

SUMMARY
Cancer cells must ensure a steady supply of nutrients to enable proliferation and evade cell death. Depriving cancer cells of the amino acid cystine can trigger the non-apoptotic cell death process of ferroptosis. Here, we report that cancer cells can evade cystine deprivation-induced ferroptosis by the uptake and catabolism of the cysteine-rich extracellular protein albumin. This protective mechanism is enhanced by mTORC1 inhibition and involves albumin degradation in the lysosome, predominantly by cathepsin B (CTSB). CTSB-dependent albumin breakdown followed by export of cystine from the lysosome via the transporter cystinosin fuels the synthesis of glutathione, which suppresses lethal lipid peroxidation. Notably, when cancer cells are grown under non-adherent conditions as spheroids mTORC1 pathway activity is reduced and albumin supplementation alone affords considerable protection against ferroptosis. These results identify the catabolism of extracellular protein within the lysosome as a mechanism that can inhibit ferroptosis in cancer cells.
Graphical Abstract
eTOC:
Mechanisms that regulate ferroptosis sensitivity and resistance remain poorly defined. Armenta et al demonstrate that uptake and lysosomal catabolism of extracellular protein can prevent ferroptosis in cystine-deprived cells by maintaining intracellular levels of cysteine and glutathione. This compensatory mechanism may limit the effectiveness of cancer therapies that target cystine uptake.
INTRODUCTION
For many cancer cells, the thiol-containing amino acid cysteine is conditionally essential for survival and proliferation (Combs and DeNicola, 2019; Eagle, 1959). Cysteine is a proteogenic amino acid, but also needed for the synthesis of glutathione (GSH), coenzyme A, and other important sulfur-containing metabolites. Among other functions, GSH and coenzyme A are needed to prevent ferroptosis, a non-apoptotic, oxidative form of cell death (Badgley et al., 2020; Dixon et al., 2012; Leu et al., 2019). The function of coenzyme A in ferroptosis is unclear. By contrast, GSH is a co-substrate for glutathione peroxidase 4 (GPX4), an essential enzyme that prevents ferroptosis by reducing potentially toxic membrane lipid hydroperoxides to non-toxic lipid alcohols (Friedmann Angeli et al., 2014; Ingold et al., 2018). The ferroptosis mechanism is an emerging target for anti-cancer therapy (Bartolacci et al., 2022; Hangauer et al., 2017; Jiang et al., 2021; Koppula et al., 2022; Viswanathan et al., 2017). It is therefore of interest to understand how cancer cells manage cysteine metabolism to inhibit ferroptosis.
Cysteine is typically present at low abundance within the cell and in the fluid surrounding tumors in vivo (Abu-Remaileh et al., 2017; Sullivan et al., 2019). Depriving cells of extracellular cystine, the disulfide of cysteine typically present outside the cell, potently induces ferroptosis in numerous cultured cancer cell lines (Dixon et al., 2014; Poltorack and Dixon, 2021; Yang et al., 2014). Cells can be starved of cystine by small molecules, like erastin, that inhibit the system xc- cystine/glutamate antiporter, by removing cystine from the growth medium of cultured cells, or by degrading extracellular cystine/cysteine using the engineered enzyme cyst(e)inase (Badgley et al., 2020; Cramer et al., 2017; Dixon et al., 2014; Tarangelo et al., 2018; Wang et al., 2019; Zhang et al., 2019). The mechanisms employed by cancer cells to maintain intracellular cysteine levels and prevent the onset of ferroptosis under conditions of extracellular cystine limitation are not fully understood.
Protein uptake from the extracellular environment via macropinocytosis or other endosomal processes can fuel cancer cell proliferation and resistance to anti-cancer therapies (Davidson et al., 2017; Jayashankar and Edinger, 2020; Kamphorst et al., 2015; Kim et al., 2018). The abundant serum protein albumin, in particular, can serve as a source of amino acids to support tumor growth (Commisso et al., 2013; Kamphorst et al., 2015). Catabolism of intracellular or extracellular protein in the lysosome liberates amino acids that can then be exported into the cytosol and used for the synthesis of proteins and other products (Cantin et al., 2000; Jayashankar and Edinger, 2020; Kim et al., 2018; Nofal et al., 2021; Palm, 2019; Perera et al., 2015; Wyant et al., 2017; Zhang and Commisso, 2019). Whether the catabolism of extracellular protein liberates sufficient cysteine to suppress ferroptosis and enable proliferation under cystine-poor conditions is unclear.
Protein catabolism in the lysosome is promoted by inhibition of the mechanistic target of rapamycin complex 1 (mTORC1) (Nofal et al., 2017; Palm et al., 2015; Ratto et al., 2022). We recently showed that mTOR inhibition prevents the depletion of intracellular glutathione and attenuates ferroptosis in cultured cells deprived of cystine (Conlon et al., 2021). In the present study, we tested the hypothesis that mTOR inhibition, together with the uptake and catabolism of extracellular protein, would be sufficient to compensate for the loss of transporter-mediated cystine uptake and inhibit ferroptosis. We find that uptake and lysosomal catabolism of extracellular albumin, a cysteine rich protein, is sufficient to prevent cancer cells deprived of extracellular free cystine from undergoing ferroptosis. This protective mechanism requires the lysosomal enzyme cathepsin B and the lysosomal cystine exporter cystinosin. This mechanism enables extracellular albumin to fuel ongoing GSH synthesis within the cell in the absence of cystine uptake from the environment.
RESULTS
Cystine deprivation triggers ferroptosis that can be suppressed by extracellular albumin
How cancer cells cope with limited extracellular amino acids, especially cystine, is not entirely clear. To investigate, we deprived HT-1080N fibrosarcoma cells of cystine and measured both cell proliferation and cell death over time. These cells express a live cell marker, nuclear-localized mKate2 (denoted by the superscript ‘N’) and were incubated with the dead cell dye SYTOX Green. This enabled cell proliferation and cell death to be directly quantified using the scalable time-lapse analysis of cell death kinetics (STACK) method (Forcina et al., 2017; Inde et al., 2021). Cells cultured in complete medium proliferated rapidly for four days then died, most likely due to overcrowding-induced apoptosis (Forcina et al., 2017), as this death was blocked by the pan-caspase inhibitor Q-VD-OPh (Figure 1A,B). By contrast, cells exposed to cystine-free medium did not proliferate and mostly died within 24 h in a manner that was suppressed by the ferroptosis-specific inhibitor ferrostatin-1 (Fer-1), but not by Q-VD-OPh (Figure 1A,B). Selective deprivation of three other amino acids (Met, Arg, Leu) resulted in proliferative arrest and/or a minimal amount of cell death over eight days that was not affected by Fer-1 (Figure 1A,B). Thus, cultured HT-1080 cells are specifically sensitive to the induction of ferroptosis upon deprivation of extracellular cystine.
Figure 1. Albumin promotes cell survival in response to cystine deprivation.
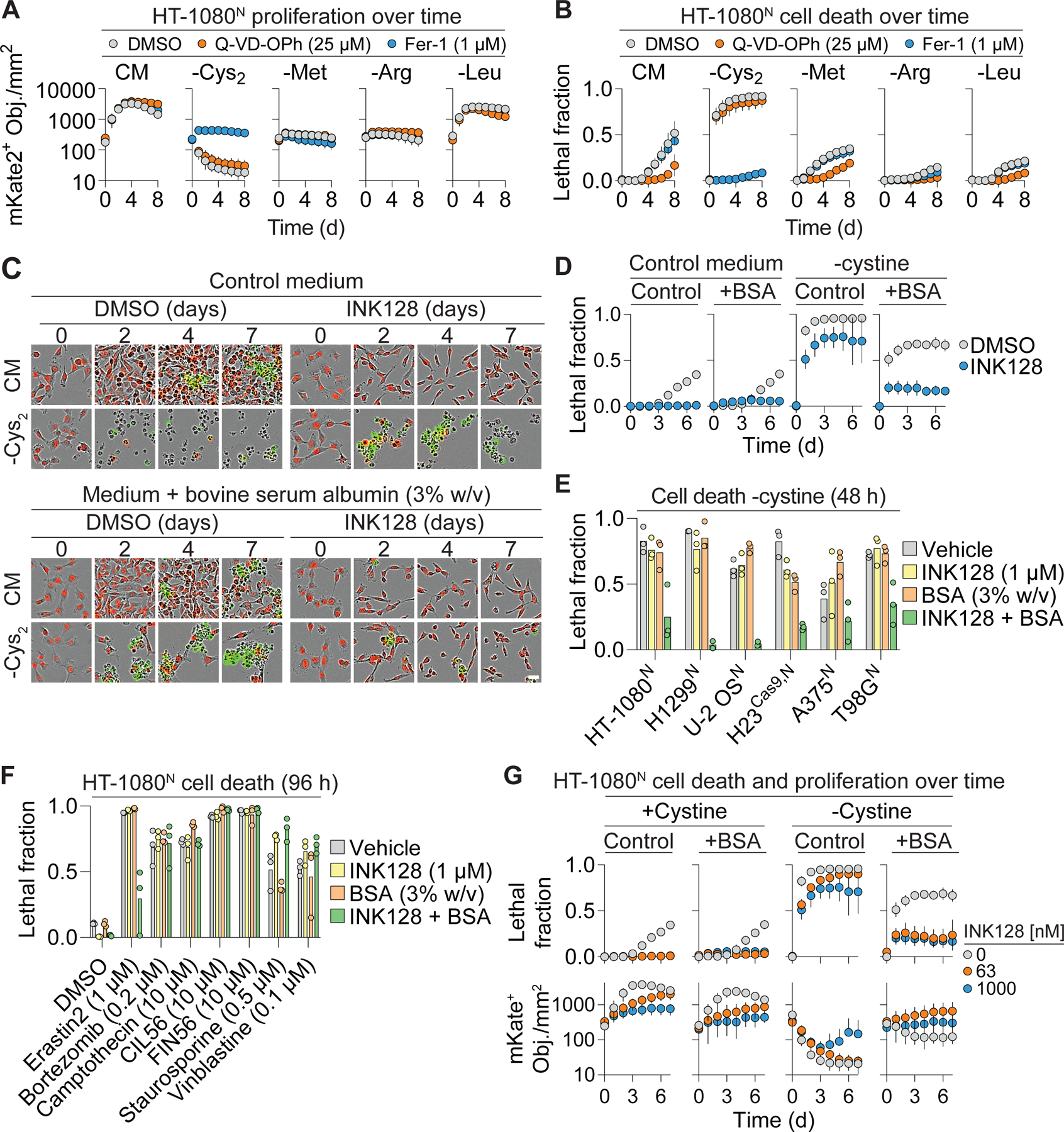
(A) Cell proliferation determined by live cell (nuclear-mKate2 positive objects; mKate2+ Obj.) counts over time in complete medium (CM) or medium lacking the indicated amino acid ± apoptosis (Q-VD-OPh) or ferroptosis (Fer-1) inhibitors. Cys2: cystine. (B) Cell death in populations from (A) determined by integration of live (mKate2+) and dead (SYTOX Green-positive) counts over time into the lethal fraction score (Forcina et al., 2017). A lethal fraction score of 0 equals no cell death and 1 equals complete population cell death. (C) Representative images from three independent experiments of HT-1080N cells cultured over time in complete medium (CM) or medium lacking cystine. Medium contains 20 nM SYTOX Green to mark dead cells. INK128: 1 µM. Scale bar = 30 µm. (D) Quantification of cell death in (C). BSA: bovine serum albumin. (E) Cell death in different cell lines in medium lacking cystine. (F) Cell death in response to different inducers of cell death. (G) Cell death and proliferation over time. Note that some of the lethal fraction data shown here for comparison purposes is also depicted in (D). These data were from the same experiment. Results in (A, B, D, and G) are mean ± SD from three independent experiments. Datapoints from independent experiments are shown in (E) and (F). See also Figure S1 and Figure S2.
Cystine deprivation in vivo can slow tumor growth but does not appear to cause significant tumor regression (Badgley et al., 2020; Wang et al., 2019; Zhang et al., 2019). This suggests that compensatory resistance mechanisms may exist that limit ferroptosis in response to cysteine deprivation. In addition to transporter-mediated uptake of free amino acids, cancer cells can internalize and catabolize extracellular protein, which can contain extensive endogenous and bound cysteine (Eagle et al., 1960; Palm, 2019; Zhang and Commisso, 2019). The catabolism of ingested protein can be increased by mTOR inhibition (Nofal et al., 2017; Palm et al., 2015). Moreover, we recently found that mTOR inhibition inhibits ferroptosis in response to cystine deprivation (Conlon et al., 2021). Accordingly, we hypothesized that providing cells with additional extracellular cysteine-containing protein, together with mTOR inhibition, would be sufficient to prevent ferroptosis in cells deprived of free extracellular cystine. As an extracellular protein source we first tested the abundant serum protein albumin at a final concentration (3% w/v) mimicking that found in human serum (Choi et al., 2004; Keyser et al., 1981). Consistent with our recent results (Conlon et al., 2021), treatment with the ATP-competitive mTOR inhibitor INK128 alone was sufficient to attenuate cystine deprivation-induced cell death, albeit to a limited extent over seven days (Figure 1C,D). Incubating cells in albumin-enriched medium alone likewise partially inhibited cell death over time in response to cystine deprivation (Figure 1C,D). By contrast, the combination of albumin and mTOR inhibitor (A+I) potently suppressed cell death under cystine-deprived conditions for at least seven days (Figure 1C,D). In addition to HT-1080N cells, A+I treatment suppressed cell death in cystine-deprived H1299N and H23Cas9,N non-small cell lung carcinoma cells, U-2 OSN osteosarcoma, A375N melanoma, T98GN glioblastoma, and PaTu 8988T pancreatic adenocarcinoma cell lines, consistent with a broadly generalizable effect (Figure 1E, S1A).
We investigated the nature of the A+I protective effect further. The ability of A+I treatment to inhibit ferroptosis in cells deprived of cystine was concentration-dependent for both albumin and INK128, indicating a titratable effect (Figure S1B,C). This protective effect was also observed when using structurally distinct mTOR inhibitors (Torin 1, rapamycin) in combination with albumin, suggesting that the protective mechanism was not linked to INK128 per se (Figure S1D,E). The mTOR kinase is present in two distinct signaling complexes, mTORC1 and mTORC2. Short hairpin RNA (shRNA) targeting the mTORC1 subunit RPTOR, but not the mTORC2 complex subunit RICTOR, could substitute for pharmacological mTOR inhibition in protecting cells from cystine deprivation-induced death when combined with extracellular albumin (Figure S1F,G). These results argued that our pharmacological mTOR inhibitor treatments were acting in an on-target manner and indicated that mTORC1 inhibition was required for albumin to protect against ferroptosis.
