

Abstract
Endothelial mechanobiology is a key consideration in the progression of vascular dysfunction, including atherosclerosis. However mechanistic connections between the clinically associated physical stimuli, vessel stiffness and shear stress, and how they interact to modulate plaque progression remain incompletely characterized. Vessel-chip systems are excellent candidates for modeling vascular mechanobiology as they may be engineered from the ground up, guided by the mechanical parameters present in human arteries and veins, to recapitulate key features of the vasculature. Here, we report extensive validation of a vessel-chip model of endothelial yes-associated protein (YAP) mechanobiology, a protein sensitive to both matrix stiffness and shearing forces and, importantly, implicated in atherosclerotic progression. Our model captures the established endothelial mechanoresponse, with endothelial alignment, elongation, reduction of adhesion molecules, and YAP cytoplasmic retention under high laminar shear. Conversely, we observed disturbed morphology, inflammation, and nuclear partitioning under low, high, and high oscillatory shear. Examining targets of YAP transcriptional co-activation, connective tissue growth factor (CTGF) is strongly downregulated by high laminar shear, whereas it is strongly upregulated by low shear or oscillatory flow. Ankyrin repeat domain 1 (ANKRD1) is only upregulated by high oscillatory shear. Verteporfin inhibition of YAP reduced the expression of CTGF but did not affect ANKRD1. Lastly, substrate stiffness modulated the endothelial shear mechanoresponse. Under high shear, softer substrates showed the lowest nuclear localization of YAP whereas stiffer substrates increased nuclear localization. Low shear strongly increased nuclear localization of YAP across stiffnesses. Together, we have validated a model of endothelial mechanobiology and describe a clinically relevant biological connection between matrix stiffness, shear stress, and endothelial activation via YAP mechanobiology.
Introduction
Mechanobiology is a discipline of physiology which focuses on how physical environments and mechanical stimuli influence cell behavior and downstream biochemical responses.1,2 Endothelial cells (ECs) form 3-dimensional lumenized structures and respond to physical cues such as varying matrix stiffnesses and fluid shear stresses which play a vital role in modulating endothelial homeostasis, preventing atherosclerotic progression and adverse cardiovascular events.3 Perturbation of these atheroprotective physical inputs results in EC activation and inflammation. However, flow patterns and magnitudes which induce EC activation (and atherosclerotic progression) are termed disturbed flow4–6 and contain varying parameters depending on the site occurrence, such as insufficient magnitude (low shear) or stark directional changes (oscillatory or recirculating flow). At sites of disturbed flow, atherosclerotic progression is exacerbated.7
Matrix stiffening is the other primary physical parameter clinically correlated with adverse cardiovascular events, with sites of arterial stiffening associated strongly with atherosclerosis.8,9 Furthermore, computational studies on fluid dynamics in the coronary arteries have associated sites of high wall stiffness and low or disturbed shear as primary predictors of where atherosclerotic plaques developed.10–12 Together, these correlations indicate that aberrant physical cues from substrate stiffness and shear promote vascular disease. However, the interaction of these vascular parameters in modulating signaling pathways and biological response are difficult to elucidate with current experimental models.13
Recent work on endothelial mechanobiology has highlighted the Yorkie associated homologue proteins YAP (yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif; collectively referred to as YAP/TAZ or YAP alone) as mechanosensitive, biological sensors which are regulated in endothelial cells by mechanical cues like substrate stiffness14 as well as shear stresses.15–17 Extensively studied in the context of cell growth and apoptosis via the Hippo pathway,18 a variety of biochemical and mechanical cues regulate YAP/TAZ activity15–24 apart from Hippo. Unidirectional, high, laminar shear stress; soft substrates; and cell confluence serve to deactivate YAP activity and cause cytoplasmic retention. Conversely, disturbed flow, stiff matrices, and lack of cellular confluence serve to activate YAP and induce nuclear translocation. Importantly, the cell confluence pathways are tied to Hippo signaling, while substrate stiffness and shear stress (mechanobiology pathways) that affect YAP activity are not.18–21 Highly relevant to EC YAP mechanobiology and vascular homeostasis is that activation of YAP (nuclear partitioning) via any of these physical cues is associated strongly with EC activation, vascular inflammation, and atherosclerotic progression.3,7,8,11,15,16,25,26 As a result, YAP/TAZ activity has been implicated in atherosclerotic progression as a key biological event and specific downstream targets of its activity contribute to plaque progression.27,28 Altogether, this paradigm highlights YAP/TAZ as major endothelial mechanical and biochemical sensors important for maintaining homeostasis. Notably, YAP/TAZ responds to both stiffness and shear, suggesting that the connection between the clinical paradigms of stiffness and endothelial dysfunction or inflammation may be explained in part via dysregulated YAP activity, with shear stress and substrate stiffness interacting to generate the biological response. Thus, we hypothesized that increasing substrate stiffness affects the EC shear mechanoresponse by upregulating YAP activity, connecting vascular stiffness with endothelial activation (causing atherosclerotic progression).
Testing this hypothesis and precisely controlling for all the physical parameters to study YAP activity is non-trivial,18,20–22 and systems to study YAP mechanobiology are in clear demand.29 Furthermore, there are relatively few studies which precisely isolate the interactions of these parameters in vessels to establish a mechanistic connection between them,30 and elucidating this further has several implications in cardiovascular medicine. Thus, to explore whether there exists a connection between the physical parameters of stiffness and shear stress via EC YAP mechanobiology, we employed organ-on-a-chip technology. Organ-on-a-chip (organ-chips) systems have emerged as biomimetic in vitro models of human physiology designed to reproduce physiologically-relevant environments and to replicate tissue- and organ-level functions.31–35 Work from our group and others has shown that organ-chips may be an exceptional tool to isolate and elucidate the biological effects of a range of cellular, chemical, and physical parameters.32–34,36 These vessel-chips facilitate the study of endothelial pathology by supporting system interrogation wherein each degree of freedom may be allowed to vary individually.37,38 Currently, there are few engineered systems which are reported for the study of YAP/TAZ,39,40 which focus either on the development of the system, cyclic strain, or developmental biology. Several microfluidic shearing systems exist which are focused on studying ECs with design and development ongoing,41–44 however, these systems do not currently incorporate relevant, interacting physiological forces (e.g. stiffness and shear stress) to study how they collaboratively influence endothelial biology. In this work, we employ a vessel-chip to study the interaction of clinically relevant physical parameters (stiffness and shear) to yield novel biological insights into endothelial mechanobiology.
Our vessel-chip model integrates three-dimensional (3D) luminal geometry, dynamic range of uniform and oscillatory shear stress, and variable matrix stiffness that replicates the physiological and pathophysiological range of these parameters in the human vasculature10,11,45–47 facilitating the study of how these parameters interact and influence the EC mechanoresponse. We extensively validate our model from a biological perspective to show that it captures the in vivo behavior of YAP/TAZ accurately in previously studied regimes while additionally exploring shear stress patterns not commonly addressed. We further tie the YAP behavior in the system to established biological paradigms, demonstrating EC inflammation under regimes which upregulate YAP activity, relevant modulation of gene expression, and confirm the findings via pharmacologic inhibition of YAP. Lastly, we vary shear stress and substrate stiffness together to explore how convoluting physical inputs affects ECs, in which we demonstrate that substrate stiffening results in upshifted YAP nuclear partitioning under high shear, connecting the two physical paradigms. We discuss several novel biological findings during validation and the implications of YAP in EC mechanobiology throughout. Holistically, this work demonstrates use of engineered organ-chip systems as tools to construct biology in a “bottom-up” fashion resulting in novel biological findings previously inaccessible via conventional methodology.
Materials and methods
Channel fabrication
Vessel-chips were fabricated according to our previous work.48 Briefly, positive channels were designed in Solidworks®, acquired from OutputCity Inc., and photolithographically printed onto silicon wafers (University Wafer Corp.). The upper chamber of vessel-on-a-chip devices were fabricated via soft lithography using polydimethylsiloxane (PDMS, Dow Corning) and then bonded to a borosilicate glass (BSG) slide or cover slip (75 mm × 25 mm) which was either spin-coated with self-consistent PDMS (uniform channel) or left uncoated for stiff BSG controls (stiffness studies). Bonding was performed following O2 plasma treatment (Thierry Zepto, Diener Plasma) in a dedicated clean room.
Stiffness measurements
Substrate stiffness was varied by changing the crosslinker concentration of the PDMS formulations.49 Compressive data was collected for the combination of PDMS with 5%, 7.5%, and 10% by weight of crosslinker. First, samples were punched into 10 mm diameter discs using a biopsy punch and heights measured using digital calipers. Samples were compressed to not greater than 20% of the measured height at a strain rate of 1 mm min−1 utilizing an ADMET MTEST Quattro eXpert 7600 single column testing system with a 25 lb load cell. The raw data collected was processed into MS-Excel to calculate the compressive moduli as the slope of the linear region of the strain vs. stress graph. For atomic force microscopy validation studies on ECM functionalized and non-functionalized PDMS thin films, a Catalyst Biomicroscope (Bruker) was employed. MLCT conical probes (0.01 N m−1; Bruker Nano) were used to measure elastic moduli pre- and post-ECM functionalization. The AFM scanning frequency used was 1 Hz in PBS. All data was processed using Nanoscope Analysis 1.50r1 (Bruker). Sneddon’s conical fitting was used on thin films, taking from 0–10% (ref. 50) of the full AFM force curve for at least 15 technical replicates. Iterative outlier analysis was used to remove any technical replicates which deviated strongly from the spread, and the group was averaged to yield the stiffness of the PDMS film.
Cell culture
Human umbilical venous endothelial cells (HUVECs, Lonza) were cultured in endothelial growth media (EGM-2, Lonza) using an endothelial cell supplement EGM-2-M (Lonza). Cell culture washes were performed using pH 7.2 phosphate buffered saline (PBS, Gibco), and cell passaging was performed using 1× 0.25% Trypsin–EDTA (Gibco). HUVECs used were all between p4 and p6, passage matched for each experiment.
