depending on the availability of IL-2 and IL-6 in the microenvironment34,35. Some studies suggest that αV integrins expressed by mucosal DCs are crucial to activate Tregs and to promote their local release of TGF-β36–38. Yet, whether activated Tregs detach from DCs and roam to target antigen primed effectors for engaging in direct T-T interactions, in other words performing infectious tolerance, remains elusive39. While some studies suggested that Tregs use TGF-β1 as an active suppressor mechanism at mucosal sites to suppress antigen-specific effector T cells for food and commensal antigens, other studies demonstrated that TGF-β1 deficient Tregs are functional and mice with Treg-specific deletion of TGF-β1 are phenotypically normal31,40,41. The differences between the studies may reflect differences in the bacterial flora in the animal rooms. Mice with a Treg-specific deletion of GARP did not develop spontaneous autoimmune disease, while other studies demonstrated enhanced susceptibility to colitis42,43. In a xeno-GVHD model, antibody mediated blockade of GARP reversed the suppressive effects of co-transferred Tregs without affecting their numbers44. Taken together, TGF-β1 produced by Treg does not appear to be a major Treg suppressor mechanism. In a manner similar to IL-10 it may be required in certain situations characterized by high levels of inflammation to help “put out the fire.”.
IL-35 is a member of IL-12 family of cytokines. While this family is composed of cytokines with proinflammatory activity, IL-35 displays immunosuppressive properties. It is a heterodimer of the IL-12 p35 subunit and Epstein-Barr virus-induced gene 3 (Ebi3) that binds to IL-12Rβ2 and gp130 respectively45. Although it presents an unconventional signaling pattern with the ability to transmit signals through IL-12Rβ2 or IL-27R homodimers, it exerts optimal immunoregulatory function through the heterodimeric receptor46. While IL-35 has been claimed to be secreted by Tregs, co-expression of Ebi3 and p35 has also been detected in peripheral B cells, γδ T cells, CD8+ T cells, and placental trophoblasts47,48. While Ebi3 expression is readily detected in Treg, it has been difficult to detect mRNA for p3549. Detailed studies of the role of IL-35 in Treg biology have been hampered by the lack to reagents and the ability to produce recombinant IL-35. While Ebi3−/− mice do not display an autoimmune phenotype50, Ebi3−/− Tregs failed to protect from colitis in a cell transfer model of colitis suggesting that Tregs produce IL-35 when there is an active breach of peripheral tolerance51. More recently, IL-35 expressing Tregs were found to promote T cell exhaustion in the tumor microenvironment further supporting a local, niche-specific immunoregulatory role for IL-3552.
Counteractive mechanisms of suppression
IL-2 depletion
IL-2 is the major cytokine produced by T cells to promote their survival, proliferation and immunoregulation. IL-2 exerts its activities in autocrine and paracrine fashion through its receptors IL-2Rα (CD25), IL-2Rβ (CD122) and common gamma chain (γc) receptor (CD132) shared with IL-7 and IL-1553. Medium affinity IL2-Rβ and γc heterodimers are expressed by mostly CD8 memory cells and NK cells in the steady state, and activation signals transiently induce the expression of high affinity heterotrimeric receptor complex on T cells, B cells, DCs. IL-2 signals propagate through the intracellular domains of IL-2Rβ and γc via JAK1 and JAK3 binding. This results in the phosphorylation of STAT5 that serves as the molecular switch to implement biological functions of IL-254,55. Phosphorylated STAT5 (pSTAT5) activates gene expression of a series of lineage specific transcription factors such as Foxp3, Tbet, GATA3 thus promoting Treg, Th1, Th2 functions while it represses IL-17A production and Bcl-6 expression, thus inhibiting Th17 and Tfh programs56,57. Additionally, high levels of IL-2 favor the development of short-lived effector cells, while low levels of IL-2 signaling promote memory T cell formation58,59. Although Tregs are unable to produce IL-2, their development, homeostasis and functional stability are heavily dependent on it. To acquire a mature Treg phenotype with CD25 and Foxp3 expression, thymic Treg precursors rely on IL-2Rβ for IL-2 signals provided by thymic medulla60. In the periphery, they require IL-2 produced by effector T cells to maintain their homeostasis and function. Binding of pSTAT5 to the promoter and the noncoding intronic regulatory element CNS2 is required for Foxp3 induction and stability61.
