


Abstract
The C-terminal domain (CTD) downstream from residue 235 of Escherichia coli RNA polymerase α subunit is involved in recognition of the promoter UP element. Here we have demonstrated, by DNase I and hydroxyl radical mapping, the presence of two UP element subsites on the promoter D of phage T7, each located half and one-and-a-half helix turns, respectively, upstream from the promoter –35 element. This non-typical UP element retained its αCTD-binding capability when transferred into the genetic environment of the rrnBP1 basic promoter, leading to transcription stimulation as high as the typical rrnBP1 UP element. Chemical protease FeBABE conjugated to αCTD S309C efficiently attacked the T7D UP element but not the rrnBP1 UP element. After alanine scanning, most of the amino acid residues that were involved in rrnBP1 interaction were also found to be involved in T7D UP element recognition, but alanine substitution at three residues had the opposite effect on the transcription activation between rrnBP1 and T7D promoters. Mutation E286A stimulated T7D transcription but inhibited rrnBP1 RNA synthesis, while L290A and K304A stimulated transcription from rrnBP1 but not the T7D promoter. Taken together, we conclude that although the overall sets of amino acid residues responsible for interaction with the two UP elements overlap, the mode of αCTD interaction with T7D UP element is different from that with rrnBP1 UP element, involving different residues on helices III and IV.
INTRODUCTION
The basic characteristics of nucleotide sequences that allow Escherichia coli RNA polymerase to distinguish the promoter from non-promoter DNA have been, and remain, the subject of intensive studies. Two specific functional domains (regions 2.4 and 4.2) of the σ70 subunit, the promoter recognition unit of RNA polymerase, interact with two canonical sequence elements (signals –10 and –35), each consisting of six nucleotide pairs in length, of the promoter (1). The level of sequence conservation of canonical hexamers is, however, usually moderate (7–9 bp matches within a total of 12 bp) (2). This difference in the sequences between various promoters is related to differential gene expression and efficient regulation. For many promoters, additional sequence elements are involved to increase the affinity and/or specificity of selection by the RNA polymerase. For instance, dinucleotide TG located 1 bp upstream from the –10 element in the ‘extended –10’ promoters makes an additional contact with the domain 2.5 of the σ70 subunit (3).
The ‘UP element’ located upstream from the promoter –35 element interacts with the C-terminal domain of α subunit, hereafter called αCTD, and enhances transcription (4–6). Structural studies of UP element–αCTD interaction indicated that αCTD associates with the DNA minor groove within A/T-rich sequences (7–9). The typical AT-rich UP element could be found only in a limited set of promoters, most of which are highly expressed in growing cells (10), even though physical interactions between the RNA polymerase and the upstream promoter DNA are registered in the majority of promoters tested (11,12). Based on the statistical analysis, a set of frequently occurring sequence motifs, which are different from the rrnBP1-type UP element, has been identified in the corresponding region (13). We then performed several lines of study to examine possible involvement of these non-typical UP elements in transcription regulation and to identify structural modules of the RNA polymerase involved in their recognition. Based on the multipoint monitoring of transcription complexes with T7D and uxuAB (14), we found that in the promoter open complexes, these non-typical UP elements are located close to the C-terminal end of the αCTD helix IV instead of the helix I for typical UP element recognition. These findings together raised the possibility of an alternative mode for αCTD–DNA interaction. This study was undertaken in order to characterize both DNA and protein elements involved in this interaction.
MATERIALS AND METHODS
Construction and purification of mutant α subunits
Thirty point, one deletion and one single-Cys mutant derivatives of the rpoA gene were used in this study (Table 1). The expression plasmids for four mutant rpoA genes, C54AC131AC176AC269AS309C, R265A, K297A and K298A, were constructed previously (6), while the pGEMA series expression plasmids for the other 22 mutants were constructed using a set of the rpoA mutant genes provided by R. Gourse (Table 1). The other five point mutants, P293A, N294A, E302A, K304A and L312A, were prepared in this study by the single-strand template mutagenesis method (15) using pGEMA as a parent plasmid. All the mutant constructions (both newly prepared and recloned) were checked by DNA sequencing performed with a DSQ-1000L DNA sequencer (Shimadzu, Japan). Mutant α proteins were purified according to Fujita and Ishihama (16) (see Table 1 for details) from over-expressed cell lysates using HPLC system with a Poros HQ/H (4.6 × 100 mm; PerSeptive Biosystems) column. As a control the α subunit with deleted CTD up to residue 235 (α235) was prepared by the standard procedure (16). The purity of all α proteins was >95% as judged by SDS–polyacrylamide electrophoresis (PAGE).
Table 1. Mutant α subunits.
| Mutation | Conservationa | Fraction | Source |
|---|---|---|---|
| 1. R265A | + | Supernatant | Ref. 6 |
| 2. R284A | + | Precipitate | R. Gourse |
| 3. T285A | + | Supernatant | R. Gourse |
| 4. E286A | + | Precipitate | R. Gourse |
| 5. V287A | +(E)b | Precipitate | R. Gourse |
| 6. E288A | + | Precipitate | R. Gourse |
| 7. L289A | + | Supernatant | R. Gourse |
| 8. L290A | Precipitate | R. Gourse | |
| 9. K291A | + | Supernatant | R. Gourse |
| 10. T292A | +(I) | Precipitate | R. Gourse |
| 11. P293A | Precipitate | This work | |
| 12. N294A | + | Precipitate | This work |
| 13. L295A | +(F) | Supernatant | R. Gourse |
| 14. G296A | + | Supernatant | R. Gourse |
| 15. K297A | Supernatant | Ref. 6 | |
| 16. K298A | + | Supernatant | Ref. 6 |
| 17. S299A | + | Supernatant | R. Gourse |
| 18. L300A | Precipitate | R. Gourse | |
| 19. T301A | +(K) | Precipitate | R. Gourse |
| 20. E302A | Precipitate | This work | |
| 21. I303A | + | Supernatant | R. Gourse |
| 22. K304A | Supernatant | This work | |
| 23. D305A | Precipitate | R. Gourse | |
| 24. V306A | Precipitate | R. Gourse | |
| 25. L307A | + | Supernatant | R. Gourse |
| 26. S309A | +(K) | Precipitate | R. Gourse |
| 27. R310A | + | Precipitate | R. Gourse |
| 28. G311A | Precipitate | R. Gourse | |
| 29. L312A | + | Supernatant | This work |
| 30. R317A | Precipitate | R. Gourse | |
| 31. Δ235 | Supernatant | Ref. 16 |
a+ indicates the amino acid residues conserved among RNA polymerase α subunits from prokaryots and chloroplasts.
bThe residues shown in parenthesis incidate the amino acid residues conserved in organisms other than E.coli.
