

Abstract
NahE is a hydratase-aldolase that converts o-substituted trans-benzylidenepyruvates (H, OH, or CO2−) to benzaldehyde, salicylaldehyde, or 2-carboxybenzaldehyde, respectively, and pyruvate. The enzyme is in a bacterial degradative pathway for naphthalene, which is a toxic and persistent environmental contaminant. Sequence, crystallographic, and mutagenic analysis identified the enzyme as a member of the N-acetylneuraminate lyase (NAL) subgroup in the aldolase superfamily. As such, it has a conserved lysine (Lys183) and tyrosine (Tyr155), for Schiff base formation, as well as a GXXGE motif for binding of the pyruvoyl carboxylate group. A crystal structure of the selenomethionine derivative of NahE shows these active site elements along with nearby residues that might be involved in the mechanism and/or specificity. Mutations of five active site amino acids (Thr65, Trp128, Tyr155, Asn157, and Asn281) were constructed and kinetic parameters measured in order to assess the effect(s) on catalysis. The results show that the two Trp128 mutants (Phe and Tyr) have the least effect on catalysis, whereas amino acids with bulky side chains at Thr65 (Val) and Asn281 (Leu) have the greatest effect. Changing Tyr155 to Phe and Asn157 to Ala also hinders catalysis, and the effects fall in between these extremes. These observations are put into a structural context using a crystal structure of the Schiff base of the reaction intermediate. Trapping experiments with substrate, Na(CN)BH3, and wild type enzyme and selected mutants mostly paralleled the kinetic analysis, and identified two salicylaldehyde-modified lysines: the active site lysine (Lys183) and one outside the active site (Lys279). The latter could be responsible for the observed inhibition of NahE by salicylaldehyde. Together, the results provide new insights into the NahE-catalyzed reaction.
Keywords: hydratase-aldolase, enzyme kinetics, active-site mutants, Schiff base, naphthalene catabolic pathway
Graphical Abstract
INTRODUCTION
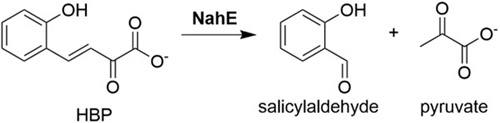
A key reaction in bacterial catabolic pathways for polycyclic aromatic hydrocarbons (PAHs) is catalyzed by a bifunctional hydratase-aldolase.1-6 In the naphthalene catabolic pathway, trans-o-hydroxybenzylidenepyruvate hydratase-aldolase (designated NahE) converts trans-o-hydroxybenzylidenepyruvate (1, R = OH, Scheme 1) to salicylaldehyde (2) and pyruvate (3).1,5,6 The enzyme can also turnover substrates where R = H or CO2− (4 or 6) to afford benzaldehyde (5) or 2-carboxybenzaldehyde (7), respectively, along with pyruvate. NahE processes these substrates through the Schiff base of 1, 4, or 63,5,6 that allows for water addition at C4 (8-10) to set up a retro-aldol reaction to yield products (2, 5, or 7) and the Schiff base of 3. Subsequent hydrolysis generates 3 and completes the reaction.
Scheme 1.

The Hydratase-Aldolase Reactions in PAH Catabolic Pathways.
There is significant interest in repurposing this set of hydratase-aldolases for two reasons.7-13 First, PAHs are persistent environmental contaminants that cause human health problems either directly or indirectly (via reactive metabolites).7,8 PAHs are also toxic to other organisms, most notably marine and other aquatic wild life. Studies of the PAH degradative pathways as well as the individual hydratase-aldolases and their specificities can contribute to the development of technologies for the removal of PAHs from the environment.9 Second, there is a strong desire to replace toxic reagents and solvents in chemical processes with enzymes.10 The properties of NahE have received much scrutiny over the past 20 years as one such biocatalyst, most recently for the synthesis of substituted quinolines.11-13
Sequence analysis identified NahE as an N-acetylneuraminate lyase (NAL) subgroup member in the Class I aldolase superfamily.14 Members of this subfamily show a conserved (β/α)8-barrel structure, two strictly conserved active site residues (tyrosine and lysine) that form a Schiff base with substrate, and a GXXGE motif that is associated with the binding of the pyruvoyl carboxylate group.14-19 Much of the remaining portion of the active site is tailored to accommodate the individual reactions catalyzed by NAL subfamily members. For the NahE-catalyzed reaction, this involves binding of the o-substituent and the addition of water at C4 (of 1, 4 or 6) as the respective Schiff bases to set up a retro-aldol fission of 8-10.
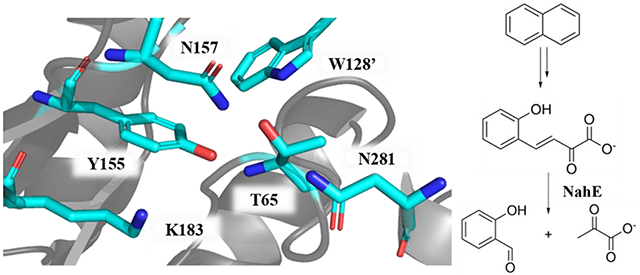
Sequence and crystallographic analysis of the selenomethionine derivative of NahE show Lys183 and Tyr155 along with the GXXGE motif (Gly64, Thr65, Phe66, Gly67, Glu68) (Figure 1, PDB code 6DAO).5,6 Previous work confirmed the presence of a Schiff base in the reaction and the critical nature of Lys183 in the formation of the Schiff base.5,6 In the presence of substrate and Na(CN)BH3, NahE is inactivated by covalent modification of Lys183. In addition, the K183A mutant is nearly inactive, confirming its importance in the mechanism.6
Figure 1.

Close-up of the active site of the selenomethionine derivative of NahE, showing Lys183 and the proximity of Tyr155.5 The other residues under investigation (Thr65, W128, Tyr155, Asn157, Asn281) are also shown. Thr65 is shown as part of the GTFGE loop. The one letter amino acid codes are used in the Figure for simplicity (PDB code 6DAO).
Likewise, roles for other active site residues (Thr65, W128, Tyr155, N157, N281) can be assigned based on sequence analysis and studies of subfamily members, most notably, PhdJ, which is the related hydratase-aldolase in the phenanthrene degradative pathway.5,6 In this study, Thr65, W128, Tyr155, N157, and N281 were subjected to mutagenesis and the kinetic properties of these mutants were characterized. The results of trapping experiments using one substrate (i.e., 1 in Scheme 1), enzyme (wild type and selected mutants), and Na(CN)BH3, followed by mass spectral identification and quantification, are mostly consistent with the kinetic analysis. Interestingly, the doubly salicylaldehyde-labeled enzyme is one of the predominant species for the Y155F mutant. In-depth analysis shows that two lysine residues (Lys183 in the active site and Lys-279 outside the active site on the surface of the protein) are covalently modified by salicylaldehyde that is released from the processing of substrate (HBP, 1, Scheme 1). This observation might be the basis for the inhibition of NahE activity by salicylaldehyde. Finally, the results are put into a structural context using a crystal structure of the Schiff base of the intermediate in the reaction (8 in Scheme 1), and a mechanism proposed.
EXPERIMENTAL PROCEDURES
Materials.