We considered potential alternative explanations for the ability of A+I to inhibit ferroptosis in response to cystine deprivation, including general suppression of cell death, cell cycle arrest, induction of autophagy, and altered iron homeostasis. A+I treatment did not appear to be a general suppressor of cell death, as this treatment prevented cell death in response to the small molecule system xc- inhibitor erastin2, which blocks cystine uptake (Dixon et al., 2014), but not in response to small molecule inhibitors of the proteasome, topoisomerase function, microtubule integrity, or other essential functions (Figure 1F). The protective effect of the A+I combination was not recapitulated in HT-1080 cells by the combination of albumin and the CDK4/6 inhibitor palbociclib, suggesting that proliferative arrest caused by mTORC1 inhibition was insufficient to account for the protective effects of albumin co-treatment (Figure S2A,B). A+I treatment protected equally well against ferroptosis in Control and ATG7 gene-disrupted (i.e., “knockout”, KO) PaTu 8988T cell lines, suggesting that autophagy was not likely to be essential for this effect (Figure S2C,D). Finally, A+I treatment did not increase the expression of the anti-ferroptotic proteins GPX4 or FSP1 in HT-1080 cells (Bersuker et al., 2019; Doll et al., 2019; Yang et al., 2014), or decrease intracellular iron levels, as assessed by expression of the transferrin receptor (TFRC) or IRP2, which are induced by iron starvation (Figure S2E). These results argued against several alternative mechanisms of ferroptosis resistance in A+I-treated cells.
In response to amino acid deprivation, the combination of albumin with low doses of mTOR inhibitor can enable some amount of cell proliferation (Palm et al., 2015). In line with this finding, the combination of albumin with low dose INK128 (63 nM) not only inhibited ferroptosis but was compatible with some cell proliferation in cystine-free conditions (Figure 1G). These results supported a hypothesis that extracellular protein specifically inhibited ferroptosis and enabled cell proliferation when cystine was not present, through a mechanism that was enhanced by mTORC1 inhibition. These results also further argued against a protective mechanism that involved iron deprivation, as this would be expected to arrest cell proliferation.
Albumin fuels GSH synthesis to suppress ferroptosis
Ferroptosis is caused by compromised defense against the accumulation of toxic lipid hydroperoxides. Given our results, we hypothesized that A+I treatment would prevent membrane lipid peroxidation in cystine-deprived cells. Consistent with this hypothesis, oxidation of the lipid peroxidation sensor C11 BODIPY 581/591 was strongly suppressed by the A+I combination (Figure 2A). Albumin is reported to act as a direct antioxidant and can also bind other molecules known to inhibit lipid peroxidation, like oleic acid (Carballal et al., 2003; Magtanong et al., 2019; Neuzil et al., 1993), suggesting two plausible alternative mechanisms that could explain reduced level of lipid peroxidation and ferroptosis. Indeed, albumin treatment alone weakly inhibited ferroptosis in response to low doses of the GPX4 inhibitors FIN56 or ML162 (Shimada et al., 2016) (Figure S2F). However, this protective effect was lost at high doses of ML162 and FIN56 and, unlike in cystine-deprived cells, not enhanced by mTOR inhibition (Figure 1F, 2B, S2F). Thus, while albumin may partially inhibit lipid peroxidation and ferroptosis through a direct antioxidant or related effect, the A+I combination strongly suppresses ferroptosis only in response to cystine deprivation.
Figure 2. Albumin increases glutathione levels during cysteine deprivation.
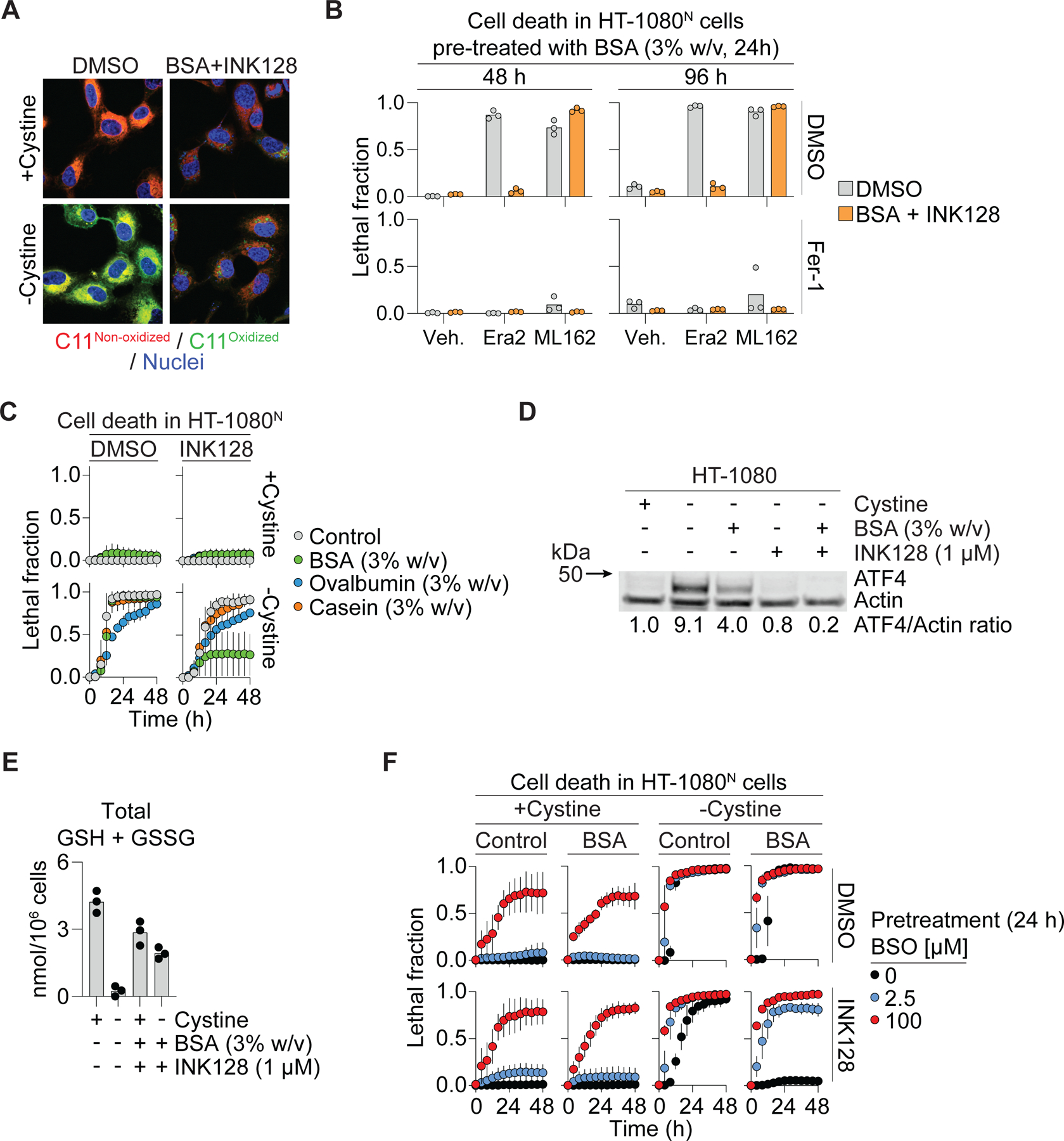
(A) C11 BODIPY 581/591 (C11) oxidation assessed by confocal microscopy in HT-1080 cells. BSA: bovine serum albumin (3% w/v). INK128: 1 µM. Scale bar = 20 µm. (B) Cell death in HT-1080N cells pre-treated with BSA (24 h) then treated as indicated at time 0 and examined 48 and 96 h later. Era2 (erastin2): 1 µM. ML162: 2 µM. Fer-1 (ferrostatin-1): 1 µM. (C) Cell death over time. INK128: 1 µM. (D) Protein levels following 10 h treatment. Blot is representative of three independent experiments. Mean ATF4/Actin protein level ratios (normalized to the +cystine condition) determined from densitometry of three independent blots are indicated. (E) Total glutathione (GSH + GSSG) following 8 h treatment. INK128: 1 µM. (F) Cell death following 24 h pretreatment ± BSO (buthionine sulfoximine) then treated as indicated alongside BSO. BSA: 3% w/v. Results in (B) and (E) show datapoints from independent experiments. Results in (C) and (F) are mean ± SD from three independent experiments. See also Figure S3.
Collectively, results obtained to this point bolstered our working model that A+I treatment blocked ferroptosis in response to cystine deprivation by providing an alternate cysteine source. This model was further supported by several additional lines of evidence. First, compared to other proteins, albumin is both highly abundant in serum and rich in cysteine residues (Geyer et al., 2016) (Figure S3A). Indeed, albumin (35 cysteines/molecule) was a more potent inhibitor of ferroptosis than the less cysteine-rich molecules ovalbumin (6 cysteines/molecule) or casein (1 cysteine/molecule) when added to cells in the same proportion (Figure 2C). Second, albumin treatment modulated a canonical biochemical marker of amino acid starvation, accumulation of activating transcription factor 4 (ATF4) (Conlon et al., 2021; Wortel et al., 2017): accumulation of ATF4 in response to 10 h cystine deprivation was inhibited by the addition of extracellular albumin (Figure 2D). Note that cells were harvested at 10 h since to wait longer would mean that cystine-deprived control cells would start to die. It is likely that the partial inhibition of ATF4 accumulation by albumin treatment alone is transient, consistent with the partial suppression of cell death (Figure 1D). Also, mTOR inhibition decreases ATF4 mRNA transcription (Park et al., 2017), reducing the pool of ATF4 mRNA available to translate, likely accounting for the blunting effect of INK128 and combined A+I treatment on ATF4 levels. Some evidence suggests that ATF4 may accumulate in response to oxidative stress (Wortel et al., 2017), which in the context of ferroptosis would be downstream of cysteine deprivation per se. However, we found that suppressing lipid peroxide accumulation using ferrostatin-1 did not prevent ATF4 protein from accumulating in response to cystine deprivation, or stop albumin from blunting this phenotype, suggesting that ATF4 accumulation was linked to cysteine deprivation and not oxidative stress (Figure S3B). Third, in cells deprived of cystine, A+I treatment largely preserved levels of the downstream cysteine-containing metabolite glutathione (Figure 2E). This was functionally important, as the ability of A+I to prevent ferroptosis in cystine-deprived HT-1080N, A375N, H1299N and T98GN cancer cell lines was blunted to a variable degree by inhibition of de novo glutathione synthesis using buthionine sulfoximine (BSO) (Figure 2F, S3C,D).
Collectively, these results suggested that extracellular albumin combined with mTOR inhibition could block toxic lipid hydroperoxide accumulation and the induction of ferroptosis in response to cystine deprivation by maintaining the intracellular levels of glutathione and possibly other sulfur-containing metabolites (Badgley et al., 2020; Leu et al., 2019). We recently showed that stabilization of the transcription factor p53 also led to conservation of intracellular glutathione and ferroptosis inhibition under conditions of cystine deprivation (Tarangelo et al., 2018; Tarangelo et al., 2022). We therefore examined whether p53 stabilization contributed to the protective effects of A+I treatment in response to cystine deprivation. In fact, neither cystine deprivation nor A+I treatment stabilized p53 in either HT-1080 or U-2 OS cells (Figure S3E). A+I treatment was also capable of fully suppressing ferroptosis in cystine-deprived H1299 cells, which are p53 deficient (Figure 1E, S3E). Thus, p53 stabilization was not necessary for A+I treatment to protect cells from ferroptosis following cystine deprivation.
Lysosomal protease function is required for albumin to suppress ferroptosis
Extracellular albumin is ingested by cancer cells and catabolized within the lysosome to liberate free amino acids, a mechanism that can help cancer cells to survive in nutrient-scarce environments (Commisso et al., 2013; Kamphorst et al., 2015; Nofal et al., 2021; Wyant et al., 2017). We hypothesized that lysosome-dependent proteolytic degradation of ingested albumin liberated free cysteine to stabilize intracellular GSH levels, suppress toxic lipid hydroperoxide accumulation, and inhibit ferroptosis in cystine-deprived cells. We tested this hypothesis by perturbing lysosome activity in different ways. Lysosomal protease activity can be inhibited pharmacologically using deacidifying agents (e.g., chloroquine) or using a cocktail of the protease inhibitors pepstatin A, leupeptin, and E-64 (Palm et al., 2015; Poole and Ohkuma, 1981). Consistent with our hypothesis, co-treatment with chloroquine or the protease inhibitor cocktail prevented A+I treatment from suppressing ferroptosis in cystine-deprived cells (Figure 3A). We confirmed that chloroquine and protease inhibitor cocktail treatment inhibited lysosomal albumin catabolism, as assessed by dequenching of the probe DQ-BSA (Commisso et al., 2013; Reis et al., 1998) (Figure 3B,C, S4A). Both chloroquine and the protease inhibitor cocktail likewise resulted in accumulation of LC3B, a protein constitutively degraded in the lysosome, although this was more pronounced with chloroquine than with protease inhibitors (Figure S4B). Concordant with these results, ATF4 accumulation, which was blunted in cystine-deprived cells by albumin, INK128 or A+I treatment, reverted back to higher levels upon co-addition of chloroquine or the protease inhibitor cocktail (Figure 3D). Phenotypically, we generalized these results to additional cancer cell lines, showing that chloroquine and/or protease inhibitor cocktail partially reverted the anti-ferroptotic effects of A+I treatment in A375N, H1299N and T98GN cells (Figure 3E,F). Of note, the protective effects of A+I treatment was not reverted by chloroquine or the protease inhibitor cocktail in U-2 OSN cells, indicating that non-lysosomal mechanisms of protection may also be possible in some instances (Figure 3E,F). However, in most cell lines lysosome function appeared to be required for A+I treatment to protect against ferroptosis.
Figure 3. Lysosomal function is necessary for albumin to protect from ferroptosis.