The expression of the IL2-R on Tregs can also play an important role in Treg suppression. IL-2 production by effector T cells has long been proposed as a major target for Treg mediated suppression. Early in vitro studies demonstrated that once activated, Tregs downregulated IL-2 transcription by effector T cells in a contact dependent manner62,63. An alternative view is that IL-2R οn Tregs senses the local IL-2 gradient and positions Tregs around potential zones of immune activation and then acts like a sink to deplete IL-2 and drive T cells into an apoptotic state due to cytokine withdrawal. This was initially confirmed by the resistance of T cells lacking proapoptotic molecule Bim to Treg mediated suppression64. It was also demonstrated that in an inflammatory bowel disease model induced by the adoptive transfer of Bim−/− CD4+CD45RBhigh T cells into a lymphopenic host, co-transfer of Tregs failed to suppress Bim−/− T cells, while later reports that used the same experimental setting failed to show any significant difference in Treg suppression of WT and Bim−/− T cells65. Furthermore, T cells lacking other key molecules that are implicated in cytokine withdrawal associated programmed cell death were found as susceptible to Treg-mediated suppression as WT T cells, suggesting that induction of apoptosis may not be an outcome of Treg mediated IL-2 depletion. Thus, the IL-2 depletion model of suppression mechanism remains controversial, especially in vivo. Indeed, as Tregs do suppress the production of IL-2, the source of the IL-2 that must be depleted remains obscure.
Recently, the cell extrinsic role of Treg IL-2Rα was readdressed in elaborate mouse models of Treg specific loss of the IL-2R and gain of constitutive STAT5 activation66. Treg-specific deletion of IL-2Rα and IL-2Rβ both resulted in a similar systemic autoimmune disease that was more severe than that is seen in global deletions of the same receptors possibly due to defective IL-2 signaling and impaired T effector function in the global knock outs. In healthy female mice that are heterozygous for IL-2Rβ-sufficient and IL-2Rβ-deficient Tregs due to random X chromosome inactivation, IL-2Rβ−/− Tregs were less represented in the Treg pool, displayed lower levels of Foxp3 and Treg associated markers, without any defect in the percentage of activated Tregs perhaps due to ongoing inflammation. Yet, these Tregs were unable to suppress effector T cells upon adoptive transfer in vivo. When either mice with a Treg specific deletion of the IL-2Rα or IL-2Rβ were transduced with a gain of function mutation for constitutive activation of STAT5, thymic development and suppressor function of Tregs were rescued despite ongoing CD8+ T cell mediated autoimmunity. This indicated an indispensable cell-extrinsic role for Treg IL-2Rα in suppressing CD8+ T cell activation, but not CD4+ T cell activation. For CD4+ T cells, intact STAT5 signaling in IL-2Rα deficient Tregs seemed to be sufficient66. Lack of IL-2Rα may alter the thymic selection of Treg repertoire differently and thus may confound the interpretation of effects of IL-2Rα deficiency on post-thymic mature Tregs.
In a mouse model where the IL-2Rα was inducibly deleted from Tregs after thymic development, IL-2Rα−/− Tregs were found to maintain a Foxp3+ pool despite a reduced level of Foxp3 expression. They also displayed reduced CTLA-4 expression and provided decreased protection in an EAE model67. However, in such a model it is difficult to extrapolate how the effector T cells are affected as the deletion is temporary and rapidly compensated by the unperturbed recent thymic immigrants. Lastly, detailed imaging of naïve lymphoid tissue showed that while resting Tregs are located inside the T cell zone, Tregs expressing pSTAT5 are located at T-B border as multiple heterogenous clusters68. These clusters were found in close proximity to IL-2 producing CD4+ T cells in both germ free and single pathogen free mice suggesting that IL-2 production is triggered by presentation of self-antigens on MHCII. It was also demonstrated that clusters failed to form when TCR expression is disrupted in the Treg compartment, while Tregs with unperturbed TCR made tight contacts with DCs and expressed pSTAT5. These suggest while TCR-pMHCII interaction activates Tregs and also positions them in close proximity to CD4+ T cells. Initially it was claimed that STAT5 signaling in Tregs is indicative of active suppression via IL-2 removal. Because activated Tregs suppress CD4+ T cells using mechanisms other than IL-2 removal, STAT5 activity should rather be interpreted as a marker of IL-2R signaling, indicating activation of suppressive ability, but not the actual suppression.
