

Abstract
Background
Amongst risk alleles associated with late-onset Alzheimer’s disease (AD), those that converged on the regulation of microglia activity have emerged as central to disease progression. Yet, how canonical amyloid-β (Aβ) and tau pathologies regulate microglia subtypes during the progression of AD remains poorly understood.
Methods
We use single-cell RNA-sequencing to profile microglia subtypes from mice exhibiting both Aβ and tau pathologies across disease progression. We identify novel microglia subtypes that are induced in response to both Aβ and tau pathologies in a disease-stage-specific manner. To validate the observation in AD mouse models, we also generated a snRNA-Seq dataset from the human superior frontal gyrus (SFG) and entorhinal cortex (ERC) at different Braak stages.
Results
We show that during early-stage disease, interferon signaling induces a subtype of microglia termed Early-stage AD-Associated Microglia (EADAM) in response to both Aβ and tau pathologies. During late-stage disease, a second microglia subtype termed Late-stage AD-Associated Microglia (LADAM) is detected. While similar microglia subtypes are observed in other models of neurodegenerative disease, the magnitude and composition of gene signatures found in EADAM and LADAM are distinct, suggesting the necessity of both Aβ and tau pathologies to elicit their emergence. Importantly, the pattern of EADAM- and LADAM-associated gene expression is observed in microglia from AD brains, during the early (Braak II)- or late (Braak VI/V)- stage of the disease, respectively. Furthermore, we show that several Siglec genes are selectively expressed in either EADAM or LADAM. Siglecg is expressed in white-matter-associated LADAM, and expression of Siglec-10, the human orthologue of Siglecg, is progressively elevated in an AD-stage-dependent manner but not shown in non-AD tauopathy.
Conclusions
Using scRNA-Seq in mouse models bearing amyloid-β and/or tau pathologies, we identify novel microglia subtypes induced by the combination of Aβ and tau pathologies in a disease stage-specific manner. Our findings suggest that both Aβ and tau pathologies are required for the disease stage-specific induction of EADAM and LADAM. In addition, we revealed Siglecs as biomarkers of AD progression and potential therapeutic targets.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13024-022-00589-x.
Keywords: Amyloid-β (Aβ), Tau, Microglia, Alzheimer’s disease (AD), Sialic acid-binding immunoglobulin-type lectin (Siglec)
Background
Neuroinflammation is increasingly recognized as a key regulator of disease progression in neurodegenerative disorders [1, 2], including Alzheimer’s disease (AD), which is the most common cause of dementia [3]. Recent studies suggest that neuroinflammation serves as a mechanistic link between the development of amyloid-β (Aβ) plaques and tau neurofibrillary tangles, and the canonical pathologies of AD that are thought to drive synapse loss and neuronal death [3]. In addition to well-characterized AD susceptibility genes such as APP, PSEN1 and PSEN2 (early-onset familial AD [4]), and APOE (late-onset AD [5, 6]), several additional risk alleles for late-onset AD were identified in genes regulating immunomodulation, including TREM2 [7, 8], phosphoinositide phospholipase Cγ2 (PLCG2) [9] and CD33 [10, 11].
As the brain’s resident innate immune cells, microglia play multifunctional roles in brain health and the progression of neurodegenerative diseases such as AD [1, 2, 12]. Depending on the disease stage, microglia may protect against neurodegenerative proteinopathy and/or contribute to inflammatory damage [2, 13]. Microglia maintain brain health by clearing cellular debris, including Aβ plaques and tau aggregates [13]. Microglial activation correlates positively with cognition and gray matter volume in humans, indicating that microglia can be protective, at least during early-stage AD [14]. However, microglia also secrete proinflammatory cytokines and can directly contribute to tau pathology [14, 15] and its subsequent neurotoxicity [1, 2]. This dichotomous role of microglia in maintaining this balance between phagocytosis/clearance and pro-inflammatory mediator release is thought to be an essential and potentially targetable determinant of AD progression [16].
Recent studies have identified subtypes of microglia that display a dynamic range of responses and functions [17], emphasizing the ability of microglia to serve a variety of important physiological roles [12]. Transcriptomic analyses of bulk tissues revealed disease-associated changes in microglia associated with AD [18, 19]. Subsequent single-cell RNA-Sequencing (scRNA-Seq) approaches, however, were necessary to identify disease context-dependent microglial subtypes. scRNA-Seq has shed light on the spatial and developmental heterogeneity of microglia and provides a high-resolution view of the transcriptional landscape of microglia subtypes during development and disease progression [17, 20–23]. Since AD is a chronic disease with decades-long prodromal stages, understanding the disease stage-specific impacts of microglia subtypes is necessary to clarify the dual nature of microglia activation. The identification of disease-associated microglia (DAM), a unique TREM2-dependent subtype that expresses CD11c and is localized near Aβ plaques in an amyloidosis mouse model [20], supports this notion. Despite these advances, a critical unresolved question is whether disease stage-specific microglia subtypes exist that are activated in response to both Aβ and tau pathologies. These microglia subtypes would represent novel therapeutic targets for modifying disease progression.
Here, we used an AD mouse model (Tau4RΔK-AP mice), in which wild-type tau is converted into tau aggregates to drive neuron loss in a neuritic plaque-dependent manner [24]. This animal model serves as an excellent mouse model to profile microglia subtypes across different stages of AD-like disease progression. To assess the requirement for both Aβ and tau pathologies to induce disease stage-specific microglia subtypes, control mice accumulating either Aβ plaques (APP;PS1 mice) or tau tangles (Tau4RΔK mice) were also profiled. Using scRNA-Seq approaches, we found that microglia respond to the development of Aβ and tau pathologies in a disease-stage-specific manner. During early-stage disease in 6-month-old Tau4RΔK-AP, but not APP;PS1 or Tau4RΔK mice, the presence of both Aβ and tau pathologies induced a novel microglia subtype we termed Early-stage AD-Associated Microglia (EADAM), which is distinct from DAM and express multiple interferon-regulated genes. We found that the signature genes in EADAM were also associated with a subgroup of microglia in the brains of early (Braak II) stages of AD. In late-stage disease (12-month-old Tau4RΔK-AP mice), we found that another novel microglia subtype emerged in response to tau pathology that we termed Late-stage AD-Associated Microglia (LADAM), which expresses MHC and S100 family genes. We further observed a unique subtype of LADAM located near white matter, which is molecularly similar to a previously identified white matter-associated microglial subtype (WAM) that is increased during aging [25] and undergoes additional molecular transitions with Aβ and tau pathologies.
LADAM microglia, including WAM-like LADAM, were observed in the late (Braak VI), but not early (Braak II) stages of AD. Corroborating these findings, we found that sialic acid-binding immunoglobulin-like lectin (Siglec) family members are associated with specific subsets of microglia that are activated in a disease stage-specific manner in both mouse models of AD and AD patients. For example, Siglec-F is upregulated in response to Aβ pathology and is selectively expressed in Aβ-associated DAMs, while Siglec-G is upregulated in white-matter-associated LADAM in late-stage AD. These findings are consistent with a model whereby Aβ or tau pathologies stimulate the activation of microglia in different patterns. Both Aβ and tau pathologies are necessary to induce the emergence of EADAM and LADAM, and these findings provide important implications for the identification of novel molecular targets and therapeutic strategies for the treatment of AD.
Methods
Mice
Tau4RΔK-AP (APPswe;PS1ΔE9;CamKII-tTA;TetO-TauRDDK),Tau4RΔK (CamKII-tTA;TetO-TauRDΔK) and APP;PS1 (APPswe;PS1ΔE9) transgenic mice were generated as described previously. Tau4RΔK were generated by crossbreeding TetO-TauRDΔK transgenic mice carrying mutant Tau fragment with regulatory element moPrP-tetP promoter [26] with CamKII-tTA mice to bring the Tau transgene under the control of the tet-off CamKII promoter [27]. Tau4RΔK mice were crossbred with APPswe;PS1ΔE9 mice [28] to generate Tau4RΔK-AP mice that develop both Tau pathology and Aβ amyloidosis. Because of sex differences observed in Tau4RΔK-AP mice, only female mice were used in this study. Brains were collected from the mice at 6-month-old and 12-month-old for scRNA-Seq, histological and immunohistological studies. Among the three replicates (both 6-month-old and 12-month-old), two sets of the APPswe;PS1ΔE9 and control mice also contain CamKII-tTA driver. Compared with the one set APPswe;PS1ΔE9 and wild-type mice without CamKII-tTA driver, we did not observe a significant difference in microglia in our scRNA-Seq data. To suppress the potential impacts of TTA expression during postnatal development [29], mice were fed with Teklad Global 18% Protein Rodent Diet containing 200 mg/Kg doxycycline hydrochloride (Envigo Teklad Diets, Madison WI) during gestation and before weaning.
CaMKIIαCreERT2;Tdp-43F/F mice with loxP sites flanking Tdp-43 exon 3 were generated as described previously [30]. Oral tamoxifen citrate was administered in the feed (Harlan Teklad) at an average of 40 mg/kg/day for 4 weeks beginning at 6- or 9 months of age to induce recombination in excitatory forebrain neurons. Mice were singly housed during this period to monitor tamoxifen-feed intake. Afterward, the mice were returned to their original cage grouping with their littermates. Brain tissues were collected two months after the tamoxifen treatment for scRNA-Seq analysis.
All mice were housed in a climate-controlled facility (14-h dark and 10-h light cycle) with ad libitum access to food and water managed by Research Animal Resources at Johns Hopkins University. All animal procedures were in accordance strictly with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Johns Hopkins University Animal Care and Use Committee.
scRNA-Seq cell preparation
Mouse cortices were collected and dissociated using a previously published protocol [31, 32]. Basically, cerebral cortices and hippocampi were dissected into Hibernate-A media with a 2% B-27 and GlutaMAX supplement (0.5 mM final). Tissues were dissociated in papain (Worthington) and debris was removed using OptiPrep density gradient media following cell dissociation. Cells were then processed immediately for scRNA-Seq.
Study design and participant
The postmortem tissues used in the present study were provided by the Johns Hopkins Brain Resource Center. For histological analysis, formalin-fixed paraffin-embedded (FFPE) tissue Sects. (10 μm) of the inferior parietal region were obtained from 36 pathologically confirmed AD cases with Braak neurofibrillary stages II-VI and controls. Details of human cases used for histology are available in Table S10. The cohort (n = 68) included 35 males and 33 females ages 51 to 96 years (x = 75.84), 62 Whites, and 5 African-Americans. The average postmortem interval was 23 h. We also examined FFPE hippocampal samples from patients with non-AD tauopathies for histology (Table S10).
In addition to the FFPE sections, we also examined frozen samples from the superior frontal gyrus and entorhinal cortex in AD cases for snRNA-Seq (n = 8), as well as 2 samples from patients with non-AD tauopathy. Details of human cases used for snRNA-Seq are available in Table S11. All AD subjects had been prospectively recruited, clinically characterized by the Johns Hopkins Alzheimer’s Disease Research Center (ADRC), and underwent neuropathologic postmortem examination excluding Lewy body disease or non-AD tauopathies. The clinical and autopsy components of this study were approved by the Johns Hopkins Medicine IRB.
Isolation of nuclei from the frozen human brain for snRNA-Seq
Flash-frozen brain tissue was processed for snRNA-Seq following a modified 10 × Genomics protocol. Briefly, lysis buffer containing (10 mM Tris–HCl pH7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40, 0.01% Digitonin, 1 U/ul RNase inhibitor, 1% BSA) was added into a tube containing brain micropunch (~ 100 μm) and incubated on ice for 15 min with gentle pestle grinding (5 strokes every 3 min). Wash buffer (10 mM Tris–HCl pH7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.2 U/ul RNase inhibitor, 1% BSA) was added and filtered through a 50 μm filter. Debris was removed using OptiPrep density gradient media and nuclei morphology was accessed under the light microscope. Nuclei were then immediately processed for snRNA-Seq.
scRNA-Seq/snRNA-Seq generation
Cells or nuclei were loaded into the 10 × Genomics Chromium Single Cell System (10 × Genomics) and libraries were generated using v3.1 chemistry following the manufacturer’s instructions. Three biological replicates, where each replicates consisted of littermate mice, were used for both 6-month-old and 12-month-old scRNA-Seq runs. Two technical replicates were used for TDPKO scRNA-Seq. Two biological replicates, where each biological replicate had a technical replicate, were used for human snRNA-Seq. Libraries were sequenced on Illumina NovaSeq6000. scRNA-Seq data were first processed through the Cell Ranger (v.3.1.0, 10 × Genomics) with default parameters, aligned to the mm10 genome (refdata-cellranger-mm10-3.0.0), and matrix files were used for subsequent bioinformatic analysis. snRNA-Seq data were first processed through the Cell Ranger (v.5.0.0, 10 × Genomics) with ‘include-introns’, aligned to the GRCh38 genome (refdata-gex-GRCh38-2020-A), and matrix files were used for subsequent bioinformatic analysis.
scRNA-Seq data analysis
Processing scRNA-Seq datasets
Seurat v3.15 [33] was used to process matrix files, first by selecting cells with more than 500 genes, 1000 UMI, and less than 50% mitochondrial genes and 20% ribosomal genes. Datasets were then normalized using Seurat ‘scTransform’ function with regressing the number of genes and UMIs using ‘vars.to.regress’, and Harmony v1.0 [34] was used to adjust for batch variation by treating individual scRNA-Seq run as a variance group.
The top 20 variables obtained from Harmony analysis were used for UMAP dimensional reduction. The Louvain clustering algorithm was used to first identify main cell types with default resolution and the top 20 reduction variables obtained from Harmony analysis. Individual cell types were first identified by cross-referencing previous scRNA-Seq datasets [32, 35], ASCOT database [36], as well as other datasets obtained from brain myeloid and microglia [21, 37, 38]. Doublet cells—cells that express both cell-type-specific genes (~ 5% of the overall datasets) and unhealthy cells (when mitochondrial/ribosomal genes were expressed at much higher levels compared to other clusters) were excluded from further analysis. This was performed in individual ages, across all genotypes. Individual microglia were subsetted and processed as described above. Any doublet cells (~ 2%) that couldn’t be previously identified were removed for further analysis. WAM [25], EADAM, or LADAM genes (top 50 genes from differential gene expression lists) were superimposed using Seurat AddModuleScore function to calculate module score.