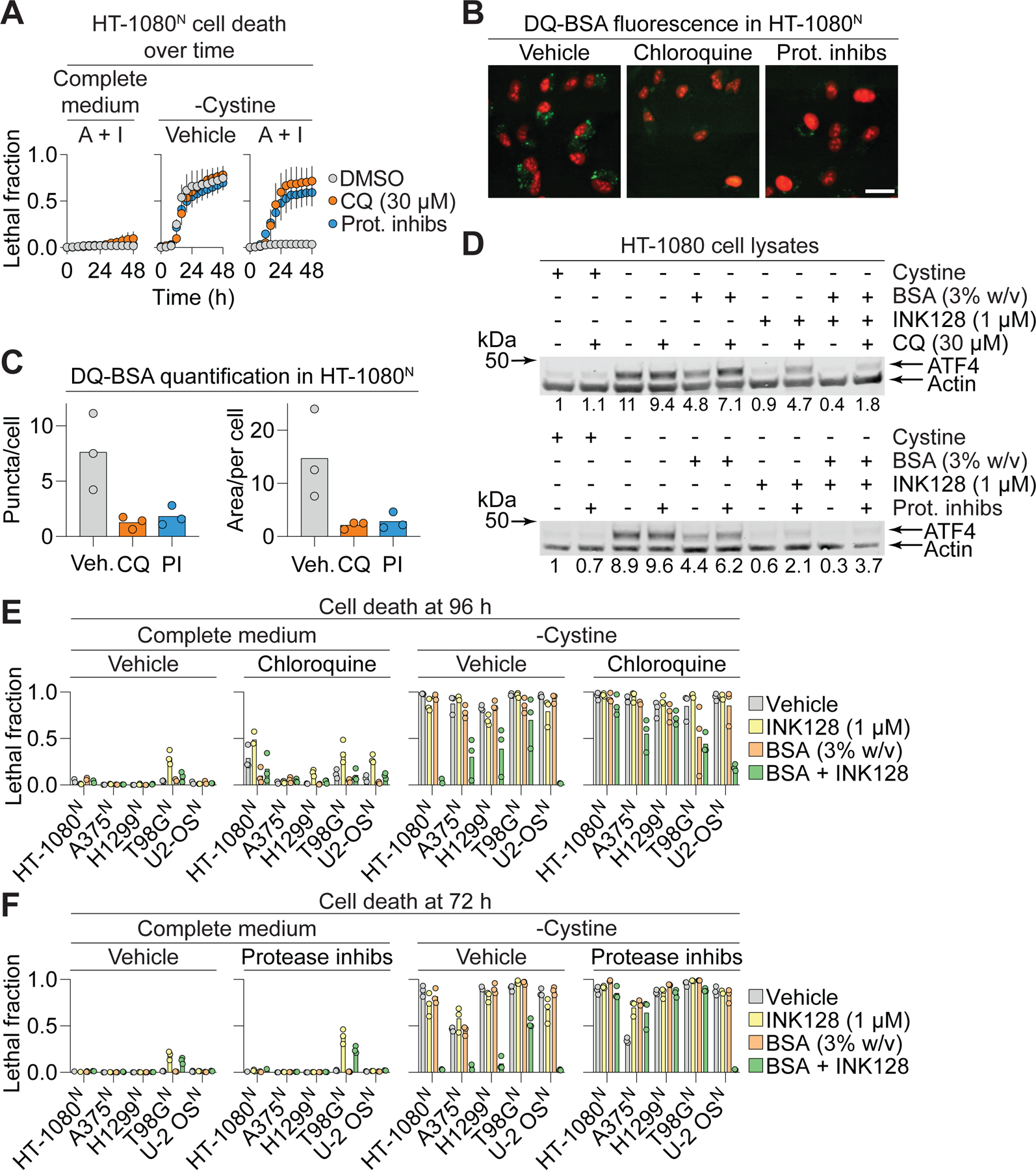
(A) Cell death over time. A+I: bovine serum albumin (BSA, 3% w/v) + INK128 (1 µM). Prot. inhibs: protease inhibitor cocktail (leupeptin: 10 µM, pepstatin A: 2 µM, E-64: 2 µM). Data represent mean ± SD from three independent experiments. (B) DQ-BSA fluorescence assessed by fluorescence microscopy in HT-1080N cells treated. Chloroquine (30 µM), protease inhibitor cocktail (Prot. inhibs, as in (A)). Images are representative of three independent experiments. Scale bar = 30 µm. (C) Quantification of DQ-BSA puncta or area (in square pixels) per cell. Datapoints represent counts of puncta from all images within a condition divided by number of cells in those images, from three independent experiments (minimum 30 cells/experiment and condition). (D) ATF4 protein levels in response to different treatment conditions (10 h). CQ: chloroquine. Blots are representative of three independent experiments. Mean ATF4/Actin protein level ratios (normalized to the +cystine condition) determined from densitometry of three independent blots are indicated. (E) Cell death in various cell lines ± chloroquine (30 µM). (F) Cell death in various cell lines ± protease inhibitor cocktail as in (A). Datapoints from independent experiments are shown in (E) and (F). See also Figure S4.
CTSB promotes albumin catabolism to suppress ferroptosis
We next sought to identify specific lysosomal protease(s) involved in albumin catabolism within the lysosome, with the hypothesis that one or more specific cathepsin enzymes would be needed for this process. For example, a recent study suggested that cathepsin L may be one important enzyme required for albumin degradation (Nofal et al., 2021). For our studies we used pancreatic cancer cell lines, which can internalize albumin avidly (Commisso et al., 2013), and detectably express at least 12 different lysosomal cathepsins genes (Figure S5A). To first examine albumin breakdown within the lysosome we assessed DQ-BSA dequenching. In both PaTu 8988T and MIA PaCa2 pancreatic adenocarcinoma cell lines, dequenched DQ-BSA colocalized with a lysosomal marker (LysoTracker dye), and treatment with the mTOR inhibitor Torin 1 increased lysosome number and dequenching of DQ-BSA, consistent with expectations and previous reports (Commisso et al., 2013; Nofal et al., 2017; Palm et al., 2015) (Figure 4A). In both cell lines, DQ-BSA dequenching was suppressed by the cathepsin B (CTSB) inhibitor CA074 methyl ester but not by the cathepsin L and S inhibitor R11-OEt (van der Linden et al., 2016) (Figure S5B). Of importance, the CTSB inhibitor itself did not affect macropinocytic uptake, as determined by the internalization of TMR-dextran (Figure S5B).
Figure 4. CTSB is required for ferroptosis suppression by extracellular albumin.
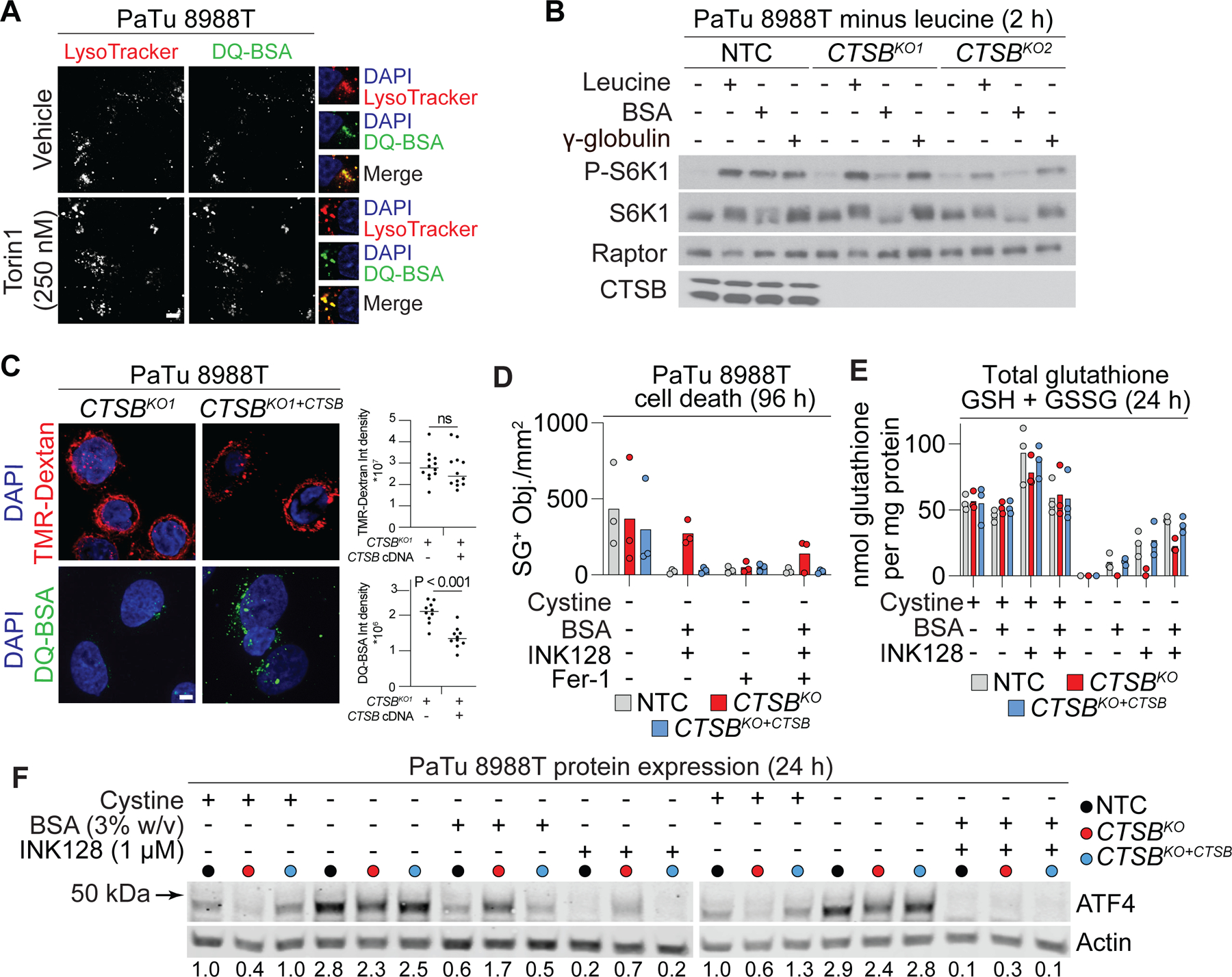
(A) LysoTracker Red, a marker for lysosomes, and self-quenched bovine serum albumin (DQ-BSA) fluorescence in PaTu 8988T cells. Scale bar = 5 µm. (B) Activity of the mTORC1 signaling pathway in PaTu 8988T cells starved of leucine for 2 h, then restimulated with medium containing leucine (Leu), bovine serum albumin (BSA, 5% w/v), or γ-globulin (3% w/v) for 4 h. Raptor was used as a loading control. (C) Analysis of lysosomal uptake and degradation of self-quenched bovine serum albumin (DQ-BSA) in PaTu 8988T cells. The macropinocytic cargo 70 kDa tetramethylrhodamine dextran (TMR-dextran) is used to assess any effect on uptake. On the right, each datapoint represents ≥ 3 fields of view with ≥ 10 cells in total, with the median indicated by the horizontal bar. Scale bar = 5 μm. CTSBKO1: cathepsin B knockout, CTSBKO1+CTSB: CTSB knockout reconstituted with CTSB cDNA. (D) Cell death determined by counting SYTOX Green-positive (SG+) dead cells. BSA: 3% w/v, INK128: 1 µM, Fer-1 (ferrostatin-1): 1 µM. NTC: non-targeting CRISPR control. (E) Total glutathione (GSH + GSSG) measured using Ellman’s reagent following 24 h of treatment. INK128: 1 µM, BSA: 3% w/v. (F) Protein levels determined by western blot. Blots are representative of three independent experiments. Mean ATF4/Actin protein level ratios (normalized to the +cystine condition) determined from densitometry of three independent blots are indicated. See also Figure S5.
Next, as a screening strategy to identify cathepsins involved in albumin catabolism, we examined the ability of extracellular albumin to restore mTOR pathway activity in cells treated with different inhibitors. Here, we used leucine deprivation as a condition to inhibit mTOR activity basally (Wyant et al., 2017), as cystine deprivation does not potently inhibit mTOR activity itself over short timescales (Conlon et al., 2021). Strikingly, the CTSB inhibitor CA074 methyl ester prevented extracellular albumin from re-activating the mTOR pathway in leucine- and serum-deprived cells, consistent with this enzyme being required for albumin catabolism (Wyant et al., 2017) (Figure S5C). Indeed, CRISPR/Cas9-mediated disruption of CTSB in PaTu 8988T cells prevented albumin, but not γ-globulin, from reactivating the mTOR signaling pathway upon leucine starvation (Figure 4B). By contrast, genetic disruption of CTSL and CTSD had no effect on the ability of extracellular albumin to reactivate the mTOR pathway in leucine-deprived media (Figure S5A,D). Thus, CTSB appeared to be a major albumin proteolytic enzyme in the pancreatic cancer cell lines examined here.
Based on these results, we examined PaTu 8988T non-targeting control (NTC) cells, CTSB gene-disrupted (KO1) cells, or CTSBKO1 cells where wild-type CTSB was reconstituted by gene overexpression (KO1+CTSB) (Figure S5E). With respect to albumin catabolism, we observed that CTSB reconstitution restored the dequenching of DQ-BSA that was lost in CTSBKO1 cells (Figure 4C). Turning to cell death, we examined how CTSB impacted the ability of A+I treatment to prevent ferroptosis in response to cystine deprivation. NTC, CTSBKO1, and CTSBKO1+CTSB cells were all killed by cystine deprivation; however, while non-targeting control and reconstituted CTSBKO1+CTSB cells were fully protected under these conditions by A+I treatment, CTSBKO1 cells were not (Figure 4D). Under conditions of cystine deprivation and A+I treatment, NTC and CTSBKO1+CTSB cells also retained higher levels of intracellular glutathione than CTSBKO1 cells, as detected using Ellman’s reagent or liquid chromatography coupled to mass spectrometry (Figure 4E, S5F). Albumin supplementation alone also more effectively reduced ATF4 protein accumulation and suppressed C11 BODIPY 581/591 oxidation in cystine-deprived NTC and CTSBKO1+CTSB cells than in CTSBKO1 cells (Figure 4F, S5G). Together, these results suggested that lysosomal CTSB function was necessary for extracellular albumin to fully suppress ferroptosis in cells deprived of extracellular cystine.
CTNS export is required for albumin to suppress ferroptosis
Cysteine is exported from the lysosome in the form of cystine by the transporter cystinosin (CTNS) (Gahl et al., 2002). Our results suggested that lysosomal albumin catabolism by CTSB liberated free cysteine that could be used in subsequent GSH synthesis and thus to suppress toxic lipid hydroperoxide accumulation. However, cysteine generated in this manner in the lysosome needs to be exported to the cytosol. Thus, we hypothesized that CTNS function would be necessary for A+I treatment to protect cells against ferroptosis. To test this hypothesis, we generated two independent clonal CTNS gene-disrupted (KO) cell lines which, due to the absence of a suitable antibody, we confirmed to be mutant by genomic DNA sequencing (see Methods). When cultured in regular medium, unmodified Control cells and CTNSKO1/2 cell lines had similar mTOR pathway activity, proliferation rates, and cell death in response to cystine deprivation (Figure 5A, S6). However, when deprived of cystine, Control cells but not CTNSKO1/2 cells were protected against ferroptosis by the A+I combination (Figure 5A). Thus, CTNS appeared to be required for A+I to inhibit ferroptosis in response to cystine deprivation.
Figure 5. Cystinosin is required for ferroptosis suppression by extracellular albumin.