Preparation of FeBABE-tethered α subunit
Conjugation of iron(S)-1-(p-bromoacetamidobenzyl)-ethylenediaminetetraacetate (FeBABE) to a single Cys residue at position 309 was carried out as described previously (14,17). In brief, 0.25 ml of 40 µM solution of the single Cys mutant α dissolved in 20 mM MOPS (pH 8.0 at 37°C), 10 mM MgCl2, 0.2 M KCl, 0.1 mM EDTA and 6 M urea was mixed with 3.4 µl of 30 mM FeBABE solution in dimethyl sulfoxide. The reaction was carried out at 37°C for 1 h and then terminated by adding 0.25 ml of 1 M Tris–HCl (pH 8.0). Unconjugated FeBABE was removed by gel filtration through a Sephadex G-50 column (1 × 8 cm) in the same buffer. The level of modification, estimated by measuring unmodified Cys (18), was found to be 53%. The effect of chemical modification on the transcription activity was <20%, estimated as described previously (14).
Reconstitution of holoenzymes
The β, β′ and σ subunits were purified from over-expressed E.coli as described previously (16). The reconstitution of core enzyme was carried out using the purified β and β′ subunits, and either wild-type or mutant α subunits according to the standard procedure (16). Assembled RNA polymerases were separated from unassembled subunits and subassemblies by chromatography through a heparin–agarose column (HiTrapTM, Pharmacia) using a HPLC system. Proteins were eluted with a linear gradient of 0–1.5 M NaCl in 0.01 M Tris–HCl (pH 8.0), 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT) and 5% glycerol, giving the protein peaks in the order of α dimer, α2β and core enzyme with the increase in NaCl concentration. Peak fractions containing the reconstituted core enzyme were pooled, dialyzed against the storage buffer (10 mM Tris–HCl pH 7.8, 10 mM MgCl2, 0.2 M KCl, 0.1 mM EDTA, 1 mM DTT and 50% glycerol) and stored at –30 or –80°C until use. For reconstitution of RNA polymerase containing FeBABE-tethered α subunit, DTT was removed from all protein solutions and reaction mixtures for reconstitution.
Holoenzyme was prepared by adding purified σ70 to the reconstituted core enzyme at a molar ratio of 4:1.
Transcription assay
Transcription activity was tested under the conditions of single-round transcription as described previously (19,20). In brief, the formation of open complexes was performed in preincubation mixture which contained 35 µl of 50 mM Tris–HCl (pH 8.0 at 37°C), 3 mM Mg acetate, 50 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT, 25 µg/ml of nuclease-free bovine serum albumin, template DNA and RNA polymerase. Transcription was carried out under template excess conditions so as to detect changes in the enzyme activity with maximum sensitivity. The templates used were: a 355 bp fragment containing promoter T7D (T7D transcript, 224 bases) (14); plasmid pWR52 carrying promoters rrnBP1 and RNA-I (4). The mixture was incubated for 30 min at 37°C for open complex formation with T7D or promoter. Open complex formation with rrnBP1 was carried out at 22°C and in the presence of ATP and CTP for stabilization of the open complexes formed (4). Mixed transcription assays performed in the presence of T7D promoter fragment and pWR52 were carried out at both 37 and 22°C in the presence of ATP and CTP.
Transcription was initiated by adding 15 µl of a prewarmed substrate–heparin mixture in the same buffer. The final concentration was: 160 µM for ATP, GTP and CTP; 50 µM for [α-32P]UTP (2 µCi per reaction) and 200 µg/ml heparin. RNA synthesis was allowed for 15 min at 37 or 22°C and terminated by adding 50 µl of stopping solution (40 mM EDTA and 200 µg/ml yeast tRNA). RNA was precipitated with ethanol and analyzed by 5–8% PAGE in the presence of 8 M urea. Gels were exposed to imaging plates and the plates were analyzed with a BioImage Analyzer BAS2000 (Fuji, Tokyo).
DNase I footprinting assays
Open complex formation between T7D or hybrid promoter constructions (see Fig. 3) and wild-type or mutant RNA polymerases was carried out by the same procedure as in the case of transcription assay. Templates used for topological studies were generated by PCR using 32P-end-labeled primers and either DNA of phage T7 (wild-type T7D promoter) or plasmid pWR52 (rrnBP1 and its derivatives containing T7D-rrnBP1 hybrid promoters). PCR products were separated by 5% PAGE and promoter-containing fragments were eluted by a standard procedure. To identify the RNA polymerase contact region on both strands of the T7D template with high fidelity, we used a short fragment of 213 bp in length (–131/+82). All fragments bearing the hybrid promoter constructions have 77 bp long upstream and 82 bp long downstream sequences with respect to the transcription start point (see Fig. 3). For open complex formation, the reconstituted holoenzyme (0.15–1.0 µM) containing wild-type or mutant α subunits was mixed with 32P-end-labeled DNA fragment (15 nM) in 20 µl of the transcription buffer. DNase I (2 µl) was added after 30 min of incubation at 37°C (final concentration, 1 µg/ml) and the digestion reaction was allowed for 20 s. Cleavage was terminated by adding 22 µl of a stopping solution containing 8 M ammonium acetate, 100 µg/ml yeast tRNA and 20 mM EDTA. DNA was precipitated with ethanol and analyzed by 10% PAGE containing 6 M urea. Gels were exposed to imaging plates and the plates were analyzed with a BioImage Analyzer BAS2000 (Fuji, Tokyo).
Figure 3.

Construction of T7D-rrnBP1 hybrid promoters and location of the footprint signals. Starting from the rrnBP1 promoter, sequence elements of T7D promoter were inserted at various positions so as to generate hybrid promoters, RF, RC, RFC and DR. Large grey arrows and brackets indicate the modified sequences. Signals obtained by DNase I and hydroxyl radical footprintings, shown in Figure 4, are indicated along these hybrid promoter sequences. Symbols are the same as in Figures 2 and 4. Guanines displaying increased reactivity against DMS in transcription complex (data not shown) are indicated by dots below the sequences.
Hydroxy radical footprinting
For open complex formation, the reconstituted holoenzyme (150 nM) containing wild-type or E286A mutant α subunits was mixed with 32P-end-labeled DNA fragment (15 nM) in 40 µl of the transcription buffer and complex formation was allowed at 37°C. The procedure of hydroxyl radical treatment was performed essentially as described by Tullius and Dombroski (21). In brief, after 30 min of incubation, 0.48 µl of 100 mM sodium ascorbate was added followed by the addition of 3.2 µl of 0.15% (v/v) H2O2 and 4.8 µl of a freshly prepared mixture of 50 mM Fe(NH4)2(SO4)2 and 100 mM Na2EDTA. A digestion reaction was allowed for 2 min at 37°C, after which 48 µl of 0.3 M NaCl and 64 µl of 20 mM thiourea were added to terminate the reaction. The samples were precipitated with ethanol and analyzed as described above.