Differential gene tests on the scRNA-Seq datasets were initially performed using Seurat v3.15’FindAllMarkers’ function (test.use = “wilcox”, logfc.threshold = 0.5, min.pct = 0.2). A cluster of microglia that was distributed almost equally among all four genotypes, and showed higher expression of immediate-early genes such as Fos, Atf3, and Junb [39], and this cluster is derived from technical artifacts during the heat-activated enzymatic dissociation step [39] and these microglia were then removed for any downstream analysis.
To identify the percentage or expression level of EADAM or LADAM genes across genotypes, as well as in other mouse models of neurodegenerative disorders and human AD samples, EADAM (Table ST3) and/or LADAM (Table ST5)-enriched genes were first identified(higher than average logfc > 0.2). Cells that express higher than UMI > 3 were flagged as positive cells, and then the percentage of positive cells relative to the total number of cells in a cluster was identified. These parameters can clearly be defined over 98% of EADAM or LADAM cells in our AD scRNA-Seq dataset.
Regulons
To identify regulons controlling gene expression in different microglial populations across ages and genotypes, SCENIC [40] using python implemented pySCENIC v.0.10.0 Gene regulatory networks (using –masks_dropouts), regulons and network activity of regulons was calculated using default parameters with mm10 feather files on the microglial dataset using raw count matrix. Regulon specificity scores were ranked following the SCENIC pipeline and top regulons with a z-score higher than 2 were identified as microglia-specific.
GO pathway analysis
Top differential genes (adjusted p-value < 0.05, fold change > 0.5) in each microglial cluster were used as an input for GO pathway analysis using ClusterProfiler (v3.12.0) [41].
RNA velocity
RNA velocity [42] was utilized to understand the dynamic state of microglia, and how each genotype-specific disease-associated microglia cluster was initiated across AD progression. Kallisto v0.46.2 and bustools v2.27.9 [43, 44] python wrapper kb-python were used to obtain spliced and unspliced transcripts using –lamanno with GRCm38 mouse genome. Scanpy v1.5.1 [45] and scVelo v0.2.1 [46] were used to process the Kallisto output with default parameters, based on UMAP coordinates obtained from Seurat.
Pseudotime analysis
Monocle v3.0.2 [47] was used to perform pseudotime analysis to identify differences in gene expression across microglial disease states, where the trajectory routes used for pseudotime analysis were identified based on trajectories from RNA velocity analysis, and high-variance genes (q < 0.001) were used for pseudotime plotting.
Histology and Immunohistochemical analysis
For the histological and immunohistochemical analysis, mice were anesthetized and brains were removed and weighed. Hemibrains were fixed by submerging into 4% PFA in PBS, embedded into the paraffin, and sectioned in the sagittal plane. For histological analyses, 10 μm brain sections were stained with hematoxylin and eosin (H&E) or Cresyl violet. For immunohistochemical analysis, sections were treated with 10 mM citrate buffer (pH 6.0) by microwave in high power for 6 min for antigen retrieval; endogenous peroxidase was quenched by treating with 0.3% H2O2, and nonspecific binding of antibodies was eliminated using blocking buffer (10% normal goat serum in PBS with 0.3% Triton-X) for one hour at room temperature. The primary antibody was prepared in a blocking buffer and was applied overnight at 4 °C, followed by a secondary antibody for 30 min incubation at room temperature. For the secondary antibody and avidin-biotinylated peroxidase system, we used the Vectastain Universal Elite ABC kit (Vector Laboratories). Brain sections were stained with: antiserum against Aβ peptides 6E10 (1:1,000; SIG-39300, Covance); rabbit antiserum against phosphorylated S422 of tau (1:2,000; 44764G, Invitrogen, Carlsbad, CA); polyclonal antiserum against GFAP (1:1,000, Z0334, Dako Corporation, Carpinteria, CA); polyclonal antiserum against microglial (IBA1, 1:1,000, CP290, Biocare Medical, CA); monoclonal antibody against microglial (IBA1, 1:1000, MA5-27,728), monoclonal antibody against NeuN (MAB377, Millipore); polyclonal antiserum against Siglec-10 (1:50, HPA027093, Sigma); monoclonal Antibody Siglec-F (CD170, 1:300, 14–1702-82, ThermoFisher Scientific), polyclonal antiserum against microglial CD22 (Cy34.1, 1:200, BE0011, BioCell); monoclonal antibody against LPL (1:200, ab21356, Abcam); monoclonal antibody against Lgals3 (1:200, 14–5301-82, Invitrogen). monoclonal antibody against STAT1 (1:200, AHO0832 ThermoFisher Scientific).
For immunofluorescence, the section were detected by secondary antibodies: Alexa Flour 488 goat anti-rabbit IgG(H + L), Alexa Flour 488 goat anti-mouse IgG(H + L); Alexa Flour 488 goat anti-rat IgG(H + L); Alexa Flour 594 goat anti-rabbit IgG(H + L), or Alexa Flour 594 goat anti-rabbit IgG(H + L) (ThermoFisher Scientific). Sections were counterstained with DAPI before being examined under a Zeiss LSM 510 laser scanning fluorescence confocal microscope. Z-stack projections were made from serial scanning every 0.34 μm to reconstruct the images.
Thioflavin-T (ThioT) staining, after deparaffinized and hydrated, slides were incubated in potassium permanganate solution (0.25%) for 5 min, then the slides were transferred to 1% potassium-disulfate and oxalic acid for 5 min before incubated in 0.02% ThioT for 8 min. Slides were dehydrated and mounted with an aqueous mounting medium for fluorescence.
The quantitative score of Siglec-10 positive cells in the inferior parietal region of human brains was the average count of at least three brain sections in a power (2,000X) microscope field using the ImageJ program. Siglec-10 signals in gray and white matter in the same section were counted separately. The histological section of the human inferior parietal region includes the cerebral cortex and subjacent white matter. These two compartments can be easily separated by their cytological features. The cortex contains a large number of neurons characterized by their large size and prominent nucleus and nucleolus. By contrast, the white matter is rich in oligodendrocytes, which are small, have a compact nucleus and virtually no cytoplasm; and astrocytes. Neurons are virtually absent in the white matter.
Neurons in mouse brains were identified by NeuN staining. The quantitative scores of NeuN + , IBA1 + , SiglecG + , Lgal3 + cells were the average counts in a power (2,000X) microscope field of at least three sagittal sections at 2 mm from the midline of the mouse brains using ImageJ.
To quantify the Amyloid plaques, at least three sagittal sections at 2 mm from the midline of the mouse brains were selected. Amyloid plaque size, numbers, or area fraction were quantified based on the 6E10 immunoreactivity or ThioT staining using ImageJ.
Statistical analysis
All data were analyzed statistically by unpaired Student’s two-tailed t-test or one-way ANOVA with Tukey correction for multiple comparisons for cell counting analysis, and two-way ANOVA with Tukey’s multiple comparison test for scRNA-Seq cell distribution analysis (distribution of microglia clusters within each genotype and distribution of cell types within each genotype), using GraphPad Prism (GraphPad Software, La Jolla CA the USA). In all tests, values of p < 0.05 were considered to indicate significance.
Results
ScRNA-Seq of microglia in mice harboring Aβ plaques and/or tau deposition
To identify microglia subtypes through AD-like pathology progression, we performed scRNA-Seq in the cerebral cortex and hippocampus of transgenic mice displaying Aβ plaques and/or tau pathologies. We took advantage of our previously characterized mouse model of AD (Tau4RΔK-AP mice), which exhibits AD-like pathologies including Aβ plaques and tau tangles and results in progressive neuronal loss and brain atrophy [24]. In a cross-breeding strategy using mutant APPswe;PS1ΔE9 (APP;PS1) [28] and Tau4RΔK mice, a cohort of Tau4RΔK-AP mice was generated. Female mice were aged to either 6-month-old or 12-month-old. Six-month-old Tau4RΔK-AP mice mimic an early AD stage, characterized by low levels of Aβ plaques with minimum tau phosphorylation (Figure S1) and deposition [24] in the hippocampus but not in the cortex and no loss of neurons (Figure S1). Twelve-month-old Tau4RΔK-AP mice mimic a late disease stage, characterized by robust Aβ plaques (Figure S1), tau deposition (Figure S1), loss of neurons and brain atrophy (Figure S1). For these two time points, in addition to Tau4RΔK-AP mice, we also collected cerebral cortices and hippocampi from littermate controls (WT), APP;PS1 (Aβ plaques (S1)), and Tau4RΔK (tau deposition, neuronal loss (Figure S1), and brain atrophy (Figure S1)) mice. Compared to Tau4RΔK mice at 12 months of age, Aβ plaques accelerated tau pathogenesis and tau aggregation-dependent neuronal loss and brain atrophy (Figure S1) in 12-month-old Tau4RΔK-AP mice, as previously shown [24]. On the other hand, tau aggregation has little effect on the deposition of Aβ plaques, as well as plaques morphology, in Tau4RΔK-AP mice as compared with that of APP;PS1 mice (Figure S1). Twelve-month-old Tau4RΔK-AP mice also showed an increase in plaque numbers and areas compared to 6-month-old Tau4RΔK-AP mice but plaque size did not differ (Figure S1). We also observed an increase in microglia in the cerebral cortex, hippocampus, and corpus callosum between 6 and 12 months (Figure S2). We then subjected these tissues across 4 genotypes at both 6 months and 12 months to scRNA-Seq analysis.
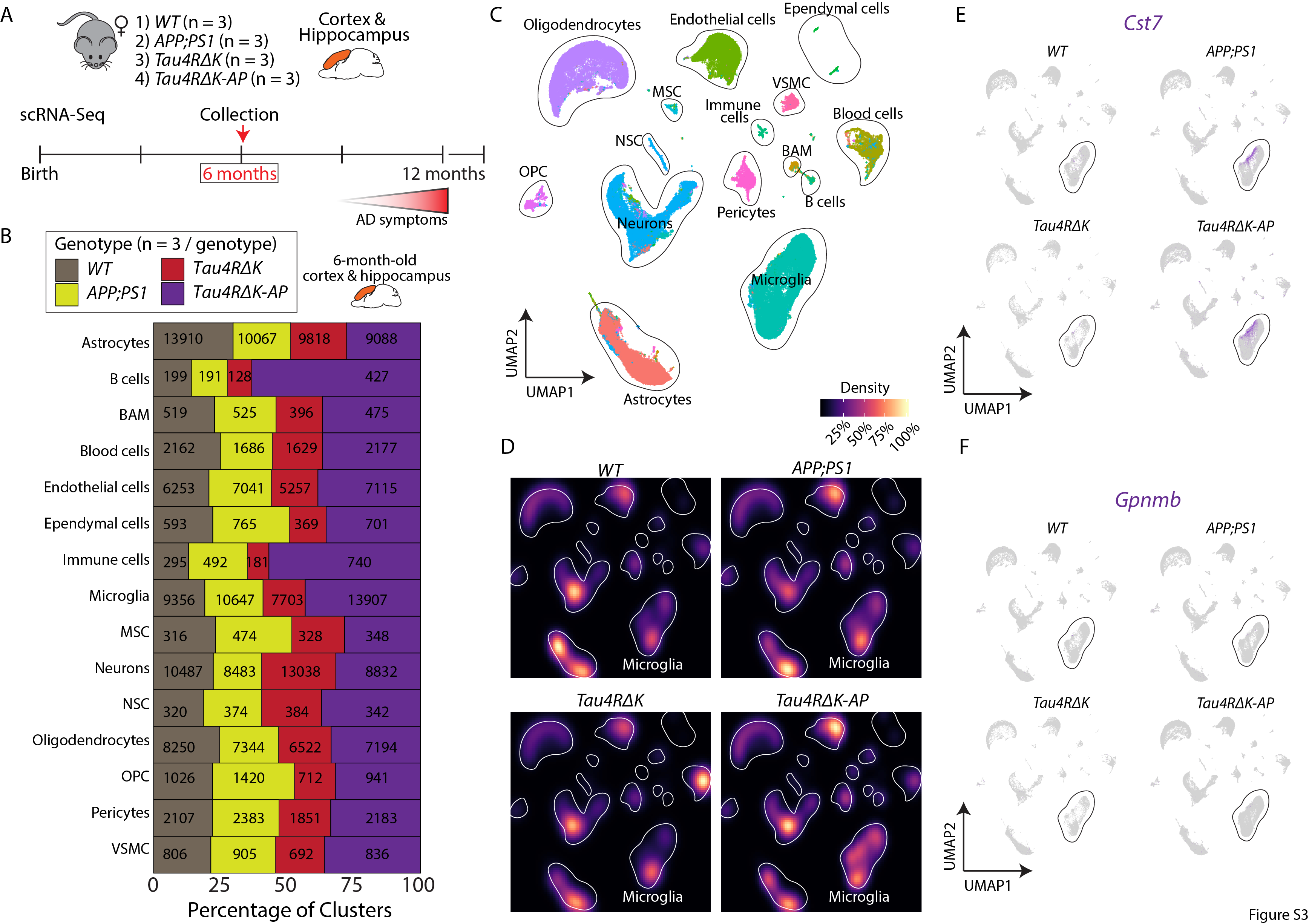
We first analyzed female 6-month-old cerebral cortices and hippocampus, across four genotypes in triplicates (Figure S3). We observed all major cell types of CNS, including neurons, astrocytes, oligodendrocytes, and microglia (Figure S3, Table S1). Previously, we confirmed the initiation of tau deposition in 6-month-old cerebral cortices [24]. We observed the absence of neuronal loss or brain atrophy in these mice, along with the presence of Aβ plaques (Figure S1). As expected, we observed a few DAM microglia in Tau4RΔK-AP brains in early-stage disease (Figures S3, S9). Analyzing three biological replicate samples, we observed that microglia in APP;PS1 and Tau4RΔK-AP mice (Figures S3, S5), expressed a high level of disease-associated microglia 1 (DAM1) marker including Cst7 (Figure S3). However, we failed to observe DAM2 markers, including Gpnmb, even in APP;PS1 and Tau4RΔK-AP mice at this age (Figure S3).