Chemicals, biochemicals, buffers, and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), Fisher Scientific Inc. (Pittsburgh, PA), Fluka Chemical Corp. (Milwaukee, WI), or EMD Millipore, Inc. (Billerica, MA). Reagents for molecular biology manipulations were obtained from sources listed in the text. trans-o-Hydroxybenzylidenepyruvate was synthesized as described elsewhere.5 The prepacked PD-10 Sephadex G-25 columns were obtained from GE Healthcare (Piscataway, NJ). The Econo-Column® chromatography columns were obtained from BioRad (Hercules, CA). The Ni-NTA resin was purchased from Fisher Scientific (Pittsburgh, PA). The Amicon stirred cell concentrators and the ultrafiltration membranes (10,000 Da, MW cutoff) were purchased from EMD Millipore Inc. Oligonucleotide primers were synthesized by Sigma-Aldrich.
Bacterial Strains and Plasmids.
Escherichia coli strain BL21Gold(DE3) was obtained from Agilent Technologies (Santa Clara, CA). The construction of the expression vector for NahE in pET24 is described elsewhere.5
General Methods.
The PCR amplification of DNA sequences was conducted in a GeneAmp 2700 thermocycler (Applied Biosystems, Carlsbad, CA). Techniques for restriction enzyme digestion, ligation, transformation, and other standard molecular biology manipulations were based on methods described elsewhere.20 DNA sequencing was performed in the DNA Sequencing Facility in the Institute for Cellular and Molecular Biology (ICMB) at the University of Texas at Austin. Electrospray ionization mass spectrometry (ESI-MS) and LC-MS/MS analysis were carried out respectively on an Orbitrap Fusion 1 or an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher Scientific, San Jose, CA) in the Proteomics Facility in the ICMB. Steady-state kinetic assays were performed on an Agilent 8453 diode-array spectrophotometer at 22 °C. Non-linear regression data analysis was performed using the program GraphPad Prism 9 (GraphPad Software, San Diego, CA). Protein concentrations were determined by the Waddell method.21 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out on denaturing gels containing 12% polyacrylamide.22
Construction of the His-tag NahE.
The expression vector for NahE in pET24 was previously constructed,5 but retains a stop codon at the end of the NahE gene (and not after the His-tag), lacks the His-tag cleavage site (ENLYFQG), and is not in frame to express the His-tag. Hence, a new expression vector was constructed to address these issues. A single PCR mixture (12 μL) was set up that contained pET24/NahE (50-100 ng/μL),5 deoxynucleoside triphosphates (dNTPs) (0.2 mM each), Thermococcus kodakarensis (KOD) DNA polymerase (0.5 units), 10× KOD PCR buffer (when diluted to 1 × buffer concentration contributes 2 mM MgSO4), both from a Novagen KOD Hot Start DNA Polymerase kit, the forward and reverse primers, (5’-gctaaatacagtaaa GAAAACCTGTATTTTCAGGGC caccaccaccaccacCACCACCACCACCAC- 3’) and (5’-GTGGTGGTGGTGGTG GCCCTGAAAATACAGGTTTTC tttactgtatttagc-3’), respectively, (0.4 μM each), and purified water up to the final reaction volume. The primers contain NahE-specific sequence (lower case), the His-tag cleavage site (bold), and plasmid-specific sequence (upper case). The PCR amplification protocol consisted of an initial 5-min denaturation cycle at 95 °C, followed by 35 cycles of 95 °C for 30 s, a gradient increase of 55 + 0.5n °C/cycle for 30 s (where n is equal to the number of cycles), 72 °C for 6 min, a 10-min elongation cycle at 72 °C, and a hold at 4 °C. The PCR reaction product size was confirmed on a 1% agarose gel. The PCR reaction was digested overnight at 37°C with DpnI (1 μL) followed by a second aliquot of DpnI (1 μL), and allowed to digest for another 2 h at 37 °C. The resulting mixture was transformed into E. coli BL21(DE3) cells using the calcium chloride heat shock method.20 The transformation mixture was plated on a Lysogeny broth (LB) plate supplemented with kanamycin (Kn, 50 μg/mL), and cells were grown overnight at 37 °C. A single colony was used to inoculate 10 mL of LB/Kn (50 μg/mL). The cultures grew overnight with shaking (200 rpm) at 37 °C. DNA was extracted from the culture using the Sigma GenElute Plasmid MiniPrep kit, eluted with nuclease-free water (50 μL), and stored at −20 °C for use as template in the mutagenesis work. The DNA was sequenced in order to verify that the sequence was correct (sole stop codon after the His-tag, His-tag cleavage site, and His-tag in frame). Only 5 of the 6 histidine residues were present. An aliquot (1 mL) of the remaining culture was used to make a glycerol cell stocks, which were stored at −80 °C.
Construction of the T65V, Y155F, N157L and N157A Mutants of His-tag NahE.
Mutations were constructed using a whole-plasmid PCR technique adapted from the QuikChange® Kit (Agilent Technologies).23 Accordingly, PCR reactions (12 μL) were set up for each of the desired mutants that contained the His-tag-NahE in pET24 (50-100 ng/μL), dNTPs (0.2 mM each), KOD DNA polymerase (0.5 units), 10 × KOD PCR buffer (when diluted to 1 × buffer concentration contributes 2 mM MgSO4), the appropriate forward and reverse primers (0.4 μM each) (Table S1), and purified water to the final reaction volume. The PCR amplification protocol consisted of an initial 5-min denaturation cycle at 95 °C, followed by 25 cycles of 95 °C for 30 s, a gradient increase of 55 + 0.5n °C/cycle for 30 s (where n is equal to the number of cycles), 72 °C for 6 min, a 10-min elongation cycle at 72 °C, and a hold at 4 °C. For each, the PCR reaction product size was confirmed on a 1% agarose gel and treated with DpnI, as described above. The resulting mixture was transformed into E. coli BL21(DE3) cells by electroporation using a Bio-Rad MicroPulser Electroporation unit. A portion (5 μL) of the transformation mixture was treated as described above and the DNA was extracted and sequenced in order to verify the intended mutations. Those with the correct mutations were stored at −20 °C. An aliquot (1 mL) of each 10 mL culture was used to make glycerol stocks, which were stored at −80 °C.
Construction of the T65S, W128Y, W128F, and N281L Mutants of His-tag NahE.
Mutations were constructed using a whole-plasmid PCR technique, described above.23 Accordingly, PCR mixtures were set up as described above using the forward and reverse primers in Table S2. The PCR amplification protocol for these mutants consisted of an initial 5-min denaturation cycle at 95 °C, followed by 30 cycles of 95 °C for 30 s, 30-s annealing at 65 °C (W128Y) or 70 °C (T65S,W128F, N281L), 72 °C for 6 min, a 10-min elongation cycle at 72 °C, and a hold at 4 °C. The PCR reaction product sizes were confirmed on a 1% agarose gel. Each PCR mixture was treated with DpnI, as described above. A portion (5 μL) of the digested mixture was transformed into E. coli BL21(DE3) cells via electroporation using a Bio-Rad MicroPulser Electroporation unit. The transformation mixture was processed as described above and the extracted DNA was submitted for sequence analysis in order to verify that the desired mutations had been introduced. Those with the correct mutations were stored at −20 °C. An aliquot (1 mL) of each 10 mL culture was used to make glycerol stocks, which were stored at −80 °C.
Growth and Expression of the His-tag NahE Mutants.