Channel functionalization and lumenization
After fabrication of the vessel-chips, each channel was treated with O2 plasma (PDC-32G, Harrick Plasma) for 5 minutes, and then an extracellular matrix (ECM) solution of 100 μg mL−1 rat tail collagen type I (Corning) and 50 μg mL−1 human fibronectin (Sigma Aldrich) was perfused through the channel and incubated for 30 minutes at 37 °C, 5% CO2. Plate controls reported were done on BSG cover slips, or 10% PDMS spin-coated glass cover slips using the same plasma treatment procedure and ECM solution. Afterwards, each channel/cover slip was then placed in an incubator at 37 °C and 5% CO2 for 30 minutes. The ECM solution was subsequently removed by perfusing EGM-2-M media through the chamber, or via washing of the cover slips. On the fully assembled vessel-chip devices, HUVECs were seeded onto the top and bottom faces of the chamber and allowed 1 hour for attachment. After the seeding period, the seeded vessel-on-a-chip devices were attached to a syringe pump (PHD ULTRA 4400, Harvard Apparatus) using a Luer curved dispenser tips (Qosina) and 20″ tubing (0.094′ ID, 0.145′z OD; Qosina) and operated in suction mode. Media reservoirs were made from 5 mL syringes (BD). To allow for lumen formation, each chamber was then placed in shearing flow overnight at a flow rate of 1 μL min−1 before each experiment was conducted (post-lumenization; t = 0). For the quasistatic control, HUVECs were seeded and cultured under extremely low shear (<2 μL per hour) using two 1 mL pipets as reservoirs for 48 hours such that cellular confluence was achieved.
Flow rates
Flow rate shear stresses were calculated numerically using the finite element method in Ansys Fluent v29R1 from Solidworks v19 models. Cell culture media (comparable to water) was used as the fluid for modeling (density: 0.998 g cm−3; viscosity: 0.001 Pa s). A flow rate of 1 μL min−1 was calculated to be 1.12 dynes per cm2 (low/venous shear), and 10 μL min−1 was calculated to be 11.2 dynes per cm2 (high/arterial shear). Values are reported as exact calculations at the center of the XZ planes (top/bottom wall shear) – distant from the corner. Disturbed flow was induced via an oscillatory flow pattern of 1 or 10 μL min−1, 2 seconds of withdrawal and 1 second of infusion (100% duty cycle, 0.33 Hz) to deliver fresh media. Each of these regimens were run for 3 hours and 6 hours after appropriate lumenization.
Verteporfin treatment
Verteporfin was purchased from Millipore Sigma (≥94% HPLC) and dissolved into a 1 mM stock in dimethyl sulfoxide (DMSO; Millipore Sigma) and stored at −20 °C until use. The stock solution was diluted into 2 mL of EGM-2-M media for the corresponding experiment. Each concentration was supplemented with DMSO to ensure each flow chamber received the same amount of DMSO regardless of dilution. Verteporfin was introduced into the chamber in a dose-dependent manner. DMSO controls were media and the total DMSO volume required for dilution only.
RNA isolation and gene expression
HUVECs in each channel were trypsinized and cells pelleted for RNA extraction and lysed with 10 minutes of detachment. Note that YAP is rapidly inactivated upon trypsinization and gene expression is thus dependent on the half-life of the mRNA;51 cells must be lysed quickly and RNA extraction begun immediately. RNA was extracted using an Arturus™ PicoPure™ RNA Extraction kit (Applied Biosystems, Thermo Fisher Scientific). The cDNA was synthesized from the extracted RNA using a 5× iScript Reverse Transcriptase Supermix (Bio-Rad Laboratories, Inc.). The cDNA was then diluted to a volume of 100 μL for qPCR. For each qPCR reaction, a volume of 20 μL was used consisting of 10 μL of Power SYBR® Green Master Mix (Applied Biosystems, Thermo Fisher Scientific), 7 μL of H2O (DNAse/RNAse free, molecular grade), 1 μL of the gene specific primer, and 2 μL of the sample cDNA. All primers were purchased as validated 20× SYBR® Green assays for glutaraldehyde 3-phosphate dehydrogenase (GAPDH), endothelial nitric oxide synthase (eNOS), connective tissue growth factor (CTGF), ankyrin repeat domain 1 (ANKRD1), intercellular adhesion molecule 1 (ICAM-1), vascular adhesion molecule 1 (VCAM-1), vascular endothelial growth factor receptor 2 (VEGFR2), and interleukin 6 (IL-6), (Bio-Rad Laboratories, Inc.). RT-qPCR was performed on a QuantStudio 12 K Flex (Applied Biosystems, Life Technologies) with the following pre-set settings: 96-Fast Well Plate 0.1 mL, Relative Quantification (−ΔΔCt), SYBR® Green Reporter, and Standard Run Time. All gene expression results were reported as a fold change with respect to the denoted control and the housekeeping gene, GAPDH.52
Immunohistochemistry
Each sample was fixed with 16% formaldehyde (Thermo Fisher Scientific) for 20 minutes at 4 °C and then blocked and permeabilized with 2% bovine serum albumin (BSA, Sigma Aldrich) and 0.1% Triton X-100 (Thermo Fisher Scientific) in PBS (blocking buffer) for 1 hour at 4 °C. Channels were then stained with either rabbit anti-YAP (Abcam), rabbit anti-VCAM1 (Abcam), rabbit anti-vWF (Dako), or mouse anti-CD144 (BD Pharmigen) in blocking buffer (1 : 100) overnight at 4 °C. Secondary staining was performed with donkey anti-rabbit Alexa Fluor™ 555 (Invitrogen Molecular Probes, Thermo Fisher Scientific) for 1 hour at room temperature. Actin was stained with phalloidin Alexa Fluor™ 488 (Invitrogen Molecular Probes, Thermo Fisher Scientific). Nuclei were stained with 4′,6′-diamidine-2′-phenylindole dihydrochloride (DAPI, Roche Diagnostics).
Imaging and analysis
Image acquisition was performed on an FLUOVIEW FV3000 confocal microscope (Olympus Corporation). Analysis was performed on the associated software (cellSens, Olympus Corporation) for YAP partitioning and circularity index by denoting regions of interest and extracting the raw fluorescent values and measurements. YAP partitioning was calculated as the nuclear YAP signal divided by the total cellular YAP signal for 60 cells. Circularity index was calculated via 4π(area/perimeter2) for 60 cells. Actin alignment images were “grayscaled” in GIMP or ImageJ, and analysis was performed using the directionality function in Fiji (ImageJ) using Fourier component analysis for 0° to 90°. The values were reported in vector notation of the form (r, θ), where r corresponds to the relative density (magnitude) of observations for a given angle calculated from the directionality function, and θ (phase) is the angle itself. Thus, angular preferences are visualized as radial spikes or peaks, and random organization is a radially homogenous distribution. Five or greater fields of view were taken for each experimental condition in the study of actin alignment (indicated). Color mapping was performed using OrientationJ.53
Statistics and data visualization
All data was analyzed using either an appropriate Student’s t-test or ANOVA with post hoc Tukey’s/Scheff’s test. Statistical significance was set as p < 0.05, however all p-values are reported as their exact calculated value. All data visualization (except bar graphs) was performed in Python 3.7 using Matplotlib54 with Seaborn packages. All scripts are available upon request; examples scripts are included in ESI.†
Results and discussion
Design of the microfluidic vessel-chip system
In order to design a microfluidic culture environment conducive to studying endothelial mechanobiology, we first engineered the key physiologic parameters directly into the system and developed protocols to study ECs under different shear patterns. We employed the anatomy of a typical vessel-chip consisting of a microfluidic channel with a rectangular cross section providing a 3D culture environment for lumen formation (Fig. 1a–c), where HUVECs cultured within the chamber formed a circular monolayer around the inner walls of the chamber (Fig. 1d) after 18 hour incubation under a low shear rate (1 μL min−1; Fig. 1e). Next, to determine the shear regimes studied during the experimental portion of the protocol (t = 0; Fig. 1e), we used shear stresses and patterns which are known to be protective or inflammatory. To this end, we imposed a low shear (inflammatory) regime, a high shear regime (protective), and oscillatory versions of both (inflammatory).5 As the HUVEC response to shear has been extensively characterized,55–57 in that they respond favorably to arterial range shear (10–30 dynes per cm2),27 we selected a flow rate of 10 μL min−1 (11.2 dynes per cm2) as our model of high shear. Conversely, low shear regimes (1–3 dynes per cm2) are activating for endothelial cells – thus we selected a flow rate of 1 μL min−1 (1.12 dynes per cm2) as our low shear regime (Fig. 1f). To then model disturbed flow, we imposed a flow reversal pattern (oscillatory flow) to induce EC activation (Fig. 1g), based on flow patterns known to cause activation.4,5,58 Within our model, in order to deliver media over the course of the experiment, a net positive flow vector was required, and thus the oscillatory regime displayed a frequency of 0.3 Hz (2 second withdrawal, 1 second infusion). 3D fluid modeling confirmed conventional laminar regimes within the cuboidal chamber and presence of a flow reversal in the oscillatory regimes which remained entirely laminar (Fig. 1h and i). It is beneficial to note that in the context of the model, disturbed flow is isolated to being comprised of only flow reversal. While from a fluid mechanics perspective this is trivial, biologically the scenarios which activate ECs are complex, and several fundamental flow patterns may activate ECs, of which flow reversal is one. Other patterns such as recirculating flow also induce activation, and are classified as disturbed flow, and additionally disturbed flow need not be laminar.6,17,41,59 Lastly, to complete our protocol development, our experiment duration (t = 0, Fig. 1e) was set to follow the onset and stabilization of EC gene expression under shear, starting within an hour and stabilizing by 6 hours.60 Thus, we selected an experimental duration of 6 hours. We confirmed this time point using gene expression of various shear sensitive genes (Fig. S1†).
Fig. 1.