Removal of inflammatory mediators and metabolic regulation
Release of intracellular purines, particularly ATP, is used as a way of communication between immune cells. While some stimuli such as pathogen associated molecular patterns stimulate a controlled release of ATP through specialized channels, excessive inflammation and tissue damage can cause an uncontrolled release of intracellular content including ATP69. Elevated extracellular ATP (eATP) concentration serves as a danger signal and induces immune response via inflammasome activation. While low levels of eATP activates CD4+ and CD8+ T cells, increasing levels become toxic and may drive cells to apoptosis through the very receptors that activate them70,71. There are mechanisms evolved to cope with rising extracellular ATP such as the expression of membrane ectonucleotidases CD39 and CD73. CD39 metabolizes ATP into non-toxic AMP while CD73 catabolizes AMP into adenosine. CD39 and CD73 are expressed by innate and adaptive immune cells including DCs, macrophages, neutrophils, B and T cells72. Among T cells, Tregs express high levels upon activation72,73. Adenosine can bind to receptor A2A receptors that are expressed by many cells including DCs and effector T cells and binding of adenosine to A2AR results in inhibition of T cell activation74. However, generation of extracellular adenosine is not restricted to Tregs and in fact, adenosine can be generated by many cells including innate immune cells and endothelium75,76. Thus, the effect of adenosine produced by Tregs may not be significant, as Tregs constitute a small portion of immune cells at inflammatory sites. Instead, CD39-CD73 activity may play a role in tuning Treg activity in inflammatory environments and/or providing Tregs protection from apoptosis. Another possibility is that the upregulation of CD39-CD73 expression may not indicate active use of ATP degradation as a suppression mechanism, but may rather demonstrate that Tregs have high suppressive ability. To this end, CD39-CD73 mediated ATP removal and adenosine production may have a limited role in Treg suppression and represent a context and niche dependent mechanism.
Another metabolic strategy used by Tregs is the disruption of optimal milieu for effector T cells to thrive and function77. This effect is mediated via reverse signaling through CD80-CD86 upon engagement of CTLA-4 in APCs. This interaction results in the induction of the enzyme indolamine-2,3-dioxygenase (IDO) that catabolizes the essential amino acid tryptophan into kynurenine. Kynurenine binds aryl hydrocarbon receptors in APCs, thus helping them assume a tolerogenic phenotype78,79. Depletion of tryptophan in the environment is also sensed by the effector T cells and Tregs. It induces cell cycle arrest of T cells and increases T cell apoptosis by inhibiting the mechanistic target of rapamycin complex 1 (mTORC1), and inducing a stress response that activates the integrated stress response kinase GCN280,81. Furthermore, trythophan depletion increases the conversion of effector T cells into iTregs and stabilizes Treg phenotype via reducing Akt phosphorylation82,83. IDO activity has been detected in both innate and adaptive cells of the immune system as well as in solid tissue in niches such as intestinal lamina propria and tumor microenvironment12,84,85. Therefore, Treg mediated regulation of IDO activity is possibly limited.
Single cell transcriptomic analyses of tissue-resident and lymphoid Tregs identified unique TCR landscapes and transcription profiles in tissue-resident Treg subsets dictated by their anatomic localization rather than ontogeny86–88. Tissue resident Tregs may also utilize unique suppressor mechanisms tailored for their environment. Visceral adipose tissue (VAT) associated Tregs express PPARγ and play an important role in reducing insulin resistance associated with inflammation of fat tissue89. Recently it was found that both human and mouse Tregs express the enzyme 15-hydroxyprostaglandin dehydrogenase (HPGD), that catabolizes prostaglandin PGE2 into 15-keto-PGE2 and HPGD activity is especially pronounced in VAT associated Tregs due to PPARγ expression. 15-keto-PGE2 acts on effector T cells and reduce their proliferation. While Treg-specific deletion of HPGD did not result in autoimmunity, the mice developed a metabolic syndrome over time characterized by decreased insulin sensitivity and impaired glucose homeostasis. Fat tissue of these mice acquired an inflammatory phenotype with age that is characterized with larger adipocytes, infiltration of NK cells and inflammatory macrophages90. Overall these findings support the concept that tissue resident Tregs use site-specific suppressor mechanisms that are not used by their lymphoid counterparts. Further study of the metabolic regulation of tissue Tregs may reveal other unique suppressor mechanisms that potentially can be manipulated for treatment of organ-specific autoimmune diseases.
Tregs as robbers of surface molecules.