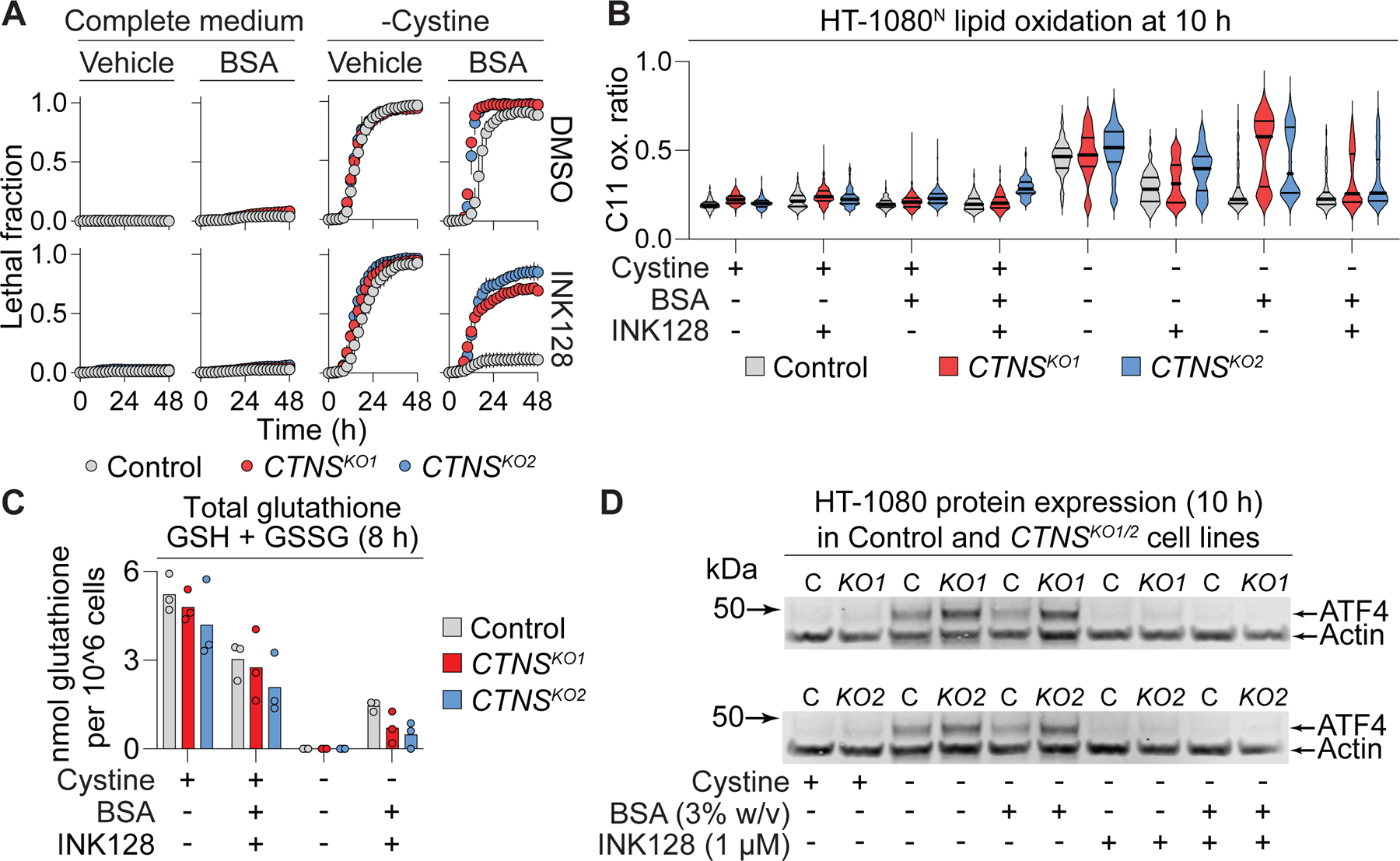
(A) Cell death in HT-1080N Control or cystinosin (CTNS) gene-disrupted (knockout, KO) cell lines. INK128: 1 µM, bovine serum albumin (BSA): 3% w/v. Data represent mean ± SD from three independent experiments. (B) C11 BODIPY 581/591 (C11) oxidation ratio (oxidized C11/[reduced C11 + oxidized C11]). INK128: 1 µM, BSA: 3% w/v. Data acquired from 59–512 individual cells per condition were analyzed. Representative data from one of three independent experiments is shown. (C) Total glutathione (GSH + GSSG) measured using Ellman’s reagent. INK128: 1 µM, BSA: 3% w/v. (D) Protein levels determined by western blot. C: Control, KO: knockout. Blots are representative of three independent experiments. See also Figure S6.
Functionally, cystine deprivation for 10 h increased lipid oxidation in both Control and CTNSKO1/2 cell lines (Figure 5B). Consistent with our viability results, A+I suppressed C11 oxidation following 10 h of cystine deprivation more effectively in Control cells than in CTNSKO1/2 cell lines (Figure 5B). However, observed differences in C11 oxidation were small at this timepoint; it was not possible to examine C11 oxidation at later timepoints following cystine deprivation as Control cells would be killed under these conditions. Tellingly, A+I treatment less effectively maintained intracellular glutathione levels in cystine-deprived CTNSKO1/2 cells compared to Control cells (Figure 5C). Additionally, ATF4 accumulation in response to cysteine deprivation was higher in CTNSKO1/2 cells compared to Control cells, and less effectively suppressed by albumin supplementation (Figure 5D). These data suggested that CTNS function was required for A+I treatment to support GSH synthesis and suppress ferroptosis in response to deprivation of extracellular cystine.
Albumin protects cell spheroids and patient-derived glioma cells from ferroptosis
Compared to cells grown in monolayers, cells grown under detached conditions more closely recapitulate the in vivo tumor microenvironment (Dixon et al., 2014; Friedrich et al., 2009; Jiang et al., 2016; Schafer et al., 2009). Accordingly, we examined whether extracellular albumin combined with mTOR inhibition could inhibit ferroptosis in HT-1080N spheroids established over the course of three days in ultralow adherence vessels (Figure 6A). Since it was not possible to replace the growth medium without disturbing the spheroids, we instead induced cystine deprivation using the small molecule system xc- inhibitor erastin2. Unexpectedly, co-treatment with albumin alone was sufficient to fully maintain HT-1080N spheroid morphology and overall viability, as assessed by total spheroid ATP levels, even without the addition of mTOR inhibitor (Figure 6A,B). Based on previous findings (Barbone et al., 2008; Riedl et al., 2017), we hypothesized that spheroid growth may reduce mTOR signaling and obviate the need for exogenous mTOR inhibition. Indeed, HT-1080N cells grown as spheroids had reduced RPS6 phosphorylation compared to HT-1080N cells growth in monolayers (Figure 6C). Similar results for ferroptosis inhibition and mTOR pathway activity were obtained in A375N cells grown as spheroids versus in monolayer, suggesting a generalizable effect (Figure S7A-C).
Figure 6. Albumin promotes spheroid viability in response to cystine deprivation.
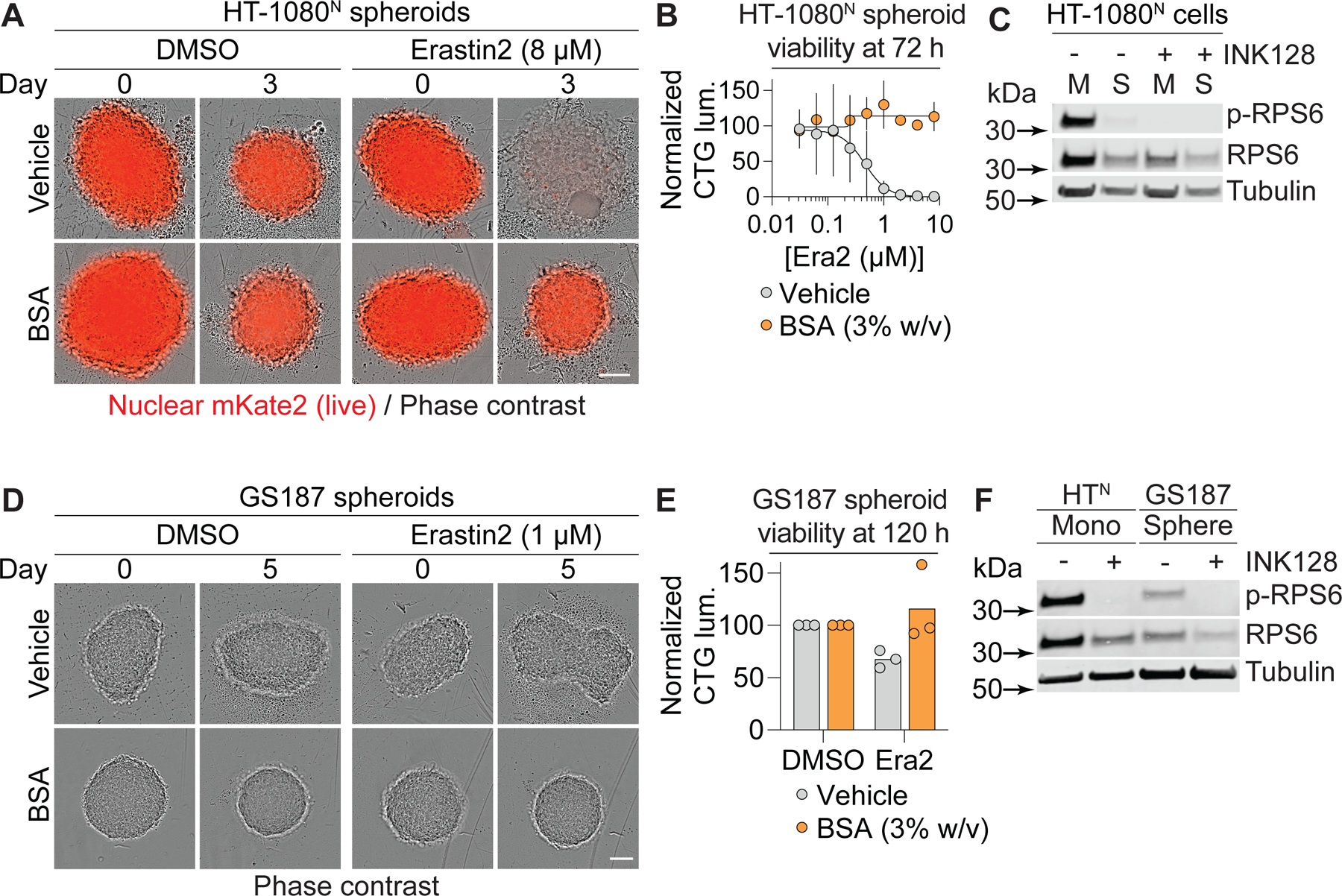
(A) HT-1080N spheroids established over three days (day 0) prior to treatment for three days (Day 3). BSA: bovine serum albumin, 3% w/v. Scale bar = 100 µm. (B) Viability of HT-1080N spheroids as established and treated as in (A) determined using CellTiter-Glo. (C) mTOR pathway activity assessed by western blot following 24 h treatment. M: monolayer, S: spheroid. INK128: 1 µM. (D) Gliomaspheres (line: GS187) established over three days (day 0) prior to treatment for five days. BSA: 3% w/v. Scale bar = 100 µm. (E) Viability of GS187 spheroids as established and treated as in (D) determined using CellTiter-Glo. (F) mTOR pathway activity assessed by western blot in HT-1080N cells grown in monolayer (control) or GS187 cells grown as spheroids and treated ± INK128 (1 µM) for 24 h. In (A) and (D), images are representative of three independent experiments. In (B) and (E), data are mean ± SD from three independent experiments. In (C) and (F), blots are representative of three independent experiments. p-RPS6 is phosphorylated at Ser235/236. See also Figure S7.
To further extend our results, we examined human primary patient-derived gliomaspheres (Mai et al., 2017). In response to erastin2 treatment, we observed a small but quantitative reduction in gliomasphere viability that was reverted by co-incubation with albumin alone (Figure 6D,E). Compared to HT-1080N cells grown in monolayers, which we used for the purposes of comparison, gliomaspheres had decreased RPS6 phosphorylation, suggestive of reduced mTORC1 pathway activity (Figure 6F). Thus, the viability of primary cancer spheroids deprived of cystine can be supported by exogenous albumin alone, as observed in established cancer cell lines grown under similar conditions.
DISCUSSION
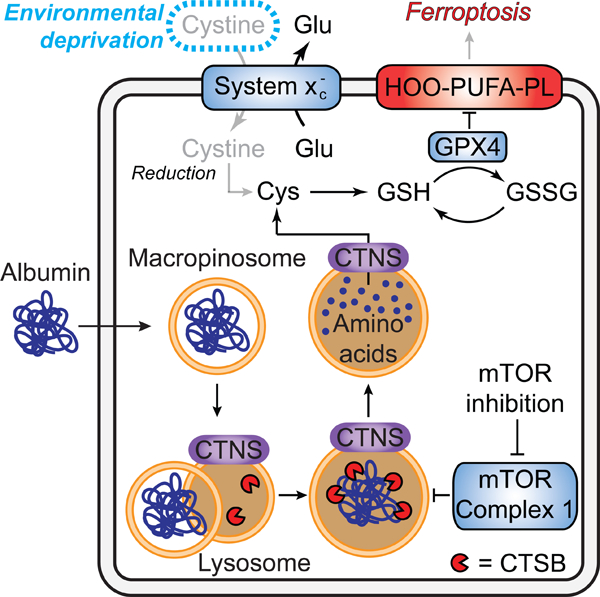
Cysteine is required to enable cell proliferation and ward off ferroptosis in many cancer cells in vitro and in vivo (Badgley et al., 2020; Combs and DeNicola, 2019; Dixon et al., 2014). Here, we define a mechanism that is active in many cancer cells that enables these cells to evade ferroptosis when extracellular cystine is absent: the uptake and lysosomal catabolism of extracellular albumin. Albumin is both highly abundant in serum and remarkably cysteine-rich (Figure S3A). Cysteine is usually maintained at a low level within the cell compared to other amino acids (Abu-Remaileh et al., 2017). Based on our findings, we propose that serum albumin acts as an important extracellular cysteine reservoir, accessible to cells that can internalize and catabolize this protein. Along with direct uptake of cysteine (Meira et al., 2021), and induction of the cysteine-generating transsulfuration pathway (Zhu et al., 2019), the uptake and catabolism of extracellular albumin, or other proteins obtained from serum or from dead cell debris (Kim et al., 2018), are likely to be important mechanisms used by cancer cells to maintain intracellular cysteine levels when extracellular free cystine is limiting (Figure 7).
Figure 7. Model for how exogenous protein protects from ferroptosis.