The glycerol cell stock of each mutant was used to inoculate 10 mL of LB/Kn (50 μg/mL) in a 50-mL sterile conical tube. This starter culture was grown overnight at 37 °C with shaking (200 rpm). A portion (1 mL) of the starter culture was used to inoculate 1 L of sterile LB/Kn (50 μg/mL) in 2L flasks. The cultures were grown at 37 °C with shaking (200 rpm) until the OD600 was ~ 0.5. The cultures were chilled on ice for 10 min and then induced with isopropyl β-D-thiogalactoside (IPTG) at a final concentration of 0.75 mM and expressed for 20 h at 20°C with shaking (200 rpm). After centrifugation (11000g × 30 min), the cell pellet from 1 L of culture (except T65V) was resuspended in 10 mL of 50 mM Na2HPO4 buffer, pH 7.2 (Buffer A). The typical yield was ~4 g/L mixture. For the T65A mutant, the cell pellets from 3-1 L cultures were combined and resuspended in Buffer A. The resuspensions were flash frozen and stored at −80 °C.
Purification of His-tag NahE Mutants.
Cell resuspensions were thawed and made 1 mM in phenylmethylsulfonyl fluoride (PMSF). Cells were disrupted by sonication (two cycles of 3 min at 1-s pulses, 40% duty cycle/50% output) using a model W-385 Ultrasonic Processor Sonicator (Heat Systems-Ultrasonics, Inc., Newtown, CT). The cell debris was pelleted (25400g × 45 min), and the lysate was loaded onto a hand-packed Ni-NTA column (5 mL bed volume) pre-equilibrated with Buffer A. The bound proteins were washed with 40 mL of Buffer A made 20 mM in imidazole followed by a second wash of 40 mL of Buffer A made 60 mM in imidazole. The proteins were then eluted using a linear gradient of Buffer A made 60 mM in imidazole to Buffer A made 100 mM imidazole (80 mL total volume). The target proteins were eluted with Buffer A made 120 mM in imidazole (~40 mL) until they were recovered from the column. All His-tag NahE mutants except T65V, W128Y, W128F, and Y155F were eluted at 25 °C and flash frozen as the eluent. The others were eluted while on ice, covered, and then stored at 4 °C overnight. The samples appeared near-homogeneous, as determined by SDS-PAGE. The samples were concentrated to ~0.5 mL using a 50-mL Amicon Stirred Cell (membrane cutoff 10000 Da) and loaded onto a PD-10 column equilibrated in Buffer A made 200 mM in NaCl, aliquoted, flash frozen, and stored at −80 °C until examined by mass spectroscopy and kinetic analysis.
Purification of the His-tag Y155F Mutant of NahE.
The His-tag Y155F mutant of NahE used for salicylaldehyde labeling, proteolytic digestion, and mass spectral analysis was purified as follows. Cells (2.8 g) were thawed in 20 mM Na2HPO4 buffer (pH 7.2, 15 mL) and made 1 mM in PMSF and 6-aminocaproic acid. The cells were disrupted by sonication and the mixture was centrifuged as described above. The supernatant was loaded on a hand-packed DEAE anion exchange column (~2×13 cm) equilibrated in the 20 mM Na2HPO4 buffer. The column was washed with 50-mL of buffer followed by a linear gradient (0-0.5 M Na2SO4 in 20 mM Na2HPO4 buffer, 100 mL total). Fractions (5 mL) were collected and assayed with trans-benzylidenepyruvate (4, BP, Scheme 1). The enzyme elutes at ~0.25 M Na2SO4. The most active fractions were combined, concentrated to 10 mL using a 50-mL Amicon stirred cell concentrator equipped with 10 kDa membrane, and loaded on a hand-packed Ni-NTA column (~1×3 cm) pre-equilibrated with 20 mM Na2HPO4 buffer. The column was washed with 20 mM Na2HPO4 buffer (~50 mL) followed by a linear gradient (0-120 mM imidazole in 20 mM Na2HPO4 buffer, 100 mL total). The protein did not elute so an aliquot (~25 mL) of 150 mM imidazole was used. Fractions were collected into test tubes containing 5 mL of 20 mM Na2HPO4 buffer (for a total of 10 mL). The protein elutes in fractions 2 and 3, as assayed in very diluted aliquots. The two fractions were pooled (20 mL) and diluted with 20 mM Na2HPO4 buffer (30 mL) and exchanged into buffer using the 50-mL Amicon stirred cell concentrator in order to dilute the imidazole concentration under 1 mM. (The Y155F mutant does not appear to be stable in high concentrations of imidazole.) The resulting solution was concentrated to 1 mL and loaded on a Sephadex G-75 column (~0.5×40 cm). Fractions (1 mL) were eluted with the 20 mM Na2HPO4 buffer and analyzed by SDS-PAGE. The most homogeneous fractions were combined and concentrated to 2.3 mg/mL and analyzed by mass spectrometry.
Steady State Kinetics of NahE-His Variants with HBP.
The assays were carried out at 22 °C in 50 mM Na2HPO4 buffer (pH 7.2) using trans-o-hydroxybenzylidenepyruvate (1, HBP, Scheme 1) as substrate. Various stock solutions of HBP (167–14097 μM) were prepared by dissolving an appropriate amount of the crystalline material in absolute ethanol. Subsequent dilutions were made up in 50 mM Na2HPO4 buffer (pH 7.2). The final substrate concentrations ranged from 1-300 μM. Stock solutions of enzyme (0.0106, 4.65, 0.079, 0.2536, 0.089, 7.49, 16.58, 5.65, 9.12 mg/mL) were made up for WT, T65V, T65S, W128F, W128Y, Y155F, N157A, N157L, and N281L, respectively. These stock solutions correspond to final enzyme concentrations of 0.0083, 12.18, 0.0103, 0.02, 0.0163, 4.9, 2.17, 0.74, 23.9 μM for WT, T65V, T65S, W128F, W128Y, Y155F, N157A, N157L, and N281L, respectively.
The conversion of HBP to pyruvate and salicylaldehyde was monitored by following the formation of salicylaldehyde at 255 nm. Both HBP and salicylaldehyde absorb at 255 nm and the decrease in the absorbance of HBP (ε = 3590 M−1cm−1) occurs simultaneously with the increase in the absorbance of salicylaldehyde (ε = 16130 M−1cm−1). Therefore, the two extinction coefficients were added together (20080 M−1cm−1) and this extinction coefficient was used to convert the change in absorbance to change in concentration. The corrected absorbances were plotted versus time. Absorbances were collected in triplicate and the standard deviations are based on the replicate measurements. For each concentration of HBP, the initial rates were calculated based on the slope determined from the change in absorbance over time. The data were fit to a linear equation in order to determine the slope of the line which is the initial rate. Initial rates were determined, plotted against the substrate concentration, and fit to the Michaelis–Menten equation using GraphPad Prism 9. The high concentration of N281L necessary to measure activity precluded saturation with substrate so the kcat/Km was determined by a linear fit of the plot of vo vs the concentration of substrate using GraphPad Prism 9.24 For all proteins, the kinetic parameters are based on the monomer molecular mass. The kinetic parameters are collected in Table 3.
Table 3.
Steady-state Kinetic Parameters for His-tag NahE and Mutants with HBPa
| |||
|---|---|---|---|
| Enzyme | kcat(S−1) | Km (μM) | kcat/Km (M−1s−1) |
| WT | 8.0 ± 0.4 | 4 ± 1 | (2.2 ± 0.6) × 106 |
| T65S | (8.5 ± 0.2) × 10−3 | 1.7 ± 0.3 | 5000 ± 1000 |
| T65V | (4.0 ± 0.5) × 10−3 | 21 ± 8 | 190 ± 80 |
| W128Y | 5.0 ± 0.2 | 7 ± 1 | (7 ± 0.1) × 105 |
| W128F | 8 ± 0.4 | 8 ± 2 | (1 ± 0.2) × 106 |
| Y155F | (4.0 ± 0.2) × 10−3 | 4 ± 1 | 1000 ± 200 |
| N157L | (6 ± 0.5) × 10−1 | 60 ±10 | (1 ± 0.2) × 104 |
| N157A | (1.3 ± 0.3) × 10−1 | 8 ± 1 | (1.6 ± 0.1) × 104 |
| N281L | -- | -- | 90 ± 10b |
The kinetic parameters were determined by the assay described in the text.