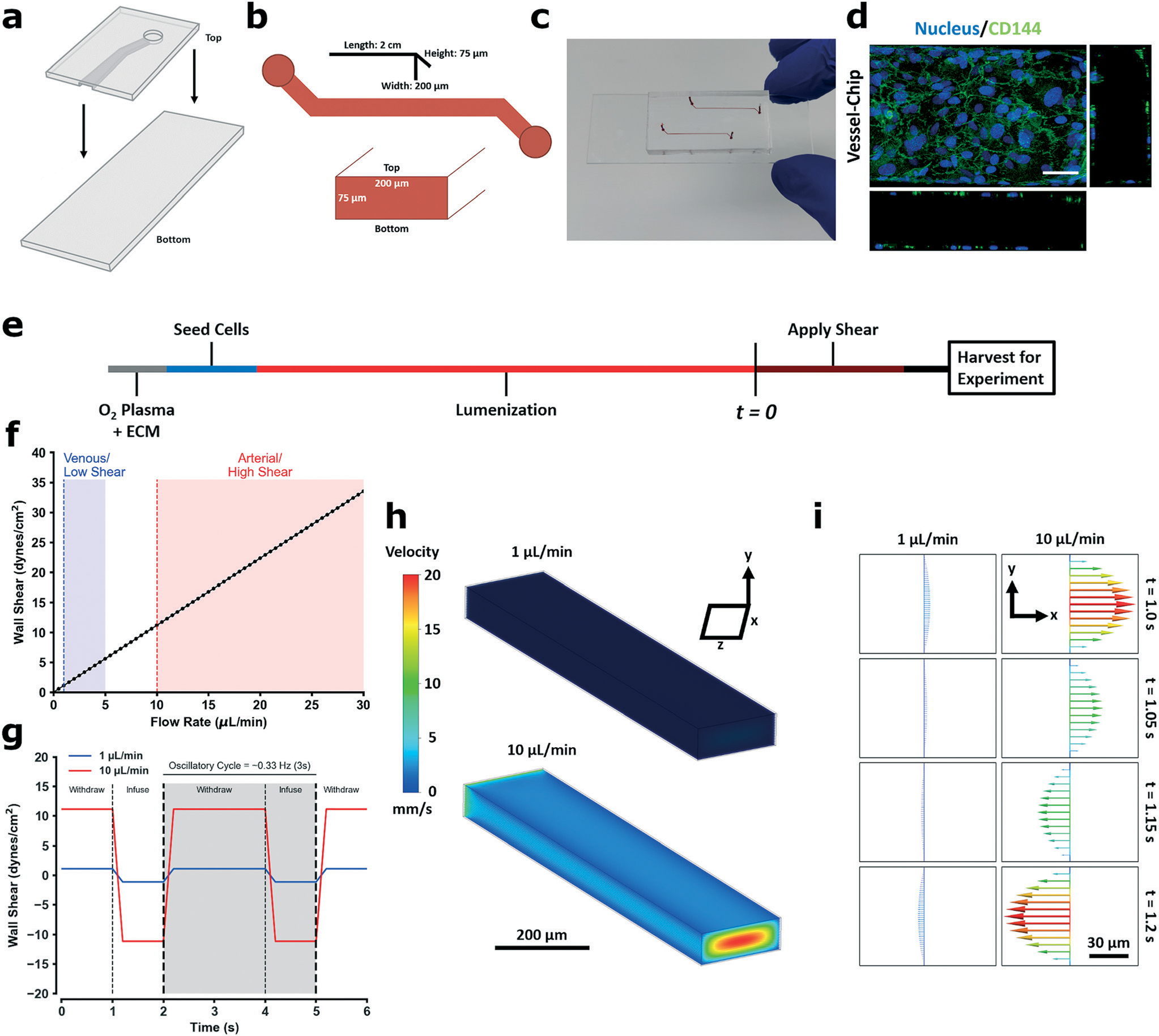
Fabrication of the vessel-chip microfluidic device for endothelial mechanobiology and preliminary quantification. a Details the parts of the device bonded together to form a microfluidic channel. The PDMS upper is fabricated via soft-lithography from a positive master mold. The bottom is a glass slide which is left untreated (stiff control) or spin-coated with PDMS to make a fully coated device. The upper and the lower parts are both O2 plasma treated and then bonded together and placed under weight (500 g or greater) for 30 minutes. b Lays out the dimensions of the channel. Each channel is 2 cm long, has a height of 75 μm, and a width of 200 μm. The PDMS upper comprises the “top” face of the channel, while the bottom glass slide is the “bottom;” these faces are referred to when seeding cells in the device as both faces are seeded prior to experimentation. c Are pictures of a fully fabricated device. d Is a representative confocal maximum Z-image of a HUVEC lumen formed on the device using this protocol (flow rate: 10 μL min−1). e Is a graphical schematic of the cell culture protocol used for each experiment. After bonding, devices are O2 plasma treated again and perfused with an ECM matrix (100 μg mL−1 collagen type I + 50 μg mL−1 fibronectin) and incubated at 37 °C, 5% CO2 for 30 minutes in a cell culture incubator. After the incubation was done, endothelial media (EGM-2) was perfused through the channel to remove excess matrix protein. HUVECs were then seeded on each face. To seed a face, cells were suspended in media and perfused through the channel. The channel was then placed in an incubator for 1 hour to let the cells attach, either right side up to seed the bottom face, or upside down to seed the top face (2 hours total seeding time). After seeding and cell attachment was complete, the channels were placed under perfusion (1 μL min−1) for 18 hours to allow for cell proliferation and lumen formation (lumenization). After the 18 hour lumenization was complete, different shear stresses and patterns were applied for the time required for each experiment. 3-Dimensional computation fluid analysis of the applied wall shear stress based on the flow rate with corresponding physiological ranges are shown in f, while g details the oscillatory flow pattern used to induce activation (0.3 Hz cycle, 2 seconds withdraw, 1 second infuse). Flow velocity heatmap profiles are shown in h for the unidirectional regimes and the corresponding flow rate, and i details the velocity profile during an oscillatory cycle, showing the 1-dimensional change in flow direction and magnitude.
Endothelial YAP/TAZ mechanobiology in the vessel-chip
Our next goal was to establish that our vessel-chip model recapitulated known patterns of YAP/TAZ biology in endothelial cells: cytoplasmic retention under high, unidirectional (atheroprotective) laminar shear and nuclear partitioning when exposed to disturbed flow.15,16 We began by setting a quasistatic control baseline for HUVECs cultured on the chip (flow <2 μL per hour), as cells cultured in the vessel-chip require perfusion to remain alive and fully static state is not relevant. At these quasistatic conditions, partial YAP nuclear partitioning was observed (nuclear YAP/total YAP; Fig. S2a and b†). HUVECs elongated and aligned weakly along the flow vector, both attributable to culture under flow (Fig. S2c and d†). This baseline established, we moved to study and compare physiologic/pathologic conditions. We examined YAP partitioning via immunofluorescence after 6 hours of the different flow regimes detailed earlier (Fig. 2a and b). In all disturbed flow regimes (low shear 1 μL min−1 and both oscillatory regimes), we observed strong nuclear partitioning of YAP, while under the high atheroprotective regime, YAP was retained strongly in the cytoplasm (Fig. 2c). We note that the quasistatic control YAP partitioning values were statistically identical to that of the oscillatory regimes (p = 0.9), indicating the strong influence of protective shear on YAP cytoplasmic retention and importance of fluid flow. Altogether, all parameters which conventionally classify as disturbed flow strongly induced YAP nuclear partitioning. Although this observation is consistent with previous studies15,16 reporting that disturbed flow results in YAP activation in ECs, we have added some greater resolution to the mechanosensing paradigm, as few studies have explored differences between unidirectional and oscillatory flow at low levels of shear stress. We observed that the flow pattern appears to be the dominant determinant of YAP localization in high shear regimes (10 μL min−1 unidirectional vs. oscillatory). By contrast, when the shear stress is low, ECs are comparably activated by unidirectional or oscillatory flow.
Fig. 2.