Dimethylsulfate modification
Dimethylsulfide (DMS) (1 µl) was dissolved in 19 µl of DMS-buffer (50 mM sodium cacodylate and 1 mM EDTA) immediately before use, and an aliquot of 1 µl was added to the 32P-end-labeled DNA fragments (15 nM) in 20 µl of the standard transcription buffer. Modification was allowed for 20 s and terminated by the addition of 5.25 µl of 1.5 M sodium acetate, 1 M β-mercaptoethanol and 100 µg/ml of yeast tRNA. For the strand-cleavage reaction at methylated guanine residues, DNA was ethanol precipitated, dried, resuspended in 100 µl of 1 M piperidine, heated at 90°C for 30 min and lyophilized for dryness. Dried DNA samples were dissolved in 20 µl of water and lyophilized again before loading on to the sequencing gel (22). For methylation of both adenine and guanine residues, DMS-treated samples were resuspended in 20 µl of cold water and treated with 200 mM perchloric acid (23). Samples were kept on ice for 30 min, precipitated with ethanol, treated with piperidine and analyzed as described above.
FeBABE-mediated cleavage of DNA
The DNA cleavage reaction induced by protein-bound FeBABE was carried out as described previously (14,17). In brief, the mixture of 32P-end-labeled DNA fragments (6 nM) and FeBABE-conjugated RNA polymerase (20 nM) was incubated at 22 or 37°C for 10 min in 90 µl of DNA cleavage buffer (40 mM HEPES pH 8.0, 10 mM MgCl2, 0.1 M KCl, 0.1 mM EDTA and 5% glycerol). To remove non-specifically bound RNA polymerase, salmon sperm DNA (final concentration, 40 µg/ml) was added at 30 s before the start of the DNA cleavage reaction. Open complex formation with rrnBP1 was carried out in the presence of 160 µM of ATP and CTP. The cleavage reaction was initiated by the addition of sodium ascorbate (pH 7.0; final concentration 2 mM) and hydrogen peroxide (final concentration 1 mM) and then allowed to proceed for 20 min before quenching with 0.1 M thiourea and 100 µg/ml sonicated salmon sperm DNA. After dilution with 10 mM Tris–HCl (pH 7.4)–0.1 mM EDTA buffer up to a total volume of 200 µl, DNA was extracted with phenol/chloroform, precipitated with ethanol and analyzed by electrophoresis on 8% polyacrylamide gel containing 8 M urea. Gels were exposed to imaging plates and plates were visualized with BAS2000 BioImage analyzer. The templates used were 213 bp DNA fragments, bearing either T7D or rrnBP1 promoter region (from –131 to +82). Both fragments were prepared by polymerase chain reaction using either phage T7-DNA or plasmid pSLUP (14).
RESULTS
RNA polymerase α subunit contact sites on the T7D promoter
Previously we analyzed the topology of binary complexes between RNA polymerase and promoter T7D by the spectroscopic observation of a fluorescent probe, fluoresceinmonomercur acetate and contact-dependent DNA cleavage with FeBABE probe, both tethered to single C269 or C309 (14). These analyses indicated that C269, which is involved in interaction with the rrnBP1 promoter UP element (5,6,24), does not closely approach the surface of T7D promoter DNA. Instead the T7D promoter interacts with the α subunit surface including C309, which is separated from C269 by ∼25 Å (24).
When FeBABE was conjugated to C309 of the α subunit, two subsites are attacked by hydroxyl radicals generated by the FeBABE (Fig. 1, right panel) (14). Strong cleavage was registered 3–6 bp upstream from the –35 element (phosphodiester bonds –40/–39/–38/–37 on the top strand and –41/–40/–39 on the bottom strand), and weaker digestion was observed approximately one helix turn upstream (bonds –49/–48/–47 on the top strand and –53/–52/–51/–50 on the bottom strands) (for the location see Fig. 6). The DNA backbone of rrnBP1 was, however, not cleaved by FeBABE conjugated at position C309 (Fig. 1, left panel), thus confirming the topological difference of the open transcription complexes formed with these two templates.
Figure 1.
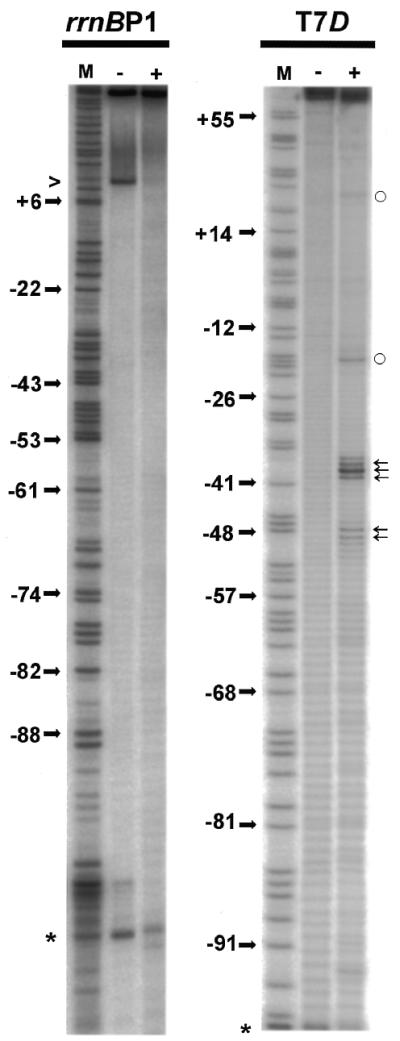
Site-specific DNA cleavage within open complexes formed by the holoenzyme carrying FeBABE-tethered S309C mutant α subunit. DNA fragments (213 bp) containing either T7D or rrnBP1 promoter (from –131 to +82) were 32P-labeled at position –131 of the top strand. Open complexes were formed with the RNA polymerase containing of FeBABE conjugated S309C mutant α subunit. The DNA cleavage reaction was performed as described in Materials and Methods. Lane (–) represents DNA (6 nM) treated with reducing agents in the absence of RNA polymerase while lane (+) shows DNA extracted from the open complex (formed in the presence of 20 µM modified RNA polymerase) treated with reducing agents. Lane M represents a T-sequencing ladder of the primer extension reaction. Phosphodiester bonds specifically cleaved by S309C-conjugated FeBABE are indicated by arrows on the right. The numbers on the left indicate some positions of thymines. Open circles on right indicate bonds, which became hyperreactive to radicals upon open complex formation. A band generated by spontaneous nicking is marked by caret on the left. Primers (when used) are indicated by asterisk on the left.
Figure 6.

