

Abstract
Histidine phosphorylation is an important and ubiquitous post-translational modification. Histidine undergoes phosphorylation on either of the nitrogens in its imidazole side chain, giving rise to 1- and 3- phosphohistidine (pHis) isomers, each having a phosphoramidate linkage that is labile at high temperatures and low pH, in contrast with stable phosphomonoester protein modifications. While all organisms routinely use pHis as an enzyme intermediate, prokaryotes, lower eukaryotes and plants also use it for signal transduction. However, research to uncover additional roles for pHis in higher eukaryotes is still at a nascent stage. Since the discovery of pHis in 1962, progress in this field has been relatively slow, in part due to a lack of the tools and techniques necessary to study this labile modification. However, in the past ten years the development of phosphoproteomic techniques to detect phosphohistidine (pHis), and methods to synthesize stable pHis analogues, which enabled the development of anti-phosphohistidine (pHis) antibodies, have accelerated our understanding. Recent studies that employed anti-pHis antibodies and other advanced techniques have contributed to a rapid expansion in our knowledge of histidine phosphorylation. In this review, we examine the varied roles of pHis-containing proteins from a chemical and structural perspective, and present an overview of recent developments in pHis proteomics and antibody development.
Covalent post-translational modifications (PTMs) of different amino acids in proteins increase the chemical complexity of the proteome, and, thereby, diversify cellular functions. Reversible phosphorylation is the most abundant PTM, and is involved in protein–protein interactions, protein trafficking, immune responses, cell cycle division, etc. [1]. Deregulated protein phosphorylation is implicated in many diseases. Of the nine amino acids that can be phosphorylated in biological systems, Ser, Thr and Tyr undergo O-linked phosphorylation to form a phosphomonoester linkage that is stable at high temperatures, and under acidic or basic conditions. Their chemical stability makes pSer, pThr and pTyr amenable to study using conventional biochemical techniques, and they constitute most of the current phosphoproteome, and they are called canonical phosphorylations. In contrast, the phosphorylated forms of the three basic amino acids, His, Arg and Lys, which undergo N-linked phosphorylation to form a phosphoramidate linkage, are hydrolyzed at high temperatures and in acidic conditions [2]. The phosphorylated forms Asp and Glu, which form acyl linkages and, Cys, which forms a thioester linkage upon phosphorylation, exhibit similar lability. pHis, pLys, pArg, pAsp, pGlu and pCys are known as non-canonical or cryptic phosphorylations, because of their functional anonymity and apparently limited occurrence in the proteome. However, recent advances in analytical tools and high sensitivity workflows have led to the realization that non-canonical phosphorylation, especially of His, Lys and Arg, may be more common than previously suspected and have a wide variety of functions. Phosphorylation of these three amino acids has even been predicted to be important in the prebiotic chemical evolution where their chemistry was utilized to synthesize biomolecules and their acid-labile nature was exploited as an on/off switch to regulate the intracellular microenvironment [3].
Among the His, Lys and Arg non-canonical phosphorylations, phosphohistidine (pHis) is the most common and well studied. pHis was discovered in 1962 in mitochondrial protein fractions of bovine liver by Boyer et al. [4]. Subsequent studies showed that the protein harboring pHis was succinyl Co-A synthetase (SCS), a key enzyme in the TCA cycle that generates GTP. Boyer and colleagues worked at the interface of chemistry and biology to contribute to the initial understanding of the chemical properties of the 1- and 3-isoforms of monomeric pHis, both of which are found in proteins [5,6]. Although the first protein with pHis was discovered in eukaryotes, the pHis field is dominated by instances from prokaryotes. Indeed, the long-standing dogma was that pHis biology is only important in prokaryotes, lower eukaryotes and plants [7]. Not until more recently did an interest in the role of pHis in metazoans begin to build, largely due to the development of new tools and techniques for detecting and analyzing pHis in proteins. Several excellent reviews have discussed the role of pHis in prokaryotes, and some more recent articles have covered pHis in eukaryotes and the design and synthesis of pHis analogues, which have allowed the development of anti-pHis antibodies [2,7–14]. Here, we aim to bring together the present understanding of the biochemical aspects of pHis PTM biology and present an overview of the structures of pHis-containing proteins available in the PDB structure database, many of which have not been considered previously. Such a structural perspective is important to understand the functions of histidine phosphorylation of proteins. We then focus on recent advances in phosphoproteomics and pHis-specific antibody development and the lessons learnt about how 1-pHis and 3-pHis are selectively recognized based on the recently solved structures of a series of pHis monoclonal antibodies by comparison with the known structures of other phospho-specific antibodies. We conclude by presenting perspectives on where the pHis field will go in the next few years.