Using scRNA-Seq, we also identified cell clusters corresponding to major subtypes of neurons, glia, and immune cells (Figure S4, Table S2) in 12-month-old mice of all four genotypes, analyzing three biological replicates. Consistent with our previous observation, AD-like pathologies in Tau4RΔK-AP mice led to gliosis and neuronal loss. An increase in the microglia population along with a corresponding decrease in the number of neurons was observed in the Tau4RΔK-AP mice (Figures S1, S4). Compared to the other three control mice (WT, APP;PS1, and Tau4RΔK mice), Tau4RΔK-AP mice also showed an increase in immune cells as well (Figure S4).
APP;PS1, Tau4RΔK and Tau4RΔK-AP mice showed a marked increase in the number and proportion of microglia as compared to WT mice, (Figures S2, S4, S5). Known DAM1 marker genes such as Cst7, and DAM2 genes such as Gpnmb, were strongly expressed in both APP;PS1 and Tau4RΔK-AP mice (Figure S4). These results establish that these scRNA-Seq datasets have sufficient quantitative power to identify microglia subtypes that are influenced by Aβ and/or tau deposition in a disease stage-specific manner.
Identification of a novel Early-stage AD-Associated Microglia (EADAM) induced by both Aβ plaques and tau deposition
We then subsetted and profiled microglia from the cerebral cortex and hippocampus of 6-month-old Tau4RΔK-AP mice, along with the three control mouse lines. Homeostatic microglia expressing genes such as Tmem119 and P2ry12, were predominantly detected across all genotypes (Fig. 1A, B), but two additional microglia clusters were also observed (Fig. 1A, Table S3). The first microglia cluster expressed classic DAM1-like markers, including Apoe, Cst7, and Lpl (Fig. 1B). The second microglia cluster expressed DAM1-like markers but also displayed a unique gene module consisting of interferon-related genes, such as Ifitm3, Ifit27l2a, and Ifit3, as well as Stat1 (Fig. 1B). Since this cluster was predominantly composed of Tau4RΔK-AP microglia and detected during the early and presymptomatic stage of the disease (Fig. 1C), we thus identify this cluster as Early-stage AD-Associated Microglia (EADAM). Our regulon and pathway analysis confirmed the association of EADAM with interferon-regulating transcription factor Irf2/7/9 (Fig. 1D) and linkage to the GO term ‘Response to Interferon-beta’ (Fig. 1E).
Fig. 1.

The microglia subtype EADAM is observed in the Tau4RΔK-AP mice in the 6-month-old cortex. A UMAP plot showing microglia clusters—Homeostatic, DAM, and EADAM, across 4 genotypes in 6-month-old cortex (n = 3/genotype), and pie graphs showing the distribution of 3 microglia clusters across genotypes (n = 3/genotype). B Violin plots showing top cluster genes of 3 microglia clusters. C Bar graphs showing the distribution of 3 microglia clusters within each genotype (n = 3/genotype). Note the significant increase in the EADAM cluster in Tau4RΔK-AP mice. D Regulon analysis with SCENIC [40], showing key regulons in each microglia cluster. E GO analysis of top differential genes reveals a biological function in each microglial cluster. F UMAP plots of 7-month-old 5XFAD mice in Zhou et al. 2020 [23] (top) and a small cluster of cells (red circle) expressing a few EADAM-enriched markers (bottom). G Heatmap showing expression of EADAM-enriched genes across genotypes. * P < 0.05, ** P < 0.005, *** P < 0.0001
A small EADAM-like cluster is also detected in APP;PS1 microglia (Fig. 1A, C). This observation is consistent with the previous report that the accumulation of Aβ plaques leads to the activation of the interferon pathway [48–51]. A small cluster of cells that resemble EADAM-like microglia, expressing Irf7 and other EADAM-enriched genes, such as Ifit3, Isg15, Ifi27l2a, were also detected in 7-month-old 5xFAD snRNA-Seq data [23] (Fig. 1F).
We next compared differential gene expressions between EADAM populations across four genotypes. Despite a significant contribution of APP;PS1 microglia to EADAM cluster, the overall level of EADAM-enriched genes was expressed at a much lower level in APP;PS1 microglia than that of Tau4RΔK-AP EADAM (Fig. 1F, Table S4). On the other hand, the expression of interferon pathway-related genes was not observed in Tau4RΔK microglia, suggesting that while the accumulation of Aβ plaques leads to activation of the interferon pathway, both Aβ plaques and tau aggregation are necessary to fully induce EADAM gene modules in Tau4RΔK-AP microglia. In summary, our data identify a novel microglia subtype termed EADAM that is induced by a combination of Aβ plaques and tau deposition during early-stage AD.
Identification of Late-stage AD-Associated Microglia (LADAM)
To further clarify the influence of Aβ plaques and tau tangles in microglia pathophysiology during late-stage AD, we unbiasedly subdivided subsetted microglia populations into five distinguishable clusters across all four genotypes (WT, APP;PS1, Tau4RΔK, and Tau4RΔK-AP) in 12-month-old mice. A few notable differences were observed in 12-month-old mice compared to 6-month-old mice. The DAM cluster was further divided into two clusters—DAM1 and DAM2. The DAM1 cluster expressed genes such as Tyrobp, Ctsd, C1qa (Fig. 2A, B, Table S5), whereas DAM2 expressed DAM1-enriched genes and additional disease-related genes, such as Gpnmb, Cst7, and Spp1 (Fig. 2B, Table S5). The induction of DAM2 in response to Aβ plaques has been documented in a variety of APP-based mouse models of AD [20]. Indeed, both clusters were highly represented in animal models with Aβ pathologies (APP;PS1 and Tau4RΔK-AP mice, Fig. 2A, C), but the DAM2 abundance in mouse models with only tau pathologies (Tau4RΔK mice) was low (Fig. 2A, C, Table S6). These observations are consistent with the view that these DAM clusters arise mainly as a result of Aβ plaques. Similar to situations in the EADAM population of 6-month-old mice, Tau4RΔK-AP mice generally have higher levels of DAM-enriched genes than that of APP;PS1 mice (Fig. 2F). Since Aβ plaques level in Tau4RΔK-AP mice is similar to that of APP;PS1 mice (Figure S1), this observation indicates that while Aβ plaques can induce DAM2, tau deposition and/or neuronal loss could further enhance DAM-enriched genes in Tau4RΔK-AP mice.
Fig. 2.

The microglia subtype LADAM is observed in the Tau4RΔK-AP mice in the 12-month-old cortex. A UMAP plot showing microglia clusters—Homeostatic, DAM1, DAM2, EADAM, and LADAM, across 4 genotypes in 12-month-old cortex (n = 3/genotype), and pie graphs showing the distribution of 4 microglia clusters across genotypes (n = 3/genotype). B Violin plots showing top cluster genes of 3 microglia clusters. C Bar graphs showing the distribution of 5 microglia clusters within each genotype (n = 3/genotype). Note the significant increase in the LADAM cluster in Tau4RΔK-AP mice. D Regulon analysis with SCENIC [40], showing key regulons in each microglia cluster. E GO analysis of top differential genes reveals a biological function in each microglia cluster. F Heatmap showing expression of DAM-enriched genes across genotypes. G Heatmap showing expression of LADAM-enriched genes across genotypes. * P < 0.05, ** P < 0.005, *** P < 0.0001
A diminished EADAM cluster was still detected in 12-month-old Tau4RΔK-AP mice. Interestingly, EADAM-like microglia were also detected in 12-month-old Tau4RΔK mice, although they were absent at 6 months of age (Fig. 2C). In addition to the widespread tau aggregation, Tau4RΔK mice already show a neuronal loss at 12-month-old [24], suggesting that while the presence of tau aggregation alone may not induce EADAM formation, cell death-induced inflammation may also induce EADAM-like gene expression in microglia.
We also observed a unique microglia cluster that was not detected in 6-month-old mice (Fig. 2A). Cells in this cluster expressed MHC Class II genes, such as Cd74, H2-A2, H2-Eb1, and H2-Ab1, and additional markers like Lgals3 (Fig. 2B, Table S4). We termed these microglia Late-stage AD-Associated Microglia (LADAM), which were found in Tau4RΔK-AP, as well as Tau4RΔK, and APP;PS1 samples (Fig. 2A, C). Regulon analysis identified key transcription factors associated with gene regulatory networks across all microglial clusters. Notably, while Hif1a and Eomes are selectively expressed in the DAM2, Runx3 and Irf4/7 are expressed in LADAM (Fig. 2D), and GO analysis on LADAM shows enrichment for ‘MHC Class II’ (Fig. 2E). In addition to MHC class II genes, we also identified enriched expressions of S100a family genes, including S100a4, S100a6, and S100a10 (Fig. 2G).
Although LADAM was observed in Tau4RΔK-AP,Tau4RΔK, and APP;PS1 samples, we found that many LADAM-specific genes (e.g. MHC Class II genes) are more highly expressed in Tau4RΔK than in APP;PS1 microglia (Fig. 2G), suggesting that this microglia subtype might be induced by the development of tauopathies, such as tau tangles or tau phosphorylation. Moreover, relative to Tau4RΔK microglia, we found that LADAM gene signatures were more pronounced in Tau4RΔK-AP microglia (Fig. 2G, Table S5), which exhibited Aβ plaques, more robust tau pathologies and neuronal loss. These datasets show that tau pathologies are sufficient to induce the emergence of LADAM and that their amplification is Aβ plaque-dependent. However, since MHC class II genes that are expressed in LADAM, such as Cd74, H2-A2, H2-Eb1, and H2-Ab1, have also been previously observed in microglia in both the CK-p25 neurodegeneration mouse model [52, 53] and some mouse models of late-stage amyloidosis [48–50], some of LADAM gene modules may be induced at least in part by neuronal death.
Requirement of both Aβ plaques and tau deposition to elicit the emergence of disease stage-dependent microglia subtypes
To clarify how the emergence of EADAM, DAM2, and LADAM is regulated by Aβ plaques and tau deposition, we merged datasets obtained from microglia of both 6-month-old and 12-month-old samples (Fig. 3A). RNA velocity analysis shows that DAM1 is likely to give rise to, or to be closely related to, both DAM2 (Aβ plaques driven) and EADAM (Aβ plaque and tau deposition driven), whereas the convergence of DAM2 (Aβ plaque-driven and tau deposition-enhanced) and EADAM may ultimately lead to induction of LADAM, as the result of tau deposition/cell loss-driven and Aβ plaque-formation (Fig. 3B). As shown above, EADAM is composed mostly of cells derived from 6-month-old Tau4RΔK-AP mice (Fig. 3B). The DAM2 clusters were enriched in 12-month-old APP;PS1, and Tau4RΔK-AP mice (Fig. 3B), and LADAM clusters were enriched in 12-month-old APP;PS1, Tau4RΔK, and Tau4RΔK-AP mice (Fig. 3B).
Fig. 3.

The emergence of disease stage-dependent microglia subtypes coincides with Aβ and tau deposition. A UMAP plot showing microglia clusters in the 6- and 12-month-old cortex (all genotypes) (left) and UMAP plot with RNA velocity (right) showing 2 potential transitions between DAM1 and LADAM, with transitions into EADAM and/or DAM2 in between. B UMAP plot showing the density of the captured microglia clusters across genotypes in the 12-month-old cortex. C Pseudotime analysis showing gene expression changes between DAM1, DAM2, and LADAM. D Pseudotime analysis showing gene expression changes between DAM1, EADAM, and LADAM
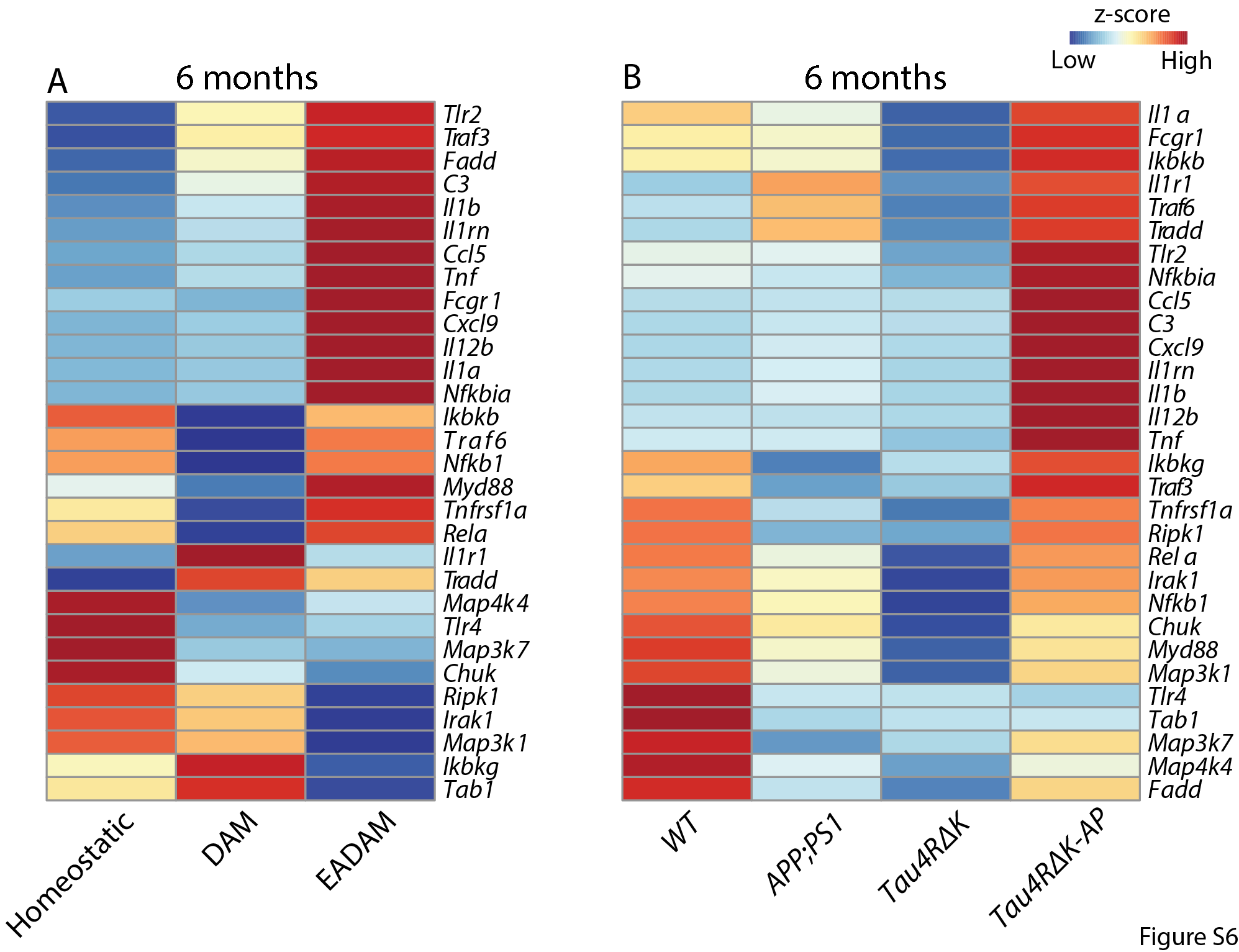
Furthermore, RNA velocity coupled with pseudotime analysis (Fig. 3C, D) identified gene expression changes occurring during the temporal progression from DAM1 to DAM2 to LADAM, with changes in multiple inflammation-related genes, such as Ccl6, Csf1, Cd34 (Fig. 3C); as well as during the temporal progression from DAM1 to EADAM to LADAM, with changes in interferon genes, such as Ifiti family members (Fig. 3D). We have also identified NF-kB pathway genes, which are important in tau seeding and spreading, are enriched in EADAM and Tau4RΔK-AP mice at 6-month-old (Figure S6) as previously shown [54].