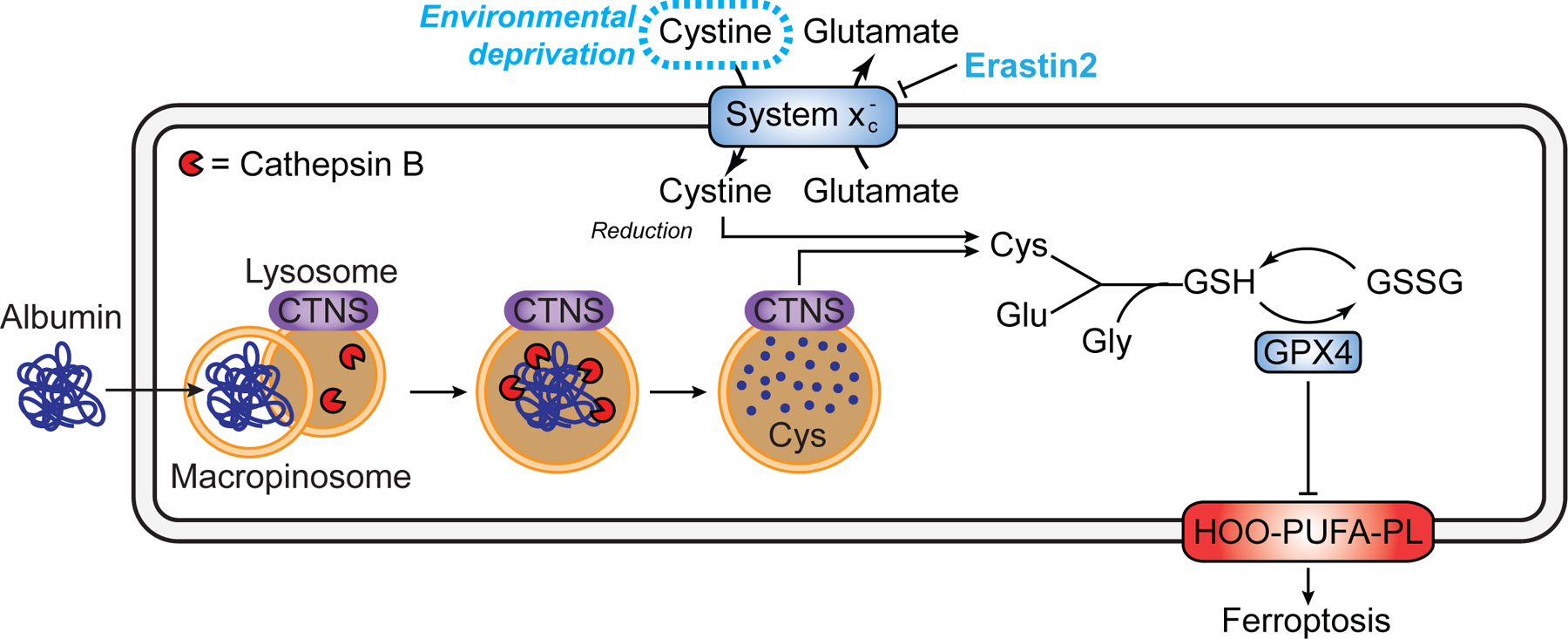
CTNS, cystinosin. Note that while in some cases albumin likely enters the cell via macropinocytosis other routes of entry and trafficking to lysosomes is possible.
Our results suggest that albumin catabolism within the lysosome by CTSB, and potentially other proteases (Nofal et al., 2021), is essential for inhibiting ferroptosis in cystine-deprived cells. Protein catabolism in the lysosome is strongly amplified by concomitant inhibition of mTORC1 (Nofal et al., 2017; Palm et al., 2015; Ratto et al., 2022), a result which we confirm here. We recently reported that mTORC1 inhibition alone is sufficient to attenuate ferroptosis in cystine-deprived cells, and proposed that this involves slowing of mTORC1-dependent mRNA translation, thereby enabling diversion of available cysteine towards GSH synthesis (Conlon et al., 2021). However, effects on mRNA translation only appeared to partially account for the ability of mTORC1 inhibition to inhibit ferroptosis in these experiments. Given our present findings, we speculate that the catabolism of albumin or other proteins present in standard growth medium containing 10% serum may also contribute to the ability of mTOR inhibition alone to protect from ferroptosis, at least under our growth conditions.
Our results indicate that protection from ferroptosis by extracellular protein requires export of newly liberated cystine from the lysosome into the cytosol via the transporter CTNS, at least in HT-1080 cells. This is consistent with the overall model that lysosomal albumin catabolism feeds cysteine to the cytosolic GSH biosynthetic machinery to maintain GPX4 activity and suppress lethal lipid peroxidation under conditions of extracellular cystine limitation. By contrast, disruption of CTNS has little effect on basal ferroptosis sensitivity when albumin is not present, suggesting that cystine stored in the lysosome (Abu-Remaileh et al., 2017) does not normally contribute to ferroptosis regulation. Intriguingly, in D. melanogaster, CTNS-dependent cystine export can help stimulate mTORC1 activity through a complex mechanism involving regulation of CoA metabolism and Krebs cycle activity (Jouandin et al., 2022). Speculatively, in metabolically stressed cancer cells, reduced lysosomal cystine export via CTNS could attenuate mTORC1 signaling and thereby help prime cells to use extracellular albumin to restore intracellular amino acid homeostasis.
Albumin can be internalized into cancer cells through macropinocytosis and other endocytic processes including receptor-mediated uptake (Commisso et al., 2013; Kamphorst et al., 2015; Kim et al., 2018). Macropinocytosis is stimulated by oncogenic activation of the RAS pathway (Commisso et al., 2013). We observed that extracellular protein prevented ferroptosis in cell lines with (e.g., HT-1080) and without (e.g., T98G, U-2 OS) RAS pathway mutations. It is possible that macropinocytosis is active in non-RAS pathway mutant cell lines for other reasons, or that other internalization processes can contribute to protein uptake in these cells. Nevertheless, albumin taken into the cell by whatever mechanism appears in most cases to be routed to the lysosome, where catabolism of this protein can supply the cell with cysteine needed to synthesize GSH and thereby block lipid peroxidation and the onset of ferroptosis.
Limitations of the study
The combination of albumin and mTOR inhibition appears to suppress ferroptosis in most cystine-deprived cancer cells lines by enhancing lysosomal protein catabolism. However, in U-2 OS cells, the protective effect of A+I treatment was not lost upon pharmacological inhibition of lysosomal function. It is possible that other mechanisms of albumin-mediated protection against ferroptosis, such as a direct antioxidant effect or the facilitated import of anti-ferroptotic molecules are more important mediators or ferroptosis resistance in some cell lines. This study also does not address the potential role of albumin in maintaining intracellular cysteine homeostasis in non-cancer cells. Interestingly, Slc7a11 knockout mice lacking system xc- function are viable and exhibit only minor phenotypes, especially in comparison to animals lacking Gpx4 (Friedmann Angeli et al., 2014; Sato et al., 2005). Along with direct cysteine uptake (Meira et al., 2021), it is possible that the uptake and catabolism of albumin or other cysteine-rich extracellular proteins reduces the dependence of cells in the body on system xc--mediated cystine uptake for survival.
STAR◆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott Dixon (sjdixon@stanford.edu).
Materials Availability
Plasmids, cell lines and other materials generated in this study will be shared by the lead contact upon request.
Data and Code Availability
Uncropped western blot images reported in have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
One ImageJ script used to analyze DQ-BSA puncta and one R program used to analyze cysteine abundance in proteins have been deposited to Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| GAPDH | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Phospho-4EB-P1 (Thr37/46) | Cell Signaling Technology | Cat# 9459; RRID:AB_330985 |
| Phospho-RPS6 (Ser235/236) | Cell Signaling Technology | Cat# 4858; RRID:AB_916156 |
| RPS6 | Cell Signaling Technology | Cat# 2217; RRID:AB_331355 |
| Alpha-tubulin, clone DM1A | Fisher Scientific | Cat# MS581P1 |
| FSP1 | Proteintech | Cat# 20886–1-AP, RRID:AB_2878756 |
| GPX4 | Abcam | Cat# ab125066, RRID:AB_10973901 |
| CTSL | Abcam | Cat# ab200738 |
| TFRC | Thermo Fisher Scientific | Cat# 13–6800, RRID:AB_2533029 |
| Beta-Actin | Santa Cruz Biotechnology | Cat# sc-47778, RRID:AB_626632 |
| p53 | Santa Cruz Biotechnology | Cat# sc-126, RRID:AB_628082 |
| Rb | BD Biosciences | Cat# 554136, RRID:AB_395259 |
| Raptor | Millipore Sigma | Cat# 09–217, RRID:AB_612103 |
| Phospho-S6K1 (Thr389) | Cell Signaling Technology | Cat# 9234, RRID:AB_2269803 |
| S6K1 | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Phospho-RPS6 (Ser235/236) | Cell Signaling Technology | Cat# 4858, RRID:AB_916156 |
| RPS6 | Cell Signaling Technology | Cat# 2217, RRID:AB_331355 |
| Phospho-4E-BP1 (Thr37/46) | Cell Signaling Technology | Cat# 9459; RRID:AB_330985 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644, RRID:AB_2097841 |
| Phospho-Rb | Cell Signaling Technology | Cat# 8516, RRID:AB_11178658 |
| Phospho-Akt (Thr308) | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| Akt | Cell Signaling Technology | Cat# 9272, RRID:AB_329827 |
| ULK1 | Cell Signaling Technology | Cat# 6439, RRID:AB_11178933 |
| CTSB | Cell Signaling Technology | Cat# 31718, RRID:AB_2687580 |
| CTSD | Cell Signaling Technology | Cat# 2284, RRID:AB_10694258 |
| ATF-4 | Cell Signaling Technology | Cat# 11815, RRID:AB_2616025 |
| IRP2 | Cell Signaling Technology | Cat# 37135, RRID:AB_2799110 |
| ATG7 | Cell Signaling Technology | Cat# 2631, RRID:AB_2227783 |
| GAPDH | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| LC3B | Cell Signaling Technology | Cat# 2775, RRID:AB_915950 |
| 680LT Donkey-anti-mouse | LI-COR | Cat# 926–68022, RRID:AB_10715072 |
| 680LT Donkey-anti-rabbit | LI-COR | Cat# 926–68023, RRID:AB_10706167 |
| 800CW Donkey-anti-mouse | LI-COR | Cat# 926–32212, RRID:AB_621847 |
| 800CW Donkey-anti-rabbit | LI-COR | Cat# 926–32213, RRID:AB_621848 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYTOX Green | Molecular Probes | Cat# S7020 |
| INK128 | Selleck Chemical | Cat# S2811 |
| Bovine serum albumin | Gemini Bio-Products | Cat# 700–100P |
| Bovine serum albumin | Sigma Aldrich | Cat# A3294 |
| Dimethyl Sulfoxide | Sigma-Aldrich | Cat# 276855, CAS: 67–68-5 |
| Ferrostatin-1 | Sigma-Aldrich | Cat# SML0583, CAS: 347174–05-4 |
| Q-VD-OPh | Fisher Scientific | Cat# OPH00101M |
| Torin 1 | Cayman Chemical | Cat# 10997 |
| Rapamycin | Fisher Scientific | Cat# BP2963–1 |
| Erastin2 | Custom synthesis | N/A |
| Bortezomib | Fisher Scientific | Cat# NC0587961, CAS: 179324–69-7 |
| Camptothecin | Fisher Scientific | Cat# AC276721000, CAS: 7689–03-4 |
| CIL56 | Shimada et al, 2016 | N/A |
| FIN56 | Shimada et al, 2016 | N/A |
| Staurosporine | Sigma-Aldrich | Cat# S6942, CAS: 62996–74-1 |
| Vinblastine | Selleck Chemical | Cat# S1248, CAS: 143–67-9 |
| Palbociclib | Selleck Chemical | Cat# S1116 |
| Deferoxamine mesylate | Cayman Chemical | Cat# 14595, CAS: 138–14-7 |
| C11 BODIPY 581/591 (4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3- undecanoic acid | Molecular Probes | Cat# D3861 |
| Methanol | Sigma-Aldrich | Cat# 34860, CAS: 67–56-1 |
| Hoechst 33258 | Thermo Fisher Scientific | Cat# H3569 |
| ML162 | Custom synthesis | |
| Pepstatin A | Sigma-Aldrich | Cat# P5318 |
| Leupeptin Hydrochloride | Sigma-Aldrich | Cat# L9783 |
| E-64 | Sigma-Aldrich | Cat# E3132 |
| Chloroquine Diphosphate | Sigma-Aldrich | Cat# C6628 |
| DQ-BSA | Invitrogen | Cat# D1205 |
| Ovalbumin | A.G. Scientific | Cat# O-2577 |
| Casein | EMD Millipore | Cat# 218680 |
| Glutathione quantification assay kit | Cayman Chemical | Cat# 703002 |
| Buthionine sulfoxamine | Thermo Fisher Scientific | Cat# AC23552–001 |
| Nutlin-3 | Selleck Chemical | Cat# S1061 |
| CA074-Me | Gift from Matthew Bogyo, Stanford University School of Medicine | |
| R11-OEt | Gift from Matthew Bogyo, Stanford University School of Medicine | |
| LysoTracker Red | Invitrogen | Cat# L7528 |
| TMR-Dextran | Invitrogen | Cat# D12050 |
| γ-globulin | Fisher scientific | Cat# ICN19147805 |
| Deposited Data | ||
| Unprocessed western blots | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |
| Experimental Models: Cell Lines | ||
| Human: HT-1080 | ATCC | Cat# CCL-121, RRID:CVCL_0317 |
| Human: HT-1080N | Forcina et al, 2017 | N/A |
| Human: H1299N | Tarangelo et al, 2018 | N/A |
| Human: U-2 OSN | Forcina et al, 2017 | N/A |
| Human: H23Cas9,N | Inde et al, 2021 | N/A |
| Human: A375N | Conlon et al, 2021 | N/A |
| Human: T98GN | Forcina et al, 2017 | N/A |
| Human: PaTu 8988T | Gift from Rushika Perera, University of California San Francisco | N/A |
| Human: PaTu 8988 ATG7 KO1 | This paper | N/A |
| Human: PaTu 8988 ATG7 KO2 | This paper | N/A |
| Human: PaTu 8988 ATG7 KO3 | This paper | N/A |
| Human: MIA PaCa2 | Gift from Rushika Perera, University of California San Francisco | N/A |
| Human: PaTu 8988 CTSB KO1 | This paper | N/A |
| Human: PaTu 8988 CTSB KO2 | This paper | N/A |
| Human: PaTu 8988 CTSD KO1 | This paper | N/A |
| Human: PaTu 8988 CTSD KO2 | This paper | N/A |
| Human: PaTu 8988 CTSL KO1 | This paper | N/A |
| Human: PaTu 8988 CTSL KO2 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| See Table S1. | ||
| Recombinant DNA | ||
| IncuCyte NucLight Red Lentivirus Reagent (EF-1 α, Puro) | Essen BioScience | Cat# 4625 |
| shScramble | Addgene | Cat# 1864 |
| shRPTOR | Addgene | Cat# 1858 |
| shRICTOR | Addgene | Cat# 1853 |
| Software and Algorithms | ||
| GraphPad Prism 9.0.1 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| Microsoft Excel 16.45 | Microsoft Corporation | N/A |
| ImageJ 1.52q | Schneider et al, 2012 | https://imagej.nih.gov/ij |
| R | https://www.r-project.org/ | Version R-4.0.5 |
| RStudio | https://www.rstudio.com/products/rstudio/download/ | Version 1.4.1106 |
| Other | ||
| ImageJ DQ-BSA processing script | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |
| R program for protein cysteine abundance calculation | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell lines and culture conditions
Most cell lines were obtained originally from primary vendors, then immediately expanded and frozen in multiple aliquots. Cell lines were validated based on known morphology, growth rates, and ferroptosis sensitivity. Low passage cells (< 30 passages) were used for all experiments. HT-1080 cells (sex: male) were obtained from ATCC (Cat# CCL-121, Manassas, VA). The polyclonal nuclear localized mKate2-expressing HT-1080 (HT-1080N), A375N (sex: male), H1299N (sex: male), T98GN (sex: male), H23Cas9,N (sex: male), and U-2 OSN (sex: female) cell lines were described previously (Conlon et al., 2021; Forcina et al., 2017; Inde et al., 2021; Tarangelo et al., 2018). PaTu 8988T (sex: female) and MIA PaCa2 (sex: male) cells were the kind gift of Rushika Perera (UCSF). HT-1080, HT-1080N, A375N, H1299N, T98GN, PaTu 8988T and MIA PaCa2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Cat# MT-10–013-CV, Thermo Fisher Scientific). U-2 OSN cells were cultured in McCoy’s 5A medium (Cat# MT10050CV, Thermo Fisher Scientific). H23Cas9,N cells were cultured in RPMI 1640 medium (Cat# SH30027FS, Thermo Fisher Scientific). All growth media were supplemented with 10% fetal bovine serum (FBS, Cat# 26140–079, Gibco) and 0.5 U/mL Pen/Strep (P/S, Cat# 15070–063, Gibco). HT-1080 medium was additionally supplemented with 1x non-essential amino acids (NEAAs, Cat# 11140–050, Gibco). HBSS (Cat# 14025134). During routine passaging and cell seeding prior to experiments, cells were trypsinized and counted with a Cellometer Auto T4 cell counter (Nexelcom). Trypsin (Cat# 25200072) was from Gibco and PBS (Cat# 97062–338) from VWR.