Biochemical and functional versatility of phosphohistidine
The five-membered imidazole side chain of histidine has two phosphorylatable nitrogens, N1 and N3, giving rise to two phosphoisomers, 1-pHis (or pros-pHis) and 3-pHis (or tele-pHis), respectively (Figure 1A). Some of the literature and the PDB (protein data bank) use Nδ1 and Nε2 to refer to N1 and N3 positions on imidazole ring of His, respectively. The physical properties of these two isomers are quite different. The phosphoramidate bond in free 1-pHis is relatively unstable compared with that in 3-pHis, which is attributable to its proximity to the α-amino group making the phosphoryl group more electrophilic and a better leaving group. However, both pHis isomers are labile at high temperatures and acidic pH, with half-lives in order of seconds (18–25 s) in presence of 1 M HCl and 46°C [6]. Whereas 1-pHis has a half-life of 1 and 34 min at pH 7 and pH 8, respectively, 3-pHis has a half-life of 78 min at pH >7 at 46°C [6,13]. Note that the half-life of the monomeric pHis amino acid is quite different from that of the pHis in peptides and proteins; for example, the half-life of pHis in histone H4 was estimated to be 2 h [15]. This means that the local peptide environment influences the stability of pHis. A third form pHis has been reported in studies of chemically phosphorylated histidine — 1,3-diphosphohistidine, where both the 1- and 3- positions are phosphorylated (Figure 1A) [5]. Upon standing, this doubly phosphorylated His compound decomposes to 3-pHis. Although 1,3-diphosphohistidine has been observed in peptides, it does not appear to have any biological significance, as it is not reported to occur in proteins [16,17]. The free energy ΔG°) of hydrolysis of the phosphoramidate bond in pHis is −12 to −14 kcal/mol, compared with −6.5 to −9.5 kcal/mol for phosphomonoesters, meaning that pHis is thermodynamically less stable than pTyr/pSer/pThr in proteins [18].
Figure 1. Chemistry and biochemical functions of phosphohistidine.

(A) Histidine undergoes phosphorylation on N1 and N3 to form 1-pHis and 3-pHis, respectively. 1,3 diphosphohistidine can also be formed upon His phosphorylation in vitro. (B) Some metabolic enzymes use a catalytic His to accept a phosphoryl group from a donor before transferring it onto an acceptor, thus acting as enzyme intermediate (upper panel). Histidine kinase (HK) undergoes phosphorylation on the active site His to generate pHis and then transfers the phosphoryl group onto a His in a substrate. pHis phosphatases act by hydrolyzing pHis present either in substrates or HKs (lower panel). Created with biorender.com.
The side chain of His has pKa close to physiological pH. This property allows His to act as a nucleophile, a general acid or a general base in many enzymatic reactions; unsurprisingly, His is present at the active site of ~50% of enzymes [19]. Not only His itself, but also the N-phosphorylated and N-methylated His are post translationally modified forms that represent an alternate, elegant way for His to participate in enzymatic reactions and couple it to regulatory functions of the proteins [4,20].
Recent literature suggests that a wide range of proteins undergo pHis post-translational modification, and based on the functional and mechanistic aspects, these proteins can be broadly classified under four categories, (a) proteins that use pHis as an enzyme intermediate, (b) histidine kinases (HK) with protein kinase activity, (c) HK substrates and (d) pHis phosphatases (Figure 1B).