The kcat/Km was determined by a linear fit of vo vs [HBP], as described in the text.
Covalent Modification of His-tag NahE and Mutants by HBP in the Presence of Na(CN)BH3.
Each reaction mixture was made up in 50 mM NaH2PO4 buffer (pH 7.2) containing enzyme (~1 mg/mL solution) and Na(CN)BH3 (30 μL of a 33.4 mg/mL solution made up in 100 mM Na2HPO4). The amounts of enzyme are as follows: wild type, 108 μL of a 9.3 mg/mL solution; W128F, 109 μL of a 9.2 mg/mL solution; N157A, 121 μL of a 8.3 mg/mL solution; T65A, 200 μL of a 5 mg/mL solution; Y155F, 111 μL of a 8.0 mg/mL solution; and N281L, 200 μL of a 5 mg/mL solution. The final volumes were ~120 μL (before addition of aliquots of HBP). The reaction mixtures were allowed to incubate for 5 min before addition of HBP. Aliquots from a stock solution of HBP (10 μl from a 3 mg/mL solution) were added to each reaction mixture so that the final concentration was 6 eq of HBP to enzyme. Reactions were carried out at room temperature for 90 min. Samples were prepared as described elsewhere and subjected to ESI-MS analysis.25
Covalent Modification of the Y155F Mutant by Salicylaldehyde, Proteolytic Digestion, and Mass Spectrometric Analysis.
The Y155F enzyme (220 μL of 2.3 mg/mL solution) was added to a solution of 50 mM NaH2PO4 buffer (pH 7.2) containing Na(CN)BH3 (30 μL of a 33.4 mg/mL solution made up in 100 mM Na2HPO4, pH 8). Subsequently, 6 equivalents (based on monomer molecular mass) of salicylaldehyde (6.4 μL of a 1.5 mg/mL) were added and the mixture was allowed to incubate overnight at 5 °C overnight. This sample and the HPD-treated Y155F (previous section) were exchanged into 10 mM (NH4)2CO3 buffer (pH ~8) using a PD-10 Sephadex G-25 column, fractions (300 μL) were collected, protein was located using the Bradford assay and appropriate fractions combined (900 μL). An aliquot (50 μL for HBP and 100 μL for salicylaldehyde) was removed and submitted for mass spectral analysis to confirm labeling. Subsequently, aliquots (50 μL for HBP and 100 μL for salicylaldehyde) of the samples were treated with sequencing grade endoproteinase Glu-C from S. aureus V8 (protease V8) using a modification of a literature procedure.26 Accordingly, a portion (50 μg) of protease V8 was resuspended in 10 mM (NH4)2CO3 buffer, pH 8 (20 μL) and then 5 μL of this protease V8 solution was added to each sample. The samples were incubated at 37 °C for 60 h and submitted for liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis using a Dionex LC and Orbitrap Fusion Tribrid mass spectrometer with a 30 min run time. The sample was injected and desalted using the high flow protein trap LC and then eluted into the MS. The data were processed using Proteome Discover (PD) 2.2 and the proteome software Scaffold 5.
Crystallization.
NahE for crystallographic studies was purified as described previously.5 Diffraction-quality crystals of apo enzyme were obtained in 0.1 M MES (pH 6.5-8.0) and 21-26% PEG 2000 monomethyl ether (MME), in sitting drop and incubated at room temperature.5 The crystals were set up in sitting drop and incubated at room temperature. To obtain NahE in complex with the Schiff base of 4-hydroxy-4-(2-hydroxyphenyl)-2-oxobutanoate (designated NahE-HI where HI stands for hydroxy intermediate, 8 in Scheme 1), an apo crystal of NahE was soaked in the mother liquor containing trans-o-hydroxybenzylidenepyruvate (HBP, 1 in Scheme 1, 5 mM) for ~30 min, prior to cryoprotection with 25% glycerol and vitrification in liquid nitrogen. X-ray diffraction data were collected at Advanced Light Source beamline 5.0.2 (ALS, Berkeley, CA).
Data collection, Processing, Structure Determination and Refinement.
Data were indexed, integrated, and scaled using HKL-200027. A molecular replacement solution was identified using Phaser28 with the NahE apo structure as a model for molecular replacement to identify the solution (PDB code 6DAO). Structures were refined using Phenix Refine29. Manual model building was done using Coot30. Model structures were evaluated during and after refinement using Molprobity.31 The data collection and refinement statistics are summarized in Table 1. Figures were prepared using PyMol (The PyMOL Molecular Graphics system, Version 1.8 Schrodinger, LLC).32
Table 1.
X-ray Crystallography Data Collection and Refinement Statistics
| Data Collection | |
| Space Group | P 32 2 1 |
| Cell Dimensions | |
| a, b, c (Å) | 88.0, 88.0, 141.6 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 2.10 (2.15-2.10)* |
| Rsym/Rpim | 0.129 (0.854)/0.042 (0.269)* |
| CC ½ ϒ | 0.886 |
| I / σ | 20.6 (2.3)* |
| Completeness (%) | 100 (100)* |
| Redundancy | 10.8 (11.0)* |
| Refinement | |
| Resolution (Å) | 44.0-2.10 (2.18-2.10)* |
| Unique reflections | 37400 (3661)* |
| Rwork | 0.2022 (0.2373)* |
| Rfree± | 0.2608 (0.3270)* |
| No. atoms | |
| Protein | 5058 |
| Water | 180 |
| B-factors (Å2) | |
| Protein | 29.9 |
| Water | 28.5 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 0.883 |
| Ramachandran plot | |
| Favored | 97.80% |
| Allowed | 2.20% |
| Outliers | 0.00% |
| Molprobity score ^ | 4.51 (98th percentile) |
Values for the corresponding parameters in the outermost shell in parenthesis.
CC1/2 is the Pearson correlation coefficient for a random half of the data, the two numbers represent the lowest and highest resolution shell, respectively.
Rfree is the Rwork calculated for about 10% of the reflections randomly selected and omitted from refinement.
MolProbity score is calculated by combining clashscore with rotamer and Ramachandran percentage and scaled based on X-ray resolution. The percentage is calculated with 100th percentile as the best and 0th percentile as the worst among structures of comparable resolution.
RESULTS
Expression and Purification of the His-tag NahE Mutants.
Eight mutants of the His-tag NahE were constructed by a whole-plasmid PCR technique23 and the expressed proteins purified to near homogeneity by immobilized metal ion chromatography (using nickel). The yields ranged from 4-15 mg (per liter of culture). The His-tag was not removed. All mutations were verified by DNA sequencing and the average molecular masses of six out of eight were determined in the course of experiments described later (Table 2).
Table 2.
Calculated and Observed Masses for His-tagged NahE and Mutantsa
| Enzyme | Calculated Mass (Da) | Observed Mass (Da) |
|---|---|---|
| WT | 38,174.60 | 38,175.35 |
| T65V | 38,172.63 | 38,172.86 |
| W128F | 38,135.57 | 38,135.22 |
| Y155F | 38,158.61 | 38,157.94 |
| N157L | 38,173.66 | 38,173.65 |
| N157A | 38,131.58 | 38,130.18 |
| N281L | 38,173.66 | 38,173.98 |
Average molecular masses where standard deviations are presented in Table S3.