Location of the footprint signals along the T7D and rrnBP1 promoter sequences. Signals obtained by DNase I and hydroxy-radical footprintings are indicated along the T7D and rrnBP1 promoter sequences. Symbols are the same as shown in Figures 2 and 4. Half-ellipses indicate guanines protected against modification by AcHAQO (data not shown). Thin arrows on T7D sequence indicate phosphodiester bonds specifically cleaved by FeBABE attached to C309 of αCTD (Fig. 1) (14), while thick arrows on rrnBP1 indicate those cleaved by FeBABE attached to C269 of αCTD (17). Horizontal brackets indicate the putative α-subunit binding subsites.
To locate precisely the α subunit contact site(s) on T7D promoter, we first performed the DNase I protection assay for its open complex formed with the RNA polymerase holoenzyme (Eσ70). Upstream from the basic promoter, at least two protected regions were identified (Fig. 2, DNase lanes), which represent the αCTD contact sites on this promoter (see below). The promoter-proximal αCTD-contact region between –42 and –37 on the top strand is separated from the promoter –35 element by DNase I-hypersensitive sites, which were observed at –37 to –35 on the bottom strand (Fig. 2, DNase lanes; see also Fig. 6). Upstream from this promoter-proximal αCTD-contact region, DNase I-hypersensitive sites appeared between –49 and –46 on the bottom strand. Upstream from these DNase I-hypersensitive sites, the promoter-distal αCTD-contact region could be detected as the weakly protected region on the top strand between –52 and –47 (Fig. 2, DNase lanes; see also Fig. 6). Thus both α-subunit binding sites are separated from each other (each site is hereafter defined as the UP element subsite) and also from the σ-subunit contact region by the DNase I-hypersensitive sites on the bottom strand.
Figure 2.

DNase I and hydroxyl-radical footprints of T7D and rrnBP1 promoters by wild-type and α-E286A mutant RNA polymerases. T7D DNA fragment (213 bp from –131 to +82) was 32P-labeled at position –131 of the top strand or at position +82 of the bottom strand, while promoter rrnBP1 (159 bp from –77 to +82) was 32P-labeled at position –77 of the top strand. DNase I and FeEDTA-generated hydroxyl-radical footprintings were carried out as described in Materials and Methods. Lanes shown in each panel represent: G, guanine-specific sequencing ladder; –, free DNA in the absence of RNA polymerase (15 nM); w.t., open complex with wild-type RNA polymerase (150 nM); E286A, open complex with mutant RNA polymerase (150 nM) containing E286 α subunit. Numbers on the left indicate positions of some guanines. Phosphodiester bonds protected against DNase I cleavage are indicated by black squares while DNase I hyperreactive bonds are indicated by large arrowheads [DNase panels]. Phosphodiester bonds additionally protected by the RNA polymerase containing E286A mutant α subunit are marked by gray squares, while bonds protected against hydroxyradical attack are marked by open squares, Small arrows on left indicate the phosphodiester bonds, which were cleaved by both DNase I and hydroxyl radicals. For assignment, difference in the migration rate between products generated by enzymatic and chemical reactions was taken into account. Superimposed scans of the lanes containing products of the hydroxyl radical reaction are aligned along the gels.
In parallel, we also carried out the hydroxyl radical protection assays. The RNA polymerase Eσ70 holoenzyme-T7D open complex was exposed to hydroxyl radicals generated from EDTA-chelated Fe(III) in the presence of H2O2 (Fig. 2, FeEDTA lanes). The two upstream regions protected against DNase I digestion were also protected against hydroxyl radical attack (Fig. 2, FeEDTA lanes; for summary see Fig. 6). Since these regions also include the sites specifically cleaved by FeBABE tethered to C309 (Fig. 1) (14), we concluded that the helix IV of αCTD is in close contact with the T7D UP element.
Because of the presence of DNase I-insensitive A/T runs in the upstream region of rrnBP1, the DNase footprint does not allow precise localization of the α subunit-binding sites (Fig. 2) (4,6,10,25). In contrast, the hydroxy radical footprint allows more accurate mapping of the phosphodiester bonds protected by αCTD (10,25). Two αCTD-binding sub-sites were identified for rrnBP1 (Fig. 2; for summary see Fig. 6). The promoter-distal subsite is located between –55/–47 on the top and –57/–51 on the bottom strands (Figs 2 and 6). NMR studies indicate that the αCTD is not fixed at a single position within AT-rich sequences, but interacts with flanking sequences (8). The hydroxyl radical protection pattern in the T7D UP element region is apparently different from that of rrnBP1. The result of footprinting experiments indicates two αCTD-binding subsites for the T7D promoter, even though the promoter-distal subsite is not so strong.
Topological characterization of RNA polymerase complexes formed with T7D/rrnBP1 hybrid promoters
The UP element of rrnBP1 retains the αCTD-binding activity when transferred into another genetic environment (25). To test this possibility for the T7D UP element, we transferred it into the genetic environment of rrnBP1, of which the dependence on the presence of UP element is well characterized. For this purpose, four mutant promoters were constructed as shown in Figure 3. The promoter-proximal UP element sequence (–41)TCCTC(–37) of the rrnBP1 promoter was replaced by AGGCG, the sequence of the corresponding region of T7D promoter, to generate a synthetic promoter RF. A single T to C substitution mutation of rrnBP1 at position –50 leads to change the rrnBP1 UP element sequence (–51)ATTTTAA(–45) to the T7D type sequence ACTTTAA to generate a synthetic promoter RC. Synthetic promoter RFC carries both of these mutations in RF and RC promoters. Synthetic promoter DR is a chimeric promoter consisting of upstream T7D sequence (–77 to –37) and downstream rrnBP1 sequence (–33 to +82).
The promoter activity of all these chimeric constructs was very close to the activity of the wild-type rrnBP1 promoter (RF, 112 ± 15%; RC, 102 ± 10%; RFC, 119 ± 5%; and DR, 90 ± 10%), as measured by using single-round transcription assays. This immediately indicates that the minimum unit of T7D UP element is as active as that of the rrnBP1 UP element. The interpretation was directly examined by DNase I protection and hydroxy radical cleavage footprinting assays. Figure 4 shows the footprinting patterns for the top strand. In the case of RF promoter, phosphodiester bonds –42/–41/–40 and –39/–38 were protected from DNase I digestion (Fig. 4, DNase panel) and bonds –42/–41/–40 were protected from hydroxyl radical cleavage by Fe(III)-EDTA (Fig. 4, FeEDTA panel). Thus, we conclude that the UP element sequence of T7D retains its α subunit-binding activity when transferred to the rrnBP1 genetic environment. Phosphodiester bond –44/–43 displayed hyper-reactivity to DNase I digestion. Except for these changes, no significant differences were registered between the intact rrnBP1 and the hybrid promoter RF within the range examined.