In a manner similar to effector T cells, Tregs must be activated through their T cell receptor (TCR) to function. This process requires a full-on synapse with antigen presenting cells (APC), similar to a T cell that needed to turn on effector functions91. The Treg-APC synapse has long been considered as merely a permissive step for all the subsequent actions of Tregs. However, recently it was discovered that Treg-APC contact might actually dictate the fate of Treg mediated inhibition. Tregs were shown to bind DC with high avidity and engulf parts of DC membrane containing pMHCII and costimulatory ligands via a process similar to trogocytosis92. Depending on the initial antigen load, this was shown to cause a significant loss of DC surface pMHCII and thus a complete abrogation of effector T cell priming in vivo. Surprisingly, when APCs present two antigens, Tregs specific for one of them capture only its cognate antigen leaving the presentation of the other antigen intact. In an antigen limiting environment in vivo, this was shown to result in complete antigen-specific inhibition of effector T cell proliferation. Although the B7 family of costimulatory ligands, such as CD80, CD86 and ICOSL are also acquired by antigen-specific Tregs, APCs that are stripped off of a particular pMHCII were able to sustain the proliferation of other effector T cells that recognize another antigen presented on APC, suggesting that APC function was not globally impaired. It is likely that APCs replenish the costimulatory ligands on cell surface because the mRNA transcripts of the B7 family molecules continue being detected after Treg contact93. Although pMHCII capture might not work as an efficient suppression mechanism in antigen abundant system such as infection, where there is a constant supply of high doses of antigen (Fig. 2), it may present an ideal way to suppress the spontaneous presentation of self-antigens on MHCII, before detection by a rare self-antigen specific clonal escapee. Because thymic Treg output constitutes a more self-skewed repertoire than that of effector T cells, the likelihood of a self-specific Treg to come across its antigen is theoretically higher. This numeric advantage and unique pMHCII stripping together may jointly endow Tregs with the ability to constantly prune self-antigen-MHCII complexes and thereby prevent autoimmunity. This model is further supported by the mouse models of Treg-selective TCR ablation suggesting that continuous pMHCII-TCR interactions are indispensable for Treg-mediated suppression of autoimmunity94,95.
Figure 2.
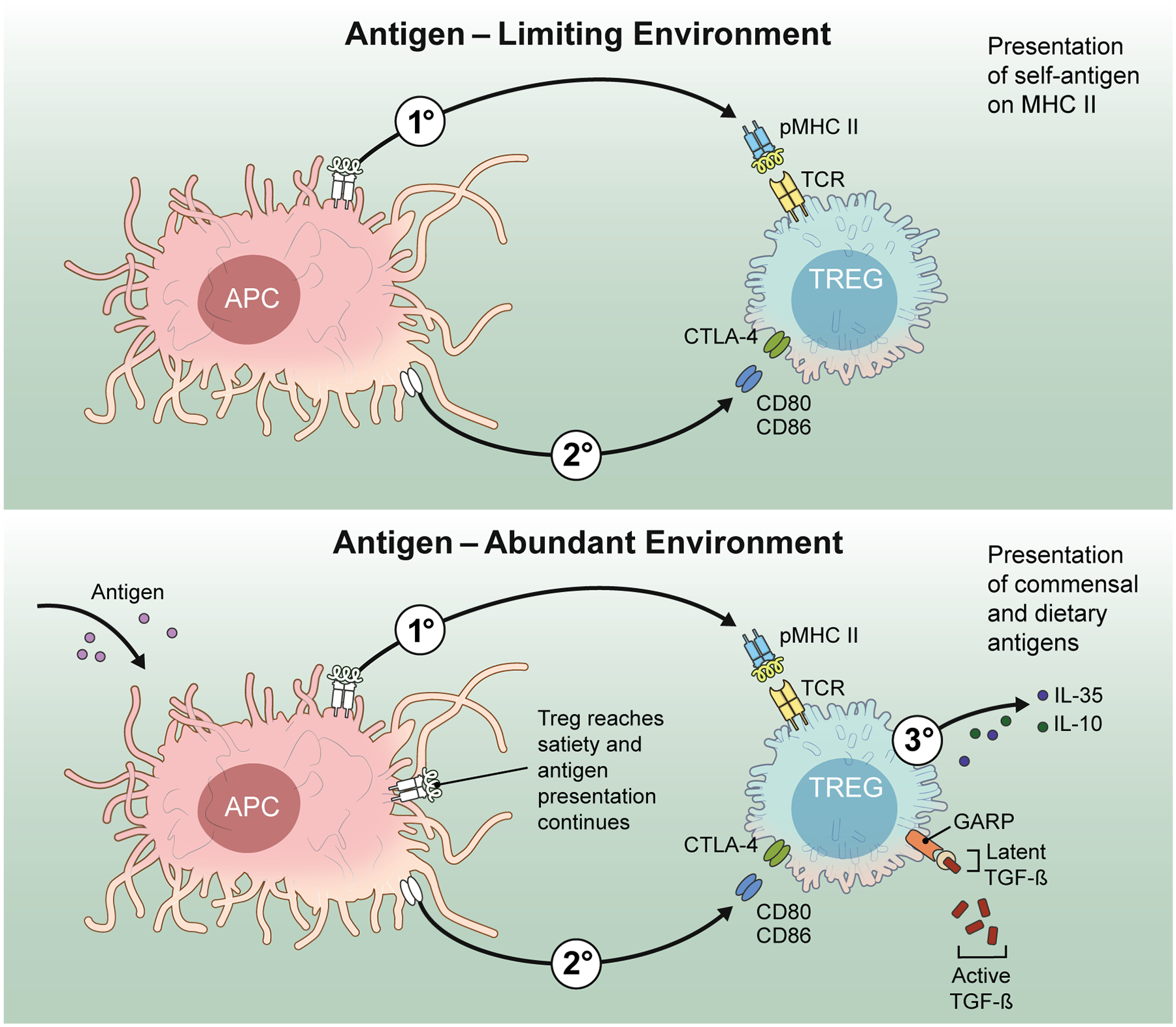
The “stealing” of pMHCII complexes from DC surfaces functions most effectively under limiting antigen conditions (left panel). The process can be helped by simultaneous removal of costimulatory molecules. In contrast, in the presence of high concentrations of antigen, the theft of pMHCII complexes is unlikely to be an effective suppressive mechanisms (right panel). More active mechanisms such as the secretion of inhibitory cytokines must be called into action.