Steady-state Kinetics of His-tag NahE with HBP.
The kinetic parameters for wild type enzyme and eight mutants were measured and are collected in Table 3. The kinetic plots are shown in Figure S1. Kinetic parameters for the wild type enzyme were previously reported (kcat ~11.7 s−1, Km ~1.2 μM, and kcat/Km ~1 × 107 M−1s−1),5 and those reported here are comparable. Two mutants (W128Y and W128F) have kinetic parameters that are comparable to those of wild type where kcat is ~1.6-fold lower or comparable and Km is ~2-fold higher. (Although Km has no meaning in a multi-step enzymatic reaction, it is presented for comparative purposes.) The overall kcat/Km was ~2-3-fold lower. The most detrimental effect occurred with the N281L mutation (kcat/Km is reduced more than 24,000-fold). The N281L mutant could not be saturated (and kcat and Km could not be measured) so the kcat/Km was determined by a linear fit of the data, as described in the experimental.24 The T65S mutant showed a ~940-fold decrease in kcat and a ~2-fold decrease in Km. The overall kcat/Km decreased ~440-fold. In contrast, the T65V mutant showed an almost ~2000 decrease in kcat and an almost ~5-fold increase in Km. The overall kcat/Km decreased ~12,000-fold. The Y155F mutant showed a ~2000-fold decrease in kcat and a comparable Km. The overall kcat/Km decreased ~2200-fold. Changing Asn157 to a leucine (N157L) gave a ~13-fold decrease in kcat and 15-fold increase in Km. The overall kcat/Km decreased ~220-fold. Changing Asn157 to alanine was less detrimental, and gave a ~61 -fold decrease in kcat and 1.8-fold increase in Km. The overall kcat/Km decreased ~140-fold.
Mass Spectral Analysis of His-tag NahE and NahE Mutants in the Presence of HBP and Na(CN)BH3.
The wild type NahE and six mutants (T65V, W128F, Y155F, N157L, N157A, and N281L) were incubated in individual reactions with trans-o-hydroxybenzylidenepyruvate (HBP) (6:1 eq of substrate to enzyme) and Na(CN)BH3. After 90 min, they were passed through a PD-10 Sephadex G-25 column and analyzed by mass spectrometry, as described here and elsewhere.25 The masses of the products (and approximate quantities) are collected in Table 4 and are shown in Figures S2-S4 and in Tables S3 and S4. In Table 4, the first two columns show the masses of the unlabeled protein along with the fractional abundance. The second two columns show the masses of the protein modified by HBP (and reduced) along with the fractional abundance. The last two columns show the masses of the protein modified by salicylaldehyde (and reduced) along with the fractional abundance.
Table 4.
Mass Spectral Analysis of His-tag NahE and Mutants after Incubation with HBP and Na(CN)BH3a
| Enzyme | Unlabeled (Da) |
Frac. Abund. (%) |
HBP (Da) |
Frac. Abund. (%) |
Salicylaldehyde (Da) |
Frac. Abund. (%) |
|---|---|---|---|---|---|---|
| WT | 38,174 | 65 | -- | -- | 38,277 | 21 |
| T65Vb | 38,173 | 19 | 38,347 | 31 | 38,274 | 19 |
| W128Fb | 38,135 | 25 | 38,311 | 7.5 | 38,239 | 44 |
| Y155Fb,c | 38,158 | 44 | -- | -- | -- | -- |
| N157Lb | 38,172 | 22 | 38,352 | 46 | 38,278 | 2.3 |
| N157A | 38,130 | 72 | 38,308 | 16 | -- | -- |
| N281L | 38,174 | 53 | 38,351 | 9 | 38,276 | 18 |
A full list of the masses can be found in Tables S3 and S4. The masses in Table 4 are rounded off for clarity.
Four mutants show double labeling with salicylaldehyde (T65V: 38,387 Da, 13.2%; W128F: 38,345 Da, 6%; Y155F: 38,362 Da, 30%; N157L: 38,393, 6%).
The Y155F mutant showed a small amount of labeling with pyruvate (38,233 Da, 2.2%).
The potential products in the course of the NahE-catalyzed reaction are shown in Scheme 2 [after reduction of the Schiff base by Na(CN)BH3]. The reported masses (in Tables 4 and S4) vary depending on the enzyme, and were determined on the Dionex LC and Orbitrap Fusion 1 mass spectrometer. The intermediate-labeled enzyme (8 in Scheme 1) is not anticipated to be stable under the conditions of the mass spectral analysis (and was not observed). Moreover, the HBP-labeled enzyme might include intermediate-labeled enzyme that underwent dehydration. We and others have shown that salicylaldehyde inhibits NahE activity with an IC50 ~20 μM.33,34 The inhibition does not appear to be competitive because increasing the concentrations of HBP does not overcome inhibition. This observation suggests that salicylaldehyde is a potential inhibitor or regulator (as well as the product of the reaction) at another site. In the experiments below, the sites of labeling for the singly and doubly labeled salicylaldehyde-enzyme were identified using the Y155F mutant (because it had the highest abundance of doubly labeled protein).
Scheme 2.

Potential Products shown as the reduced Schiff Bases
For the wild type enzyme, the most abundant species were unlabeled enzyme (38,174 Da) and salicylaldehyde-labeled enzyme (38,277 Da) where unlabeled enzyme predominates. For the T65V enzyme, there were three species: unlabeled enzyme (38,173 Da), HBP-labeled enzyme (38,347), and salicylaldehyde-labeled enzyme (38,274 Da). The HBP-labeled enzyme predominates, which is consistent with the poor kcat/Km (~280 M−1S−1). For the W128F enzyme, the predominant species was the salicylaldehyde-labeled enzyme (38,239 Da) followed by unlabeled enzyme (38,135 Da) and a small amount of the HBP-labeled enzyme (38,311 Da). With the exception of the small amount of HBP-labeled enzyme, the results were roughly comparable to those of wild type. For both the T65V and W128F, there wer smaller amounts of the doubly salicylaldehyde-labeled enzyme (more for the T65V mutant).
For the Y155F enzyme, the unlabeled enzyme (38,158 Da) and the doubly salicylaldehyde-labeled enzyme (38,362 Da) were the predominant species. There was also a small amount of the pyruvate-labeled enzyme (38,233 Da). Loss of the active site tyrosine may slow down the rate of hydrolysis of the Schiff base of pyruvate so a small amount accumulates. For the N157L and N157A enzymes, there was mostly unlabeled enzyme (38,172 Da and 38,130 Da, respectively) and HBP-labeled enzyme (38,352 Da and 38,308 Da, respectively). For the N157L protein, there was a small amount of the salicylaldehyde-labeled enzyme (38,278) along with more doubly salicylaldehyde-labeled enzyme (38,393 Da). The N157L protein showed significantly more HBP-labeled enzyme than the other mutants. This might be the result of a significantly impaired enzyme. Finally, for the N281L enzyme, there was unlabeled enzyme (38,174 Da), HBP-labeled enzyme (38,351 Da) and salicylaldehyde-labeled enzyme (38,276 Da). It is surprising that there is not more HBP-labeled enzyme, but this might be a function of not being able to saturate the enzyme.