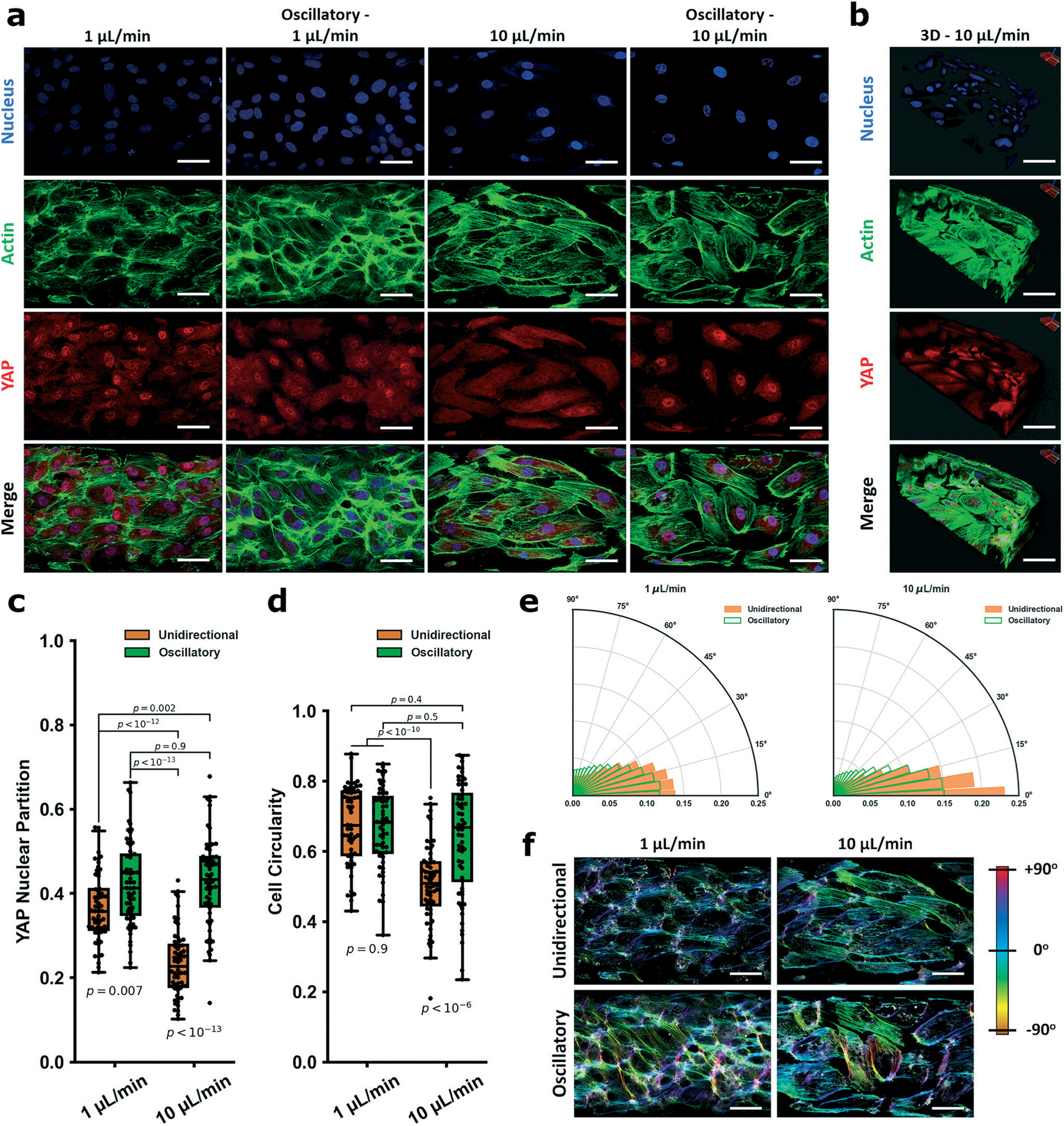
YAP mechanobiological model validation of the vessel-chip system for studying endothelial mechanobiology. In a are representative fields of view within the vessel-on-a-chip device for 1 μL min−1, oscillatory 1 μL min−1, 10 μL min−1, and oscillatory 10 μL min−1 flow. Cells were stained for nuclei (DAPI, blue), actin (phalloidin, green,) and YAP (red). Oscillatory flow was induced by alternating between withdrawal (2 seconds) and infuse (1 second). b Is a Z-stack image of a section of a vessel-on-a-chip showing lumen formation (10 μL min−1). Scale bars in a and b are all 50 μm. YAP partitions for each group are: 1 μL min−1: 0.37 ± 0.08; oscillatory 1 μL min−1: 0.42 ± 0.1; 10 μL min−1: 0.23 ± 0.08; oscillatory 10 μL min−1: 0.43 ± 0.11 (c). The cellular morphology (circularity) is quantified in d. Circularity is defined as 4π(area/perimeter2) and is valued from 0 to 1. Indices closer to 1 indicate more circular cell morphology, while elongated cells have indices closer to 0. CSI measured for the flow fields are: 1 μL min−1: 0.67 ± 0.11; oscillatory 1 μL min−1: 0.67 ± 0.11; 10 μL min−1: 0.51 ± 0.11; oscillatory 10 μL min−1: 0.64 ± 0.16. Directionality assays based on actin alignment detailing how HUVECs align along the flow vector within the fluidic chamber under the different shear regimens/patterns are shown in e. Data is visualized in the form of (r, θ) – r is the normalized density of actin filaments aligned at each angle, θ. Higher values of r for a given θ indicate increasing alignment. For 10 μL min−1, HUVECs aligned strongly along the flow vector, while the effect was evident but not as strong in the 1 μL min−1 regime. HUVECs in both oscillatory flow fields showed a markedly decreased alignment distribution around the flow vector. Color maps showing the gradient of actin alignment (from the representative actin images in a) are visualized in f using OrientationJ. For YAP partition experiments and CSI experiments, n = 60 cells. Alignment polar histogram plots are comprised of an average of 5–6 fields of view, directionality calculated from 0° to 90° binned 15 times in ImageJ. Statistics performed using a single factor ANOVA with post hoc Tukey’s test for significance. Each p-value between groups is reported natively as calculated, significance taken at p < 0.05. All values reported as mean ± standard deviation.
Within our vessel-chip model, we additionally observed common morphology changes associated with disturbed flow. HUVECs exposed to high shear became elongated, while in all disturbed flow regimes cells remained rounded (Fig. 2d). Although EC exposed to both unidirectional regimes aligned primarily along the 0° flow vector, those exposed to high shear displayed a much more prominent alignment peak (frequency peak: 1 μL min−1 = 0.13 vs. 10 μL min−1 = 0.23). HUVECs exposed to oscillatory flow regimes all displayed a blunted alignment (Fig. 2e). Representative color mapping of actin alignment (from Fig. 2a) demonstrates the wide distribution of angles seen in the oscillatory regimes (Fig. 2f). Taken together, our data suggests that the key observations of in vivo YAP behavior are captured within the vessel-chip system, while additionally facilitating novel findings pertaining to the relative influence that shear stress magnitude and pattern have on endothelial activation. Moreover, morphological characteristics of ECs subject to (disturbed) flow were readily captured and shown to be consistent with current understanding.61,62
Endothelial activation and inflammation in the vessel-chip
In the context of vascular mechanobiology, drawing connections between the detrimental mechanoresponse from disturbed flow (YAP nuclear localization and increased activity) and the clinical implications (atherosclerotic progression and vascular disease) would be vital to capture within our model to ensure the mechanobiological changes parallel alterations in endothelial activation. Specifically, does nuclear YAP partitioning (with disturbed flow) parallel endothelial activation and inflammation? Accordingly, we selected an atheroprotective regime (unidirectional high shear) and the corresponding disturbed flow pattern (oscillatory high shear), and assessed the relative level of two molecules known to be induced under disturbed flow and integral to atherosclerotic progression: vascular cell adhesion molecule 1 (VCAM-1) and von Willebrand factor (vWF).5,63–68 We observed reduced levels of both VCAM-1 and vWF when HUVECs were subject to unidirectional high shear, whereas high oscillatory shear greatly increased the levels of both VCAM-1 and vWF (Fig. 3a–d), illustrating the connection between the EC mechanoresponse and EC inflammatory activation.
Fig. 3.
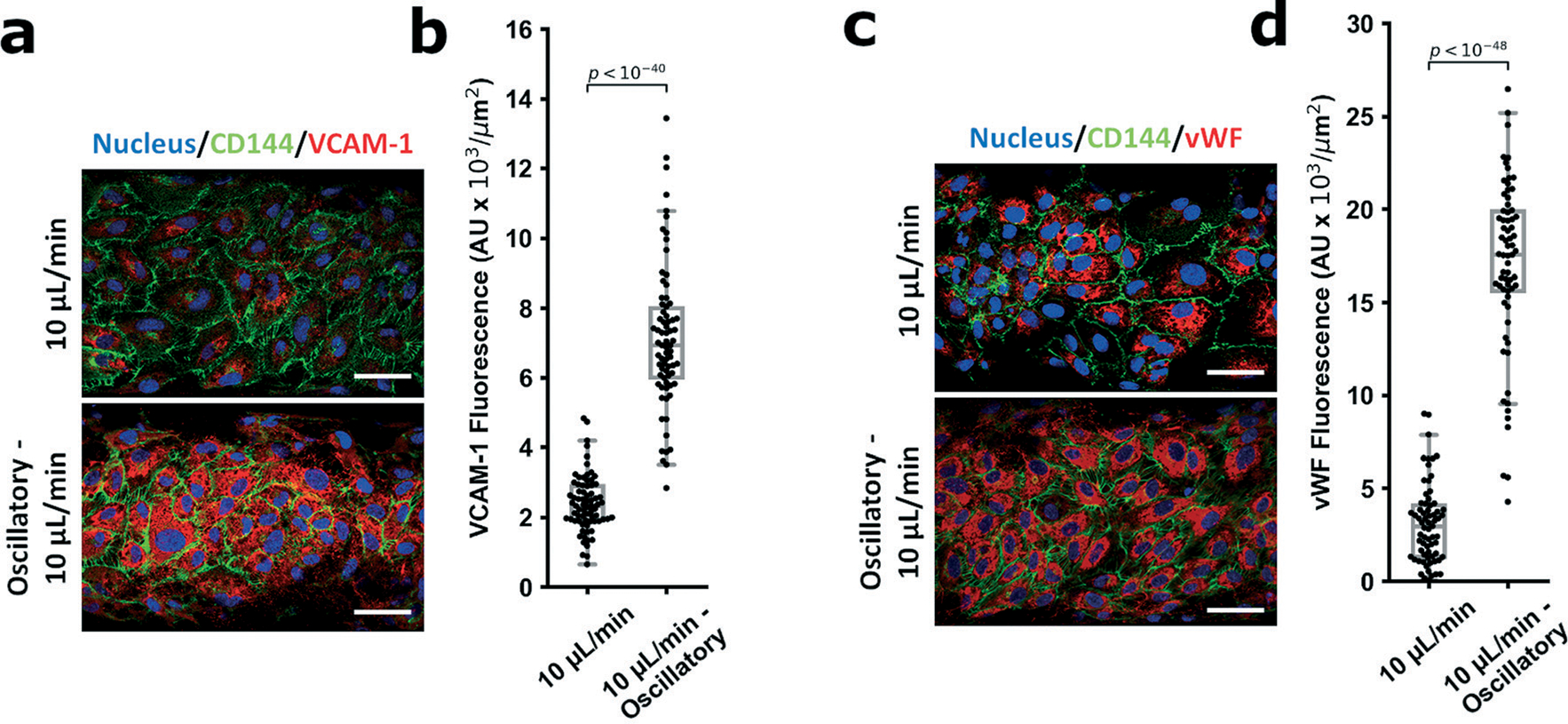
Endothelial activation studies between the different shear regimes. In order to establish the system parameters causing activation of YAP correlate to endothelial activation, 2 markers of endothelial inflammation (VCAM-1 and vWF) were examined under the laminar and oscillatory flow conditions. For both markers, oscillatory flow patterns resulted in marked intensity increases for VCAM-1 (a and b, fluorescence: 10 μL min−1 − 2.4 ± 0.8 AU × 103 μm2, oscillatory 10 μL min−1 − 7.1 ± 2 AU × 103 μm2) and vWF (c and d, fluorescence: 10 μL min−1 − 3.1 ± 2 AU × 103 μm2, oscillatory 10 μL min−1 − 17 ± 5 AU × 103 μm2). For VCAM-1, n = 74 cells; for vWF, n = 69 cells. Statistical analysis was performed using an unpaired Student’s t-test, significance taken at p < 0.05. Data is reported as the normalized intensity (to cellular area), mean ± standard deviation.