CTLA-4 is another important molecule implicated in the contact-dependent inhibition of APC function by Tregs. It is the most well-known inhibitory T cell coreceptor homologous to CD28 with shared binding partners, CD80 and CD86. Unlike constitutive surface expression of CD28, CTLA-4 expression is induced upon TCR stimulation in effector T cells96. CTLA-4 has been considered to play a cell intrinsic role in limiting T cell activation to prevent immunopathology. Because CTLA-4 expression is temporally distinct from TCR and CD28 signals, it had been thought that it shuts down the activating signals later in activation by outcompeting CD28 at the synapse by its higher affinity for ligands. More recently, it was described to remove CD80/CD86 from the synapse via transendocytosis in both effector T cell and Treg97,98. Because Tregs express CTLA-4 constitutively this has been proposed as a major suppression mechanism by which Tregs prevent autoimmunity98–100. Germline deficiency of CTLA-4 results in lethal autoimmunity 3–4 weeks after birth101. This lethal phenotype indicates that CTLA-4 plays an important role in immune tolerance. However, the role of CTLA-4 in Treg compartment remains obscure. While Treg specific deletion of CTLA-4 from birth results in fatal autoimmunity with slightly later onset than in the global knockout, deletion of Treg CTLA-4 in adult mice does not reduce their suppressive function and in fact enhances it and renders the mice resistant to EAE102. This raises the possibility that CTLA-4 plays a role in the thymic development of Tregs and somehow renders the Tregs with germline CTLA-4 deficiency less functional102,103. Yet, the role of CTLA-4 in immunotolerance is intertwined and more studies are needed to fully dissect out how Tregs use CTLA-4 for suppression.
CTLA-4 is not specifically expressed on Tregs, therefore all the regulatory effects of CTLA-4 cannot be attributed to them. Yet, the constitutive expression of CTLA-4 in Tregs vs. inducible expression in effector T cells may provide a time frame for Tregs to prevent unwanted immune responses. Therefore, Tregs may use it in addition to pMHCII removal to ensure the downmodulation of costimulatory ability of the APC (Fig. 2). These dual effects may serve as a safeguard to deprive effector T cells of second signals when antigen removal by Tregs is incomplete due to overwhelming antigen load and/or low affinity of Treg TCR for antigen. Alternatively, incomplete antigen removal may deliberately be used to direct effector T cells to antigen expressing APCs which lack co-stimulatory ability thus resulting in T cell anergy. In addition, the immune events that take place at epithelial barriers and in infectious settings are inherently antigen abundant due to continuous exposure to commensal microorganisms, dietary antigens and pathogens. In mucosal tissue, there are multiple factors, such as TGF-β, dietary vitamin A, short-chain fatty acids etc. that have evolved to ensure a tolerogenic environment favoring a high (p)Treg/effector T cell ratio. Tregs may also use captured pMHCII to form synapses with antigen-specific effector T cells and render them tolerant and/or convert them to pTregs via membrane-bound TGF-β. Overall the role of Treg in infectious settings is complex as they must allow a strong anti-pathogen response and depletion of pMHCII is unlikely to be operative in the presence of high amounts of antigen. Later in the course of infection, when the level of pathogens has been reduced, pMHCII depletion may be a highly effective mechanism for the control of immunopathology.