Figure 4.

DNase I and hydroxyl radical footprints of T7D-rrnBP1 hybrid promoters by wild-type RNA polymerase. DNase I (top panels) and FeEDTA-generated hydroxyl-radical (bottom panels) footprintings by wild-type RNA polymerase were carried out as in Figure 1. All the templates used were 159 bp long with 32P label at position –77 of the top strand. The length of the forward primers was 53, 39, 53 and 59 nt for RF, RC, RFC and DR templates, respectively (see Fig. 3 for sequence). Gels were calibrated by G+A sequencing ladder (G+A lanes). All other designations are as indicated in Figures 1 and 2.
For the synthetic promoter RC with a single base substitution T to C at –50, the change in both DNase I protection and hydroxy radical cleavage patterns was obviously more than those with RF. Less protected against nuclease cleavage was phosphodiester bond –48/–47, while unprotected against hydroxyl radical were bonds –49/–48/–47. Pronounced protection was observed in the region –49 to –53 within the αCTD-binding site of T7D. Taking these observations altogether we concluded that the T-to-C transition at –50 in the A/T run of rrnBP1 preserves the local contact with the αCTD, but leads to decrease in interaction with the RNA polymerase at the neighboring regions.
The RFC template carrying both the T7D UP sequence (as in RF) and the T-to-C change at –50 (as in RC) had a significant influence on both DNase I cleavage and hydroxy radical footprinting patterns, including the same effects that were observed with the RF and RC probes, even though the DNase I-hyper-reactivity at –44/–43 bond and the hydroxyl radical protection at –49/–48 bond were less than those observed with RF and RC, respectively. The footprinting patterns of the chimeric promoter DR consisting of upstream T7D and downstream rrnBP1 patterns were similar to those of RFC. Moreover, the DNase I cleavage and hydroxy radical footprinting patterns of the upstream region from the promoter –35 element for both RFC and DR were essentially the same with those observed with the native T7D promoter. Taken the data of functional and topological analyses together we concluded that the T7D UP element (and even its individual subsites) retains the αCTD-binding activity even when transferred into another genetic environment.
Identification of amino acid residues on αCTD involved in T7D UP element recognition: construction of αCTD mutants
The site-specific cleavage of the promoter T7D UP element by C309-tethered FeBABE indicates that the functionally important amino acid residue(s) should be located within the distance of ∼20 Å from S309. αCTD is composed of four α helices and S309 is located at the C-terminal-proximal end of α-helix IV (24). The Ala scanning was then performed for all residues from 284 to 312 including α-helices III (286–292) and IV (297–309). In addition we checked the α mutants with Ala substitutions in position 317 that are required for activator-mediated transcription (26,27) and position 265 (helix I) that is critical for rrnBP1 UP element recognition (5,6,24). Functional significance of helix II (amino acids residues 278–283) was not tested because this structural module is located on the opposite side from the S309 surface of the protein globule. A set of the mutant α subunit genes was prepared, starting from the wild-type rpoA in the overexpression plasmid pGEMA. The α235 derivative lacking the entire CTD downstream from position 235 was used as a control lacking the UP element recognition sequence.
All these mutant α subunits were over-expressed, purified and reconstituted into the mutant holoenzymes. Among 31 mutant α subunits examined, 14 were recovered from the soluble fraction of whole cell lysates, while 17 were from precipitates (Table 1). The efficiencies of core enzyme assembly from the mutant α subunits were, however, essentially the same as that from wild-type α subunit. The functional activities of wild-type and mutant RNA polymerases were analyzed using a single-round transcription assay. High concentrations of the templates were used to facilitate quantitative comparison of the enzyme activity among mutant RNA polymerases.
Identification of amino acid residues on αCTD involved in T7D UP element recognition: transcriptional activity of the mutant enzymes at rrnBP1
In order to test the roles of the mutated residues in recognition of the well-characterized rrnBP1 UP element, we first analyzed efficiency of mutant enzymes in rrnBP1 transcription. Mixed transcription assays were carried out using plasmid pWR52, possessing both the test promoter rrnBP1 and the reference promoter RNA-1 (4), as template (Fig. 5A). The activity of rrnBP1 transcription by the mutant RNA polymerase containing α235, devoid of the C-terminal 94 residues, was <20% of the level of wild-type RNA polymerase (Fig. 5B), which is in line with previous data (4). A similar level of inhibition was observed for the point mutant R265A, which plays a key role in rrnBP1 UP element-dependent transcription (5,6). The inhibitory effect on UP element-dependent transcription was also observed for substitutions in the loop region between helices III and IV (L295A, G296A) and in the N-terminal part of helix IV (K298A and S299A). The results are generally in agreement with our previous observations (6), even though the inhibitory effect of K297A was less pronounced than when plasmid pRLG862 was used as template (6). In addition we observed reliable inhibition in the case of I303A mutant. The side chain of this hydrophobic amino acid residue is deeply buried in the structure of αCTD and is located near L295, L290 and I300, probably indicating that a stiff hydrophobic core is required to create the optimal configuration providing the UP element contact surface. Substitutions in the other positions gave little effect, except that L290A exhibited a slight stimulatory effect.
Figure 5.

Transcription in vitro of rrnBP1 and T7D promoters by wild-type and mutant RNA polymerases. (A) Mixed transcription in vitro of truncated T7D template (6 nM) and plasmid pWR52 (9 nM) carrying both rrnBP1 and RNA I promoters was carried out under the standard single-round conditions using wild-type or the indicated mutant RNA polymerases (each 24 nM). RNA products were analyzed by urea–PAGE. (B) Gels of transcripts formed in the mixed transcription assay were traced and the band intensities of T7D (open bars) and rrnBP1 (filled bars) transcripts were normalized based on the yield of RNA-I transcript. The T7D and rrnBP1 transcription levels by the wild-type RNA polymerase were set as 100%. The activities of the mutant RNA polymerases were estimated as the relative values to that of wild-type enzyme. The assays were repeated three times in the presence of rrnBP1 alone and three times in the presence of both rrnBP1 and T7D. Statistical deviations are shown in each bar. (C) The relative activity of rrnBP1 and T7D transcription was calculated, using the data shown in (B), for the wild-type and mutant RNA polymerases.
Identification of amino acid residues on αCTD involved in T7D UP element recognition: transcription of T7D by mutant RNA polymerases
Next the transcription efficiency of T7D promoter by the mutant enzymes was examined using the same linear template (from –131 to +224) that was used in the assay of T7D UP element–αCTD interaction (14) in the simultaneous presence of pWR52 (Fig. 5A). The levels of T7 and rrnB transcripts were normalized after correction for the recovery of RNA-I used as an internal control. For confirmation of the results, we also carried out a second set of mixed transcription using the same 335 bp wild-type T7D promoter and a 118 bp long mutant T7D promoter containing the native T7D promoter sequence from –35 to +64 plus an upstream substitution with 19 bp long G/C-rich sequence. The rrnBP1/T7D ratios calculated from the transcription data, shown in Figure 5B, are plotted in Figure 5C.