-
a
Enzymes that utilize pHis: Proteins that use pHis as an enzyme intermediate are mostly metabolic enzymes and function by shuffling phosphate between small molecules (Figure 3D and 4A). A generalized mechanism involves a His at the active site of an enzyme that carries out a nucleophilic attack on the substrate phosphorous atom to form an intermediary covalent phosphoryl-His-enzyme complex (either 1- or 3-pHis). A general acid/base in the catalytic site then mediates the hydrolysis of the phosphoramidate linkage using water/hydroxide ion as a nucleophile to reform the enzyme and release products (Figure 1B).
Figure 3. Role of pHis in prokaryotes.
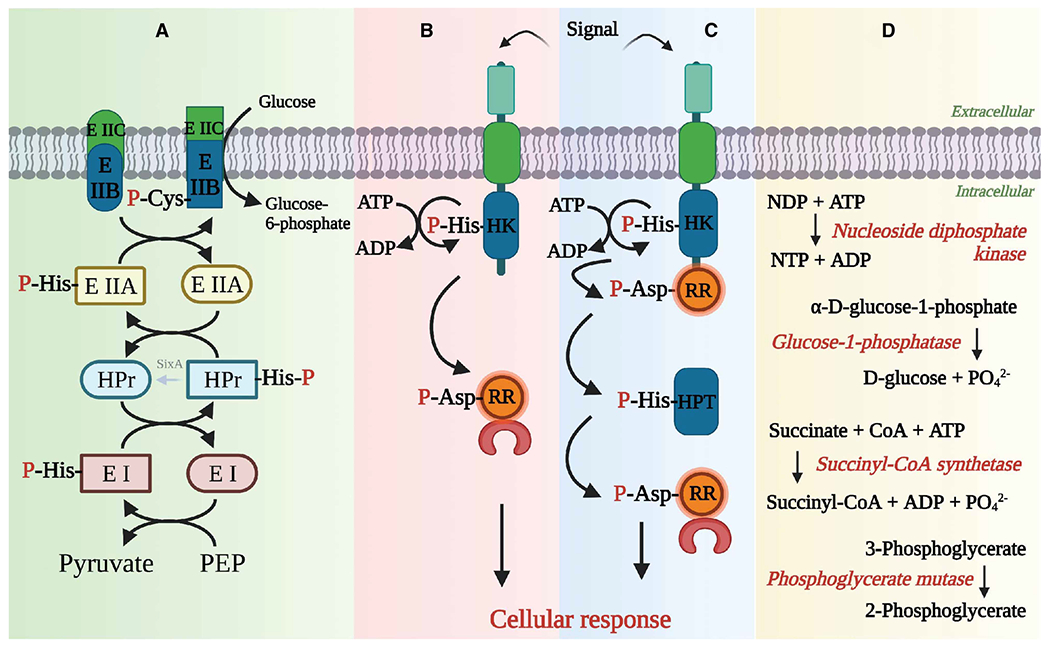
(A) Multi-component phosphotransferase systems use His to relay a phosphoryl group from a high energy PEP molecule to a sugar molecule through a series of enzymes, such as EI, HPr and EII. (B) Two component systems (TCS) are the major signal transduction systems present in prokaryotes, lower eukaryotes and plants. A simple TCS module has a HK that senses the signal, which triggers phosphorylation of its catalytic His, and this phosphoryl group is then transferred onto an Asp in response regulator (RR) protein, which is involved in the cellular response to the signal. (C) Multistep phospho-relay systems are similar to TCS, but have more than one pair of HK and RR proteins. The phosphate is relayed through the pathway in a tandem manner. (D) pHis also acts as an enzyme intermediate in some of the prokaryotic metabolic enzymes. Some representative enzymes are noted here. Created with biorender.com.
Figure 4. Role of pHis in mammals. pHis has multifaceted roles to play in eukaryotes, especially mammals.

(A) pHis acts as an enzyme intermediate in enzyme superfamilies like histidine acid phosphatases and NDPKs, and enzymes from the acyl CoA synthetase family, phospholipase D family, etc. (B) NME1 and NME2 are the only known HKs in mammals. # Histone H4 HK is another reported mammalian HK that phosphorylates histone H4 but it has not been isolated or characterized so far. (C) There are several substrates for HKs, which accept a phosphoryl group from NME1/2, but for some the phosphate donor is not yet determined. (D) Phosphohistidine phosphatases (pHis phosphatases) hydrolyze the phosphoramidate bond in phospho-HK and its pHis substrates. PHPT1 dephosphorylates several HK substrates, while PGAM5 can dephosphorylate NME2. LHPP is known to hydrolyze the phosphoramidate bond, but its substrates remain undetermined. Created with biorender.com.