Conclusion
Tregs are the swiss army knife of immune tolerance with a hidden toolkit that is only revealed after TCR signaling. While the TCR stimulus licenses Tregs to suppress via multiple mechanisms, pMHCII depletion dampens the presentation of cognate antigen without disabling other APC functions and provides important clues about what additional mechanisms should be in place for handling the situation optimally. Here, we propose a three tier regulation by Tregs with TCR binding and capture of pMHCII being tier one, tuning costimulation being tier two, and use of active suppression mechanisms being tier three (Fig. 2).
Although mouse models for the global and Treg specific deletion of key mechanistic targets have been immensely helpful, we still need more elaborate study designs to distinguish altered function in “otherwise normal” Tregs from the “inherently defective” Tregs. This is likely to require a more rigorous effort to watch normal Tregs and catch the mechanisms in action. However, further progress might require hiring a detective instead of an immunologist to identify additional “master thieves” of the immune system.
Acknowledgment
The authors are funded by the Division of Intramural Research, NIAID, NIH.
References
- 1.Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394(6695):744–751. [DOI] [PubMed] [Google Scholar]
- 2.Andersen KG, Nissen JK, Betz AG. Comparative Genomics Reveals Key Gain-of-Function Events in Foxp3 during Regulatory T Cell Evolution. Front Immunol. 2012;3:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shanmugasundaram R, Selvaraj RK. Regulatory T cell properties of chicken CD4+CD25+ cells. J Immunol. 2011;186(4):1997–2002. [DOI] [PubMed] [Google Scholar]
- 4.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. [DOI] [PubMed] [Google Scholar]
- 5.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. [DOI] [PubMed] [Google Scholar]
- 6.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. [DOI] [PubMed] [Google Scholar]
- 7.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–197. [DOI] [PubMed] [Google Scholar]
- 8.Ali N, Zirak B, Rodriguez RS, et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell. 2017;169(6):1119–1129 e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rieckmann M, Delgobo M, Gaal C, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. 2019;129(11):4922–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burzyn D, Kuswanto W, Kolodin D, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cipolletta D, Feuerer M, Li A, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whibley N, Tucci A, Powrie F. Regulatory T cell adaptation in the intestine and skin. Nat Immunol. 2019;20(4):386–396. [DOI] [PubMed] [Google Scholar]
- 13.Magnuson AM, Kiner E, Ergun A, et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc Natl Acad Sci U S A. 2018;115(45):E10672–E10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shevach EM. Foxp3(+) T Regulatory Cells: Still Many Unanswered Questions-A Perspective After 20 Years of Study. Front Immunol. 2018;9:1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol. 2019;10:3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32(1):23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piazzon MC, Lutfalla G, Forlenza M. IL10, A Tale of an Evolutionarily Conserved Cytokine across Vertebrates. Crit Rev Immunol. 2016;36(2):99–129. [DOI] [PubMed] [Google Scholar]
- 18.Paul G, Khare V, Gasche C. Inflamed gut mucosa: downstream of interleukin-10. Eur J Clin Invest. 2012;42(1):95–109. [DOI] [PubMed] [Google Scholar]
- 19.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28(4):546–558. [DOI] [PubMed] [Google Scholar]
- 20.Newfeld SJ, Wisotzkey RG, Kumar S. Molecular evolution of a developmental pathway: phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics. 1999;152(2):783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Worthington JJ, Fenton TM, Czajkowska BI, Klementowicz JE, Travis MA. Regulation of TGFbeta in the immune system: an emerging role for integrins and dendritic cells. Immunobiology. 2012;217(12):1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143(1):3–9. [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9(6):632–640. [DOI] [PubMed] [Google Scholar]
- 25.Oh SA, Liu M, Nixon BG, et al. Foxp3-independent mechanism by which TGF-beta controls peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2017;114(36):E7536–E7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akkaya B, Holstein AH, Isaac C, et al. Ex-vivo iTreg differentiation revisited: Convenient alternatives to existing strategies. J Immunol Methods. 2017;441:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura K, Kitani A, Fuss I, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004;172(2):834–842. [DOI] [PubMed] [Google Scholar]
- 28.Oida T, Xu L, Weiner HL, Kitani A, Strober W. TGF-beta-mediated suppression by CD4+CD25+ T cells is facilitated by CTLA-4 signaling. J Immunol. 2006;177(4):2331–2339. [DOI] [PubMed] [Google Scholar]