The mutant RNA polymerase containing α235 retained 30–45% the activity of wild-type RNA polymerase (Fig. 5A and B), confirming that the T7D promoter also requires αCTD for maximum transcription. Substitutions R265A, L295A, G296A, K298A, S299A and I303A, which all strongly inhibited rrnBP1 transcription, also inhibited T7D RNA synthesis. However, a well-pronounced difference was observed for some mutations in response to rrnBP1-type and T7D-type UP elements. Mutation E286A activated T7D transcription but slightly inhibited rrnBP1 RNA synthesis. On the other hand, L290A activated rrnBP1 transcription, but inhibited T7D RNA synthesis. Several mutations within helix IV only affected transcription from T7D and the inhibitory effect of L289A, I303 and L307A was also stronger for T7D template.
The influence of these mutations on the αCTD interaction with the T7D promoter was directly analyzed by DNase I footprinting. Compared with the wild-type RNA polymerase, the mutant enzyme containing E286A α, which activates T7D transcription, protected additional bonds –53/–52 on the top strand and –53/–54 on the bottom strand on T7D template (Fig. 2), consistent with its high T7D-binding activity. With other species of the mutant RNA polymerase showing inhibitory effects on T7D transcription, such as L289A, L290A, I303A and L307A, 2–4-fold higher concentrations were required to give comparable footprints with the wild-type enzyme even though the DNA protection patterns were not specifically changed by mutations (data not shown).
Although both αCTD deletion and Ala substitution at R265, the most critical residues for rrnBP1 UP element recognition, resulted in marked reduction in recognition of both rrnBP1 and T7D UP elements (see Fig. 5), the effect was more severe for rrnBP1 transcription, leading to the rrnBP1/T7D ratio of <1 (Fig. 5C). Likewise, Ala substitution at G296, K298 and S299, near the N-terminal end of helix IV, resulted in more severe reduction in recognition of the rrnBP1 UP element, while L289A had a greater influence on T7D transcription. The decrease in the rrnBP1/T7D ratio for E286A was attributed to the activation in T7D UP element recognition (Fig. 5B), while the ratio increase for L290A was due to the activation of rrnBP1 UP element recognition (Fig. 5B). A number of mutations within helix IV tend to have a bigger effect on T7D transcription than on rrnBP1 (Fig. 5C). These observations together indicate that in addition to the known recognition surface (the helix I and the loop between helices III and IV) for rrnBP1-type UP element, at least some residues on the surface of helices III and IV are involved in T7D UP element binding and transcription activation.
To ascertain the level of reliability for the promoter-specific differences observed above, we applied the Student’s t-test using standard SigmaPlot software for the normalized transcription data per reference promoter (a total of six experiments for each promoter) obtained in the mixed or separate transcription assays of T7D or rrnBP1 promoters. A reliable difference between T7D and rrnBP1 activation was confirmed for 13 point substitutions, among which the high confidence level (P < 0.0005) was observed for L290A, E286A, R265A, K304A, G296A, L289A and K298A (decreasing in this order). A >99.5% confidence level (0.0005 < P < 0.005) was for L312A and S299A, while >95% (P < 0.05) was for mutations K297A, L307A, L300A and I303A. At least three mutations from this set (E286A, L290A and K304A) gave opposite effects on the T7D and rrnBP1 transcription activation. The t-test data confirmed that even though the amino acid residues involved in interaction with T7D and rrnBP1 UP elements compose highly overlapping sets, the functional manifestation is reliably different in some cases.
DISCUSSION
The C-terminal domain of RNA polymerase α subunit plays a major role in transcription activation by both class-I transcription factors and DNA UP elements (28,29). αCTD domains involved in the transcription activation have been analyzed in detail by making mutants defective in transcription activation, footprintings with various reagents (4,6,14), mapping DNA cleavage sites by αCTD-tethered chemical nucleases (14,17) and structural analysis of αCTD–DNA complexes (8,24). Taken together the functional modules have been identified: D261 determinant (Ht2 region) for σ region 4 interaction; D265 determinant (helix 1 and loop between helices 3 and 4) for UP element recognition; and E273 determinant (C terminus of helix 1) and V287 determinant (helix 3) for CRP activation region 1 (AR1).
Mutations in helices III and IV affected transcription activation by T7D UP element (this paper) and the FeBABE conjugation at S309C led to the introduction of cleavage at the T7D UP element (14); together these indicate the involvement of a helix III–hairpin–helix IV structural module in T7D UP element recognition. Involvement of αCTD helices III and IV in transcription activation has been identified in certain cases: L307P inactivates transcription from katG (30); mutations L289A and L307A inhibit RNA synthesis from promoter uhp (31); L289A inactivates MerR activator-dependent transcription of merPTPCAD (32); and E302, D305 and A308 are required for FNR-dependent transcription from class-I promoters (26). Most of these amino acid residues are hydrophobic and therefore may influence the structure of the protein far away from their localization, or may participate in the protein–protein interaction with regulatory factors or other RNA-polymerase subunits. However, at least part of the amino acid residues, which were detected as involved in interaction with T7D UP element, are exposed on the protein surface and could be in direct contact with DNA.
Side chains of V287, L290, L300, L307, A308, L312 and L314 form a hydrophobic cluster on the surface of αCTD. Substitution of L290, L300, L307 and L312 with less hydrophobic alanine led to reduction in transcription activation at T7D. E286 and K304, neighboring in the three-dimensional structure, are located within the same cluster. Substitution E286A obviously increases overall compactness in this region. As summarized in Figure 6, the specific stimulatory effect of this modification on T7D transcription as well as a pronounced influence on the local footprinting pattern indicate the significance of hydrophobic interaction for this promoter activation. Functional manifestation of K304A may be compromised by the same positive effect and negative input conditioned by elimination of positively charged side chain from the DNA-binding site.
The DNA-binding domain of the α subunit has been annotated as a duplicated helix–hairpin–helix (HhH) module (33). Helices III and IV form a perfect HhH fold, which is separated from another less perfect HhH module by helix II. Single HhH modules are usually unstable and require hydrophobic cores for stabilization. This requirement underlies the observed transcription dependence on these hydrophobic amino acid residues, which are deeply buried in the protein globule (L289, I303).