Most of these enzymes belong to superfamilies/families of enzymes, such as acyl-CoA synthetases, phospholipase D, and histidine acid phosphatases. The phosphoryl transfer in these families of proteins occurs via a 3-pHis intermediate. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB), phosphoglycerate mutase 1 (PGAM1) and glucose-1-phosphatase, all members of the histidine acid phosphatase superfamily, are involved in the glycolysis and gluconeogenesis pathways (Figure 2B) [21]. Other notable proteins from this superfamily are prostate acid phosphatase, STS-1 (UBASH3B/TULA-2) and STS-2 (UBASH3A/TULA-1), which have phosphotyrosine phosphatase activity but act through a 3-pHis enzyme intermediate [21]. Succinyl Co-A synthetase (SCS or SUCLG1), ATP citrate lyase (ACLY) and ADP-forming acetyl Co-A synthetases, involved in the TCA cycle, and lipid and amino acid biosynthesis, respectively, belong to the acyl-CoA synthetase family [22]. Phospholipase D and tyrosyl-DNA phosphodiesterase I (TDP1), members of the phospholipase D superfamily, are involved in lipid biosynthesis and single-strand DNA break repair, respectively [23,24]. Glucose-6-phosphatase is in the PAP2 (Type 2 phosphatidic acid phosphatase) family of enzymes, and also uses His as a nucleophilic center to hydrolyze glucose-6-phosphate to glucose in the gluconeogenesis and glycogenolysis pathways, thus regulating cellular glucose levels [25].
Figure 2. Catalytic mechanisms of phosphohistidine proteins.

(A) NDPK catalyzes transfer of a phosphoryl group from ATP to NDP in a nucleophilic substitution reaction. The catalytic mechanism involves synchronous shift of a proton from N1 of His122 to oxygen of the γ-phosphate and phosphoryl transfer of the γ-phosphate of ATP onto N1 of His122 to form 1-pHis (residues are numbered based on Dictyostelium discoideum NDPK). In the next step, 1-pHis is hydrolyzed by nucleophilic attack of NDP and NTP is released as the final product.
(B) 2,3-Bisphosphoglycerate (2,3-BPG) is the substrate of PGAM1 (residues are numbered based on human PGAM1) which is converted into either 2-phosphoglycerate (2-PG) or 3-PG. His11 nucleophilically attacks 2,3-BPG to extract a phosphoryl group to form pHis, which is subsequently hydrolyzed by a water-derived hydroxyl ion with the help of Glu89 (general base) present at the active site. Arg10, Gly12, Arg62, His186 are the conserved residues of histidine acid phosphatase family and they interact electrostatically with the catalytic residues, substrates and products. (C) PHPT1 is a pHis phosphatase (residues are numbered based on human PHPT1) that uses catalytic His53 as a general base to extract a proton from a water molecule thus activating it to nucleophilically attack the phosphate of a pHis protein to hydrolyze.
Nicotinamide phosphoribosyltransferase (NAMPT) is another metabolic enzyme that utilizes a pHis intermediate, but in a slightly different way. NAMPT catalyzes the condensation of nicotinamide with 5′-phosphoribosyl-1-pyrophosphate (PRPP) to form nicotinamide mononucleotide (NMN), the precursor in the NAD+ biosynthesis pathway. ATP-mediated phosphorylation of NAMPT on the active site His to form a 1-pHis enzyme intermediate increases the catalytic efficiency of the enzyme by 1000 fold, favoring the formation of NMN [26]. The nucleoside diphosphate kinases (NDPK) catalyze the transfer of a phosphoryl group from ATP to non-adenosine nucleoside diphosphates through a 1-pHis intermediate thus maintaining nucleotide triphosphate homeostasis (Figure 2A) [27,28]. The NDPKs form an eponymous superfamily of proteins that share ~30% sequence homology, and are omnipresent in all kingdoms of life; these enzymes are called NDKs in prokaryotes and lower eukaryotes and NME/NDPK proteins in higher eukaryotes. There are 10 members in the mammalian NME family of proteins (NME1–10) that differ in terms of sequence identity, catalytic activity and cellular localization [29].