Two observations are of note. First, the products are mostly consistent with the kinetic data. The exceptions are the T65V and N281L enzymes. Even though they are the “poorest” enzymes, as assessed by the kcat/Km values, they still generate product (salicylaldehyde). The T65V enzyme has mostly HBP-labeled enzyme and the N281L enzyme has mostly unlabeled enzyme (in additional to a small amount of salicylaldehyde-labeled enzyme). This observation might be related to the inability to saturate the enzyme (as noted above). The second observation is the quantities of singly or doubly salicylaldehyde-labeled enzyme generated for all enzymes except the N157A enzyme. The highest quantity of doubly salicylaldehyde-labeled is observed for the Y155F protein. It is consistent with the observation that salicylaldehyde inhibits enzyme activity, suggesting that once salicylaldehyde is generated, it does not readily dissociate from the active site or binds elsewhere.
Covalent Modification of Lys279 by Salicylaldehyde in the Y155F Mutant-catalyzed Reaction using HBP.
In order to determine the sites of covalent modification by salicylaldehyde in the doubly-labeled Y155F mutant, the enzyme was incubated with HBP at pH 7.2, as described in the experimental section. After incubation overnight, the enzyme was recovered and analyzed by MS. The major signals were found at 38,158 Da and 38,362 Da, corresponding to unlabeled protein and doubly labeled protein (Tables 4 and S4).
In a separate experiment, the Y155F mutant was incubated with salicylaldehyde under the same conditions. After incubation overnight, the enzyme was recovered and analyzed by MS. The major signals are found at 38,160 Da and 38,264 Da, corresponding to unlabeled protein and labeled protein. The mass difference is 104 Da, which is consistent with the side chain of Lys modified by salicylaldehyde and reduced. The reduced product now has one less proton on the epsilon nitrogen giving a mass of 106 Da. (For a protein of this size, the mass difference of 2 Da is within the experimental error.) The identities of the other signals are not known.
After it was confirmed that the enzyme(s) was labeled, the HBP- and salicylaldehyde-treated Y155F proteins were digested with protease V8, and the resulting mixtures were analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). For the HBP-treated sample, the majority of label was localized to peptide fragments 173-191 (MSKIPQVVTAKYLGIGMLD) and 277-285 (FSKYNIGLE). The first fragment (with the active site Lys183) showed a mass of 2169.1584 Da (calcd, 2169.1578 Da). The second fragment (with Lys279) showed a mass of 1175.5870 Da (calcd, 1175.5863 Da). In the same samples, the unmodified fragments (173-191 and 277-285) showed masses of 2063.1158 Da (calc. 2063.1159 Da) and 1069.5446 Da (calcd, 1069.5444 Da). The mass differences were 106.0424 Da and 106.0426 Da (calcd, 106.0419 Da) corresponding to modification by salicylaldehyde. Hence, both Lys183 (in the active site) and Lys279 (outside the active site) were modified by salicylaldehyde that is generated by the enzymatic processing of HBP.
For the salicylaldehyde-treated sample, the label was localized to the peptide fragments 173-191 and 173-193. Both modified and unmodified protein were in the sample. The presence of a small amount of phosphate buffer alters the specificity of V8 so that one potential cleavage site is missed leaving two additional amino acids (in fragment 173-193). The 173-191 and 173-193 fragments for unmodified protein in the sample showed masses of 2063.1161 Da (calcd, 2063.1159 Da) and 2291.2265 Da (calcd, 2291.2269 Da), respectively. The 173-191 and 173-193 fragments for modified protein in the sample showed masses of 2169.1584 Da (calcd, 2169.1578 Da) and 2397.2685 Da (calcd, 2397.2688 Da), respectively. The mass differences were 106.0423 Da and 106.0420 Da (106.0419 Da). These observations suggest that Lys279 is not readily modified (even though the crystal structure shows that it is on the surface).
Crystal Structure of NahE in complex with the Schiff base of 4-hydroxy-4-(2-hydroxyphenyl)-2-oxobutanoate (designated NahE-HI).
In order to put the results into a structural context, we incubated the substrate with the preformed NahE apo crystals and collected X-ray diffraction data (Table 1). Using wild type protein as a model for molecular replacement (PDC code 6DAO), strong positive density extended out of the lysine side chain amino group, indicating the covalent attachment of substrate onto active site lysine (Figure 2A). The structure of substrate (HBP) is mostly consistent with the additional density, except for a “blob” of strong positive density next to the phenyl ring that indicates the intermediate formation (i.e., HI) captured in the structure (Figures 2A,B). The hydroxy group forms hydrogen bonds with the side chain hydroxyl group of Tyr155 (2.8 Å) and amide of Asn157 (3.3 Å) (Figure 2C). The hydroxy group of salicylaldehyde portion forms hydrogen bonds with the carbonyl group of Glu285 (distance of 2.65 Å) and the side chain of Asn281 (3.4 Å). These interactions orientate the ligand and place the benzyl ring hydrophobically sandwiched by Phe269, Phe274, and Phe277 on one side and Trp224 on the other (Figure 2D). Mutations of Tyr155, Asn157, and Asn281 can potentially destabilize the intermediate. Thr65 is on the loop whose backbone amides interact with the intermediate's carboxylate group (Figure 2E). The Ser and Val mutations of Thr65 can alter the loop configuration and interfere with substrate and intermediate stability. Conservative mutations of Trp128 did not affect structural integrity or activity because it is located at the surface of the protein and extends outward towards solvent (Figure 2E).
Figure 2.

Reaction intermediate of NahE captured by X-ray crystallography. (A) The density maps of Lys183 modified by the Schiff base intermediate. The omit map (Fo - Fc) of Lys183 is shown in green mesh with a contour of 3 rmsd. The 2 Fo − Fc map is shown in charcoal mesh contoured to 1.5 rmsd. (B) 2 Fo − Fc map of the active site with the intermediate formation on the Lys183 contoured to 1.5 rmsd. (C) The hydrophilic interactions of intermediate with the active site. The hydrogen bonds are shown in orange dash lines with the distance (Å) labeled on the side. (D) The hydrophobic interaction formed by the modified Lys183 at the active site. (E) The amino acid residues explored in this study. The one letter amino acid codes are used in the Figure for simplicity.
DISCUSSION
NahE and PhdJ are bifunctional hydratase-aldolases that catalyze key reactions in bacterial degradative pathways for naphthalene and phenanthrene, respectively. Because of the importance they could play in the development of bioremediation technologies and biocatalysts for use in chemical processes, the mechanisms and individual specificities have garnered significant interest. Both are members of the N-acetylneuraminate lyase (NAL) subgroup of the aldolase superfamily.14,18 Sequence and crystallographic analysis along with mutagenesis and Na(CN)BH3 trapping experiments identified the major catalytic and binding elements.5,6,34 The results implicate a Schiff base mechanism in the initial hydration reaction and subsequent retro-aldol bond cleavage. Formation of the Schiff base involves a conserved Lys and Tyr (mediated by Lys183/Tyr155 in NahE and Lys180/Tyr152 in PhdJ). In addition, there is a GXXGE motif (Gly64, Thr65, Phe66, Gly67, Glu68 in NahE and Gly61, Thr62, Phe63, Gly64, Glu65 in PhdJ) that is involved in binding of the carboxylate group of the pyruvoyl moiety of substrate. In order to identify other residues involved in binding and catalysis, five active site residues were targeted for initial mutagenesis work.
Two mutants (W128Y and W128F) have kinetic parameters and activities that are nearly comparable to those of the wild type. Trp128 is contributed from an adjacent monomer (as indicated by the prime in Figure 1). It is on a loop that inserts into another monomer and might stabilize the tetramer. Tyrosine and phenylalanine are conservative mutations and the unchanged kinetic parameters suggest that these changes do not affect structural integrity or catalysis. Replacing Trp128 with a less conservative mutation could provide further insight.