A microglia cluster(s) that express either interferon genes and/or MHC Class II genes have been identified in other neurodegenerative models, such as CK-p25 [53] and Trem2 deficient [55, 56] FTDP-17-linked tau model (P301L) [57] crossed with PS2APP [58] mice. However, none of the disease models showed the development of separated EADAM and LADAM clusters in a stage-dependent manner. This led to the hypothesis that, while potential conservation of EADAM- or LADAM-like subtypes might occur across both neurodegeneration and neuroinflammation, a combination of both Aβ plaques and tau deposition is critical for the induction of disease-stage specific microglia subtypes in AD.
To test this hypothesis, we then used another neurodegenerative mouse model CamKIICreERT2;Tardbplox/lox (TDP43 KO), where a tamoxifen induction leads to conditional deletion of TDP-43 in the forebrain of the mice, resulting in the selective vulnerability of hippocampal CA3 neurons [30]. We generated scRNA-Seq data from the cerebral cortex and hippocampus of 2 different TDP43 KO samples, one being induced with 4-OHT at 6 months and collected at 9 months (TDP43 KO6−>9) and the other being induced with 4-OHT at 12 months and collected at 15 months (TDP43 KO12−>15) (Fig. 4A-C). While both TDP43 KO mice showed DAM1-like clusters, TDP43 KO6−>9 showed both EADAM- and LADAM-like clusters that respectively selectively express interferon genes or MHC Class II genes (Fig. 4A-C, Table S7). TDP43 KO12−>15 also showed an EADAM-like cluster (Fig. 4A-C, Table S8), supporting the view that EADAM- or LADAM-like subtypes might occur across neurodegenerative.
Fig. 4.

Analysis of EADAM and LADAM markers in other mouse models of neurodegenerative disease. A UMAP plot showing microglia clusters in the 6 and 12-month-old cortex (all genotypes) (Fig. 3A left panel). B UMAP plot showing microglia clusters in CamKIICreERT2;Tardbplox/lox (TDP43 KO), 4-OHT induction at 6 months and collected at 9 months. N = 2. C UMAP plot showing microglia clusters in CamKIICreERT2;Tardbplox/lox (TDP43 KO), 4-OHT induction at 12 months and collected at 15 months. N = 2. D Heatmap plot showing expression of EADAM-enriched genes across AD genotypes and in TDP43 KO. E Heatmap plot showing expression of LADAM-enriched genes across AD genotypes and in TDP43 KO
We then integrated datasets from AD mice with those from TDP43 KO mice (Fig. 4D, E). As mentioned above, both 6-month-old APP;PS1 and Tau4RΔK-AP microglia showed a similar expression pattern of EADAM-enriched genes (Fig. 4D), but Tau4RΔK-AP microglia displayed much higher expression levels of these genes than did APP;PS1 microglia (Fig. 4D). 12-month-old Tau4RΔK and Tau4RΔK-AP microglia showed overall lower expression levels, but broadly similar EADAM expression patterns (Fig. 4D). Both TDP43 KO microglia showed a much weaker level of EADAM-enriched genes, similar to the 6-month-old APP;PS1 microglia (Fig. 4D).
LADAM-enriched genes are composed of DAM2-enriched genes, MHC Class II genes, and S100a family genes. While the LADAM-like cluster in TDP43 KO6−>9 microglia expressed MHC Class II genes, the level of expression was much lower than any of APP;PS1, Tau4RΔK and Tau4RΔK-AP microglia (Fig. 4E). We also failed to detect any DAM2-like clusters, or either S100a family genes and Siglecg in TDP43 KO6−>9 microglia.
A similar observation was also made in CK-p25 microglia [53] (Figure S6). While the integration of this dataset was not possible due to the use of different single-cell formats (SMART-Seq was used to generate the CK-p25 dataset), we noted that both EADAM-like interferon genes and LADAM-like MHC Class II genes were more highly expressed in CK-p25 microglia than the control (Figure S7). However, both EADAM-like and LADAM-like microglia were intermingled, rather than forming separate clusters. We also failed to detect any S100a family genes and Siglecg expression in the CK-p25 microglia dataset, similar to our observations in the TDP43 KO dataset (Figure S7).
This indicates that Aβ plaque or tau pathology independently induces specific gene signatures in microglia like EADAM and LADAM, and similar microglia clusters can also be observed in other neurodegeneration models. Neuroinflammation and/or cell loss that occurs in neurodegeneration diseases could be a potential trigger for microglia to develop EADAM- or LADAM-like gene expression profiles. However, the combination of both Aβ plaque and tau pathology results in separated clusters of EADAM and LADAM at different stages of the disease, much higher expression levels of the signature genes, and induction of additional molecular markers in LADAM.
We have also compared our dataset with previous findings from human snRNA-Seq [59], which have identified gene expression profiles that are associated with AD pathology. The two main identified populations from the previous study were AD1-microglia, which show a strong correlation with Aβ, and AD2 microglia, which show a correlation with tau-loading. We compared the enriched genes of AD1- and AD2-microglia onto our dataset (Figure S8), and we observed a strong correlation of AD1-enriched genes in APP;PS1 and Tau4RΔK-AP at 6 months, and a strong correlation of AD2-enriched genes in Tau4RΔK (Figure S8).
Furthermore, we performed immunostaining with various markers of microglia subtypes which we have identified from our scRNA-Seq dataset. To first confirm the DAM activation, we stained the brain slices with LPL, an AD risk factor identified as a DAM-enriched gene (Fig. 1). LPL-positive microglia are detected in APP;PS1 and Tau4RΔK-AP mice at 6 months of age and the signals are further increased at 12 months of age (Figure S9). On the other hand, no LPL signals were detected in Tau4RΔK and WT mice, suggesting that the DAM were more specifically activated by Aβ pathologies in agreement with previous findings. A similar pattern of activation was also detected in CD22 (Siglec-2) (Figure S9).
One of the most upregulated genes that are strongly related to microglial DAM phenotype is Lgals3 (Figs. 1, 2), a carbohydrate-binding protein [53, 60–62]. Homeostatic microglia do not express Lgals3 (Figs. 1, 2) [20, 60], but Lgals3 expression is significantly upregulated in microglia in response to Aβ plaques both in humans and mice [63, 64]. Furthermore, Lgals3 is associated with activated microglia, and Lgals3 is mainly expressed and released in the damaged brain by reactive microglia [65]. To further confirm the DAM activation in AD mouse models, we immunostained brain slices with anti-Lgals3 antibodies (Figure S10, S11). While Lgals3 was enriched around the Aβ plaques in APP;PS1 and Tau4RΔK-AP mice (Figures S10, S11), we have also observed robust Lgals3 staining at the corpus callosum in Tau4RΔK, with much stronger expression in Tau4RΔK-AP mice (Figures S10, S11). The Lgals3 signal was co-localized with microglia marker IBA1, but not astrocyte marker GFAP, supporting the notion that Lgals3 was specifically activated in microglia by AD pathologies. We have identified a robust Lgals3 expression in LADAM in 12-month-old Tau4RΔK mice (Fig. 2), and Lgals3 levels were associated with the severity of tau aggregation (Figure S11), suggesting that Lgals3 could also be induced by Tauopathy in a sub-population of LADAM that reside near the corpus callosum.In addition, we have validated that EADAM marker, STAT1, is robustly expressed in microglia in Tau4RΔK-AP mice at 6-month-old and 12-month-old (Figure S12), but not expressed in astrocytes, which is consistent with our scRNA-Seq finding (Fig. 1). STAT1 was not strongly expressed in glial cells in wild-type mice, APP;PS1 and Tau4RΔK mice, indicating that neither Aβ nor tau pathology alone could strongly induce the STAT1 expression.
Identification of stage-specific EADAM and LADAM signatures in AD samples
We next generated snRNA-Seq dataset from the human superior frontal gyrus (SFG) at Braak stage 2, which is devoid of tau pathology, and entorhinal cortex (ERC) at Braak stage 2, where AD pathology is first detected and shares a pathological resemblance to our 6-month-old Tau4RΔK-AP mice (Fig. 5A-C). We also generated snRNA-Seq from the superior frontal gyrus (SFG) at Braak stages 4 and 6, which represent late stages of AD and share a pathological resemblance to the 12-month-old Tau4RΔK-AP mice. As a control, we generated a snRNA-Seq dataset from the ERC and SFG of a non-AD neurodegenerative disorder, primary-age-related-tauopathy (PART) (Fig. 5A-C).
Fig. 5.

6-month-old EADAM at and 12-month-old LADAM clusters in Tau4RΔK-AP mice resemble microglial subtypes seen in snRNA-Seq from AD samples. A UMAP plot showing microglia clusters in the 6 and 12-month-old cortex (all genotypes) from Fig. 3A. EADAM and LADAM-enriched gene homologs were studied in human snRNA-Seq. B Schematic showing snRNA-Seq from human postmortem samples, primary age-related tauopathy (PART) entorhinal cortex (ERC), superior frontal gyrus (SFG); Stage 2 ERC, SFG; Stage 4 SFG; Stage 6 SFG. C UMAP plot showing the distribution of subsetted microglia from human snRNA-Seq. N = 2 (PART, 1102 cells), n = 2 (Stage 2 SFG, 750 cells), n = 2 (Stage 2 ERC, 2645 cells), n = 4 (Stage 4 and 6 SFG, 1692 cells). D Violin plots showing expression of EADAM-like interferon family members. E Violin plots showing expressions of LADAM-like MHC and S100 family members. F UMAP plot showing microglia from human snRNA-Seq dataset from Morabito et al., 2021. N = 7 (Control, 1384 cells), n = 11 (AD, 2742 cells). G Violin plots showing IFN, MHC, and S100 family members in the dataset from Morabito et al., 2021
We initially checked the expression pattern of EADAM- and LADAM-enriched gene homologs in the human snRNA-Seq dataset (Figure S13), and observed moderate conservation in the gene expression pattern. Most EADAM-enriched genes were seen at Braak stage 2 SFG and ERC (Figure S13), while LADAM-enriched genes were seen at Braak stage 4 and 6 SFG (Figure S13). Some inconsistency of expression patterns might be due to the absence of AD-specific microglia clusters in the human snRNA-Seq dataset, despite having a high resolution of 6,000 cells. This observation has been previously shown in other human snRNA-Seq datasets from brains affected by AD or other neurodegenerative diseases [66–68]. The difference between the human and animal models might be due to the fact that AD and other neurodegenerative diseases typically progress slowly, over many years, and all microglia populations may eventually end up transitioning to disease-associated states. This timing is different in virtually all animal models that were designed to undergo neurodegeneration much more rapidly.
While the presence of EADAM- and LADAM-like microglial subtypes in AD demonstrated important similarities with our mouse models, we also wanted to analyze the expression of MHC and interferon-induced genes. We observed consistent changes in the expression of interferon-regulated genes. These include the transcription factors and co-regulators IRF2BP2, IRF2, and IRF9, which showed higher expression at Braak stage 2 ERC than SFG, and whose expression levels were decreased in Braak stage 4/6 SFG (Fig. 5D). There also was a similar trend in the expression of other interferon-regulated genes, including IFI44, IFI44L (Fig. 5D).
We also observed a similar trend in MHC genes, such as HLA-A, HLA-B, and HLA-DPA1, which showed a higher expression level in PART ERC and SFG, Braak stage 2 ERC, and in Braak stage 4/6 SFG than Braak stage 2 SFG (Fig. 5E). More importantly, S100 genes, such as S100A4, S100A11, were exclusively detected in Braak stage 4/6 SFG but were not detected in PART or other AD snRNA-Seq (Fig. 5E). This observation was also conserved in other high-quality human AD snRNA-Seq from the prefrontal cortex [66] (Fig. 5F, G), where interferon-regulated, MHC, and S100 family genes were more highly expressed in AD samples than in the control (Fig. 5F, G). These findings thus establish the emergence of EADAM-related genes in early AD-stage and LADAM-related genes in late AD-stage and suggest that these microglia subtypes could be induced by both Aβ plaques and tau deposition in a disease stage-specific manner.
Siglecs may serve as specific biomarkers to track Alzheimer’s disease stage-specific microglia subtypes
Our analysis identifies multiple microglial genes that are selectively expressed in late-stage AD, such as the Braak-stage-dependent expression of SIGLEC10, a human homolog of Siglecg (Fig. 5E). Since several of the Siglec family genes are thought to be relevant for AD progression [69], we focused on the expression of Siglec gene family at different microglia clusters. Homeostatic microglia expressed Mag, Siglece, Cd33, Siglech (Figure S14), and EADAM expressed similar Siglec genes as homeostatic microglia but also expressed Siglec1 (Figure S14). Both DAM2 and LADAM clusters expressed high levels of Siglecf (Figure S14). Both Siglec1 and Siglecg were enriched in the LADAM. Siglec-G expression was robustly detected near the corpus callosum at 12-month-old Tau4RΔK-AP mice (Figure S15). Siglecf, in particular, was enriched in both APP;PS1, and Tau4RΔK-AP mice (Figure S14) and showed increased expression in late-stage disease (Figure S14). These data suggested that Siglec genes are expressed in a pathology and/or stage-dependent manner in microglia.