-
b
Enzymes that utilize pHis to transfer a phosphoryl group onto histidine residues in proteins: The second category of proteins are histidine kinases that transfer a phosphoryl group onto His or sometimes another amino acid in a second protein. Out of 10 members in the NDPK or NME family of proteins in eukaryotes, NME1 (NDPK-A) and NME2 (NDPK-B) are reported to have a moonlighting function as protein-histidine kinases (HK); in fact, these are the only HKs known in eukaryotes so far [30] (Figure 4B). They catalyze the transfer of a phosphoryl group from the active site 1-pHis onto a histidine (or sometimes onto a Ser as in KSR1 or Asp as in Aldolase C) present in the substrate proteins [31,32]. They are involved in DNA replication, damage and repair, cellular differentiation, development and signal transduction [33]. There is a scanty literature on a histone H4 histidine kinase (HHK) as the second type of HK in eukaryotes. Though its occurrence was reported in regenerating rat liver, human and rat liver, hepatocellular carcinoma cell lines and thymus, this enzyme has not yet been isolated and characterized [34–36].
In the case of prokaryotes, one class of HKs form part of multi-component phosphotransferase systems that are involved in carbohydrate transport, carbon and nitrogen metabolism and potassium homeostasis [37,38]. As the name suggests, these systems are comprised of multiple proteins that relay the phosphoryl group derived from phosphoenolpyruvate (PEP) from one member onto the next (Figure 3A). For example, the PEP:sugar phosphotransferase system has five proteins (EI, HPr, EIIA, EIIB and EIIC), of which EI, HPr, EIIA and EIIB are involved in a cascade, with the phosphoryl group from PEP being relayed onto a hexose sugar using pHis as an enzyme-intermediate (interestingly, pCys is used as the phosphoenzyme intermediate in case of certain sugar specific-EIIBs), while EIIC is a permease that aids the translocation of the sugar across the plasma membranes. EIIB, which is usually fused to EIIC, catalyzes the transfer of phosphoryl group onto the sugar thus preventing its export out of the cell [39]. EI, HPr and some of the EIIA proteins act as the HKs, transferring the phosphoryl group onto His in the next protein in the cascade.
Prokaryotes, lower eukaryotes and plants also have a big repertoire of ‘histidine’ kinases that are part of two-component (TCS) or multi-step phosphorelay signal transduction systems. Although the conventional definition of histidine kinases refers to the proteins that phosphorylate His residues in other proteins, exceptions to the rule are HKs in TCS and multi-step phosphorelays. They catalyze autophosphorylation of a His residue using ATP as the substrate and transfer this phosphoryl group onto an Asp in other proteins instead. While TCSs are involved in chemotaxis, sporulation, quorum sensing, antibiotic resistance, etc. in bacteria [40–43], a multi-step phosphorelay signaling mechanism was usually adopted by lower eukaryotes, including yeast, for osmotic adaptation, oxidative stress, light perception, morphogenetic switch etc. [44–47], and by plants for regulation of ethylene and cytokinin hormone signaling, osmotic stress and drought response [48,49]. A prototypical TCS consists of two proteins namely, a multi-domain HK, which spans the plasma membrane and acts as a receptor, and a response regulator (Figure 3B) [8]. An external signal (small molecule ligand, peptide or light, etc.) in the environment is sensed and transduced into the cells via the HK membrane domains. Upon sensing a signal, the catalytic His on the homodimeric HK undergoes either cis or trans autophosphorylation using the γ-phosphate donated by ATP bound to the HK ATP-binding domain. The phosphoryl group from the HK pHis is then transferred onto an Asp in the cognate response regulator (RR) protein, which is usually a transcription factor, that is then able to up-regulate or down-regulate the transcription of effector genes. In a multi-step phosphorelay signal transduction system, there is more than one HK-RR pair involved in the tandem phosphorelay with an intermediate histidine phosphotransferase (HPT) enzyme (Figure 3C) [50]. Interestingly, no TCS or multi-step phosphorelay systems are found in higher eukaryotes including mammals.