The most detrimental effect on activity (as assessed by kcat/Km) occurs with the N281L mutation for which only a kcat/Km value could be measured: the enzyme could not be saturated with substrate. The structure shows that Asn281 (along with Glu285) plays a role in anchoring the o-hydroxy group of substrate.5,6 Substrates without the o-hydroxy group have greatly reduced kcat/Km values (600 M−1 s−1 when R = H in Scheme 1 and 1.2 × 103 M−1 s−1 when R = CO2−).5 Replacing Asn281 with leucine might prevent substrate from binding altogether due to the steric bulk (or skew the binding) and result in loss of hydrogen bond capability. A more conservative mutation (e.g., serine or histidine ) could prove more insight.
The backbone amide proton of Thr65 (along with that of Phe66) is proposed to bind the carboxylate group of HBP based on the crystal structure of NahE with the bound intermediate along with the sequence conservation in the subfamily (Figure 2).5,6,14,18 Amide protons are not affected by mutation of the side chain. However, replacing the threonine with a valine has a severe effect on activity (kcat/Km decrease 10,000-fold). The bulk would likely disrupt the GXXGE loop so as to hinder substrate binding and adversely affect the positioning of the substrate and intermediate (and disrupt interactions with stabilizing residues). The T65S mutant is a conservative change such that the disruption of the loop is not as great and the consequences listed above can occur, but not to the same degree. Hence, the effect on the kinetic parameters is not as severe.
Tyr155 and Asn157 are proximal to each other suggesting that Asn157 might assist Tyr155. The decrease in activity for the Y155F mutant is consistent with its role in Schiff base formation and hydrolysis as well as activation of the water molecule for addition at C4. Tyr155 is proximal to Lys183 (in two structures) and is highly conserved in the NAL subgroup members, all consistent with a role in Schiff base formation.5,6,14,18 In addition to assisting Tyr155, Asn157 might play a role in a hydrogen bond network (like Asn154 in PhdJ). In both enzymes, the crystal structures show several water molecules (along with a helix insert that confers hydrophobic character). One water molecule is anchored by Asn157 (in NahE) and one is anchored by Asn154 in PhdJ. The water molecule is strategically placed for addition to C4 of substrate (see Scheme 1). Changing the Asn side chain to an alanine or a leucine will disrupt the position of the water molecule and hinder catalysis.
These observations can be formulated into the mechanism shown in Scheme 3 that parallels that proposed for PhdJ.5 In step 1, the Schiff base is formed between Lys-183 and substrate (assisted by Tyr155) and an unknown BH that facilitates departure of the hydroxy group of the carbinolamine.35 This could be a water molecule. In step 2, hydration occurs at C4 (where the water molecule is activated Tyr155, as shown) and assisted in placement by Asn-157. The crystal structure shows a hydrogen bond between the side chain amide and the OH at C4. Tyr155 also initiates bond cleavage to release salicylaldehyde and the Schiff base of pyruvate. In Step 3, the Schiff base of pyruvate undergoes hydrolysis (mediated by Lys183 and Tyr155). Converting tyrosine to a phenylalanine (i.e., the Y155F mutant) slows down the rate of hydrolysis, as suggested by the accumulation of pyruvate-labeled enzyme (in the trapping experiments collected in Table 4).
Scheme 3.

One possible mechanism for NahE.
Finally, salicylaldehyde inhibits product formation in the NahE-catalyzed reaction.33,34 It was first reported that increasing the concentration of salicylaldehyde decreased the Vmax for the reaction using 8.33 It was later reported that increasing the concentration of 8 does not overcome the inhibition.34 Both observations suggest that salicylaldehyde binds at another site and might be an allosteric regulator. In order to identify the second site, we examined the inhibition in more detail.
The Y155F mutant gave the highest quantity of doubly salicylaldehyde-labeled enzyme. Hence, two experiments were carried out with this mutant. In one, the mutant enzyme was incubated with 8 (6 eq) and Na(CN)BH3 overnight. The trapped product was isolated and subjected to protease V8 digestion. Subsequent mass spectral analysis showed that two lysines were covalently modified by salicylaldehyde: the active site lysine (Lys183) and one outside the active site (Lys279). The lysines are separated by a distance of 19.6 Å. It appears that the salicylaldehyde is generated at the active site in the course of the reaction and diffuses out of the active site to modify Lys279. These were the only two lysines modified in the protein to any significant degree. In the second experiment, the enzyme was treated with salicylaldehyde and Na(CN)BH3 and allowed to incubate overnight. The trapped product was isolated and subjected to protease V8 digestion. In this case, Lys183 was covalently modified, but Lys279 was not. This result argues against diffusion of salicylaldehyde from the active site into solution and then returning to the enzyme and modifying an exposed lysine. The significance of the modification of Lys279 is unknown, and whether it represents a potential allosteric site or just some random event. The role of Lys279 is under investigation.
Supplementary Material
Highlights:
Five active site residues in NahE were examined by site-directed mutagenesis and kinetic parameters for eight mutants were determined.
The NahE- and mutant-catalyzed reactions were incubated with substrate and Na(CN)BH3 and the products identified and quantified by mass spectrometry. The results mostly paralleled the kinetic analysis.
A crystal structure of the Schiff base of the intermediate in the NahE-catalyzed reaction suggests a structural basis for the impaired catalysis of the mutant-catalyzed reactions and new insights into the wild type-catalyzed reaction.
Salicylaldehyde released from the Y155F-catalyzed reaction using trans-o-hydroxybenzylidenepyruvate covalently modifies Lys183 as well as Lys279 (19.6 Å from Lys183). Modification of Lys279 could be responsible for the observed inhibition of the wild-type enzyme.
ACKNOWLEDGEMENTS
The protein mass spectrometry analysis was conducted in the Institute for Cellular and Molecular Biology Protein and Metabolite Analysis Facility at the University of Texas at Austin.
Funding
This research was supported by the National Institutes of Health Grant GM-129331 and the Robert A. Welch Foundation Grant 2125. Protein identification was provided by the UT Austin Center for Biomedical Research Support Biological Mass Spectrometry Facility (RRID:SCR_021728).
ABBREVIATIONS
- BP
benzylidenepyruvate
- DEAE
diethylaminoethyl
- dNTPs
deoxynucleoside triphosphates
- ESI-MS
electrospray ionization mass spectrometry
- HBP
trans-o-hydroxybenzylidenepyruvate
- ICMB
Institute for Cellular and Molecular Biology
- IPTG
isopropyl β-D-thiogalactoside
- Kn
kanamycin
- KOD
Thermococcus kodakarensis
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LB
Lysogeny broth
- MALDI-MS
matrix-assisted laser desorption/ionization-mass spectrometry
- MS
mass spectrometry
- NahE
hydratase-aldolase in the naphthalene degradative pathway
- NahE-HI
NahE-hydroxy intermediate
- NMR
nuclear magnetic resonance
- NAL
N-acetylneuraminate lyase
- Ni-NTA
nickel-nitriloacetic acid
- PhdJ
hydratase-aldolase in the phenanthrene degradative pathway
- PMSF
phenylmethylsulfonyl fluoride
- PAH
polycyclic aromatic hydrocarbon
- PCR
polymerase chain reaction
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Code
The atomic coordinates and structure factors have been deposited in the Protein Data Bank as entry 8DO5 for the NahE bound intermediate.