- 29.Piccirillo CA, Letterio JJ, Thornton AM, et al. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med. 2002;196(2):237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM. Regulation of the expression of GARP/latent TGF-beta1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J Immunol. 2013;190(11):5506–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26(5):579–591. [DOI] [PubMed] [Google Scholar]
- 32.Stockis J, Dedobbeleer O, Lucas S. Role of GARP in the activation of latent TGF-beta1. Mol Biosyst. 2017;13(10):1925–1935. [DOI] [PubMed] [Google Scholar]
- 33.Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157(3):493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards JP, Thornton AM, Shevach EM. Release of active TGF-beta1 from the latent TGF-beta1/GARP complex on T regulatory cells is mediated by integrin beta8. J Immunol. 2014;193(6):2843–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards JP, Hand TW, Morais da Fonseca D, Glass DD, Belkaid Y, Shevach EM. The GARP/Latent TGF-beta1 complex on Treg cells modulates the induction of peripherally derived Treg cells during oral tolerance. Eur J Immunol. 2016;46(6):1480–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Travis MA, Reizis B, Melton AC, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449(7160):361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paidassi H, Acharya M, Zhang A, et al. Preferential expression of integrin alphavbeta8 promotes generation of regulatory T cells by mouse CD103+ dendritic cells. Gastroenterology. 2011;141(5):1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boucard-Jourdin M, Kugler D, Endale Ahanda ML, et al. beta8 Integrin Expression and Activation of TGF-beta by Intestinal Dendritic Cells Are Determined by Both Tissue Microenvironment and Cell Lineage. J Immunol. 2016;197(5):1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson J, Tran DQ, Pesu M, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205(9):1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34(3):396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med. 1996;183(6):2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeersch E, Lienart S, Collignon A, et al. Deletion of GARP on mouse regulatory T cells is not sufficient to inhibit the growth of transplanted tumors. Cell Immunol. 2018;332:129–133. [DOI] [PubMed] [Google Scholar]
- 43.Salem M, Wallace C, Velegraki M, et al. GARP Dampens Cancer Immunity by Sustaining Function and Accumulation of Regulatory T Cells in the Colon. Cancer Res. 2019;79(6):1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuende J, Lienart S, Dedobbeleer O, et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med. 2015;7(284):284ra256. [DOI] [PubMed] [Google Scholar]
- 45.Sawant DV, Hamilton K, Vignali DA. Interleukin-35: Expanding Its Job Profile. J Interferon Cytokine Res. 2015;35(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collison LW, Delgoffe GM, Guy CS, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13(3):290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee CC, Lin JC, Hwang WL, et al. Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat Commun. 2018;9(1):3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devergne O, Birkenbach M, Kieff E. Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc Natl Acad Sci U S A. 1997;94(22):12041–12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bardel E, Larousserie F, Charlot-Rabiega P, Coulomb-L’Hermine A, Devergne O. Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J Immunol. 2008;181(10):6898–6905. [DOI] [PubMed] [Google Scholar]
- 50.Liu JQ, Liu Z, Zhang X, et al. Increased Th17 and regulatory T cell responses in EBV-induced gene 3-deficient mice lead to marginally enhanced development of autoimmune encephalomyelitis. J Immunol. 2012;188(7):3099–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–569. [DOI] [PubMed] [Google Scholar]
- 52.Turnis ME, Sawant DV, Szymczak-Workman AL, et al. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44(2):316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–268. [DOI] [PubMed] [Google Scholar]
- 54.Ye C, Brand D, Zheng SG. Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther. 2018;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. [DOI] [PubMed] [Google Scholar]
- 56.Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12(6):551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ballesteros-Tato A, Leon B, Graf BA, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36(5):847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32(1):91–103. [DOI] [PubMed] [Google Scholar]
- 59.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toomer KH, Lui JB, Altman NH, Ban Y, Chen X, Malek TR. Essential and non-overlapping IL-2Ralpha-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat Commun. 2019;10(1):1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol. 2018;36:411–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188(2):287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164(1):183–190. [DOI] [PubMed] [Google Scholar]
- 64.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–1362. [DOI] [PubMed] [Google Scholar]