Gene expression analysis
Subsequently, we examined gene expression of ECs subject to the various fluid shear stress parameters. The first two genes examined, connective tissue growth factor (CTGF) and ankyrin repeat domain 1 (ANKRD1) are downstream targets of YAP/TAZ activity known to be upregulated by its transcriptional co-activation.15,16 The second two genes, endothelial nitric oxide synthase (eNOS) and vascular endothelial growth factor receptor 2 (VEGFR2), are shear responsive genes59,69–74 not directly tied to YAP activity. Unidirectional high shear downregulated expression of CTGF compared to unidirectional low shear, however ANKRD1 remained relatively unchanged (Fig. 4a). Under the conditions of high oscillatory shear, CTGF and ANKRD1 were both dramatically upregulated (Fig. 4b), consistent with previous reports.15,16 By contrast, at low levels of shear stress, there was no difference between unidirectional and oscillatory shear stress in expression of CTGF and ANKRD1 (Fig. 4c). Taken together, this data suggests than CTGF expression is highly sensitive to both the magnitude and pattern of shear stress. By contrast, the expression of ANKRD1 is not affected by difference in the magnitude of unidirectional shear stress, but is affected by the flow pattern at higher levels of shear stress. Notably, the sensitivity of CTGF to the magnitude of unidirectional shear stress has been previously reported.56 Changes in other known mechanosensitive genes (eNOS and VEGFR2) were modest but in the expected directions (Fig. 4d–f). Summarizing, these data confirm that our chip model replicates the known biological effects of laminar and disturbed flow (i.e. response of eNOS and VEGFR2) and provide new insights into the flow response of genes downstream of YAP/TAZ activation (i.e. CTGF and ANKRD1). Notably, CTGF is highly expressed in atherosclerotic plaques75–77 and knockdown greatly reduces plaque progression.78 This is consistent with our data that CTGF is upregulated by patterns of disturbed flow, which hemodynamic condition is associated with sites in the vasculature that are predisposed to atherosclerosis.
Fig. 4.
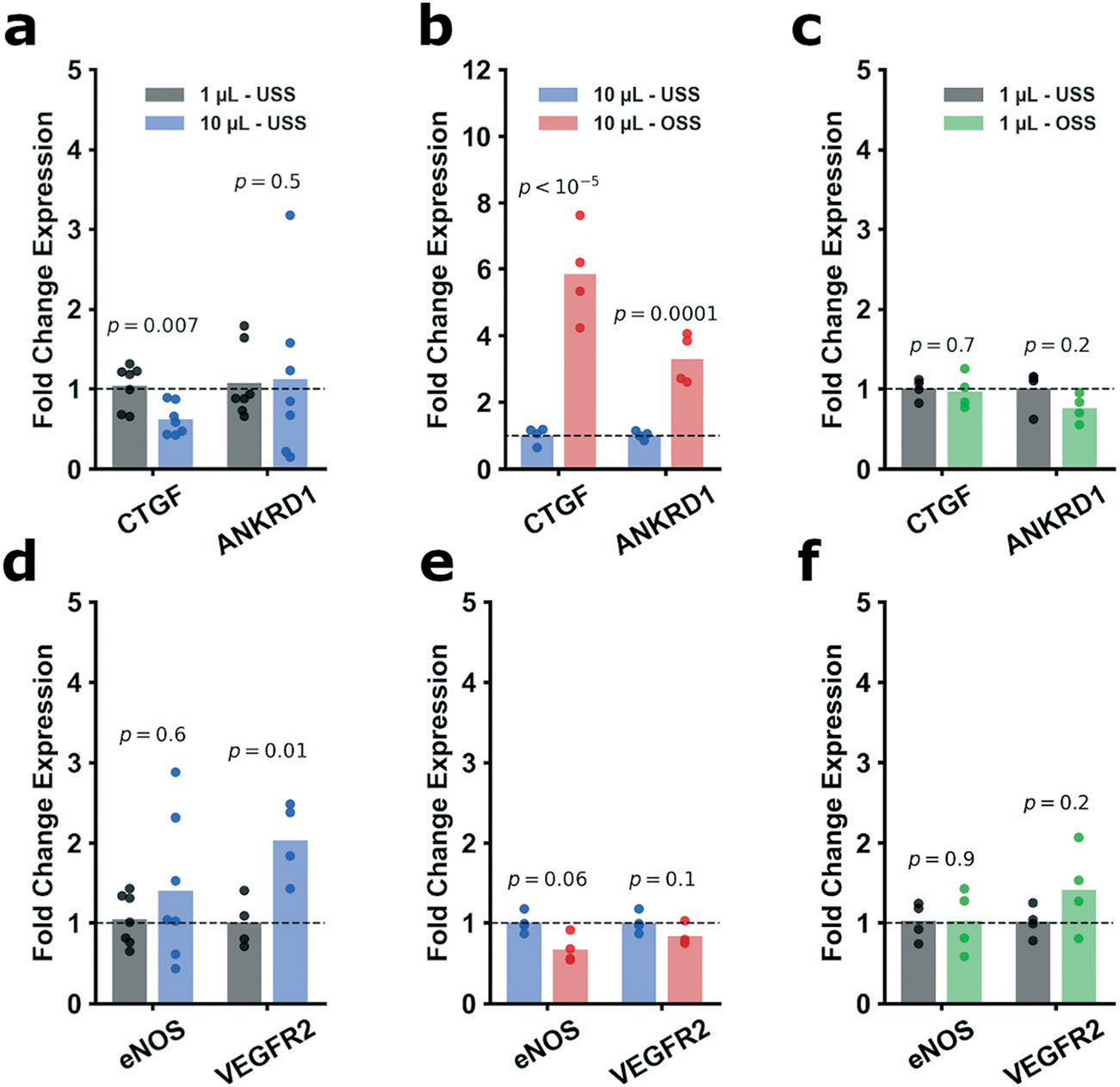
Gene expression for key mechanosensitive genes were selected as transcriptional readouts (CTGF, ANKRD1, eNOS, and VEGFR2). CTGF expression was significantly decreased from 1 μL min−1 to 10 μL min−1 (a, FC = 0.6) while ANKRD1 was not changed (FC = 1.1). However, oscillatory 10 μL min−1 vs. 10 μL min−1 displayed a marked upregulation of both CTGF and ANKRD1 (b, respectively FC = 5.9 and FC = 3.3). Conversely, expression of CTGF and ANKRD1 in the oscillatory 1 μL min−1 vs. 1 μL min−1 regime was not changed (c, FC = 0.97 and FC = 0.76). With regards to eNOS and VEGFR2, from 1 μL min−1 to 10 μL min−1, eNOS was not appreciably changed, though VEGFR2 saw modest upregulation (d, FC = 1.4 and FC = 2.0, respectively). Oscillatory 10 μL min−1 vs. 10 μL min−1 shown downward trends for both genes (e, FC = 0.67 and FC = 0.83) without reaching significance. Oscillatory 1 μL min−1 compared to 1 μL min−1 showed (f, FC = 1.0 and FC = 1.4). For a and d CTGF, ANKRD1, and eNOS, n = 7; all other genes performed n = 4. Statistical analysis was performed on the ddCts for each group using an unpaired Student’s t-test. All p-values are reported natively, significance taken at p < 0.05. Values are reported as FC. GAPDH was selected as the housekeeping gene for all experiments.
Pharmacologic inhibition of YAP activity
To confirm the importance of YAP activity on the flow-mediated regulation of mechanosensitive genes, we employed verteporfin, a potent YAP inhibitor.79–82 We assessed a dose response to verteporfin (0.125–1 μM; Fig. S4a and b†) and assessed YAP nuclear localization of treated cells subject to 10 μL min−1 oscillatory shear. We observed increasing cytoplasmic retention of YAP with increasing concentrations of verteporfin, with 1 μM verteporfin treatments resulting in cytoplasmic retention of YAP comparable to that of ECs subject to high unidirectional shear (Fig. 5a and b). Notably, concentrations above 1 μM induced excessive cellular detachment (Fig. S4c†), which is attributable to verteporfin’s ability to potently induce apoptosis and cell death at high concentrations.83 Additionally, verteporfin treatment did not rescue cell circularity/morphology nor alignment (Fig. 5c and d and S4d†). As concentrations of 1 μM demonstrated the highest tractable cytoplasmic retention, we selected 1 μM as the concentration to examine inhibition of downstream YAP gene expression. Compared the DMSO controls (required for verteporfin solubility), verteporfin strongly suppressed expression of CTGF, however, strikingly ANKRD1 expression was not affected (Fig. 5e). We note that this mirrors the finding of Wang et al.16 when they reported pharmacological inhibition of c-Jun N-terminal kinase (downstream of YAP/TAZ activity) greatly modulated CTGF expression, but minimally affected ANKRD1.16 Thus, this supports that CTGF is predominantly regulated by upstream YAP activity (and thus the EC mechanoresponse), however ANKRD1 is likely to be associated with other regulatory pathways. More generally, these pharmacological studies performed using the vessel-chip system in this manner demonstrate how chemical intervention can influence and reverse the biological response to mechanical cues, and how organ-chip systems support these preclinical and potentially translational studies in a controlled manner.
Fig. 5.