To further characterize late-stage AD-specific genes, We identified 4 subclusters within the LADAM cluster of AD mouse models (Fig. 6A). Sub-clusters 2 and 3, in particular, were most robustly and selectively expressed in Tau4RΔK-AP mice (Fig. 6B, Table S9). Cluster 2, a LADAM sub-cluster that is marked by Siglecg, also exhibited genes previously shown to be expressed in White matter-Associated Microglia (WAM), such as Lgals3, Fam20c, Vim (Fig. 6A, C) [25]. In WAM-like LADAM, we observed that this cluster also expressed S100a6, S100a4, S100a11, H2-DMb2, Runx2, and Clec10a (Fig. 6D, Table S9). While a small cluster that resembles WAM was also observed in the 6-month-old mouse cortex (Figure S6J), this small population did not express any LADAM markers in the 12-month-old cortex. Consistent with our scRNA-Seq, we did not detect high-level Siglec-G protein in the 6-month-old corpus callosum, hippocampus, and cortex (Figs. 6E, S14). Siglec-G immunoreactivity was robust in the 12-month-old corpus callosum in Tau4RΔK-AP microglia (Figs. 6, S15), as well as in the cortex and hippocampus (Figure S14). However, we failed to observe a strong Siglec-G immunoreactivity in APP;PS1 or Tau4RΔK microglia at the same age, despite increased microglia populations near the corpus callosum (Fig. 6D, S14).
Fig. 6.

A LADAM subcluster is observed in white-matter-associated microglia. A UMAP plot of 12-month-old ‘LADAM’. B Bar plot showing the distribution of 4 genotypes across 4 LADAM clusters. C UMAP plot of LADAM, showing a small cluster (C2, purple-colored cells) that resemble WAM gene modules [25]. D UMAP plots showing gene expressions that are highly enriched genes, including WAM genes (Vim and Lgals3) in the LADAM C2 cluster (left), and heatmap showing expression of WAM gene modules in C2 across genotypes (right). E Iba1 and Siglec-G immunostaining (left) and quantification (right) in 6-month-old (top) and 12-month-old (bottom) corpus callosum in WT, APP;PS1, Tau4RΔK, and Tau4RΔK-AP. F Siglec-10 immunostaining in human cortex in control, Braak stage 6, and nAD tauopathy (numbers on figure panels indicate BRC# In Table S11, immunostaining of other Braak stages are shown in Fig S15), with bar graphs showing quantification of Siglec-10 positive cells in gray (top) and white (bottom) matter. G Schematic showing human snRNA-Seq on Stage 4 and 6 SFG (left) and UMAP plot showing LADAM C2 enriched gene module (right). H UMAP plots showing expression of LADAM C2 cluster markers in human snRNA-Seq. Red boxes show high magnification views. CC = Corpus Callosum. Scale bars = 100 μm. * P < 0.05, ** P < 0.01, *** P < 0.001
To corroborate this finding in AD, we used antisera directed against Siglec-10 (the human homolog of Siglec-G) [70] to screen AD brains at multiple Braak stages (Table S10). We found Braak stage-dependent accumulation of Siglec-10 in AD brains (Figs. 6F, S15, S16), with Siglec-10 signal significantly increased from Braak stage 4 (Figs. 6F, S16) in both gray and white matters, but a slightly higher number of Siglec-10 + cells were detected in the white matter. However, Siglec-10 signal in non-AD tauopathy is similar to that of healthy controls, consistent with our snRNA-Seq data (Figs. 5G, 6F, S16). To further validate these observations, we checked snRNA-Seq datasets of human postmortem Braak stage 4 and 6 SFG (Fig. 6G, Table S11). We observed a small population of WAM-like LADAM, as seen in 12-month-old Tau4RΔK-AP mice (Fig. 6H), expressing genes like SIGLEC10, S100A6, LGALS3, FAM20C (Fig. 6H). We have also validated an upregulation of EADAM and LADAM-markers, such as Lgals3 and STAT1 in AD samples (Figure S17).
Siglecs, in particular Siglec10, are AD-enriched, as the level of Siglec-10 + cells in non-AD tauopathy was similar to that of control and low Braak stage AD (Figs. 6F, S16), indicating that Siglec-10 could potentially serve as an AD biomarker and a potential therapeutic target associated with late-stage AD. Some LADAM markers, particularly MHC II genes, were expressed in other neurodegenerative models (Figs. 4, S7), but S100 family of genes and Siglecg was unique to late-stage AD microglia and are seen in both animal models and late-stage AD patient brains. This data further supports the view that induction of the AD-specific LADAM subcluster is Aβ- and tau-dependent.
Discussion
Identification of novel disease stage-specific microglial subtypes induced by Aβ plaques and tau pathology
The identification of multiple AD risk alleles implicated in the regulation of inflammation [13, 71] and of DAM induction in response to Aβ plaques [20], strongly supports the hypothesis that microglia-driven neuroinflammation is a key regulator of AD progression. However, critical information regarding microglia subtypes that respond to disease stage-specific canonical AD pathologies of Aβ and tau, particularly during early disease stages, remains elusive. To address this issue, we took advantage of our Tau4RΔK-AP mouse model, which exhibits both Aβ plaques and tau deposition, and profiled microglia subtypes during disease progression using scRNA-Seq. To tease apart the influence of Aβ plaques from that of tau deposition, we also profiled microglial subtypes in APP;PS1 (model of Aβ plaques) and Tau4RΔK (model of tau deposition) littermate mice. We identified several disease stage-specific subtypes of microglia that are selectively induced in response to Aβ and/or tau.
We discovered the emergence of a novel microglia subtype, Early-stage AD-Associated Microglia (EADAM), that is selectively induced by both Aβ and tau pathologies but not either Aβ or tau pathologies alone, during early-stage disease before AD-like pathologies are observed. EADAM are characterized by many interferon-pathway-related genes (Ifit3, Ifit204, Ifit2) and interferon-regulating transcription factors (Irf2/7/9) as well as inflammatory chemokines (Ccl12, Cxcl10, Ccl5, Ccl2). Some of the genes in EADAM have been observed in other mouse models of amyloidosis that show early AD-like pathology [48–50], including APP;PS1 and 5xFAD mouse models. However, given that EADAM-enriched genes are expressed in much higher levels in our Tau4RΔK-AP microglia, this strongly supports the view that both Aβ and tau pathologies are required to induce disease stage-specific microglia subtypes in AD.
We also identified previously described DAM in both mouse lines in which Aβ plaques are induced (APP;PS1 and Tau4RΔK-AP) [20, 72]. However, DAM induction in Tau4RΔK mice was blunted, supporting the hypothesis that DAM are induced by Aβ plaques and/or inflammation, but not by tau pathology. While Aβ plaques alone are sufficient to induce DAM2, this subtype can be greatly amplified by both Aβ and tau pathologies, giving rise to LADAM. LADAM continues to express classic DAM genes but also upregulates additional genes, including Cd74 and MHC Class II genes. These broad LADAM marker-expressing microglia (Cd74 and MHC Class II genes) have been previously shown in some amyloidosis mouse models at late stages (known as activated response microglia) [48–50], but these genes are likely to be contributed by mild tau phosphorylation shown in these amyloidosis mouse models, based on our findings from Tau4RΔK (Fig. 2). In addition, since LADAM-like clusters and markers were observed in other neurodegeneration models, such as TDP43 KO and CK-p25 [52, 53], LADAM-enriched genes might be induced by neurodegeneration and/or inflammation in addition to tau pathologies. However, much like EADAM, LADAM-enriched genes were expressed in Tau4RΔK-AP microglia, indicating that the full set of LADAM-specific genes is induced by the combination of both Aβ and tau pathologies, and perhaps with neuronal atrophy. Furthermore, the combination of Aβ and tau pathologies induced LADAM to express unique S100a genes and Siglecg.
In particular, a sub-cluster of LADAM during late-stage disease shares a similar gene signature with previously identified white-matter-associated microglia (WAM). WAM have been previously shown to be present in 24-month-old wild-type mice [25]. Indeed, microglia near white matter tracts were detected in all four genotypes in 12-month-old mice, but only mice with Aβ plaques and tau deposition showed expression of specialized LADAM gene signatures such as Siglecg. These data indicated that Aβ plaques and tau deposition induce new molecular signatures in previously identified WAM, leading to the formation of this LADAM subcluster.
While our discovery of novel microglia subtypes has important clinical implications, the mechanisms by which EADAM and LADAM emerge during different stages of disease progression in response to Aβ plaques and tau deposition remain to be fully established. Our current study identifies potential regulators of these microglia populations in response to Aβ plaques and tau deposition. Neuronal loss or brain atrophy induced by aging and/or tau pathologies may also play an essential role in regulating this process.
In summary, our data indicated that while EADAM-related or LADAM-related gene activation represents a general response of microglia to various pathological contexts, there exists a unique gene signature that would define specific microglia subtypes that respond to specific pathology temporally during disease progression. Thus, by elucidating such “microglia response code”, one is expected to discover specific microglia subtypes relevant to the pathological context of human disease. Supporting this view, while we observed both EADAM-like and/or LADAM-like clusters in TDP43-KO or CK-p25 mouse models, there are substantial differences in expression levels of EADAM and LADAM genes in Tau4R△K-AP as compared to those of TDP43-KO or CK-p25 as well as Tau4R△K and APP;PS1 mice (Figs. 1, 2, 4, S6). We also found unique LADAM markers in Tau4R△K-AP mice that are not expressed in any other neurodegenerative models (Figs. 2, 4, S6). These data strongly support a model in which there exists an EADAM-like and LADAM-like “microglia code” harbored by microglia subtypes that respond to either a general or specific pathological context in the brain. We demonstrate for both Aβ and tau pathologies that this canonical AD pathology context selectively induces novel microglia subtypes in a disease stage-specific manner. These disease stage-dependent “microglial codes” could not only serve as biomarkers for precise diagnosis but also as the therapeutic target for stage-specific intervention of specific neurodegenerative diseases.
Identifications of disease stage-specific microglia subtypes in AD
The emergence of EADAM during the presymptomatic AD stage and detection of LADAM when AD-like pathologies are visible led to the hypothesis that AD brains may harbor similar unique microglia clusters. EADAM were observed in the entorhinal cortex during early (Braak II) but not late (Braak VI) stage disease, strongly supporting the view that EADAM first emerge in response to initial stages of Aβ plaque formation and tau deposition, corresponding to a stage of negligible or mild cognitive impairment. The microglia cluster expressing an IFN-related gene that is similar to EADAM has been shown in the FTDP-17-linked tau model (P301L) [57] crossed with PS2APP [58], but only when Trem2 is deficient [55, 56]. Since Trem2 deficiency can result in the spreading of tau aggregates [73], the FTDP-17-linked tau model (P301L) can result in mild tau aggregation, which resemble the early stages of our mouse model that is characterized by Aβ plaques stimulated pathological conversion of wild-type mouse tau to form tau tangle-like aggregates. These previous finding [56] aligns well with our finding that Aβ plaques and tau deposition are required to give rise to EADAM.
In contrast, the emergence of LADAM in both the entorhinal cortex and superior frontal gyrus did not occur until the Braak IV stage, implying that these subtypes are induced in response to more severe Aβ and tau pathologies accompanied by neuronal loss during late-stage disease. Some LADAM-enriched genes such as HLA-DRB1 and HLA-DRB5 positively correlate with AD pathology [74] and share a few similar molecular signatures (e.g. MHC Class II genes) with lipid droplet-accumulating microglia [75]. We also observed an upregulation of S100A genes in late-stage AD.
Although interferon-signaling-related genes (EADAM-enriched) and MHC and S100 family genes (LADAM-enriched) are observed during human early- and late-AD stages, respectively, not all EADAM- or LADAM-enriched genes were detected in human samples. We were also unable to identify well-defined and distinct microglial clusters in human AD snRNA-Seq, in agreement with other studies. This might reflect the much longer time course of AD progression in humans compared to mouse models, potentially leading to less well-defined microglia subtypes. The cluster-basis analysis method which is typically used for the analysis of cell states in animal models might not be the best method to define microglia subtypes in human disease states. Nonetheless, gene profiling studies conducted in appropriate animal models of AD could yield insights to facilitate the profiling of corresponding unique microglia subtypes or specific genes in AD brains, a task that can be technically challenging to interpret.
The fact that EADAM are first detected in 6-month-old Tau4RΔK-AP mice and Braak stage II AD datasets, while LADAM are first detected in 12-month-old Tau4RΔK-AP mice and Braak stage IV AD datasets, demonstrates that disease stage-dependent changes in microglial subtypes in mice with Aβ and tau pathologies mimic those seen in AD. Tau4RΔK-AP mice faithfully model not only the development of canonical AD pathologies, including the pathological conversion of wild-type mouse tau [24], but also the disease stage-specific emergence of EADAM and LADAM in response to both Aβ plaques and tau deposition, emphasizing that our mouse model will prove useful in both analyzing AD disease mechanisms and testing therapeutic strategies for treating AD.
We currently do not know the functions of these microglia subtypes, but pathway analysis (Fig. 1) of EADAM shows enrichment of ‘Response to interferon-beta. EADAM is present in Tau4RΔK-AP mice before AD-like pathology appears, and our current hypothesis is that EADAM at 6-month-old could potentially provide a protective role [76, 77]. As for LADAM, our pathway analysis (Fig. 2) shows that ‘Antigen presentation via MHC Class II’ and ‘Antigen processing and presentation’ are strongly associated with LADAM, which are common pathways associated with neurodegeneration [78]. LADAM might be involved in neuronal degeneration, as MHC Class II has been shown to be important in neuronal degeneration by microglia in some neurodegeneration [79].