GS187 gliomasoheres (sex: female) were derived from a sample isolated from a 59-year-old, newly diagnosed patient with no previous drug treatment through the UCLA Institutional Review Board (IRB) protocol 10–000655. The patient presented at clinic with headaches and neurologic complaints, MRI revealed masses in right temporal lobe and hippocampus, mass effect resulting in right uncal hemiation and 6 mm leftward shift of the septum pellucidum. The health and immune status of the patient is unknown. As previously described (Mai et al., 2017), gliomaspheres were cultured in DMEM/F-12 (Cat# 10–092-CV, Corning) supplemented with 1x B-27 supplement (Cat# 12587010, Thermo Fisher), 1x P/S, 1x GlutaMax (Cat# 35050–061, Life Technologies), heparin (5 µg/mL, Cat# H3149, Sigma), EGF (50 ng/mL, Cat# PHG0313, Fisher), and FGF-b (20 ng/mL, Cat# PHG0263, Fisher) added to the medium every 5 days and/or upon passaging. Gliomaspheres were passaged every 4–7 days into fresh media at 75,000–100,000 cells/mL following collection, centrifugation, and dissociation with TrypLE (Cat# 12605028, Thermo). One gliomasphere cell line was used for all experiments and individual cells from that cell line were randomly allocated to control or treatment groups. As is standard in cell biology, experiments were performed three times on separate days. All cell lines were grown at 37°C with 5% CO2 in humidified tissue culture incubators (Thermo Scientific).
METHOD DETAILS
Chemicals
The structure of erastin2 (compound 35MEW28) was described (Dixon et al., 2014) and was synthesized by Acme (Palo Alto, CA). ML162 and CIL56 were synthesized by Acme. SYTOX Green (Cat# S7020) was from Life Technologies. INK128 (Cat# S2811), vinblastine (Cat# S1248), palbociclib (Cat# S1116), and nutlin-3 (Cat# S1061) were from Selleck Chemicals. Bovine serum albumin (BSA, Cat# 700–100P) was from Gemini Bio-Products or Sigma Aldrich (Cat# A3294). Torin 1 (Cat# 10997) was from Cayman Chemical. Rapamycin (Cat# BP2963–1), bortezomib (Cat# NC0587961), camptothecin (Cat# AC276721000) and Q-VD-OPh (Cat# OPH00101M) were from Fisher Scientific. BSO (Cat# AC23552–0010) was from Thermo Fisher Scientific. Staurosporine (Cat# S6942), ferrostatin-1 (Cat# SML0583), chloroquine (Cat# C6628), pepstatin A (Cat# P5318), leupeptin (Cat# L9783), and E-64 (Cat# E3132) were from Sigma-Aldrich. Ovalbumin (Cat# O-2577) was from A.G. Scientific. Casein (Cat# 218680) was from EMD Millipore. DQ-BSA was from Invitrogen (Cat# D12050). Deferoxamine mesylate (DFO, Cat# 14595) was from Cayman Chemical. FIN56 was a gift from Rachid Skouta, and CA074-Me and R11-OEt were kind gifts from Matthew Bogyo. C11 BODIPY 581/591 was dissolved in anhydrous methanol and all other chemical stocks were dissolved in DMSO except chloroquine, leupeptin, and E-64, which were dissolved in H2O, and BSO, which was dissolved directly into media. All chemicals were stored at −20°C until use. DFO was warmed at 37°C immediately before use.
Amino acid deprivation and protein supplementation media
To make cystine-free DMEM, 200 µM L-methionine and 4 mM L-glutamine were added to methionine, glutamine, and cystine-free DMEM (Cat# 17–204-CL, Corning, or Cat# 21013024, Thermo Fisher Scientific), to generate ‘-cystine DMEM’. This medium was supplemented with 10% dialyzed fetal bovine serum (dFBS, Cat# 26400044, Thermo Fisher Scientific) and 0.5 U/mL P/S. For experiments using HT-1080 cells, 1x NEAAs were added. For most albumin supplementation experiments, BSA was added to DMEM or -cystine DMEM at a concentration of 3% w/v (unless otherwise indicated) and sterile filtered. Ovalbumin and casein were added to DMEM or -cystine DMEM in the same manner as BSA. For leucine starvation experiments, cells were plated in DMEM for 24 h. The following day DMEM was removed and cells were rinsed twice with PBS. Cells were then cultured in serum-free RPMI 1640 medium (US Biological life science, Cat# R8999–03A) deficient for leucine. Chemical inhibitors were added along with changing DMEM medium to serum-free and leucine-free RPMI 1640 medium. Two h later, leucine (50 mg/ml), 5% double-dialyzed BSA (Sigma Aldrich, Cat# A3294), or 3% double-dialyzed bovine γ-globulin (Fisher scientific, Cat# ICN19147805) were added for 4 h.
Cell death and proliferation tracking
Cell death was assessed at multiple time points using scalable time-lapse analysis of cell death kinetics (STACK) (Forcina et al., 2017). 5,000 HT-1080N cells/well were plated the day before the experiment into 96-well plates (Cat# 07–200-588, Corning; or Cat# 07–200-91, Thermo). For 24 h pre-treatments, 2,500 cells/well were seeded instead. For cell panel experiments, cells were seeded in their native medium. The next day, the media in each well was removed and replaced with 200 µL of media for the experiment, all containing SYTOX green (20 nM). For cell panel experiments, all cells were treated with DMEM-based media. For experiments involving amino acid deprivation, cells were first washed once with HBSS or PBS. Counts of live (mKate2 positive, mKate2+) and dead (SYTOX Green positive, SG+) cells were then counted every 2, 4, or 24 h using an Essen Incucyte Zoom Live-Cell Analysis System. SG+ counts are always presented as the maximum observed at any given timepoint during the experiment. For cell proliferation data, only mKate2+ counts were used. The following image extraction parameter values were used to count all cell lines except PaTu 8988T: for SG+ objects: Adaptive threshold adjustment 10; Edge split on; Edge sensitivity −5; Filter area min 0 µm2, max 750 µm2; Eccentricity max 0.9; for mKate2+ objects (in Nuc::mKate2 expressing cells): Adaptive threshold adjustment 1.0; Edge split on; Edge sensitivity 50; Filter area min 20 μm2, maximum 8100 μm2; Eccentricity max 1.0; and for Overlap objects: Filter area min 20 μm2, maximum 5000 μm2. For PaTu 8988T cells and their derivatives, SG+ objects were counted using these parameters: Adaptive threshold adjustment 3.0; Edge split on; Edge sensitivity 0; Filter area min 15 µm2, max 500 µm2.
Glutathione quantification via Ellman’s reagent
The day before samples were collected, 200,000 HT-1080N or PaTu 8988T cells were seeded into 6-well plates (Cat# 07–200-83, Corning). The next day, cells were washed with HBSS or PBS and treated in the conditions shown. Where applicable, HT-1080N cells were imaged in an Incucyte to determine cell number in each well. Then, cells were harvested on ice by scraping. Samples were then prepared for assessment of total intracellular glutathione (GSH+GSSG) using a glutathione quantification assay kit based on Ellman’s reagent according mostly to the manufacturer’s instructions (Cat# 703002, Cayman Chemical). To concentrate samples, cells were harvested into 250 µL MES buffer instead of 500 µL and the rest of the protocol was scaled down accordingly. When normalizing to protein content, a 30 µL aliquot was taken before deproteination to assess protein levels using the BCA assay and the volumes for the rest of the assay were adjusted accordingly. Glutathione concentrations were calculated using a glutathione standard curve and normalized to cell count (HT-1080N) or protein content (PaTu 8988T) in each sample.
Glutathione quantification via mass spectrometry
The day before the experiment, 200,000 PaTu 8988T NTC or CTSBKO cells were seeded into each well of 6-well plates. The next day, cells were washed once with PBS and treated in duplicate with 1 mL of the designated media and left for 24 h. After treatment, cells were washed on ice once in 1.5 mL/well with ice-cold 0.9% NaCl. For one duplicate, the saline was then removed, and the plates were moved to dry ice. 1 mL 80% methanol with internal standards was added to each well, cells were scraped thoroughly, and the solutions transferred to pre-chilled 1.5 mL microcentrifuge tubes. These tubes were then vortexed for 10 min at 4°C. Meanwhile, the other duplicates were harvested for BCA quantification for normalization as detailed above for western blotting. After vortexing, tubes were spun at 18,200 x g for 10 min at 4°C, and the supernatant transferred to new, pre-chilled microcentrifuge tubes and stored at −80°C until quantification via mass spectrometry as described (Abu-Remaileh et al., 2017).
C11 BODIPY 581/591 imaging
For imaging on the confocal microscope, the day before the experiment, 100,000 cells/well were seeded into 6-well plates onto 22 mm #1.5 coverslips. The next day, the medium was removed, and cells were washed once with HBSS and treated with the given media. After 10 h, the treatment media were removed, and cells were washed once with HBSS. Then, C11 BODIPY 581/591 (5 μM, Thermo Fisher Scientific, Cat# D3861) and Hoechst 33258 (1 μg/mL, Thermo Fisher Scientific, Cat# H3569) were dissolved in HBSS and 1 mL was added to each sample. Cells were incubated at 37°C for 10 min. After 10 min, the labeling mixture was aspirated off and replaced with 1 mL fresh HBSS. The coverslip was removed from each well and inverted onto a glass microscope slide with 25 μL of fresh HBSS. Imaging was performed using a Zeiss Axio Observer microscope with a confocal spinning-disk head (Yokogawa), PlanApoChromat 63x/1.4 NA oil immersion objective, and a Cascade II:512 electron-multiplying (EM) CCD camera (Photometrics). Imaging was performed on 3 independent biological replicates per treatment. Images were processed in ImageJ 2.0.0.
Other C11 imaging was conducted using a Lionheart FX automated microscope (BioTek). First, 7,500 (HT-1080) or 10,000 (PaTu 8988T) cells were seeded into each well of 96-well plates and treated the following day with designated conditions and labeled with C11 and Hoechst as described above but with PBS instead of HBSS. After labeling, fresh 1x PBS was applied to the cells. Then, the plate was immediately transferred to the Lionheart FX instrument. The plate was placed on a 37°C humidified stage, covered, and imaged in an automated mode with the following specifications: 20x, NA: 0.45 air objective; 3×3 montage at the center of each well; image acquisition using DAPI (filter cube 1225100/LED cube 1225000), GFP (12251010/1225001) and Texas Red (1225102/1225002). Images were analyzed using Gen5 v3.10 (BioTek).
DQ-BSA imaging
For live imaging on the Lionheart FX instrument, the day before the experiment, 70,000 HT-1080N cells/well were seeded into 12-well plates. The next day, cells were pre-treated with DMSO, chloroquine, or protease inhibitors in HT-1080 medium for 1 h. Then, the media were changed to include 0.1 mg/mL DQ-BSA with the same inhibitors or vehicle and incubated at 37°C for 2 h. Next, media were changed to fresh media, still with the same inhibitors or vehicle for 1 h. Finally, the plate was imaged with the Lionheart FX instrument as for C11 imaging above, but with the following specifications: 40x Plan Fluorite, NA: s0.6; 2×2 montage in at least two separate locations per well; image acquisition using GFP (DQ-BSA) and Texas Red (nuclei). Raw (stitched) .tif files were then imported to ImageJ and analyzed with a macro available upon request.
For fixed-cell imaging, cells were seeded on glass coverslips in complete DMEM for 24 h. Cells were then starved of leucine for 2 h in serum-free DMEM in the presence of vehicle or indicated inhibitor. Cells were then incubated in serum-free medium supplemented with 0.5 mg/ml TMR-Dextran (Invitrogen, Cat# D12050) or 0.1 mg/ml DQ-BSA for 4 h. Subsequently, cells were washed 3 times with PBS and fixed with 4% formaldehyde for 15 min. After fixation, cells were washed 3 times with PBS and mounted using a mounting medium with DAPI. When LysoTracker Red dye was used, 50 nM LysoTracker Red (Invitrogen, Cat# L7528) was added 1 h prior to fixation. Images were acquired on a Zeiss AxioVert200M microscope with a 63X oil immersion objective. The MetaMorph software package (Molecular Devices) was used to control the hardware and image acquisition. The excitation lasers used to capture the images were 488 nm and 561 nm. Images were processed with FIJI. Fluorescently labeled dextran or albumin was quantified using the mean fluorescence intensity determined by calculating the integrated signal from randomly chosen fields and normalized to the cell area.