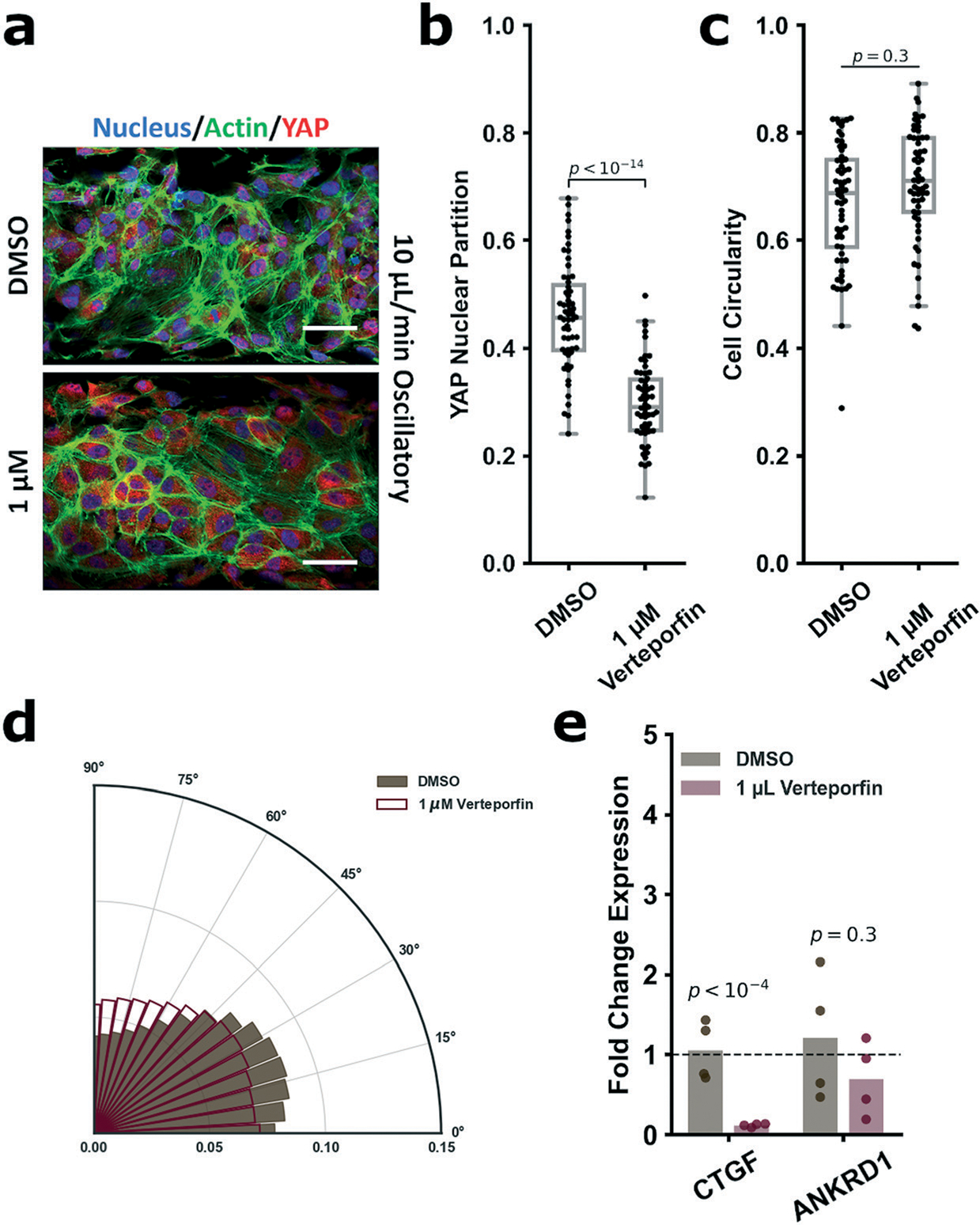
Verteporfin immunofluorescence (for nuclei (DAPI, blue), actin (phalloidin, green) and YAP (red), a) showed decreasing YAP nuclear partitioning in the 1 μM group vs. DMSO control (DMSO: 0.46 ± 0.1; 1 μM: 0.30 ± 0.1, b). Verteporfin treatment did not rescue cellular morphology or alignment under oscillatory flow (c and d). Cell circularity indices for the DMSO and 1 μM verteporfin groups were respectively 0.67 ± 0.1 and 0.71 ± 0.1. Gene expression analysis of CTGF and ANKRD1 post-verteporfin treatment (e) showed significant downregulation of CTGF (FC = 0.11) and insignificant modulation of ANKRD1 (FC = 0.70). Verteporfin treatment IF and circularity performed on n > 59 cells. YAP partition is defined as (nuclear YAP/total YAP). Circularity is defined as 4π(area/perimeter2) and is valued from 0 to 1. Alignment polar histogram plots are comprised of an average of 4–5 fields of view, directionality calculated from 0° to 90° binned 15 times in ImageJ. Data is visualized in the form of (r, θ). Higher values of r for a given θ indicate increasing alignment in that direction. Gene expression performed n = 4. Statistical analysis was performed on the calculated normalized IF values or ddCts for each group using an unpaired Student’s t-test. All p-values are reported natively, significance taken at p < 0.05. Values are reported as circularity, FC, or normalized intensity, mean ± standard deviation. GAPDH was selected as the housekeeping gene for all experiments.
Substrate stiffness impedes the shear mechanoresponse
As YAP is responsive to both substrate stiffness and shear stress, we hypothesized that stiffening the substrate matrix impedes the ability of ECs to fully respond to shear stress, upshifting YAP nuclear localization. The stiffness of the PDMS substrate was modified by varying the crosslinker concentration.49 Arterial stiffnesses range from 50 kPa up to roughly 600 kPa.45–47 Complicating this paradigm is that diseased vasculature and atherosclerotic plaques have a significantly higher range of stiffnesses, from 1 kPa up to 5 MPa.10,11 Thus, we explored a stiffness of the same order of magnitude as those reported in normal muscular arteries (300–500 kPa; such as the coronary arteries)12 as well as stiffness in the range of atherosclerotic vessels (>1 MPa). Accordingly, we reduced the crosslinker density to 7.5% (to yield 1 MPa) and to 5% (500 kPa); ECM functionalization did not alter the PDMS materials properties (Fig. S5†). We subjected endothelial cells grown on these matrices to unidirectional low or high shear and measured YAP localization (Fig. 6a and b). Consistent with our previous experiments, all low shear groups displayed elevated YAP nuclear partitioning consistent with EC activation under flow. Furthermore, all high shear groups displayed significantly decreased nuclear YAP localization. However, higher levels of stiffness attenuated the effect of high shear to reduce YAP nuclear partitioning (Fig. 6c), indicating that stiff matrices impede the protective EC shear mechanoresponse. Also, of note, there was no difference in the nuclear localization with increasing stiffness when shear stress was low. This finding indicates that low shear stress (such as that found at sites of disturbed flow in the large and medium arteries) is strongly activating and a dominant stimulator of YAP activity, EC activation, and vascular inflammation. Interestingly, high shear was sufficient to induce YAP cytoplasmic retention on borosilicate glass (Young’s modulus 64 GPa), displaying a value statistically indistinguishable from the 10% PDMS (10% PDMS = 0.32 ± 0.1; BSG = 0.32 ± 0.1; p = 0.7), which suggests the influence of matrix stiffness on the shear mechanoresponse plateaus past some highly stiff threshold (Fig. S6†). Altogether, this data suggests that ECs reside in a complex physical environment where they balance different physical cues and respond biologically to the convolution of the stimuli (Fig. 6d and e). The protective signals from high shear stress are attenuated by increasing matrix stiffness to a certain threshold, while low shear is universally activating. Notably, this data supports a biological link associating matrix stiffness and flow patterns to EC dysfunction, inflammation, and atherosclerotic progression, consistent with clinical data.
Fig. 6.
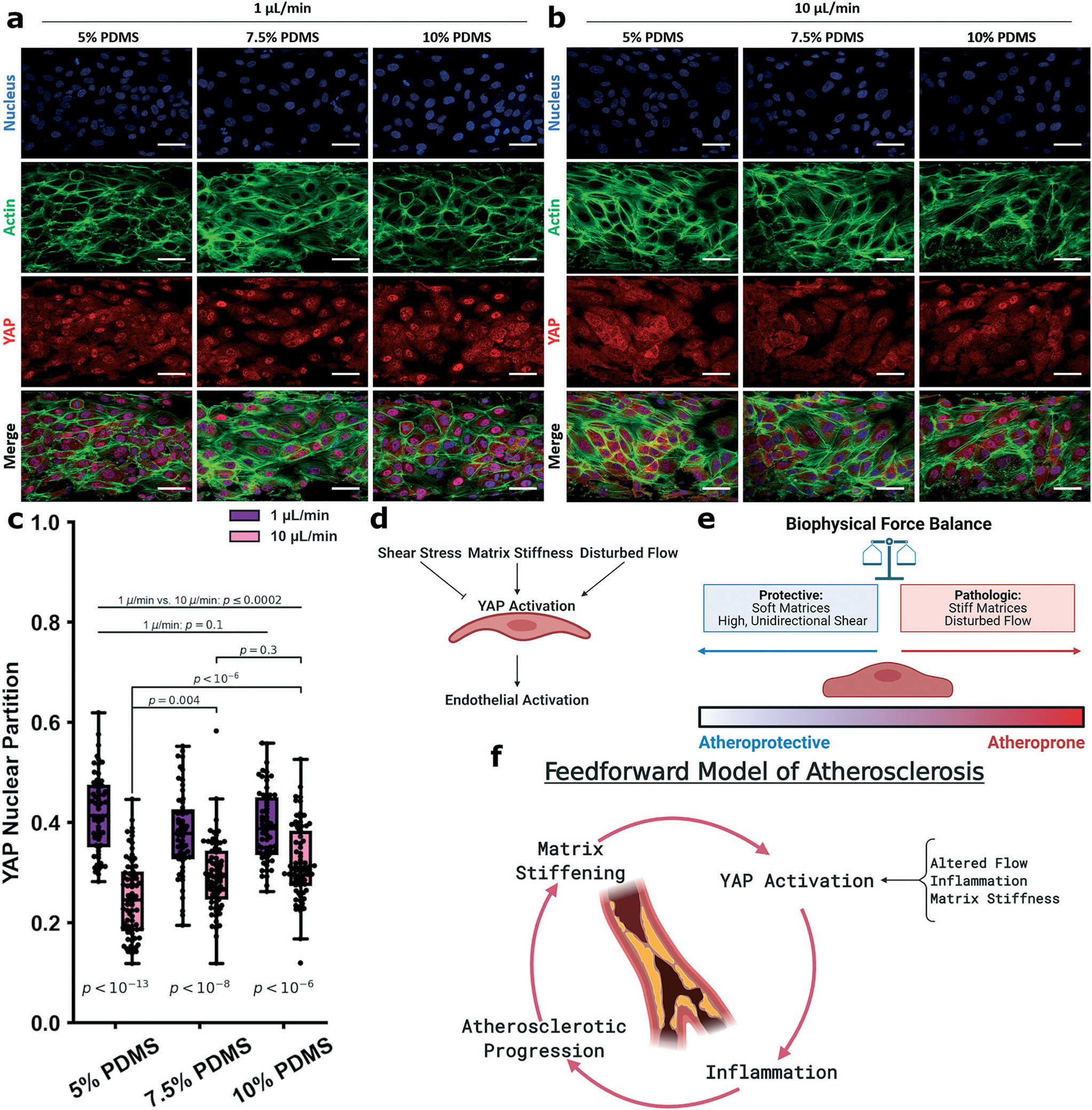
Varying device stiffness to modulate the HUVEC endothelial mechanoresponse. Experiments were performed in devices made form the varied crosslinker concentrations of 5%, 7.5%, and 10% and under the unidirectional shear regimes of 1 μL min−1 and 10 μL min−1. Cells were stained for nuclei (DAPI, blue), actin (phalloidin, green) and YAP (red), shown in a and b. Across the 1 μL min−1 groups, YAP partitioning was not affected by substrate stiffness (5%: 0.42 ± 0.1; 7.5%: 0.38 ± 0.1; 10%: 0.39 ± 0.1), while those subject to 10 μL min−1 showed increasing YAP partition as the substrate stiffened (5%: 0.25 ± 0.1; 7.5%: 0.29 ± 0.1; 10%: 0.32 ± 0.1, c). YAP nuclear partitioning between the 1 μL min−1 and 10 μL min−1 groups were significantly different between any 2 groups from different shear regimes. This suggests that ECs respond to a variety of mechanical cues which can either be protective or inflammatory (d and e). The physical regimes of stiffness and shear stress may be connected via YAP mechanobiology in a feedforward manner where an initial incident can result in propagating stiffening and YAP activity (f). For the 1 μL min−1 groups, n = 56 cells, for the 10 μL min−1 groups, n = 71–73 cells. Statistics performed using a single factor ANOVA with post hoc Tukey’s test for significance. Each p-value between groups is reported natively as calculated, significance taken at p < 0.05. All values reported as mean ± standard deviation.