Known DNA-binding HhH proteins such as RuvA (34), DNA polymerase β (35) and BAF (36) do not require specific sequences for DNA binding, and make contact with the sugar–phosphate backbone within the minor groove. The same mode of DNA interaction is utilized in the case of the rrnBP1 UP element recognition by αCTD (8), which employs helix I and a loop region between helices III and IV for structure-specific complex formation. Positively charged R265 and K298 are the main contributors to this interaction forming hydrogen bonds with negatively charged sugar–phosphate backbone on the both sides of the minor groove (8). Very similar in structure, the DNA-binding domain of Thermus thermophilus α subunit also employs the surface of helix IV and loop preceding this helix for interaction with T.thermophilus rRNA promoter. Functionally important amino acid residues (G293, G295 and S298 corresponding to E.coli N294, G296 and S299, respectively), however, do not have charged side chains and helix I including R264 corresponding to R265 in E.coli αCTD does not participate in the complex formation with the rRNA gene promoter (37). Therefore, there is a possibility for other HhH structural modules to interact with DNA without participation of helix I and even without charged amino acids as such. In fact, R265A has very faint effect on the transcription activation at λPRM promoter (38).
The main difference between E.coli and T.thermophilus rRNA promoter sequences is their base composition: extremely A(T)-rich for E.coli and extremely G(C)-rich for T.thermophilus rRNA promoter (37). Even though the A(T) content itself is not a sole determinant for providing the narrow minor groove required for efficient association of αCTD with E.coli UP element, the presence of G(C) base pairs generally increases this width. Both αCTD-binding subsites of the T7D (see Fig. 6) contain G/C base pairs. Their direct interaction with αCTD is approved by DNase I and hydroxyl radical footprinting assays (Figs 2 and 6), by site-specific FeBABE cleavage (see Fig. 1) (14) and also by AcHAQO-probing (data not shown) specifically modifying C8 of guanine residues (39). An important observation made in this paper is that introduction of C/G bp in the distal αCTD-binding site of rrnBP1 is favorable rather than unfavorable for interaction between modified site and αCTD (constructions RC and RFC; see Figs 3 and 4), which together with high transcription efficiency of all hybrid promoter constructions indicates that containing G/C base pair DNA can be a target for E.coli αCTD.
The data described herein indicate that the sets of amino acid residues important for transcription complex formation with rrnBP1 and T7D overlap, but are not completely identical (see Fig. 5). Substitutions of 13 amino acid residues show reliable different response for T7D and rrnBP1 RNA synthesis. This corresponds to the observation that interaction with these promoters differently affects spectral parameters of the fluorescent labels specifically attached either to the helix I or to the helix IV (14), while specific chemical modification of Cys269 inhibited rrnBP1 RNA synthesis having no effect on many other promoter activation (40). The possibility of explaining αCTD-mediated activation of the promoters whose upstream sequence is basically different from rrnBP1 is an important aspect of the data obtained in this study. Since rrnBP1-type UP element consensus sequence could be found only in a limited population of promoters, while the majority of promoters interact with α subunits, alternative interaction is expected to be less specific. However, some specificity still should be assumed, since there are few promoters both natural and artificial that have no contacts with RNA polymerase in the upstream region (11). Identification of the functionally important amino acid residues within helices III and IV enables identification of the alternative structural DNA module for the αCTD binding.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Rick Gourse (Univiversity of Wisconsin) for the Ala-substituted mutant rpoA genes. This work was supported by grants from the Ministry of Education, Science, Sports and Culture of Japan, the Core Research for Evolutional Science and Technology Corporation and the Russian Foundation for Basic Research (grant 00-04-48132, 01-04-97006).
REFERENCES
- 1.Gross C.A., Chan,C., Dombroski,A., Gruber,T., Sharp,M., Tupy,J. and Young,B. (1998) The functional and regulatory roles of sigma factors in transcription. Cold Spring Harb. Symp. Quant. Biol., 63, 141–155. [DOI] [PubMed] [Google Scholar]
- 2.Harley C.B. and Reynolds,R.P. (1987) Analysis of E.coli promoter sequences. Nucleic Acids Res., 15, 2343–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barne K.A., Bown,J.A., Busby,S.J. and Minchin,S.D. (1997) Region 2.5 of the Escherichia coli RNA polymerase sigma 70 subunit is responsible for the recognition of the ‘extended-10’ motif at promoters. EMBO J., 16, 4034–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross W., Gosink,K.K., Salomon,J., Igarashi,K., Zou,C., Ishihama,A., Severinov,K. and Gourse,R.L. (1993) A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science, 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- 5.Gaal T., Ross,W., Blatter,E.E., Tang,H., Jia,X., Krishnan,V.V., Assa-Munt,N., Ebright,R.H. and Gourse,R.L. (1996) DNA-binding determinants of the alpha subunit of RNA polymerase: novel DNA-binding domain architecture. Genes Dev., 10, 16–26. [DOI] [PubMed] [Google Scholar]
- 6.Murakami K., Fujita,N. and Ishihama,A. (1996) Transcription factor recognition surface on the RNA polymerase alpha subunit is involved in contact with the DNA enhancer element. EMBO J., 15, 4358–4367. [PMC free article] [PubMed] [Google Scholar]
- 7.Naryshkin N., Revyakin,A., Kim,Y., Mekler,V. and Ebright,R.H. (2000) Structural organization of the RNA polymerase-promoter open complex. Cell, 101, 601–611. [DOI] [PubMed] [Google Scholar]
- 8.Yasuno K., Yamazaki,T., Tanaka,Y., Kodama,T.S., Matsugami,A., Katahira,M., Ishihama,A. and Kyogoku,Y. (2001) Interaction of the C-terminal domain of the E.coli RNA polymerase alpha subunit with the UP element: recognizing the backbone structure in the minor groove surface. J. Mol. Biol., 306, 213–225. [DOI] [PubMed] [Google Scholar]