- 65.Szymczak-Workman AL, Delgoffe GM, Green DR, Vignali DA. Cutting edge: regulatory T cells do not mediate suppression via programmed cell death pathways. J Immunol. 2011;187(9):4416–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chinen T, Kannan AK, Levine AG, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17(11):1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fan MY, Low JS, Tanimine N, et al. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep. 2018;25(5):1204–1213 e1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature. 2015;528(7581):225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bortolotti P, Faure E, Kipnis E. Inflammasomes in Tissue Damages and Immune Disorders After Trauma. Front Immunol. 2018;9:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Borges da Silva H, Beura LK, Wang H, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature. 2018;559(7713):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Killeen ME, Ferris L, Kupetsky EA, Falo L Jr., Mathers AR. Signaling through purinergic receptors for ATP induces human cutaneous innate and adaptive Th17 responses: implications in the pathogenesis of psoriasis. J Immunol. 2013;190(8):4324–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–1232. [DOI] [PubMed] [Google Scholar]
- 74.Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem. 1997;272(41):25881–25889. [DOI] [PubMed] [Google Scholar]
- 75.Silva-Vilches C, Ring S, Mahnke K. ATP and Its Metabolite Adenosine as Regulators of Dendritic Cell Activity. Front Immunol. 2018;9:2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hasko G, Pacher P, Deitch EA, Vizi ES. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther. 2007;113(2):264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akkaya B, Roesler AS, Miozzo P, et al. Increased Mitochondrial Biogenesis and Reactive Oxygen Species Production Accompany Prolonged CD4(+) T Cell Activation. J Immunol. 2018;201(11):3294–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grohmann U, Orabona C, Fallarino F, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3(11):1097–1101. [DOI] [PubMed] [Google Scholar]
- 79.Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol. 2007;7(10):817–823. [DOI] [PubMed] [Google Scholar]
- 80.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edinger AL, Thompson CB. Antigen-presenting cells control T cell proliferation by regulating amino acid availability. Proc Natl Acad Sci U S A. 2002;99(3):1107–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen NT, Kimura A, Nakahama T, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107(46):19961–19966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yan Y, Zhang GX, Gran B, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol. 2010;185(10):5953–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hornyak L, Dobos N, Koncz G, et al. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front Immunol. 2018;9:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016;37(3):193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016;34:609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19(3):291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miragaia RJ, Gomes T, Chomka A, et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity. 2019;50(2):493–504 e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu D, Wong CK, Han JM, et al. T reg-specific insulin receptor deletion prevents diet-induced and age-associated metabolic syndrome. J Exp Med. 2020;217(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmidleithner L, Thabet Y, Schonfeld E, et al. Enzymatic Activity of HPGD in Treg Cells Suppresses Tconv Cells to Maintain Adipose Tissue Homeostasis and Prevent Metabolic Dysfunction. Immunity. 2019;50(5):1232–1248 e1214. [DOI] [PubMed] [Google Scholar]
- 91.Zhu J, Shevach EM. TCR signaling fuels T(reg) cell suppressor function. Nat Immunol. 2014;15(11):1002–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akkaya B, Oya Y, Akkaya M, et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol. 2019;20(2):218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chattopadhyay G, Shevach EM. Antigen-specific induced T regulatory cells impair dendritic cell function via an IL-10/MARCH1-dependent mechanism. J Immunol. 2013;191(12):5875–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15(11):1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vahl JC, Drees C, Heger K, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41(5):722–736. [DOI] [PubMed] [Google Scholar]
- 96.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. [DOI] [PubMed] [Google Scholar]
- 97.Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36(2):63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ovcinnikovs V, Ross EM, Petersone L, et al. CTLA-4-mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells. Sci Immunol. 2019;4(35). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hou TZ, Qureshi OS, Wang CJ, et al. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J Immunol. 2015;194(5):2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. [DOI] [PubMed] [Google Scholar]
- 102.Paterson AM, Lovitch SB, Sage PT, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med. 2015;212(10):1603–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. [DOI] [PubMed] [Google Scholar]