Conclusions
Here, our report describes the use of a vessel-chip model to arrive at novel understandings of the interaction of physiological forces which conventional methods or models may not facilitate. This type of “bottom-up” engineering approach to biology, combined with insights from current in vitro and in vivo models, may provide a more comprehensive understanding of forces acting on cells in a concerted fashion to result in a single biological response.
In the context of vascular mechanobiology, our vessel-chip model yielded several new insights into the physical cues which govern EC health and homeostasis. First, our data reveals that shear stress magnitudes and patterns are a dominant signal in EC phenotype maintenance. The most dramatic differences were observed between unidirectional and oscillatory flow patterns at high levels of shear. Interestingly, the pattern of flow (unidirectional v. oscillatory) did not appreciably modulate gene expression when ECs were subject to low levels of shear. Thus, low levels of shear (which promote atherosclerosis in the arterial circulation) dominate over the flow pattern with respect to EC signaling. Gene expression analysis across flow regimes and inhibitor studies highlighted CTGF as a highly sensitive measure of the EC mechanoresponse to shear and flow pattern. CTGF was upregulated with disturbed flow, consistent with its increased expression in atherosclerotic vessels. By contrast, ANKRD1 was only affected by the flow pattern at high levels of shear stress. Verteporfin studies confirmed that CTGF but not ANKRD1 is significantly downregulated by YAP inhibition, emphasizing the distinction between these genes.
The most significant finding of this report is the interaction of substrate stiffness with shear stress to modulate the EC YAP mechanoresponse. Here, we observed that increasing the substrate stiffness increased the levels of YAP localized to the nucleus under high shear, however low shear was strongly activating across varying stiffnesses. Our work is consistent with prior studies associating coronary wall stiffness and shear patterns with development of atherosclerosis.10–12 Indeed, the data suggests a feedforward mechanism of EC activation and atherosclerotic progression (Fig. 6f) where some inciting event (either physical or chemical6,84) induces YAP activation in ECs. This, in turns, causes endothelial activation and vascular inflammation, as reported previously.15,16 The endothelial activation and vascular inflammation then drive atherosclerotic progression, which may further stiffen the vascular architecture, activating YAP further. Kohn and colleagues observed that stiffer substrates resulted in lower eNOS production and increased RhoA activation,30 the latter of which interacts with YAP/TAZ.85 Together, their work and ours strongly suggest that matrix stiffness attenuates the endothelial response to physiological shear stress. The combination of a decrease of eNOS (also atherogenic7) and increases in RhoA and YAP/TAZ activity all serve to suggest that matrix stiffening is not only a correlative consequence, but a cause of vascular disease. Indeed, other recent reports indicate that YAP activation is associated with vessel stiffening in the context of pulmonary hypertension,86–88 though do not explore fluid shear stress as deeply.
There are model limitations and interpretation qualifiers important to discuss. Our current vessel-chip model does not incorporate strain, which is known to activate YAP activity as well.39 Furthermore, we have not incorporated a vascular smooth muscle cell layer in our model, which also contributes to vascular disease.89,90 Thus, some of the biological implications of our model findings require additional study to incorporate the interaction of these biological variables with stiffness, shear stress, and flow patterns. Also of note is that the ECM chemical composition itself influences EC behavior,91 which was not a variable considered in our work. We also assumed that the matrix is a uniform, isotropic material, however, blood vessels are composite and anisotropic.46 Lastly, our work focuses on modeling mechanobiological response in the arterial circulation (using HUVECs) which may not be entirely reflective of the venous mechanoresponse. As an example, recent work, including our own, highlights that low (~1 dyne per cm2) recirculating flow in vein leaflets is protective,92,93 rather than activating. Thus, our conclusions are only appropriately applied to arterial biology while venous mechanobiology requires more investigation.
Nonetheless, we believe the methods and resulting conclusions emphasize how engineered organ-chip models can be used in such a manner as to draw upon biological knowledge and facilitate new findings. As evidenced by the work here, organ-chip systems support biological inquiry that fills the gaps between the in vitro models and animal models. Further extension or applications of this work include system sophistication to incorporate additional geometries or cellular layers to study an additional interface, or the inclusion of iPSC-derived ECs or diseased cell models to study perturbations of the shear response induced by internal, cellular dysfunction.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the NIBIB Award # R21EB025945, NSF CAREER Award #1944322 and Texas A&M University President’s Excellence in Research Award (X-Grant) to A. J.; and by funding from the National Heart Lung and Blood Institute, 1R01HL148338 and 1R01HL133254 to J. P. C. Support provided by the consortium of the Center for Bioelectronics, Biosensors and Biochips (C3B®) and from ABTECH Scientific, Inc. The authors acknowledge the support of Texas Engineering Experiment Station (TEES) to A. J., A. K. G, and A. G.-E.: idea: B. K. W., A. J., J. P. C. and A. G.-E.; resources: A. J., A. K. G., J. P. C. and A. G.-E.; investigation: B. K. W., N. K. R. P., K. A. G., E. S. K., V. S., and J. G.; draft preparation, review, and editing: B. K. W., A. G.-E., A. J., J. P. C.
Footnotes
Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Prof. Guiseppi-Elie is founder and scientific director of ABTECH Scientific, Inc., manufacturer of microfabricated biochip devices and systems used in microfluidic systems.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc01283a
References
- 1.Ingber DE, Ann. Med, 2003, 35, 564–577. [DOI] [PubMed] [Google Scholar]
- 2.Ando J and Yamamoto K, Circ. J, 2009, 73, 1983–1992. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham KS and Gotlieb AI, Lab. Invest, 2005, 85, 9–23. [DOI] [PubMed] [Google Scholar]
- 4.Chien S, Ann. Biomed. Eng, 2008, 36, 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu J-J and Chien S, Physiol. Rev, 2011, 91, 327–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominic A, Banerjee P, Hamilton DJ, Le N-T and Abe J.-i., Redox Biol., 2020, 101614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooke JP, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 768–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Popele NM, Grobbee DE, Bots ML, Asmar R, Topouchian J, Reneman RS, Hoeks AP, van der Kuip DA, Hofman A and Witteman JC, Stroke, 2001, 32, 454–460. [DOI] [PubMed] [Google Scholar]
- 9.van Sloten TT, Schram MT, van den Hurk K, Dekker JM, Nijpels G, Henry RM and Stehouwer CD, J. Am. Coll. Cardiol, 2014, 63, 1739–1747. [DOI] [PubMed] [Google Scholar]
- 10.Ohayon J, Teppaz P, Finet G and Rioufol G, Coron. Artery Dis, 2001, 12, 655–663. [DOI] [PubMed] [Google Scholar]
- 11.Ohayon J, Yazdani SK, Malvè M, Gharib AM, Garcia A, Finet G and Pettigrew RI, in Biomechanics of Living Organs, Elsevier, 2017, pp. 193–213. [Google Scholar]
- 12.Ohayon J, Gharib AM, Garcia A, Heroux J, Yazdani SK, Malvè M, Tracqui P, Martinez M-A, Doblare M and Finet G, Am. J. Physiol. Heart Circ. Physiol, 2011, 301, H1097–H1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palombo C and Kozakova M, Vasc. Pharmacol, 2016, 77, 1–7. [DOI] [PubMed] [Google Scholar]
- 14.Totaro A, Panciera T and Piccolo S, Nat. Cell Biol, 2018, 20, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ and Chien S, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y and Huang Y, Nature, 2016, 540, 579–582. [DOI] [PubMed] [Google Scholar]
- 17.Xu S, Koroleva M, Yin M and Jin ZG, Transl. Res, 2016, 176, 18–28 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piccolo S, Dupont S and Cordenonsi M, Physiol. Rev, 2014, 94, 1287–1312. [DOI] [PubMed] [Google Scholar]
- 19.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S and Piccolo S, Cell, 2013, 154, 1047–1059. [DOI] [PubMed] [Google Scholar]
- 20.Dupont S, in The Hippo Pathway, Springer, 2019, pp. 183–202. [Google Scholar]
- 21.Dupont S, Exp. Cell Res, 2016, 343, 42–53. [DOI] [PubMed] [Google Scholar]
- 22.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N and Piccolo S, Nature, 2011, 474, 179–183. [DOI] [PubMed] [Google Scholar]