Siglec signaling in EADAM and LADAM
The identification of disease stage-enriched EADAM and LADAM in our Tau4RΔK-AP model and AD brains raise the possibility that specific signaling pathways may be important in the emergence of these microglial subtypes. CD33 (also known as Siglec-3) [80], is one of the several microglial-specific genes that have been linked to AD susceptibility via genome-wide association studies (GWAS) [81] and both mediate immune suppression by binding sialoglycan targets and attenuates phagocytosis by microglia during AD progression [80, 82]. As a result, we first focused on characterizing the expression of sialic acid-binding immunoglobulin-like lectin (Siglec) family of proteins. Human Siglecs belongs to a family of 14 distinct transmembrane proteins, many of which are expressed in overlapping subsets of immune cells and inhibit immune activation [70, 83]. In addition to Siglec-3, other members of the Siglec family might regulate microglial function in AD. Our findings showing specific enrichment of Siglec-pathway genes in EADAM and LADAM strongly support this hypothesis. Siglec-G is enriched in microglia near myelinated regions, whereas Siglec-F + microglia are more broadly expressed throughout the brain. Our data suggest that Siglec-F is dependent on Aβ deposition, that Siglec-G is dependent on tau deposition, and that Aβ and tau deposition synergistically enhance the expression of both Siglecs. The observation that Siglecg is highly expressed in LADAM while Siglec-10 (the human homolog of Siglec-G), is associated with late-stage AD pathologies exhibiting robust Aβ and Tau deposition, further supports the view that Siglec10 signaling underlies the activation of LADAM in AD. Moreover, EADAM selectively expressed both Siglec1 and Siglecf, while DAM and MHC Class II clusters both expressed Siglecg, indicating that Siglec-1, Siglec-F, and Siglec-G, respectively, represent potential biomarkers for the detection of early- and late-stage disease. Based on these findings, we hypothesize that Siglec-regulated signaling pathways may have an important function in regulating disease stage-specific microglia during AD progression. Future studies will directly address disease-associated functions of Siglec-regulated signaling pathways.
However, it is often difficult to identify Siglec orthologues between mice and humans, as this gene family evolved rapidly [83, 84]. Mice do not express Siglec-8, and mouse Siglec-3 (mSiglec‑3) has different effector domains, binding specificity, cellular distribution, and biological properties compared to those of human Siglec-3 (hSiglec‑3) [85, 86]. However, current studies suggested there is functional conservation between mice and humans in Siglec gene family. Siglec In mouse microglia, immune inhibitory Siglec-F is among the most highly up-regulated genes (27-fold) in experimental neurodegenerative proteinopathy [87]. Siglec-F, a likely functional orthologue of Siglec-8 [88], is expressed in eosinophils as well as microglia [87, 88], binds to the same sialoglycans and sialoglycan mimetics as Siglec-8 (unlike mSiglec-3) [89, 90]. Both Siglec-F and Siglec-8 also regulate eosinophilic inflammation [91, 92]. Siglec-F was upregulated on a subset of reactive microglia in models of neurodegeneration, indicating the important role of Siglec-F/Siglec-8 in regulating microglial activation during neurodegeneration [93]. In a recent study, RPTPζS3L, a sialoglycoprotein was identified as a specific ligand in the human cerebral cortex that binds CD33 as well as Siglec-8 [94]. Interestingly, RPTPζ isoform in mice binds mouse Siglec-F and cross-reacts with human CD33 and Siglec-8 [94], suggesting Siglec-F shares a similar ligand with CD33 and Siglec8 and is a potential functional counterpart of CD33 in humans. Importantly, we observed similar expression patterns of siglec signaling between mice and humans, such as Siglec-F/8 expression emerged at earlier disease stages than Siglec-G/10 in both our mouse models and in AD, indicating that our mouse model could serve as an important tool to study the functions of Siglec signaling in AD pathological stages and microglia subtypes.
Conclusions
In this study, microglia subtypes in a mouse model of AD that develop both Aβ and tau pathologies were profiled using scRNA-Seq. We found that during early-stage disease in 6-month-old Tau4RΔK-AP, the presence of both Aβ and tau pathologies induced a novel microglia subtype EADAM, which is distinct from DAM and express multiple interferon-regulated genes. In late-stage disease (12-month-old Tau4RΔK-AP mice), we found that another novel microglia subtype LADAM emerged in response to tau pathology, which expresses MHC and S100 family genes. We further observed a unique subtype of LADAM located near white matter, which is molecularly similar to a previously identified white matter-associated microglial subtype (WAM). We found that the signature genes in EADAM were also associated with a subgroup of microglia in the brains of early (Braak II) stages of AD. While LADAM microglia, including WAM-like LADAM, were observed in the late (Braak VI), but not early (Braak II) stages of AD. Corroborating these findings, we found that Siglec family members are associated with specific subsets of microglia that are activated in a disease-stage-specific manner in both mouse models of AD and AD patients. For example, Siglec-F and CD22 are upregulated in response to Aβ pathology and is selectively expressed in Aβ-associated DAMs, while Siglec-G/10 is upregulated in white-matter-associated LADAM in late-stage AD. These findings are consistent with a model whereby Aβ or tau pathologies stimulate the activation of microglia in different patterns, both Aβ and tau deposition are required to mediate the disease stage-specific induction of EADAM and LADAM, and can alter Siglec signaling across AD pathological stage and microglia subtypes. These findings may have important implications for the identification of novel molecular targets, and offer new targets for the development of pre-symptomatic biomarkers and therapeutic strategies for AD.
Our data indicated that while EADAM-related or LADAM-related gene activation represents a general response of microglia to various pathological contexts, there exists a unique gene signature that would define specific microglia subtypes that respond to specific pathology temporally during disease progression. Thus, by elucidating such “microglia response code”, one is expected to discover specific microglia subtypes relevant to the pathological context in human disease. Supporting this view, while we observed both EADAM-like and/or LADAM-like clusters in TDP43-KO or CK-p25 mouse models, there are substantial differences in expression levels of EADAM and LADAM genes in Tau4R△K-AP as compared to those of TDP43-KO or CK-p25 as well as Tau4R△K and APP;PS1 mice. We also found unique LADAM markers in Tau4R△K-AP mice that are not expressed in any other neurodegenerative models. These data strongly support a model in which there exists an EADAM-like and LADAM-like “microglia code” harbored by microglia subtypes that respond to either a general or specific pathological context in the brain. We demonstrate for both Aβ and tau pathologies that this canonical AD pathology context selectively induces novel microglia subtypes in a disease stage-specific manner. These disease stage-dependent “microglial codes” could not only serve as biomarkers for precise diagnosis but also as the therapeutic target for stage-specific intervention of specific neurodegenerative diseases.
Lastly, similar microglia cell types that share partial overlap in gene expression and low levels of expression of EADAM and LADAM markers can be detected in other mouse models of neurodegenerative disorders. While we have given unique names to AD-associated microglia in our AD models, a more standardized nomenclature for disease-associated microglial subtypes needs to be worked out by the research community in the near future.
Supplementary Information
Additional file 1: Figure S1. Histological validation of tau and Aβ pathologies in 6-month-old and 12-month-old Tau4RΔK-AP mice. A-H, Immunostaining of Aβ (6E10) (A-D), tau (pS422) (E-H) at 6-month-old (A, B, E, F,) and 12-month-old (C, D, G, H); in the Cortex (A, C, E, G), and Hippocampus (B, D, F, H), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. I, Quantification of NeuN-positive cells in the hippocampus at 6-month-old (left) and 12-month-old (right). J, Quantification of brain weight at 12-month-old. K, Quantification of Aβ deposition covered areas in the hippocampus of 12-month-old mice. L-N, Immunostaining of Thioflavin T (ThioT) at 12-month-old; in the Cortex (L), and Hippocampus (M), in APP;PS1, Tau4RΔK-AP mice. Quantification of ThioT/Plaques are shown in N. O-Q, Quantification of plaque numbers (O), plaque area (P), and plaque sizes (Q), and in the 6-month-old and 12-month-old Tau4RΔK-AP mice. Ctx = Cortex, Hippo = Hippocampus. Scale bars = 100 μm. NS = not significant, * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 2: Figure S2. Histological validation of microglia distribution in 6-month-old and 12-month-old Tau4RΔK-AP mice. A-D, Immunostaining of IBA1 at 6-month-old (A, B) and 12-month-old (C, D); in the Cortex (A, C), and Hippocampus (B, D), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. E-G, Quantification of IBA1 staining in the cortex (E), hippocampus (F), and corpus callosum (G) at 6-month-old and 12-month-old. Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. Scale bars = 100 μm. NS = not significant, * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
{kind=link}
Additional file 3: Figure S3. Distribution of cell types in 6-month-old cortex across genotypes. A, Schematic design of experiment: 4 genotypes (WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP) in the cerebral cortex at 6-month-old and 12-month-old. B, Distribution of cell types across genotypes in the 6-month-old cortex. Numbers indicate the number of captured cell types. C, UMAP plot showing captured cell types in the 6-month-old cortex (all genotypes). D, UMAP plot showing the density of the captured cell types across genotypes in the 6-month-old cortex. E, UMAP plot showing DAM marker gene Cst7. F, UMAP plot showing DAM marker gene Gpnmb. BAM = brain-associated macrophages, Ctx = Cortex, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. N = 3/genotype, WT = 56668 cells total, APP;PS1 = 52797 cells total, Tau4RΔK = 49008 cells total, Tau4RΔK-AP = 55306 cells total.
{kind=link}
Additional file 4: Figure S4. Distribution of cell types in 12-month-old cortex across genotypes. A, Schematic design of experiment: 4 genotypes (WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP) in the cerebral cortex at 6 and 12-month-old. B, Distribution of cell types across genotypes in 12-month-old cortex. Numbers indicate the number of captured cell types. C, UMAP plot showing captured cell types in the 12-month-old cortex (all genotypes). D, UMAP plot showing the density of the captured cell types for each genotype in the 12-month-old cortex. E, UMAP plot showing DAM marker gene Cst7. F, UMAP plot showing DAM marker gene Gpnmb. BAM = brain-associated macrophages, Ctx = cortex, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. N = 3/genotype, WT = 47455 cells total, APP;PS1 = 42247 cells total, Tau4RΔK = 41152 cells total, Tau4RΔK-AP = 35431 cells total.
{kind=link}
Additional file 5: Figure S5. Distribution of clusters across genotypes. A, Distribution of cell types across 6-month-old scRNA-Seq triplicates. B, Distribution of cell types across 12-month-old scRNA-Seq triplicates. BAM = brain-associated macrophages, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. R1 = Biological replicate 1, R2 = Biological replicate 2, R3 = Biological replicate 3.
{kind=link}
Additional file 6: Figure S6. Analysis of NF-kB pathway genes in 6-month-old AD mouse models. Heatmap plots showing expressions of NF-kB pathway genes at 6-month-old across microglia subtypes (A), and genotypes (B).
{kind=link}
Additional file 7: Figure S7. Analysis of EADAM and LADAM markers in other mouse models of neurodegenerative disease and AD samples. A, UMAP plot showing CK-p25 neurodegenerative scRNA-Seq microglia dataset from Mathys et al., 2017. B, UMAP plots showing EADAM and LADAM-enriched genes. Note that while IFN and MHC genes are present, S100 genes and Siglecg are absent in the dataset.
{kind=link}
Additional file 8: Figure S8. Analysis of distinct expression profiles that are associated with AD pathology in AD mouse models. Heatmap plots showing homolog expressions of Aβ-associated AD1-enriched (A), and Tau-associated AD2-enriched (B) genes from Gerrits et al., 2021 human snRNA-Seq dataset [59], in 6-month-old 12-month-old, in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP.
Additional file 9: Figure S9. Histological validation of DAM markers in AD mouse models. A-C, Immunostaining of LPL at 6-month-old (A) and 12-month-old (B, C); in the Cortex (B), and Hippocampus (A, C), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of LPL-positive cells. D-E, Immunostaining of IBA1 (Red) and LPL (Green) at 6-month-old in the cortex in APP;PS1 (D), Tau4RΔK-AP (E). White arrowheads indicate IBA1 and LPL double-positive cells. F-G, Immunostaining of CD22 (Siglec-2) at 12-month-old; in the cortex (F), and hippocampus (G), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of LPL-positive cells. Ctx = Cortex, Hippo = Hippocampus. Scale bars = 50 μm.
Additional file 10: Figure S10. Histological validation of DAM/LADAM marker in AD mouse models. A-D, Immunostaining of Lgals3 at 6-month-old (A) and 12-month-old (B-D); in the cortex (A, B), hippocampus (C), and corpus callosum (D), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of Lgals3-positive cells. E-G, Quantification of Lgals3 staining in the cortex (E), hippocampus (F), and corpus callosum (G) at 12-month-old, in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 11: Figure S11. Histological validation of LADAM marker in AD mouse models. A-C, Immunostaining of Tau(pS422) (Red) and Lgals3 (Green) at 12-month-old in the cortex (A), hippocampus (B), and corpus callosum (C) in Tau4RΔK. D, Immunostaining of GFAP (Red) and Lgals3 (Green) at 12-month-old in the corpus callosum in Tau4RΔK. E-F, Immunostaining of IBA1 (Red) and Lgals3 (Green) at 12-month-old in the corpus callosum in Tau4RΔK (E), Tau4RΔK-AP (F). Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. Scale bars = 50 μm.
Additional file 12: Figure S12. Histological validation of EADAM/LADAM markers in AD mouse models. A-B, Immunostaining of STAT1 (Red) and GFAP (Green) at 12-month-old in APP;PS1 (A), Tau4RΔK-AP (B). C-F, Immunostaining of IBA1 (Red) and STAT1 (Green) at 6-month-old (E) and 12-month-old (C, D, F) in APP;PS1 (C), Tau4RΔK (D) Tau4RΔK-AP (E, F). Scale bars = 50 μm.
{kind=link}
Additional file 13: Figure S13. Analysis of EADAM and LADAM markers in AD samples. Heatmap plots showing homolog expression of EADAM (A), and LADAM (B) genes in the human snRNA-Seq dataset.