Western blotting
For most western blots, the day before the experiment, 30,000 (72 h treatment) - 200,000 cells/well were seeded into 6-well plates (Cat# 07–200-83, Costar). The next day, if applicable, cells were treated as shown. For ATF4 time-course blots, 50,000 (48 h +cys, 10 h treatments), 100,000 (24 h), and 200,000 (48 h -cys treatments) cells were seeded and then treated to harvest all together. After treatment, the media were removed, and cells were washed twice on ice with 2 mL cold 1x PBS. Next, 40 µL cold RIPA + 0.1% SDS buffer with 5 mM NaF (Cat# S6776, Sigma-Aldrich) and 1:200 protease inhibitor cocktail P8340 (Cat# P8340, Sigma-Aldrich) was added and cells were scraped to harvest. Lysates were transferred to a 1.5 mL microcentrifuge tube and left on ice for 1 h to ensure full lysis, then stored at −80°C until needed or sonicated [1 s on, 1 s off, 60% amplitude) x 10 cycles, Fisher Scientific Model 120 Sonic Dismembrator (Thermo Fisher)] and spun down at 4°C (18,200 x g, 20 min). Cleared lysates were transferred to a new 1.5 mL tube and quantified using a BCA protein assay (Cat# 23252, Thermo Fisher Scientific) with a BSA standard curve. Equal amounts of protein were combined with 4x Bolt LDS Sample Buffer (Cat# B0007) and 10x Bolt Sample Reducing Agent (Cat# B0009) (Life Technologies), heated to 70 °C for 10 min and run on a Bolt 4–12% Bis-Tris Plus Gel (Cat# NW04120BOX, Life Technologies). Protein was then transferred to a nitrocellulose membrane using an iBlot2 transfer stack (Cat# IB23001/2, Life Technologies), which was then blocked for 1 h at room temperature using Odyssey Blocking Buffer (Cat# 927–50000, LI-COR Biotechnology) or Intercept Blocking Buffer (Cat# 927–70001, LI-COR Biotechnology), and incubated in primary antibody mixture overnight at 4 °C. Primary antibodies used were α-tubulin (Cat# MS581P1, Fisher Scientific, 1:10,000 dilution or, rarely, 1:2,000 dilution), α-TFRC (Cat# 13–6800, Thermo Fisher Scientific, 1:1000), α-β-Actin (Santa Cruz Biotechnology, Cat# sc-47778, 1:1000), α-p53 (Santa Cruz Biotechnology, Cat# sc-126, 1:200), α-Rb (BD Biosciences, Cat# 554136, 1:1000), α-Raptor (Millipore Sigma, Cat# 09–21709-217), α-FSP1 (Cat# 20886–1-AP, Proteintech), α-GPX4 (Cat# ab125066, Abcam), α-CTSL (Cat# ab200738, Abcam), α-phospho-S6K1 (Cat# 9234), α-S6K1 (Cat# 2708), α-phospho-RPS6 (Cat# 4858S, 1:1000), α-RPS6 (Cat# 2217S, 1:1000), α-phospho-4E-BP1 (Thr 37/46, Cat# 2855T, 1:1000), α−4E-BP1 (Cat# 9644S, 1:1000), α-phospho-Rb (Cat# 8516S, 1:250), α-phospho-Akt (Cat# 4056S, 1:1000), α-Akt (Cat# 9272S, 1:1000), α-ULK1 (Cat# 6439), α-CTSB (Cat# 31718S, 1:1000), α-CTSD (Cat# 2284), α-ATF-4 (Cat# 11815S, 1:1000), α-IRP2 (Cat# 37135S, 1:1000), α-ATG7 (Cat# 2631S, 1:1000), α-GAPDH (Cat# 2118S, 1:1000), or α-LC3B (Cat# 2775S, 1:1000) (Cell Signaling Technologies). The membrane was washed 3x for seven min each in TBST and then incubated in secondary antibody mixture (55 min, room temperature, 1:1 TBST and Odyssey or Intercept blocking buffer). Secondary antibodies used were donkey α-rabbit (Cat# 926–68023/32213, LI-COR Biotechnology, 1:15,000 dilution) and donkey α-mouse (Cat# 926–68022/926–32212, LI-COR Biotechnology, 1:15,000 dilution). The membrane was then washed 3x for seven min each in TBST and scanned on an Odyssey CLx Imaging System (LI-COR). Western blots were performed on three independent biological replicates unless otherwise indicated.
Western blots in Figure 4B and Fig S5C-D were conducted as follows. The day before the experiment, 400,000–500,000 PaTu 8988T or 1,000,000–1,200,000 MIA PaCa2 cells were seeded into each well of 6-well plates. The next day, cells were starved and treated as described. Cell lysates were prepared in ice-cold lysis buffer (40 mM HEPES pH 7.4,1% Triton X-100, 10 mM β-glycerol phosphate, 10 mM pyrophosphate, 2.5 mM MgCl2 with Complete EDTA-free Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail (Roche)). The soluble fractions from lysates were collected by centrifugation at 17,000 x g for 10 min in a cold centrifuge. Lysates were then quantified using a BCA protein assay and equal amounts of protein were resolved by 8–16% SDS-PAGE at 120 V. Resolved proteins were transferred for 2 h at 45 V to ethanol-pretreated PVDF membranes to be further analyzed by immunoblotting. Membranes were blocked with 5% nonfat dry milk prepared in TBST (Tris-buffered saline with Tween 20) for 1 h, then incubated overnight with primary antibodies in 5% bovine serum albumin (BSA) in TBST at 4°C. All primary antibodies were used at (1:500–1:1000) dilution. Following incubation, membranes were washed 3x for 5 min each with TBST and then incubated with the appropriate secondary antibodies diluted 1:3000 in 5% milk for 1 h at room temperature. Membranes were then washed three times with TBST before being visualized using ECL western blotting substrate.
Virus generation and use
To generate lentiviruses bearing shRNAs, plasmids encoding scramble, RPTOR, or RICTOR shRNAs (which were gifts from David Sabatini, Addgene plasmids #1864, #1858, and #1853, respectively) were co-transfected with 3rd generation lentiviral packaging plasmids (pMDLg/pRRE and pRSV-Rev, which were gifts from Didier Trono, Addgene plasmids #12251 and #12253 respectively, and pCMV-VSV-G, which was a gift from Bob Weinberg, Addgene plasmid #8454) into HEK293T cells using PolyJet (SignaGen Laboratories, Cat# SL100688) as per manufacturer instructions. Viral supernatant was harvested 48 and 72 h later, combined, filtered through a 0.45 μm PVDF filter (EMD Millipore, Cat# SLHV033RS), and stored in single-use aliquots at −80°C until use. For CTSB, CTSD, and CTSL disruption, lentiviruses were produced by co-transfecting HEK-293T cells with pLentiCRISPRv1 (with sgRNA cloned) alongside the packaging plasmids VSV-G envelope and ΔVPR. The following sense (S) and antisense (AS) oligo-nucleotides were cloned into pLentiCRISPRv1 (see Table S1 for oligonucleotide sequences). For adding back CTSB expression, retroviruses were produced by co-transfecting HEK-293T cells with pMXs-CTSB plasmid and retroviral packaging plasmids Gag-Pol and VSV-G using XtremeGene9 transfection reagent. The culture medium was changed to DMEM supplemented with 30% inactivated fetal calf serum 16 h post transfection. The virus-containing supernatant was collected 48 h post transfection and spun for 5 min at 400 x g to remove cells and then frozen at −80°C. For re-introducing CTSB, cells were seeded at a density of 2 ×106 cells/mL in DMEM containing 8 μg/mL polybrene (EMD Millipore), and then transduced with retrovirus by centrifugation at 2,200 RPM for 45 min at 37°C. After a 16–18 h incubation, cells were re-seeded into fresh medium containing blasticidin (InvivoGen, Cat# ant-bl-10p) and selected for 72 h. Stable cell lines were confirmed by the expression of CTSB by western blot.
mTORC1 and mTORC2 shRNA-mediated gene silencing
Knockdowns were performed as described (Conlon et al., 2021). Briefly, HT-1080 (immunoblot) or HT-1080N (viability) cells were seeded in either 6-well plates (immunoblot, at 30,000 cells/well) or 96-well plates (viability, at 1,000 cells/well). The next day, medium was removed and replaced with HT-1080 medium with 8 µg/mL polybrene (Sigma-Aldrich, Cat# H9268) and vehicle control or viral supernatant at an MOI of ~3. 48 h later, the media were removed and replaced with HT-1080 medium containing 2 µg/mL puromycin (Life Technologies, Cat# A11138–03). The un-transduced wells were instead treated with 1 µM INK128. 24 h later, cells in 6-well plates were harvested for immunoblots as described above, and cells in 96-well plates were washed and treated as described above for viability experiments.
CRISPR/Cas9 genome editing
HT-1080 CTNS gene disrupted cell lines were generated with the sgRNA sequence GCCAGCCTACCCGGTCTGAT. First, two oligos (see Table S1) were annealed at a concentration of 10 μM at 37°C for 30 min, followed by 95°C for 5 min, and cooled to 25°C at a rate of 5°C per min. The oligo duplex was then ligated into the plasmid pSD224 and transformed into DH5α. Plasmid DNA was extracted with a QIAGEN spin column (Cat# 27106, QIAGEN) and validated by DNA sequencing. For transfection, 150,000 HT-1080 cells were seeded into each of two wells of a 6-well plate. The next day, 1 µg plasmid (or water for mock) was mixed with 2 µL lipofectamine LTX (Life Technologies Cat# 15338–100), incubated for 15 min, and added dropwise onto cells. The next day, transfection medium was removed and replaced with fresh medium. After a 24 h recovery, GFP+ cells were single cell-sorted using a BD FACSJazz Cell Sorter (BD Biosciences) (Stanford Shared FACS Facility) into a 96-well plate containing DMEM + 30% FBS, 1x NEAAs and 0.5 U/mL P/S. Cells were incubated at 37°C until they grew into colonies. Individual colonies were expanded. DNA was harvested using the NucleoSpin Tissue kit (Clontech, Cat# 740952.250) according to manufacturer instructions and sent for sequencing. KO clones were confirmed to have homozygous frameshift mutations at the sgRNA site. For transduction to disrupt CTSB, CTSD, and CTSL, cells were seeded at a density of 1.5 ×106 cells/mL in DMEM containing 8 μg/mL polybrene (EMD Millipore), and then transduced with lentivirus by centrifugation at 2,200 RPM for 45 min at 37°C. Cells were incubated for 16–18 h, then selected with puromycin for 72 h. After transduction, cells were single-cell FACS-sorted into 96-well plates. Knockout clones were identified by immunoblotting. Control cells were generated by targeting the AAVS1 locus as described (Wang et al., 2015).
Spheroid cell culture and experiments
Cells to make spheroids were seeded in 100 µL at 10,000 cells/well in 96-well ultra-low adhesion plates (PerkinElmer, Cat #6055330) and left to establish for 72 h. Daily progress was monitored using an Incucyte. Then, 100 µL containing 2x treatment concentrations was carefully added to each well to start the experiment. Daily images were taken with an Incucyte. For spheroid western blots, multiple wells were prepared for each condition, and 20,000–30,000 cells were seeded in parallel in wells of 6-well plates to generate monolayer lysates. 72 h later, cells were treated ± 1 µM INK128. 24 h later, cells in monolayer were trypsinized, quenched, transferred to 15 mL conical tubes, and put on ice. Spheroids were collected by pipetting media including spheroids, pooling wells by condition, and transferring to 15 mL conical tubes. Cells were pelleted by centrifuge at 1000 x g, then washed with 10 mL ice-cold PBS, pelleted, washed again, then pelleted again. PBS was removed and 20–100 µL RIPA buffer with NaF and P8340 was added to each sample. Samples were then immediately sonicated, cleared, and subsequently processed as detailed above. Spheroid viability was ascertained using the CellTiter-Glo assay (Cat# G7572, Promega) according to manufacturer instructions. 100 μL medium was removed from each well and replaced with the mixed CellTiter-Glo assay reagent. Luminescence was counted using a Cytation3 plate reader (BioTek Instruments), background-subtracted using empty wells, and normalized to the DMSO control for each condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Lethal fraction calculations were performed using Microsoft Excel (Microsoft Corporation, Redmond, WA) as described (Forcina et al., 2017) with one modification to account for red/green double positive objects, i.e. dead cells still transiently expressing detectable nuclear mKate2 signal (Inde et al., 2021): the lethal fraction at a given time was calculated as: 1 - ((mKate2+ - (mKate2/SG+))/(SG+ + (mKate2+ - (mKate2+/SG+))). Densitometry on western blots was performed using ImageJ. Lionheart FX image analysis was performed using ImageJ or Gen5 v3.10 software (BioTek). Serum proteome abundance data (Geyer et al., 2016) and cysteine percent calculations were performed in RStudio, version 1.4.1106 running R version R-4.0.5. R code available upon request. Plotting was performed using GraphPad Prism 8.4/9.0 (GraphPad Software, La Jolla, CA). Figures were assembled using Adobe Illustrator 25.3.1 (Adobe Systems, San Jose, CA).
Supplementary Material
Highlights:
Extracellular albumin can block ferroptosis caused by cystine deprivation
Albumin is cysteine-rich and when catabolized can replenish GSH stores
Lysosomal albumin catabolism requires cathepsin B (CTSB)
Extracellular albumin suppresses ferroptosis in spheroid cell models
SIGNIFICANCE.
Whether disruption of cystine import can be exploited therapeutically for cancer treatment remains somewhat unclear. Numerous cancers overexpress SLC7A11 (Koppula et al., 2021), and drug-like small molecule system xc- inhibitors and extracellular cystine-degrading enzymes (e.g., cyst(e)inase) can slow tumor growth in vivo, but do not appear to cause obvious tumor regression (Cramer et al., 2017; Zhang et al., 2019). Our results, together with other recent findings (Byun et al., 2022), suggest that the uptake and catabolism of albumin from the extracellular environment could compensate for limited extracellular free cystine to inhibit ferroptosis in vivo. Cells growing in detached conditions have reduced levels of mTOR activity (Barbone et al., 2008; Riedl et al., 2017), which could favor lysosomal protein catabolism and enhance the albumin protective mechanism in vivo. Our results suggest that the induction of ferroptosis in vivo by cystine deprivation regimes may be augmented by concomitant inhibition of lysosome-mediated protein catabolism.
ACKNOWLEDGEMENTS
We thank R. Skouta, Z. Inde, I. Ulengin-Talkish, I. Foe, C. Gottlieb, K.C. Farrell, T. Stearns, M. Bogyo, and R. Perera, for equipment, reagents, and help with experiments, K. Galenkamp and C. Commisso for advice, and L. Pope and L. Magtanong for comments on the manuscript. This work was supported by the NIH (T32GM007276 to D.A.A., DP2-CA271386 to M.A.-R., and 1R01GM122923 to S.J.D.) and by the American Cancer Society (RSG-21–017-01-CCG to S.J.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
D.A.N. is a co-founder of Trethera Corporation and Katmai Pharmaceuticals and has equity in those companies and in Sofie Biosciences. M.A.-R. is a scientific advisory board member of Lycia Therapeutics. S.J.D. is a co-founder of Prothegen Inc., a member of the scientific advisory board for Ferro Therapeutics and Hillstream BioPharma, and an inventor on patents related to ferroptosis.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and one table can be found with this article online.