The authors declare no competing financial interest.
REFERENCES
- 1.Eaton RW, and Chapman PJ (1992) Bacterial metabolism of naphthalene: construction and use of recombinant bacteria to study ring cleavage of 1,2-dihydroxynaphthalene and subsequent reactions. J. Bacteriol 174, 7542–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kweon O, Kim S-J, Holland RD, Chen H, Kim D-W, Gao Y, Yu L-R, Baek S, Baek D-H, Ahn H, and Cerniglia CE (2011) Polycyclic aromatic hydrocarbon metabolic network in Mycobacterium vanbaalenii PYR-1. J. Bacteriol 193, 4326–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stingley RL, Khan AA, and Cerniglia CE (2004) Molecular characterization of a phenanthrene degradation pathway in Mycobacterium vanbaalenii PYR-1. Biochem. Biophys. Res. Commun 322, 133–146. [DOI] [PubMed] [Google Scholar]
- 4.Iwabuchi T, and Harayama S (1998) Biochemical and genetic characterization of trans-2'-carboxybenzalpyruvate hydratase-aldolase from a phenanthrene-degrading Nocardioides strain. J. Bacteriol 180, 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LeVieux JA, Medellin B, Johnson WH Jr., Erwin K, Li. W, Zhang YJ, and Whitman CP (2018). Structural characterization of the hydratase-aldolases, NahE and PhdJ: implications for specificity, catalysis, and the N-acetylneuraminate lyase subgroup of the aldolase superfamily. Biochemistry 57, 3524–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeVieux JA, Johnson WH Jr., Erwin K, Li. W, Zhang YJ, and Whitman CP (2016) The bacterial catabolism of polycyclic aromatic hydrocarbons: characterization of three hydratase/aldolase-catalyzed reactions. Perspect Sci 9, 33–41. [Google Scholar]
- 7.PAHs: An Ecotoxicological Perspective (Douben PET, Ed.) (2003) John Wiley & Sons, Ltd. [Google Scholar]
- 8.Penning TM, Burczynski ME, Hung C-F, McCoull KD, Palackal NT, and Tsuruda LS (1998) Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem. Res. Toxicol 12, 1–18. [DOI] [PubMed] [Google Scholar]
- 9.Atlas RM, and Hazen TC (2011) Oil biodegradation and bioremediation: A tale of the two worst spills in U.S. history. Environ. Sci. Technol 45, 6709–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fansher DJ, Granger R, Kaur S, and Palmer DRJ (2021) Repurposing an aldolase for the chemoenzymatic synthesis of substituted quinolines. ACS Catal. 11, 6939–6943. [Google Scholar]
- 11.Eaton RW (2000) trans-o-Hydroxybenzylidenepyruvate hydratase-aldolase as a biocatalyst. Appl. Environ. Microbiol 66, 2668–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrara S, Mapelli E, Sello G, and Di Gennaro P (2011) Characterization of the aldol condensation activity of the trans-o-hydroxybenzylidenepyruvate hydratase-aldolase (tHBP-HA) cloned from Pseudomonas fluorescens N3. Biochim. Biophys. Acta, Proteins Proteomics 1814, 622–629. [DOI] [PubMed] [Google Scholar]
- 13.Sello G, and Di Gennaro P (2013) Aldol reactions of the trans-o-Hydroxybenzylidenepyruvate hydratase-aldolase (tHBP-HA) from Pseudomonas fluorescens N3. Appl. Biochem. Biotechnol 170, 1702–1712. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence MC, Barbosa JA, Smith BJ, Hall NE, Pilling PA, Ooi HC, and Marcuccio SM (1997) Structure and mechanism of a sub-family of enzymes related to N-acetylneuraminate lyase. J. Mol. Biol 266, 381–399. [DOI] [PubMed] [Google Scholar]
- 15.Blickling S, Renner C, Laber B, Pohlenz HD, Holak TA, and Huber R (1997) Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry 36, 24–33. [DOI] [PubMed] [Google Scholar]
- 16.Theodossis A, Walden H, Westwick EJ, Connaris H, Lamble HJ, Hough DW, Danson MJ, and Taylor GL (2004) The structural basis for substrate promiscuity in 2-keto-3-deoxygluconate aldolase from the Entner-Doudoroff pathway in Sulfolobus solfataricus. J. Biol. Chem 279, 43886–43892. [DOI] [PubMed] [Google Scholar]
- 17.Taberman H, Andberg M, Parkkinen T, Janis J, Penttila M, Hakulinen N, Koivula A, and Rouvinen J (2014) Structure and function of a decarboxylating Agrobacterium tumefaciens keto-deoxy-D-galactarate dehydratase. Biochemistry 53, 8052–8060. [DOI] [PubMed] [Google Scholar]
- 18.Barbosa JA, Smith BJ, DeGori R, Ooi HC, Marcuccio SM, Campi EM, Jackson WR, Brossmer R, Sommer M, and Lawrence MC (2000) Active site modulation in the N-acetylneuraminate lyase sub-family as revealed by the structure of the inhibitor-complexed Haemophilus influenzae enzyme. J. Mol. Biol 303, 405–421. [DOI] [PubMed] [Google Scholar]
- 19.Daniels AD, Campeotto I, van der Kamp MW, Bolt AH, Trinh CH, Phillips SE, Pearson AR, Nelson A, Mulholland AJ, and Berry A (2014) Reaction mechanism of N-acetylneuraminic acid lyase revealed by a combination of crystallography, QM/MM simulation, and mutagenesis. ACS Chem. Biol 9, 1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch EF, and Maniatis T (1989) Molecular Cloning: A Laboratory Manual, 2nd ed, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 21.Waddell WJ (1956) A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med 48, 311–314. [PubMed] [Google Scholar]
- 22.Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 23.Laible M, and Boonrod K (2009) Homemade site directed mutagenesis of whole plasmids. J Vis Exp. 27, e1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SC, Johnson WH Jr., and Whitman CP (2003) The 4-oxalocrotonate tautomerase- and YwhB-catalyzed hydration of 3E-haloacrylates: implications for the evolution of new enzymatic activities. J. Am. Chem. Soc 125, 14282–14283. [DOI] [PubMed] [Google Scholar]
- 25.Wang SC, Person MD, Johnson WH Jr., and Whitman CP (2003) Reactions of trans-3-chloroacrylic acid dehalogenase with acetylene substrates: consequences of and evidence for a hydration reaction. Biochemistry 42, 8762–8773. [DOI] [PubMed] [Google Scholar]
- 26.Houmard J, and Drapeau GR (1972) Staphylococcal protease: a proteolytic enzyme specific for glutamoyl bonds. Proc. Natl. Acad. Sci. U.S.A 69, 3506–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 28.McCoy AJ (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Lano WL (2002) The PyMol molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 33.Kuhm AE, Knackmuss H-J, and Stolz A (1993) Purifcation and properties of 2’-hydroxybenzalpyruvate aldolase from a bacterium that degrades naphthalenesulfonates. J. Biol. Chem 268, 9484–9489. [PubMed] [Google Scholar]
- 34.LeVieux JA Structure-based mechanism of hydratase-aldolases in the Type 1 aldolase superfamily and structures of tautomerase superfamily members. Ph.D. Dissertation, University of Texas at Austin, Austin, TX, May 2017. [Google Scholar]
- 35.Choi KH, Lau V, Foster CE, Morris AJ, Tolan DR, and Allen KN (2006) New superfamily members identified for Schiff-base enzymes based on verification of catalytically essential residues. Biochemistry 45, 8546–8555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.