Additional file 14: Figure S14. Siglec-genes show cluster- and genotype-specific expression patterns, and histological validation of Siglec-F and Siglec-G across genotypes in 6 and 12-month-old mice. A, UMAP plot showing microglial clusters in the 6 and 12-month-old cortex (all genotypes). B, Heatmap plot showing Siglec genes expressions across microglial clusters. C, D, UMAP plot showing Siglecf expression in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP at 6-month-old (C) and 12-month-old (D) (top). Note a higher expression of Siglecf in DAM2 in APP;PS1 and Tau4RΔK-AP mice at 12-month-old. Siglec-F immunostaining was performed in the cortex in WT, Tau4RΔK, APP;PS1, Tau4RΔK-AP at 6-month-old (C) and 12-month-old (D) (bottom). E-I, Immunostaining of Siglec-F (E, F), and Siglec-G (G-I), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP; at 6-month-old (E, G) and 12-month-old (F, H, I); hippocampus (E, F, H), and the cortex (F, G, I). J, UMAP plot of 6-month-old microglia, showing a small cluster (highlighted in red) that resembles molecular genes of WAM [25]. K, Quantification of Siglec-G staining in the 6-month-old and 12-month-old cortex and hippocampus. Ctx = Cortex, Hippo = Hippocampus. Red boxes in C, D, E, F, I show high magnification views. Scale bars = 100 μm. * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 15: Figure S15. Histological validation of Siglec-G/Siglec-10 expression in microglia in AD mouse models and human postmortem samples. A-B, Immunostaining of IBA1 (Red) and Siglec-G (Green) at 12-month-old in the CC in Tau4RΔK (A), Tau4RΔK-AP (B). C, Immunostaining of IBA1 (Green) and Siglec-10 (Red) at Braak stage VI. CC = Corpus Callosum. Scale bars = 50 μm.
Additional file 16: Figure S16. Histological validation of tau and Aβ pathologies and Siglec-10 during AD progression and in nAD Tauopathy. Immunostaining of Tau (pS422) (A), Aβ (6E10) (B), and Siglec-10 (C) in Control, Braak stage 2, Braak stage 3, Braak stage 4, Braak stage 5, Braak stage 6, and in nAD Tauopathy in the cortex. Numbers indicate BRC# in Table S10. Red boxes show high magnification views. Scale bars = 100 μm.
Additional file 17: Figure S17. Histological validation of markers in AD pathology. Immunostaining of IBA1 (Red) and LPL (A, Green); Lgals3 (B, Green); STAT1 (C-D, Green) in Control (D) and AD (A-C). White arrows indicate examples of double-positive cells. Scale bars = 50 μm.
Additional file 18: Table S1. Differential gene expression as measured by scRNA-Seq in 6-month-old cortex across cell types. Table S2. Differential gene expression as measured by scRNA-Seq in 12-month-old cortex across cell types. Table S3. Differential gene expression as measured by scRNA-Seq in 6-month-old microglia subtypes. Table S4. Differential gene expression as measured by scRNA-Seq in 6-month-old microglia across genotypes. Table S5. Differential gene expression as measured by scRNA-Seq in 12-month-old microglia subtypes. Table S6. Differential gene expression as measured by scRNA-Seq in 12-month-old microglia across genotypes. Table S7. Differential gene expression as measured by scRNA-Seq in TDP43 KO6->9 microglia subtypes. Table S8. Differential gene expression as measured by scRNA-Seq in TDP43 KO12->15 microglia subtypes. Table S9. Differential gene expression as measured by scRNA-Seq in 12-month-old LADAM subtypes. Table S10. Characteristics of postmortem brains used in this study for histological analysis. Table S11. Characteristics of postmortem brains used in this study for snRNA-Seq.
Acknowledgements
We thank the Transcriptomics and Deep Sequencing Core (Johns Hopkins) for the sequencing of scRNA-Seq libraries and the Johns Hopkins ADRC Neuropathology Core for brain tissues.
Abbreviations
- SFG
Superior frontal gyrus
- ERC
Entorhinal cortex
- EADAM
Early-stage AD-Associated Microglia
- LADAM
Late-stage AD-Associated Microglia
- AD
Alzheimer’s disease
- Aβ
Amyloid-β
- scRNA-Seq
Single-cell RNA-sequencing
- Siglec
Sialic acid-binding immunoglobulin-type lectin
- FFPE
Formalin-fixed paraffin-embedded
- H&E
Hematoxylin and eosin
- APP;PS1
APPswe; PS1ΔE9
- PART
Primary-age-related-tauopathy
- ADRC
Johns Hopkins Alzheimer’s Disease Research Center
- ThioT
Thioflavin-T
Authors’ contributions
The study was designed by TL, DWK; mouse tissues were collected and characterized by TL, KJT, AW, AJL, TC, KB; JCT provided human specimens; AG, RS provided antibodies against siglecs, DWL, JPL, JCT, PCW, SB, RLS and TL prepared the manuscript. The author(s) read and approved the final manuscript.
Funding
This work was supported in part by the Maryland Stem Cell Research Fund (2019-MSCRFF-5124) to DWK, grants from the National Institute of Aging (R56AG068089) to RLS and TL, (R21AG073710) to TL, and the National Institute of Neurological Disorders and Stroke (R61NS115161 and R01NS095969) to PCW.
Availability of data and materials
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tong Li (tli1@jhmi.edu).
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Single-cell RNA-seq data have been deposited at GEO (GSE175546) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
Declarations
Ethics approval and consent to participate
All animal procedures were in accordance strictly with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Johns Hopkins University Animal Care and Use Committee.
Consent for publication
Authors consent for the publication. All authors read and approved the final manuscript.
Competing interests
S.B. receives research support from Genentech, is a co-founder and scientific advisory board member of CDI Labs LLC, and is/was a consultant for Third Rock Ventures and Tenpoint Therapeutics.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dong Won Kim, Email: kimtho87@gmail.com.
Kevin J. Tu, Email: ktu@umd.edu
Alice Wei, Email: alicewei1999@yahoo.com.
Ashley J. Lau, Email: ash.jacq.lau@gmail.com
Anabel Gonzalez-Gil, Email: alvarenga@jhmi.edu.
Tianyu Cao, Email: tcao6@jhmi.edu.
Kerstin Braunstein, Email: kbrauns2@jhmi.edu.
Jonathan P. Ling, Email: jling@jhu.edu
Juan C. Troncoso, Email: troncoso@jhmi.edu
Philip C. Wong, Email: wong@jhmi.edu
Seth Blackshaw, Email: sblack@jhmi.edu.
Ronald L. Schnaar, Email: schnaar@jhu.edu
Tong Li, Email: tli1@jhmi.edu.
References
- 1.Bohlen CJ, Friedman BA, Dejanovic B, Sheng M. Microglia in brain development, homeostasis, and neurodegeneration. Annu Rev Genet. 2019;53:263–288. doi: 10.1146/annurev-genet-112618-043515. [DOI] [PubMed] [Google Scholar]
- 2.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21:1359–1369. doi: 10.1038/s41593-018-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327–406. 10.1002/alz.12328. [DOI] [PubMed]
- 4.Bekris LM, Yu C-E, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/WNL.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 6.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278:1349–1356. doi: 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- 7.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19:622–635. doi: 10.1038/s41583-018-0057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamelin L, Lagarde J, Dorothée G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain. 2016;139:1252–1264. doi: 10.1093/brain/aww017. [DOI] [PubMed] [Google Scholar]
- 15.Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fani Maleki A, Rivest S. innate immune cells: monocytes, monocyte-derived macrophages and microglia as therapeutic targets for Alzheimer’s disease and multiple sclerosis. Front Cell Neurosci. 2019;13:355. doi: 10.3389/fncel.2019.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia Heterogeneity in the Single-cell era. Cell Rep. 2020;30:1271–1281. doi: 10.1016/j.celrep.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Xue F, Du H. TREM2 mediates microglial anti-inflammatory activations in Alzheimer’s disease: lessons learned from transcriptomics. Cells. 2021;10:321. doi: 10.3390/cells10020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wes PD, Holtman IR, Boddeke EWGM, Möller T, Eggen BJL. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia. 2016;64:197–213. doi: 10.1002/glia.22866. [DOI] [PubMed] [Google Scholar]
- 20.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. 2019;101:207–23.e10. doi: 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:131–142. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T, Braunstein KE, Zhang J, Lau A, Sibener L, Deeble C, et al. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer’s disease mouse model. Nat Commun. 2016 doi: 10.1038/ncomms12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, et al. White matter aging drives microglial diversity. Neuron. 2021;109:1100–17.e10. doi: 10.1016/j.neuron.2021.01.027. [DOI] [PubMed] [Google Scholar]
- 26.Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, et al. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 28.Jankowsky JL, Xu G, Fromholt D, Gonzales V, Borchelt DR. Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:1220–1227. doi: 10.1093/jnen/62.12.1220. [DOI] [PubMed] [Google Scholar]
- 29.Han HJ, Allen CC, Buchovecky CM, Yetman MJ, Born HA, Marin MA, et al. Strain background influences neurotoxicity and behavioral abnormalities in mice expressing the tetracycline transactivator. J Neurosci. 2012;32:10574–10586. doi: 10.1523/JNEUROSCI.0893-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaClair KD, Donde A, Ling JP, Jeong YH, Chhabra R, Martin LJ, et al. Depletion of TDP-43 decreases fibril and plaque β-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta Neuropathol. 2016;132:859–873. doi: 10.1007/s00401-016-1637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim DW, Liu K, Wang ZQ, Zhang YS, Bathini A, Brown MP, et al. Gene regulatory networks controlling differentiation, survival, and diversification of hypothalamic Lhx6-expressing GABAergic neurons. Commun Biol. 2021;4:95. doi: 10.1038/s42003-020-01616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DW, Washington PW, Wang ZQ, Lin SH, Sun C, Ismail BT, et al. The cellular and molecular landscape of hypothalamic patterning and differentiation from embryonic to late postnatal development. Nat Commun. 2020;11:4360. doi: 10.1038/s41467-020-18231-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoo S, Cha D, Kim DW, Hoang TV, Blackshaw S. Tanycyte-independent control of hypothalamic leptin signaling. Front Neurosci. 2019;13:240. doi: 10.3389/fnins.2019.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling JP, Wilks C, Charles R, Leavey PJ, Ghosh D, Jiang L, et al. ASCOT identifies key regulators of neuronal subtype-specific splicing. Nat Commun. 2020;11:137. doi: 10.1038/s41467-019-14020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun. 2021;12:1151. doi: 10.1038/s41467-021-21407-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22:1021–1035. doi: 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- 39.Marsh SE, Kamath T, Walker AJ, Dissing-Olesen L, Hammond TR, Young AMH, et al. Single cell sequencing reveals glial specific responses to tissue processing & enzymatic dissociation in mice and humans. 10.1101/2020.12.03.408542
- 40.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melsted P, Sina Booeshaghi A, Gao F, Beltrame E, Lu L, Hjorleifsson KE, et al. Modular and efficient pre-processing of single-cell RNA-seq. 10.1101/673285
- 44.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 45.Alexander Wolf F, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergen V, Lange M, Peidli S, Alexander Wolf F, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. 10.1101/820936 [DOI] [PubMed]
- 47.Qiu X, Hill A, Packer J, Lin D, Ma Y-A, Trapnell C. Single-cell mRNA quantification and differential analysis with Census. Nat Methods. 2017;14:309–315. doi: 10.1038/nmeth.4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sierksma A, Lu A, Salta E, Mancuso R, Zoco J, Blum D, et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. 10.1101/491902 [DOI] [PMC free article] [PubMed]
- 49.Frigerio CS, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019;27:1293–306.e6. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang HS, Onos KD, Choi K, Keezer KJ, Skelly DA, Carter GW, et al. Natural genetic variation determines microglia heterogeneity in wild-derived mouse models of Alzheimer’s disease. 10.1101/2020.06.02.130237 [DOI] [PMC free article] [PubMed]
- 51.Roy ER, Wang B, Wan Y-W, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020;130:1912–1930. doi: 10.1172/JCI133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bryan KJ, Zhu X, Harris PL, Perry G, Castellani RJ, Smith MA, et al. Expression of CD74 is increased in neurofibrillary tangles in Alzheimer’s disease. Mol Neurodegener. 2008;3:13. doi: 10.1186/1750-1326-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Fan L, Zhan L, Kodama L, Liu B, Chin M, et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. 10.1101/2021.02.22.432272 [DOI] [PMC free article] [PubMed]
- 55.Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee S-H, Srinivasan K, et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 Ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J Neurosci. 2020;40:1956–1974. doi: 10.1523/JNEUROSCI.1871-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S-H, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. 2021;109:1283–301.e6. doi: 10.1016/j.neuron.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Götz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 58.Ozmen L, Albientz A, Czech C, Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener Dis. 2009;6:29–36. doi: 10.1159/000170884. [DOI] [PubMed] [Google Scholar]
- 59.Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 2021;141:681–696. doi: 10.1007/s00401-021-02263-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron. 2018;97:1032–48.e5. doi: 10.1016/j.neuron.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boza-Serrano A, Ruiz R, Sanchez-Varo R, García-Revilla J, Yang Y, Jimenez-Ferrer I, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019;138:251–273. doi: 10.1007/s00401-019-02013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tao C-C, Cheng K-M, Ma Y-L, Hsu W-L, Chen Y-C, Fuh J-L, et al. Galectin-3 promotes Aβ oligomerization and Aβ toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ. 2020;27:192–209. doi: 10.1038/s41418-019-0348-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.García-Revilla J, Boza-Serrano A, Espinosa-Oliva AM, Soto MS, Deierborg T, Ruiz R, et al. Galectin-3, a rising star in modulating microglia activation under conditions of neurodegeneration. Cell Death Dis. 2022;13:628. doi: 10.1038/s41419-022-05058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet. 2021;53:1143–1155. doi: 10.1038/s41588-021-00894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pande R, Huang Y, Teeple E, Joshi P, Flores-Morales A, Latta-Mahieu M, et al. Single cell atlas of human putamen reveals disease specific changes in Synucleinopathies: Parkinson’s disease and multiple system atrophy. 10.1101/2021.05.06.442950
- 68.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s Disease. 10.1101/2020.04.04.025825 [DOI] [PMC free article] [PubMed]