- 23.Hong W and Guan KL, Semin. Cell Dev. Biol, 2012, 23, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warren JS, Xiao Y and Lamar JM, Cancers, 2018, 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heo KS, Fujiwara K and Abe J, Mol. Cells, 2014, 37, 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HL and Kim SH, Front. Cardiovasc. Med, 2019, 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diamond SL, Circ. Res, 2016, 118, 1348–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panciera T, Azzolin L, Cordenonsi M and Piccolo S, Nat. Rev. Mol. Cell Biol, 2017, 18, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brusatin G, Panciera T, Gandin A, Citron A and Piccolo S, Nat. Mater, 2018, 17, 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohn JC, Zhou DW, Bordeleau F, Zhou AL, Mason BN, Mitchell MJ, King MR and Reinhart-King CA, Biophys. J, 2015, 108, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Korolj A, Lai BFL and Radisic M, Nat. Rev. Mater, 2018, 3, 257–278. [Google Scholar]
- 32.Gold K, Gaharwar AK and Jain A, Biomaterials, 2019, 196, 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benam KH, Dauth S, Hassell B, Herland A, Jain A, Jang KJ, Karalis K, Kim HJ, MacQueen L, Mahmoodian R, Musah S, Torisawa YS, van der Meer AD, Villenave R, Yadid M, Parker KK and Ingber DE, Annu. Rev. Pathol, 2015, 10, 195–262. [DOI] [PubMed] [Google Scholar]
- 34.Bhatia SN and Ingber DE, Nat. Biotechnol, 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- 35.Bhise NS, Ribas J, Manoharan V, Zhang YS, Polini A, Massa S, Dokmeci MR and Khademhosseini A, J. Controlled Release, 2014, 190, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Meer AD, Orlova VV, ten Dijke P, van den Berg A and Mummery CL, Lab Chip, 2013, 13, 3562–3568. [DOI] [PubMed] [Google Scholar]
- 37.Jain A, van der Meer AD, Papa AL, Barrile R, Lai A, Schlechter BL, Otieno MA, Louden CS, Hamilton GA, Michelson AD, Frelinger AL 3rd and Ingber DE, Biomed. Microdevices, 2016, 18, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandian NKR, Mannino RG, Lam WA and Jain A, Curr. Opin. Biomed. Eng, 2018, 5, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lundin V, Sugden WW, Theodore LN, Sousa PM, Han A, Chou S, Wrighton PJ, Cox AG, Ingber DE and Goessling W, Dev. Cell, 2020, 52(4), 446–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vázquez-Victorio G, Peto-Gutiérrez C, Díaz-Bello B, Cano-Jorge M, Pérez-Calixto D, Jiménez-Escobar A, Espinosa-Matías S, Martínez RL, Courson R and Malaquin L, Lab Chip, 2019, 19, 3512–3525. [DOI] [PubMed] [Google Scholar]
- 41.Tovar-Lopez F, Thurgood P, Gilliam C, Nguyen N, Pirogova E, Khoshmanesh K and Baratchi S, Front. Bioeng. Biotechnol, 2019, 7, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohammed M, Thurgood P, Gilliam C, Nguyen N, Pirogova E, Peter K, Khoshmanesh K and Baratchi S, Anal. Chem, 2019, 91, 12077–12084. [DOI] [PubMed] [Google Scholar]
- 43.Inglebert M, Locatelli L, Tsvirkun D, Sinha P, Maier JA, Misbah C and Bureau L, Biomicrofluidics, 2020, 14, 024115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurgood P, Suarez SA, Pirogova E, Jex AR, Peter K, Baratchi S and Khoshmanesh K, Small, 2020, 2003612. [DOI] [PubMed] [Google Scholar]
- 45.Akhtar R, Sherratt MJ, Cruickshank JK and Derby B, Mater. Today, 2011, 14, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guimarães CF, Gasperini L, Marques AP and Reis RL, Nat. Rev. Mater, 2020, 1–20. [Google Scholar]
- 47.Ebrahimi AP, J. Vasc. Interv. Radiol, 2009, 2, 155. [PMC free article] [PubMed] [Google Scholar]
- 48.Mathur T, Singh KA, NK RP, Tsai SH, Hein TW, Gaharwar AK, Flanagan JM and Jain A, Lab Chip, 2019, 19, 2500–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mata A, Fleischman AJ and Roy S, Biomed. Microdevices, 2005, 7, 281–293. [DOI] [PubMed] [Google Scholar]
- 50.Gavara N and Chadwick RS, Nat. Nanotechnol, 2012, 7, 733–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao B, Li L, Wang L, Wang C-Y, Yu J and Guan K-L, Genes Dev, 2012, 26, 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barber RD, Harmer DW, Coleman RA and Clark BJ, Physiol. Genomics, 2005, 21, 389–395. [DOI] [PubMed] [Google Scholar]
- 53.Püspöki Z, Storath M, Sage D and Unser M, in Focus on Bio-Image Informatics, Springer, 2016, pp. 69–93. [Google Scholar]
- 54.Hunter JD, Comput. Sci. Eng, 2007, 9, 90–95. [Google Scholar]
- 55.Inoguchi H, Tanaka T, Maehara Y and Matsuda T, Biomaterials, 2007, 28, 486–495. [DOI] [PubMed] [Google Scholar]
- 56.McCormick SM, Eskin SG, McIntire LV, Teng CL, Lu C-M, Russell CG and Chittur KK, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 8955–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagel T, Resnick N, Atkinson WJ, Dewey CF and Gimbrone MA, J. Clin. Investig, 1994, 94, 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chappell DC, Varner SE, Nerem RM, Medford RM and Alexander RW, Circ. Res, 1998, 82, 532–539. [DOI] [PubMed] [Google Scholar]
- 59.Heo KS, Berk BC and Abe J, Antioxid. Redox Signaling, 2016, 25, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ajami NE, Gupta S, Maurya MR, Nguyen P, Li JY, Shyy JY, Chen Z, Chien S and Subramaniam S, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 10990–10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao S, Suciu A, Ziegler T, Moore JE Jr, Bürki E, Meister J-J and Brunner HR, Arterioscler., Thromb., Vasc. Biol, 1995, 15, 1781–1786. [DOI] [PubMed] [Google Scholar]
- 62.Potter CM, Lundberg MH, Harrington LS, Warboys CM, Warner TD, Berson RE, Moshkov AV, Gorelik J, Weinberg PD and Mitchell JA, Arterioscler., Thromb., Vasc. Biol, 2011, 31, 384–391. [DOI] [PubMed] [Google Scholar]
- 63.Doddapattar P, Dhanesha N, Chorawala MR, Tinsman C, Jain M, Nayak MK, Staber JM and Chauhan AK, Arterioscler., Thromb., Vasc. Biol, 2018, 38, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsao PS, Buitrago R, Chan JR and Cooke JP, Circulation, 1996, 94, 1682–1689. [DOI] [PubMed] [Google Scholar]
- 65.Tsao PS, Buitrago R, Chan JR and Cooke JP, Circulation, 1996, 94, 1682–1689. [DOI] [PubMed] [Google Scholar]
- 66.Hwang S-J, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, Gotto AM Jr and Boerwinkle E, Circulation, 1997, 96, 4219–4225. [DOI] [PubMed] [Google Scholar]
- 67.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos J-C, Connelly PW and Milstone DS, J. Clin. Investig, 2001, 107, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ley K and Huo Y, J. Clin. Investig, 2001, 107, 1209–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H and Schwartz MA, Nature, 2005, 437, 426–431. [DOI] [PubMed] [Google Scholar]
- 70.Tuttle JL, Nachreiner RD, Bhuller AS, Condict KW, Connors BA, Herring BP, Dalsing MC and Unthank JL, Am. J. Physiol. Heart Circ., 2001, 281, H1380–H1389. [DOI] [PubMed] [Google Scholar]
- 71.Fleming I, Fisslthaler B, Dixit M and Busse R, J. Cell Sci, 2005, 118, 4103–4111. [DOI] [PubMed] [Google Scholar]
- 72.Jin Z-G, Ueba H, Tanimoto T, Lungu AO, Frame MD and Berk BC, Circ. Res, 2003, 93, 354–363. [DOI] [PubMed] [Google Scholar]
- 73.Gee E, Milkiewicz M and Haas TL, J. Cell. Physiol, 2010, 222, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nigro P, Abe J.-i. and Berk BC, Antioxid. Redox Signaling, 2011, 15, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fan W-H, Pech M and Karnovsky MJ, Eur. J. Cell Biol, 2000, 79, 915–923. [DOI] [PubMed] [Google Scholar]
- 76.Cicha I, Yilmaz A, Klein M, Raithel D, Brigstock DR, Daniel WG, Goppelt-Struebe M and Garlichs CD, Arterioscler., Thromb., Vasc. Biol, 2005, 25, 1008–1013. [DOI] [PubMed] [Google Scholar]
- 77.Cicha I, Yilmaz A, Suzuki Y, Maeda N, Daniel WG, Goppelt-Struebe M and Garlichs CD, Clin. Hemorheol. Microcirc, 2006, 35, 203–206. [PubMed] [Google Scholar]
- 78.Niu N, Xu S, Xu Y, Little PJ and Jin Z-G, Trends Pharmacol. Sci, 2019, 40, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, Lu Y and Mills GB, Am. J. Cancer Res, 2016, 6, 27. [PMC free article] [PubMed] [Google Scholar]
- 80.Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong J, Sun X and Li J, Cancer Sci, 2017, 108, 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feng J, Gou J, Jia J, Yi T, Cui T and Li Z, OncoTargets Ther, 2016, 9, 5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brodowska K, Al-Moujahed A, Marmalidou A, zu Horste MM, Cichy J, Miller JW, Gragoudas E and Vavvas DG, Exp. Eye Res, 2014, 124, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma Y-W, Liu Y-Z and Pan J-X, Am. J. Cancer Res, 2016, 6, 2816. [PMC free article] [PubMed] [Google Scholar]
- 84.Dominic A, Hamilton D and Abe J.-i., J. Thromb. Thrombolysis, 2020, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R and Piazza S, Nat. Cell Biol, 2014, 16, 357–366. [DOI] [PubMed] [Google Scholar]
- 86.Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE and Chan SY, Cell Rep, 2015, 13, 1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y and Zhang Y-Y, J. Clin. Invest, 2016, 126, 3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ and Fredenburgh LE, Am. J. Physiol. Lung Cell Mol. Physiol, 2017, 313, L628–L647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ross R and Glomset JA, Science, 1973, 180, 1332–1339. [DOI] [PubMed] [Google Scholar]
- 90.Gomez D and Owens GK, Cardiovasc. Res, 2012, 95, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Young EW, Wheeler AR and Simmons CA, Lab Chip, 2007, 7, 1759–1766. [DOI] [PubMed] [Google Scholar]
- 92.Welsh JD, Hoofnagle MH, Bamezai S, Oxendine M, Lim L, Hall JD, Yang J, Schultz S, Engel JD and Kume T, J. Clin. Invest, 2019, 129(12), 5489–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajeeva Pandian NK, Walther BK, Suresh R, Cooke JP and Jain A, Small, 2020, 16, 2003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.