REFERENCES
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbone D, Yang TM, Morgan JR, Gaudino G, and Broaddus VC (2008). Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J Biol Chem 283, 13021–13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolacci C, Andreani C, Vale G, Berto S, Melegari M, Crouch AC, Baluya DL, Kemble G, Hodges K, Starrett J, et al. (2022). Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat Commun 13, 4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun JK, Lee S, Kang GW, Lee YR, Park SY, Song IS, Yun JW, Lee J, Choi YK, and Park KG (2022). Macropinocytosis is an alternative pathway of cysteine acquisition and mitigates sorafenib-induced ferroptosis in hepatocellular carcinoma. J Exp Clin Cancer Res 41, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin AM, Paquette B, Richter M, and Larivee P (2000). Albumin-mediated regulation of cellular glutathione and nuclear factor kappa B activation. Am J Respir Crit Care Med 162, 1539–1546. [DOI] [PubMed] [Google Scholar]
- Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, and Alvarez B (2003). Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry 42, 9906–9914. [DOI] [PubMed] [Google Scholar]
- Choi S, Choi EY, Kim DJ, Kim JH, Kim TS, and Oh SW (2004). A rapid, simple measurement of human albumin in whole blood using a fluorescence immunoassay (I). Clin Chim Acta 339, 147–156. [DOI] [PubMed] [Google Scholar]
- Combs JA, and DeNicola GM (2019). The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, Wells A, Kahanu A, Magtanong L, Watts JL, et al. (2021). A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol [DOI] [PMC free article] [PubMed]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al. (2017). Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 23, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. (2017). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Eagle H (1959). Amino acid metabolism in mammalian cell cultures. Science 130, 432–437. [DOI] [PubMed] [Google Scholar]
- Eagle H, Oyama VI, and Piez KA (1960). The reversible binding of half-cystine residues to serum protein, and its bearing on the cystine requirement of cultured mammalian cells. J Biol Chem 235, 1719–1726. [PubMed] [Google Scholar]
- Forcina GC, Conlon M, Wells A, Cao JY, and Dixon SJ (2017). Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells. Cell Syst 4, 600–610 e606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich J, Seidel C, Ebner R, and Kunz-Schughart LA (2009). Spheroid-based drug screen: considerations and practical approach. Nat Protoc 4, 309–324. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Thoene JG, and Schneider JA (2002). Cystinosis. N Engl J Med 347, 111–121. [DOI] [PubMed] [Google Scholar]
- Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, and Mann M (2016). Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst 2, 185–195. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inde Z, Rodencal J, and Dixon SJ (2021). Quantification of drug-induced fractional killing using high-throughput microscopy. STAR Protoc 2, 100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. [DOI] [PubMed] [Google Scholar]
- Jayashankar V, and Edinger AL (2020). Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat Commun 11, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol [DOI] [PMC free article] [PubMed]
- Jouandin P, Marelja Z, Shih YH, Parkhitko AA, Dambowsky M, Asara JM, Nemazanyy I, Dibble CC, Simons M, and Perrimon N (2022). Lysosomal cystine mobilization shapes the response of TORC1 and tissue growth to fasting. Science 375, eabc4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyser JW, Fifield R, and Watkins GL (1981). Standardization of immunochemical determinations of serum albumin. Clin Chem 27, 736–738. [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. (2018). PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov 8, 866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, Shi J, Liu X, Horbath A, Das M, et al. (2022). A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun 13, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P, Zhuang L, and Gan B (2021). Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Murphy ME, and George DL (2019). Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai WX, Gosa L, Daniels VW, Ta L, Tsang JE, Higgins B, Gilmore WB, Bayley NA, Harati MD, Lee JT, et al. (2017). Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat Med 23, 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira W, Daher B, Parks SK, Cormerais Y, Durivault J, Tambutte E, Pouyssegur J, and Vucetic M (2021). A Cystine-Cysteine Intercellular Shuttle Prevents Ferroptosis in xCT(KO) Pancreatic Ductal Adenocarcinoma Cells. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzil J, Gebicki JM, and Stocker R (1993). Radical-induced chain oxidation of proteins and its inhibition by chain-breaking antioxidants. Biochem J 293 (Pt 3), 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofal M, Wang T, Yang L, Jankowski CSR, Hsin-Jung Li S, Han S, Parsons L, Frese AN, Gitai Z, Anthony TG, et al. (2021). GCN2 adapts protein synthesis to scavenging-dependent growth. Cell Syst [DOI] [PMC free article] [PubMed]
- Nofal M, Zhang K, Han S, and Rabinowitz JD (2017). mTOR Inhibition Restores Amino Acid Balance in Cells Dependent on Catabolism of Extracellular Protein. Mol Cell 67, 936–946 e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W (2019). Metabolic functions of macropinocytosis. Philos Trans R Soc Lond B Biol Sci 374, 20180285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Reyna-Neyra A, Philippe L, and Thoreen CC (2017). mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep 19, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorack CD, and Dixon SJ (2021). Understanding the role of cysteine in ferroptosis: progress & paradoxes. FEBS J [DOI] [PMC free article] [PubMed]
- Poole B, and Ohkuma S (1981). Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 90, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratto E, Chowdhury SR, Siefert NS, Schneider M, Wittmann M, Helm D, and Palm W (2022). Direct control of lysosomal catabolic activity by mTORC1 through regulation of V-ATPase assembly. Nat Commun 13, 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis RC, Sorgine MH, and Coelho-Sampaio T (1998). A novel methodology for the investigation of intracellular proteolytic processing in intact cells. Eur J Cell Biol 75, 192–197. [DOI] [PubMed] [Google Scholar]
- Riedl A, Schlederer M, Pudelko K, Stadler M, Walter S, Unterleuthner D, Unger C, Kramer N, Hengstschlager M, Kenner L, et al. (2017). Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J Cell Sci 130, 203–218. [DOI] [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T, et al. (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 280, 37423–37429. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, and Brugge JS (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarangelo A, Rodencal J, Kim JT, Magtanong L, Long JZ, and Dixon SJ (2022). Nucleotide biosynthesis links glutathione metabolism to ferroptosis sensitivity. Life Sci Alliance 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden WA, Schulze CJ, Herbert AS, Krause TB, Wirchnianski AA, Dye JM, Chandran K, and Bogyo M (2016). Cysteine Cathepsin Inhibitors as Anti-Ebola Agents. ACS Infect Dis 2, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortel IMN, van der Meer LT, Kilberg MS, and van Leeuwen FN (2017). Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab 28, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, and Sabatini DM (2017). mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 171, 642–654 e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Commisso C (2019). Macropinocytosis in Cancer: A Complex Signaling Network. Trends Cancer 5, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Berisa M, Schworer S, Qin W, Cross JR, and Thompson CB (2019). Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab 30, 865–876 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Uncropped western blot images reported in have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
One ImageJ script used to analyze DQ-BSA puncta and one R program used to analyze cysteine abundance in proteins have been deposited to Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| GAPDH | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Phospho-4EB-P1 (Thr37/46) | Cell Signaling Technology | Cat# 9459; RRID:AB_330985 |
| Phospho-RPS6 (Ser235/236) | Cell Signaling Technology | Cat# 4858; RRID:AB_916156 |
| RPS6 | Cell Signaling Technology | Cat# 2217; RRID:AB_331355 |
| Alpha-tubulin, clone DM1A | Fisher Scientific | Cat# MS581P1 |
| FSP1 | Proteintech | Cat# 20886–1-AP, RRID:AB_2878756 |
| GPX4 | Abcam | Cat# ab125066, RRID:AB_10973901 |
| CTSL | Abcam | Cat# ab200738 |
| TFRC | Thermo Fisher Scientific | Cat# 13–6800, RRID:AB_2533029 |
| Beta-Actin | Santa Cruz Biotechnology | Cat# sc-47778, RRID:AB_626632 |
| p53 | Santa Cruz Biotechnology | Cat# sc-126, RRID:AB_628082 |
| Rb | BD Biosciences | Cat# 554136, RRID:AB_395259 |
| Raptor | Millipore Sigma | Cat# 09–217, RRID:AB_612103 |
| Phospho-S6K1 (Thr389) | Cell Signaling Technology | Cat# 9234, RRID:AB_2269803 |
| S6K1 | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Phospho-RPS6 (Ser235/236) | Cell Signaling Technology | Cat# 4858, RRID:AB_916156 |
| RPS6 | Cell Signaling Technology | Cat# 2217, RRID:AB_331355 |
| Phospho-4E-BP1 (Thr37/46) | Cell Signaling Technology | Cat# 9459; RRID:AB_330985 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644, RRID:AB_2097841 |
| Phospho-Rb | Cell Signaling Technology | Cat# 8516, RRID:AB_11178658 |
| Phospho-Akt (Thr308) | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| Akt | Cell Signaling Technology | Cat# 9272, RRID:AB_329827 |
| ULK1 | Cell Signaling Technology | Cat# 6439, RRID:AB_11178933 |
| CTSB | Cell Signaling Technology | Cat# 31718, RRID:AB_2687580 |
| CTSD | Cell Signaling Technology | Cat# 2284, RRID:AB_10694258 |
| ATF-4 | Cell Signaling Technology | Cat# 11815, RRID:AB_2616025 |
| IRP2 | Cell Signaling Technology | Cat# 37135, RRID:AB_2799110 |
| ATG7 | Cell Signaling Technology | Cat# 2631, RRID:AB_2227783 |
| GAPDH | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| LC3B | Cell Signaling Technology | Cat# 2775, RRID:AB_915950 |
| 680LT Donkey-anti-mouse | LI-COR | Cat# 926–68022, RRID:AB_10715072 |
| 680LT Donkey-anti-rabbit | LI-COR | Cat# 926–68023, RRID:AB_10706167 |
| 800CW Donkey-anti-mouse | LI-COR | Cat# 926–32212, RRID:AB_621847 |
| 800CW Donkey-anti-rabbit | LI-COR | Cat# 926–32213, RRID:AB_621848 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYTOX Green | Molecular Probes | Cat# S7020 |
| INK128 | Selleck Chemical | Cat# S2811 |
| Bovine serum albumin | Gemini Bio-Products | Cat# 700–100P |
| Bovine serum albumin | Sigma Aldrich | Cat# A3294 |
| Dimethyl Sulfoxide | Sigma-Aldrich | Cat# 276855, CAS: 67–68-5 |
| Ferrostatin-1 | Sigma-Aldrich | Cat# SML0583, CAS: 347174–05-4 |
| Q-VD-OPh | Fisher Scientific | Cat# OPH00101M |
| Torin 1 | Cayman Chemical | Cat# 10997 |
| Rapamycin | Fisher Scientific | Cat# BP2963–1 |
| Erastin2 | Custom synthesis | N/A |
| Bortezomib | Fisher Scientific | Cat# NC0587961, CAS: 179324–69-7 |
| Camptothecin | Fisher Scientific | Cat# AC276721000, CAS: 7689–03-4 |
| CIL56 | Shimada et al, 2016 | N/A |
| FIN56 | Shimada et al, 2016 | N/A |
| Staurosporine | Sigma-Aldrich | Cat# S6942, CAS: 62996–74-1 |
| Vinblastine | Selleck Chemical | Cat# S1248, CAS: 143–67-9 |
| Palbociclib | Selleck Chemical | Cat# S1116 |
| Deferoxamine mesylate | Cayman Chemical | Cat# 14595, CAS: 138–14-7 |
| C11 BODIPY 581/591 (4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3- undecanoic acid | Molecular Probes | Cat# D3861 |
| Methanol | Sigma-Aldrich | Cat# 34860, CAS: 67–56-1 |
| Hoechst 33258 | Thermo Fisher Scientific | Cat# H3569 |
| ML162 | Custom synthesis | |
| Pepstatin A | Sigma-Aldrich | Cat# P5318 |
| Leupeptin Hydrochloride | Sigma-Aldrich | Cat# L9783 |
| E-64 | Sigma-Aldrich | Cat# E3132 |
| Chloroquine Diphosphate | Sigma-Aldrich | Cat# C6628 |
| DQ-BSA | Invitrogen | Cat# D1205 |
| Ovalbumin | A.G. Scientific | Cat# O-2577 |
| Casein | EMD Millipore | Cat# 218680 |
| Glutathione quantification assay kit | Cayman Chemical | Cat# 703002 |
| Buthionine sulfoxamine | Thermo Fisher Scientific | Cat# AC23552–001 |
| Nutlin-3 | Selleck Chemical | Cat# S1061 |
| CA074-Me | Gift from Matthew Bogyo, Stanford University School of Medicine | |
| R11-OEt | Gift from Matthew Bogyo, Stanford University School of Medicine | |
| LysoTracker Red | Invitrogen | Cat# L7528 |
| TMR-Dextran | Invitrogen | Cat# D12050 |
| γ-globulin | Fisher scientific | Cat# ICN19147805 |
| Deposited Data | ||
| Unprocessed western blots | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |
| Experimental Models: Cell Lines | ||
| Human: HT-1080 | ATCC | Cat# CCL-121, RRID:CVCL_0317 |
| Human: HT-1080N | Forcina et al, 2017 | N/A |
| Human: H1299N | Tarangelo et al, 2018 | N/A |
| Human: U-2 OSN | Forcina et al, 2017 | N/A |
| Human: H23Cas9,N | Inde et al, 2021 | N/A |
| Human: A375N | Conlon et al, 2021 | N/A |
| Human: T98GN | Forcina et al, 2017 | N/A |
| Human: PaTu 8988T | Gift from Rushika Perera, University of California San Francisco | N/A |
| Human: PaTu 8988 ATG7 KO1 | This paper | N/A |
| Human: PaTu 8988 ATG7 KO2 | This paper | N/A |
| Human: PaTu 8988 ATG7 KO3 | This paper | N/A |
| Human: MIA PaCa2 | Gift from Rushika Perera, University of California San Francisco | N/A |
| Human: PaTu 8988 CTSB KO1 | This paper | N/A |
| Human: PaTu 8988 CTSB KO2 | This paper | N/A |
| Human: PaTu 8988 CTSD KO1 | This paper | N/A |
| Human: PaTu 8988 CTSD KO2 | This paper | N/A |
| Human: PaTu 8988 CTSL KO1 | This paper | N/A |
| Human: PaTu 8988 CTSL KO2 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| See Table S1. | ||
| Recombinant DNA | ||
| IncuCyte NucLight Red Lentivirus Reagent (EF-1 α, Puro) | Essen BioScience | Cat# 4625 |
| shScramble | Addgene | Cat# 1864 |
| shRPTOR | Addgene | Cat# 1858 |
| shRICTOR | Addgene | Cat# 1853 |
| Software and Algorithms | ||
| GraphPad Prism 9.0.1 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| Microsoft Excel 16.45 | Microsoft Corporation | N/A |
| ImageJ 1.52q | Schneider et al, 2012 | https://imagej.nih.gov/ij |
| R | https://www.r-project.org/ | Version R-4.0.5 |
| RStudio | https://www.rstudio.com/products/rstudio/download/ | Version 1.4.1106 |
| Other | ||
| ImageJ DQ-BSA processing script | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |
| R program for protein cysteine abundance calculation | Dixon, Scott (2022), “Ferroptosis Inhibition by Lysosome-Dependent Catabolism of Extracellular Protein”, Mendeley Data, V1. | DOI: 10.17632/988sm4nsdx.1 |