- 9.Ross W., Ernst,A. and Gourse,R.L. (2001) Fine structure of E.coli RNA polymerase-promoter interactions: alpha subunit binding to the UP element minor groove. Genes Dev., 15, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Estrem S.T., Ross,W., Gaal,T., Chen,Z.W., Niu,W., Ebright,R.H. and Gourse,R.L. (1999) Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Genes Dev., 13, 2134–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozoline O.N. and Tsyganov,M.A. (1995) Structure of open promoter complexes with Escherichia coli RNA polymerase as revealed by the DNase I footprinting technique: compilation analysis. Nucleic Acids Res., 23, 4533–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross W., Aiyar,S.E., Salomon,J. and Gourse,R.L. (1998) Escherichia coli promoters with UP elements of different strengths: modular structure of bacterial promoters. J. Bacteriol., 180, 5375–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozoline O.N., Deev,A.A. and Arkhipova,M.V. (1997) Non-canonical sequence elements in the promoter structure. Cluster analysis of promoters recognized by Escherichia coli RNA polymerase. Nucleic Acids Res., 25, 4703–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozoline O.N., Fujita,N. and Ishihama,A. (2000) Transcription activation mediated by the carboxyl-terminal domain of the RNA polymerase alpha-subunit. Multipoint monitoring using a fluorescent probe. J. Biol. Chem., 275, 1119–1127. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel T.A., Roberts,K.J. and Zakour,R.A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol., 154, 367–382. [DOI] [PubMed] [Google Scholar]
- 16.Fujita N. and Ishihama,A. (1996) Reconstitution of RNA polymerase. Methods Enzymol., 273, 121–130. [DOI] [PubMed] [Google Scholar]
- 17.Murakami K., Kimura,M., Owens,J.T., Meares,C.F. and Ishihama,A. (1997) The two alpha subunits of Escherichia coli RNA polymerase are assymetrically arranged and contact different halves of the DNA upstream element. Proc. Natl Acad. Sci. USA, 94, 1709–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greiner D.P., Miyake,R., Moran,J.K., Jones,A.D., Negishi,T., Ishihama,A. and Meares,C.F. (1997) Synthesis of the protein cutting reagent iron (S)-1-(p-bromoacetamidobenzyl)ethylenediaminetetraacetate and conjugation to cystein side chains. Bioconjugate Chem. 8, 44–48. [DOI] [PubMed] [Google Scholar]
- 19.Igarashi K. and Ishihama,A. (1991) Bipartite functional map of the E.coli RNA polymerase alpha subunit: involvement of the C-terminal region in transcription activation by cAMP-CRP. Cell, 65, 1015–1022. [DOI] [PubMed] [Google Scholar]
- 20.Kajitani M. and Ishihama,A. (1983) Determination of the promoter strength in the mixed transcription system: promoters of lactose, tryptophan and ribosomal protein L10 operons from Escherichia coli. Nucleic Acids Res., 11, 671–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tullius T.D. and Dombroski,B.A. (1986) Hydroxyl radical ‘footprinting’: high-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein. Proc. Natl Acad. Sci. USA, 83, 5469–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 23.Newlands J.T., Ross,W., Gosink,K.K. and Gourse,R.L. (1991) Factor-independent activation of Escherichia coli rRNA transcription. II. Characterization of complexes of rrnB P1 promoters containing or lacking the upstream activator region with Escherichia coli RNA polymerase. J. Mol. Biol., 220, 569–583. [DOI] [PubMed] [Google Scholar]
- 24.Jeon Y.H., Negishi,T., Shirakawa,M., Yamazaki,T., Fujita,N., Ishihama,A. and Kyogoku,Y. (1995) Solution structure of the activator contact domain of the RNA polymerase alpha subunit. Science, 270, 1495–1497. [DOI] [PubMed] [Google Scholar]
- 25.Attey A., Belyaeva,T., Savery,N., Hoggett,J., Fujita,N., Ishihama,A. and Busby,S. (1994) Interactions between the cyclic AMP receptor protein and the alpha subunit of RNA polymerase at the Escherichia coli galactose operon P1 promoter. Nucleic Acids Res., 22, 4375–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams S.M., Savery,N.J., Busby,S.J. and Wing,H.J. (1997) Transcription activation at class I FNR-dependent promoters: identification of the activating surface of FNR and the corresponding contact site in the C-terminal domain of the RNA polymerase alpha subunit. Nucleic Acids Res., 25, 4028–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savery N.J., Lloyd,G.S., Kainz,M., Gaal,T., Ross,W., Ebright,R.H., Gourse,R.L. and Busby,S. (1998) Transcription activation of class II CRP-dependent promoters: identification of determinants in the C-terminal domain of the RNA polymerase alpha subunit. EMBO J., 17, 3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishihama A. (1993) Protein-protein communication within the transcription apparatus. J. Bacteriol., 175, 2483–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Busby S. and Ebright,R.H. (1994) Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell, 79, 743–746. [DOI] [PubMed] [Google Scholar]
- 30.Tao K., Zou,C., Fujita,N. and Ishihama,A. (1995) Mapping of the OxyR protein contact site in the C-terminal region of RNA polymerase alpha subunit. J. Bacteriol., 177, 6740–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olekhnovich I.N. and Kadner,R.J. (1999) RNA polymerase alpha and sigma(70) subunits participate in transcription of the Escherichia coli uhpT promoter. J. Bacteriol., 181, 7266–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caslake L.F., Ashraf,S.I. and Summers,A.O. (1997) Mutations in the alpha and sigma-70 subunits of RNA polymerase affect expression of the mer operon. J. Bacteriol., 179, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao X. and Grishin,N.V. (2000) Common fold in helix-hairpin-helix proteins. Nucleic Acids Res., 28, 2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ariyoshi M., Nishino,T., Iwasaki,H., Shinagawa,H. and Morikawa,K. (2000) Crystal structure of the holliday junction DNA in complex with a single RuvA tetramer. Proc. Natl Acad. Sci. USA, 97, 8257–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelletier H., Sawaya,M.R., Wolfle,W., Wilson,S.H. and Kraut,J. (1996) Crystal structures of human DNA polymerase beta complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry, 35, 12742–12761. [DOI] [PubMed] [Google Scholar]
- 36.Umland T.C., Wei,S.-Q., Craigie,R. and Davies,D.R. (2000) Structural basis of DNA bridging by barrier-to-autointegration factor. Biochemistry, 39, 9130–9138. [DOI] [PubMed] [Google Scholar]
- 37.Wada T., Yamazaki,T. and Kyogoku,Y. (2000) The structure and the characteristic DNA binding property of the C-terminal domain of the RNA polymerase alpha subunit from Thermus thermophilus. J. Biol. Chem., 275, 16057–16063. [DOI] [PubMed] [Google Scholar]
- 38.Tang Y., Murakami,K., Ishihama,A. and DeHaseth,P.L. (1996) Upstream interactions at the lambda pRM promoter are sequence nonspecific and activate the promoter to a lesser extent than an introduced UP element of an rRNA promoter. J. Bacteriol., 178, 6945–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panigrahi G.B. and Walker,I.G. (1991) Use of monoacetyl-4-hydroxyaminoquinoline 1-Oxide to probe contacts between guanines and protein in the minor and major grooves of DNA. Interaction of Escherichia coli Integration Host Factor with its recognition site in the early promoter of transposition enhancer of bacteriophage Mu. Biochemistry, 30, 9761–9767. [DOI] [PubMed] [Google Scholar]
- 40.Ozoline O.N., Fujita,N., Murakami,K. and Ishihama,A. (1998) Monitoring of RNA polymerase-DNA UP element interaction by a fluorescent probe conjugated to alpha subunit. Eur. J. Biochem., 252, 371–381. [DOI] [PubMed] [Google Scholar]