- 69.Siddiqui SS, Matar R, Merheb M, Hodeify R, Vazhappilly CG, Marton J, et al. Siglecs in brain function and neurological disorders. Cells. 2019;8:1125. doi: 10.3390/cells8101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–172. doi: 10.1038/s41582-020-00435-y. [DOI] [PubMed] [Google Scholar]
- 72.Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12:3015. doi: 10.1038/s41467-021-23111-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, et al. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019;22:1217–1222. doi: 10.1038/s41593-019-0433-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain [Internet] Nat Neurosci. 2020;23:194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. Protective role of beta interferon in host defense against influenza A virus. J Virol. 2007;81:2025–2030. doi: 10.1128/JVI.01718-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scheu S, Ali S, Mann-Nüttel R, Richter L, Arolt V, Dannlowski U, et al. Interferon β-mediated protective functions of microglia in central nervous system autoimmunity. Int J Mol Sci. 2019;20:190. doi: 10.3390/ijms20010190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to Tango. Front Immunol. 2017;8:1905. doi: 10.3389/fimmu.2017.01905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33:9592–9600. doi: 10.1523/JNEUROSCI.5610-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Podleśny-Drabiniok A, Marcora E, Goate AM. Microglial phagocytosis: a disease-associated process emerging from Alzheimer’s disease genetics. Trends Neurosci. 2020;43:965–979. doi: 10.1016/j.tins.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Varki A, Schnaar RL, Crocker PR, et al. I-Type Lectins. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al., editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2017. [Google Scholar]
- 84.Varki A. Multiple changes in sialic acid biology during human evolution. Glycoconj J. 2009;26:231–245. doi: 10.1007/s10719-008-9183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bhattacherjee A, Rodrigues E, Jung J, Luzentales-Simpson M, Enterina JR, Galleguillos D, et al. Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun Biol. 2019;2:450. doi: 10.1038/s42003-019-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brinkman-Van der Linden ECM, Angata Angata, Reynolds SA, Powell LD, Hedrick SM, Varki A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol Cell Biol. 2003;23:4199–206. doi: 10.1128/MCB.23.12.4199-4206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lunnon K, Teeling JL, Tutt AL, Cragg MS, Glennie MJ, Perry VH. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J Immunol. 2011;186:7215–7224. doi: 10.4049/jimmunol.0903833. [DOI] [PubMed] [Google Scholar]
- 88.Bochner BS. Siglec-8 on human eosinophils and mast cells, and Siglec-F on murine eosinophils, are functionally related inhibitory receptors. Clin Exp Allergy. 2009;39:317–324. doi: 10.1111/j.1365-2222.2008.03173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu H, Gonzalez-Gil A, Wei Y, Fernandes SM, Porell RN, Vajn K, et al. Siglec-8 and Siglec-9 binding specificities and endogenous airway ligand distributions and properties. Glycobiology. 2017;27:657–668. doi: 10.1093/glycob/cwx026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nycholat CM, Duan S, Knuplez E, Worth C, Elich M, Yao A, et al. A Sulfonamide Sialoside Analogue for Targeting Siglec-8 and -F on Immune Cells. J Am Chem Soc. 2019;141:14032–14037. doi: 10.1021/jacs.9b05769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McMillan SJ, Richards HE, Crocker PR. Siglec-F-dependent negative regulation of allergen-induced eosinophilia depends critically on the experimental model. Immunol Lett. 2014;160:11–16. doi: 10.1016/j.imlet.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood. 2007;109:4280–4287. doi: 10.1182/blood-2006-08-039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morshed N, Ralvenius WT, Nott A, Watson LA, Rodriguez FH, Akay LA, et al. Phosphoproteomics identifies microglial Siglec-F inflammatory response during neurodegeneration. Mol Syst Biol. 2020;16:e9819. doi: 10.15252/msb.20209819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gonzalez-Gil A, Porell RN, Fernandes SM, Maenpaa E, August Li T, Li T, et al. Human brain sialoglycan ligand for CD33, a microglial inhibitory Siglec implicated in Alzheimer’s disease. J Biol Chem. 2022;298:101960. doi: 10.1016/j.jbc.2022.101960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Histological validation of tau and Aβ pathologies in 6-month-old and 12-month-old Tau4RΔK-AP mice. A-H, Immunostaining of Aβ (6E10) (A-D), tau (pS422) (E-H) at 6-month-old (A, B, E, F,) and 12-month-old (C, D, G, H); in the Cortex (A, C, E, G), and Hippocampus (B, D, F, H), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. I, Quantification of NeuN-positive cells in the hippocampus at 6-month-old (left) and 12-month-old (right). J, Quantification of brain weight at 12-month-old. K, Quantification of Aβ deposition covered areas in the hippocampus of 12-month-old mice. L-N, Immunostaining of Thioflavin T (ThioT) at 12-month-old; in the Cortex (L), and Hippocampus (M), in APP;PS1, Tau4RΔK-AP mice. Quantification of ThioT/Plaques are shown in N. O-Q, Quantification of plaque numbers (O), plaque area (P), and plaque sizes (Q), and in the 6-month-old and 12-month-old Tau4RΔK-AP mice. Ctx = Cortex, Hippo = Hippocampus. Scale bars = 100 μm. NS = not significant, * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 2: Figure S2. Histological validation of microglia distribution in 6-month-old and 12-month-old Tau4RΔK-AP mice. A-D, Immunostaining of IBA1 at 6-month-old (A, B) and 12-month-old (C, D); in the Cortex (A, C), and Hippocampus (B, D), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. E-G, Quantification of IBA1 staining in the cortex (E), hippocampus (F), and corpus callosum (G) at 6-month-old and 12-month-old. Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. Scale bars = 100 μm. NS = not significant, * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 3: Figure S3. Distribution of cell types in 6-month-old cortex across genotypes. A, Schematic design of experiment: 4 genotypes (WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP) in the cerebral cortex at 6-month-old and 12-month-old. B, Distribution of cell types across genotypes in the 6-month-old cortex. Numbers indicate the number of captured cell types. C, UMAP plot showing captured cell types in the 6-month-old cortex (all genotypes). D, UMAP plot showing the density of the captured cell types across genotypes in the 6-month-old cortex. E, UMAP plot showing DAM marker gene Cst7. F, UMAP plot showing DAM marker gene Gpnmb. BAM = brain-associated macrophages, Ctx = Cortex, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. N = 3/genotype, WT = 56668 cells total, APP;PS1 = 52797 cells total, Tau4RΔK = 49008 cells total, Tau4RΔK-AP = 55306 cells total.
Additional file 4: Figure S4. Distribution of cell types in 12-month-old cortex across genotypes. A, Schematic design of experiment: 4 genotypes (WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP) in the cerebral cortex at 6 and 12-month-old. B, Distribution of cell types across genotypes in 12-month-old cortex. Numbers indicate the number of captured cell types. C, UMAP plot showing captured cell types in the 12-month-old cortex (all genotypes). D, UMAP plot showing the density of the captured cell types for each genotype in the 12-month-old cortex. E, UMAP plot showing DAM marker gene Cst7. F, UMAP plot showing DAM marker gene Gpnmb. BAM = brain-associated macrophages, Ctx = cortex, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. N = 3/genotype, WT = 47455 cells total, APP;PS1 = 42247 cells total, Tau4RΔK = 41152 cells total, Tau4RΔK-AP = 35431 cells total.
Additional file 5: Figure S5. Distribution of clusters across genotypes. A, Distribution of cell types across 6-month-old scRNA-Seq triplicates. B, Distribution of cell types across 12-month-old scRNA-Seq triplicates. BAM = brain-associated macrophages, MSC = muscle stem cells, OPC = oligodendrocyte precursor cells, VSMC = vascular smooth muscle cells. R1 = Biological replicate 1, R2 = Biological replicate 2, R3 = Biological replicate 3.
Additional file 6: Figure S6. Analysis of NF-kB pathway genes in 6-month-old AD mouse models. Heatmap plots showing expressions of NF-kB pathway genes at 6-month-old across microglia subtypes (A), and genotypes (B).
Additional file 7: Figure S7. Analysis of EADAM and LADAM markers in other mouse models of neurodegenerative disease and AD samples. A, UMAP plot showing CK-p25 neurodegenerative scRNA-Seq microglia dataset from Mathys et al., 2017. B, UMAP plots showing EADAM and LADAM-enriched genes. Note that while IFN and MHC genes are present, S100 genes and Siglecg are absent in the dataset.
Additional file 8: Figure S8. Analysis of distinct expression profiles that are associated with AD pathology in AD mouse models. Heatmap plots showing homolog expressions of Aβ-associated AD1-enriched (A), and Tau-associated AD2-enriched (B) genes from Gerrits et al., 2021 human snRNA-Seq dataset [59], in 6-month-old 12-month-old, in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP.
Additional file 9: Figure S9. Histological validation of DAM markers in AD mouse models. A-C, Immunostaining of LPL at 6-month-old (A) and 12-month-old (B, C); in the Cortex (B), and Hippocampus (A, C), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of LPL-positive cells. D-E, Immunostaining of IBA1 (Red) and LPL (Green) at 6-month-old in the cortex in APP;PS1 (D), Tau4RΔK-AP (E). White arrowheads indicate IBA1 and LPL double-positive cells. F-G, Immunostaining of CD22 (Siglec-2) at 12-month-old; in the cortex (F), and hippocampus (G), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of LPL-positive cells. Ctx = Cortex, Hippo = Hippocampus. Scale bars = 50 μm.
Additional file 10: Figure S10. Histological validation of DAM/LADAM marker in AD mouse models. A-D, Immunostaining of Lgals3 at 6-month-old (A) and 12-month-old (B-D); in the cortex (A, B), hippocampus (C), and corpus callosum (D), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Black arrowheads indicate examples of Lgals3-positive cells. E-G, Quantification of Lgals3 staining in the cortex (E), hippocampus (F), and corpus callosum (G) at 12-month-old, in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP. Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 11: Figure S11. Histological validation of LADAM marker in AD mouse models. A-C, Immunostaining of Tau(pS422) (Red) and Lgals3 (Green) at 12-month-old in the cortex (A), hippocampus (B), and corpus callosum (C) in Tau4RΔK. D, Immunostaining of GFAP (Red) and Lgals3 (Green) at 12-month-old in the corpus callosum in Tau4RΔK. E-F, Immunostaining of IBA1 (Red) and Lgals3 (Green) at 12-month-old in the corpus callosum in Tau4RΔK (E), Tau4RΔK-AP (F). Ctx = Cortex, Hippo = Hippocampus, CC = Corpus Callosum. Scale bars = 50 μm.
Additional file 12: Figure S12. Histological validation of EADAM/LADAM markers in AD mouse models. A-B, Immunostaining of STAT1 (Red) and GFAP (Green) at 12-month-old in APP;PS1 (A), Tau4RΔK-AP (B). C-F, Immunostaining of IBA1 (Red) and STAT1 (Green) at 6-month-old (E) and 12-month-old (C, D, F) in APP;PS1 (C), Tau4RΔK (D) Tau4RΔK-AP (E, F). Scale bars = 50 μm.
Additional file 13: Figure S13. Analysis of EADAM and LADAM markers in AD samples. Heatmap plots showing homolog expression of EADAM (A), and LADAM (B) genes in the human snRNA-Seq dataset.
Additional file 14: Figure S14. Siglec-genes show cluster- and genotype-specific expression patterns, and histological validation of Siglec-F and Siglec-G across genotypes in 6 and 12-month-old mice. A, UMAP plot showing microglial clusters in the 6 and 12-month-old cortex (all genotypes). B, Heatmap plot showing Siglec genes expressions across microglial clusters. C, D, UMAP plot showing Siglecf expression in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP at 6-month-old (C) and 12-month-old (D) (top). Note a higher expression of Siglecf in DAM2 in APP;PS1 and Tau4RΔK-AP mice at 12-month-old. Siglec-F immunostaining was performed in the cortex in WT, Tau4RΔK, APP;PS1, Tau4RΔK-AP at 6-month-old (C) and 12-month-old (D) (bottom). E-I, Immunostaining of Siglec-F (E, F), and Siglec-G (G-I), in WT, APP;PS1, Tau4RΔK, Tau4RΔK-AP; at 6-month-old (E, G) and 12-month-old (F, H, I); hippocampus (E, F, H), and the cortex (F, G, I). J, UMAP plot of 6-month-old microglia, showing a small cluster (highlighted in red) that resembles molecular genes of WAM [25]. K, Quantification of Siglec-G staining in the 6-month-old and 12-month-old cortex and hippocampus. Ctx = Cortex, Hippo = Hippocampus. Red boxes in C, D, E, F, I show high magnification views. Scale bars = 100 μm. * P < 0.05, ** P <0.01, *** P < 0.001, **** P < 0.0001.
Additional file 15: Figure S15. Histological validation of Siglec-G/Siglec-10 expression in microglia in AD mouse models and human postmortem samples. A-B, Immunostaining of IBA1 (Red) and Siglec-G (Green) at 12-month-old in the CC in Tau4RΔK (A), Tau4RΔK-AP (B). C, Immunostaining of IBA1 (Green) and Siglec-10 (Red) at Braak stage VI. CC = Corpus Callosum. Scale bars = 50 μm.
Additional file 16: Figure S16. Histological validation of tau and Aβ pathologies and Siglec-10 during AD progression and in nAD Tauopathy. Immunostaining of Tau (pS422) (A), Aβ (6E10) (B), and Siglec-10 (C) in Control, Braak stage 2, Braak stage 3, Braak stage 4, Braak stage 5, Braak stage 6, and in nAD Tauopathy in the cortex. Numbers indicate BRC# in Table S10. Red boxes show high magnification views. Scale bars = 100 μm.
Additional file 17: Figure S17. Histological validation of markers in AD pathology. Immunostaining of IBA1 (Red) and LPL (A, Green); Lgals3 (B, Green); STAT1 (C-D, Green) in Control (D) and AD (A-C). White arrows indicate examples of double-positive cells. Scale bars = 50 μm.
Additional file 18: Table S1. Differential gene expression as measured by scRNA-Seq in 6-month-old cortex across cell types. Table S2. Differential gene expression as measured by scRNA-Seq in 12-month-old cortex across cell types. Table S3. Differential gene expression as measured by scRNA-Seq in 6-month-old microglia subtypes. Table S4. Differential gene expression as measured by scRNA-Seq in 6-month-old microglia across genotypes. Table S5. Differential gene expression as measured by scRNA-Seq in 12-month-old microglia subtypes. Table S6. Differential gene expression as measured by scRNA-Seq in 12-month-old microglia across genotypes. Table S7. Differential gene expression as measured by scRNA-Seq in TDP43 KO6->9 microglia subtypes. Table S8. Differential gene expression as measured by scRNA-Seq in TDP43 KO12->15 microglia subtypes. Table S9. Differential gene expression as measured by scRNA-Seq in 12-month-old LADAM subtypes. Table S10. Characteristics of postmortem brains used in this study for histological analysis. Table S11. Characteristics of postmortem brains used in this study for snRNA-Seq.
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tong Li (tli1@jhmi.edu).
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Single-cell RNA-seq data have been deposited at GEO (GSE175546) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.