-
c
Proteins that are phosphorylated by histidine kinases: Proteins in the third category are the substrates of HKs, being phosphorylated by the HK on a His residue. The functional span of HK substrates in eukaryotes range from metabolism, DNA replication and transcription, signal transduction, ion channel regulation and homeostasis, inflammatory responses, etc. (Figure 4C). ACLY utilizes citrate and ATP to synthesize acetyl CoA, which can be fed into the lipid biosynthesis pathway or used as a substrate for protein acetyl transferases, such as histone acetyl transferases that regulate gene expression [51,52]. ACLY uses the γ-phosphate of ATP to generate a 3-pHis760 phosphoenzyme intermediate, which is used in turn to form the citryl-phosphate enzyme intermediate used in acetyl-CoA production. In addition to direct ATP-mediated formation of pHis760, pHis118 NDPK-A (NME1) can also donate a phosphoryl group to ACLY His760 [53]. NDPK-B (NME2), the closest paralogue of NDPK-A in the NME family, catalyzes the formation of high energy phosphoramidate bond on the Gβ1 protein (His266), which is then transferred onto the GDP molecule bound to the Gα subunit to form GTP, leading to receptor-independent activation of the G protein [54]. This results in G-protein-dependent cAMP synthesis, which has implications for myocardial contractility [55].
NDPK-B/NME2 is also involved in activation of the KCa3.1 voltage-independent Ca2+ activated K+ ion channel, resulting in activation of B-cells and T-cells [56]. Chelation of Cu[II] by four His358 residues in a cytoplasmic homotetrameric helical bundle of KCa3.1 has been proposed to hold the channel in a closed state. T-cell stimulation elevates cytoplasmic calcium, which binds to calmodulin associated with the channel, resulting in a conformational change in the channel that abrogates the copper chelation and allows phosphorylation of His358 by NDPK-B, thus preventing Cu[II] re-binding and stabilizing the channel in an open state to permit K+ efflux [57]. TRPV5 (transient receptor potential-vanilloid-5) is another ion channel that is activated by phosphorylation of His by NDPK-B. Phosphorylation of His711 leads to channel opening and Ca2+ flux, which subsequently results in Ca2+ reabsorption in kidneys [58].
Annexin I is involved in the regulation of chloride ion concentration in airway epithelia where it is reported to have a phosphorylated His (His246) in its core domain that is involved in Ca2+-dependent phospholipid binding [59]. However, the functional effect of pHis on Annexin I is unexplored. Histone H4, a core subunit of the nucleosome in chromatin, is involved in DNA replication and transcriptional regulation, and it is reported to undergo phosphorylation on His. The site of phosphorylation is ambiguous, as His18 and His75 in regenerating rat liver and His75 in slime mold have been reported to undergo 1-pHis modification while His18 and His75 in Walker carcinosarcoma showed 3-pHis modification [60,61]. While the His phosphorylation of histone H4 is speculated to be mediated by HHK, its functional consequence is still unknown [36]. But based on available structural information Besant et al. suggested that phosphorylation of either of these residues might destabilize the histone–histone or histone–DNA interactions, as His18 is present on the basic tail of histone H4 that interacts with H2A–H2B of the adjacent nucleosome, and His75 is in proximity to the DNA binding region of the nucleosome core [62].
Finally, some HK substrates are either directly or indirectly metal ion-dependent proteins [57–59], and many of these proteins use His as one of the coordinating residues in the metal ion-binding site. This raises the possibility that His phosphorylation might negatively regulate metal ion binding to these proteins, and thereby serve as a general function of His phosphorylation in addition to its role as an acid/base in enzymatic reactions. In this regard, it is notable that phosphopeptide enrichment using pHis antibodies yielded several proteins that coordinate metal ions through His [12].
-
d
Proteins that dephosphorylate pHis: The fourth category of proteins are the pHis phosphatases, which dephosphorylate pHis proteins by hydrolyzing the phosphoramidate bond (Figure 3D). Because of its high chemical lability, pHis was originally assumed to undergo facile spontaneous dephosphorylation in biological systems, until the first mammalian pHis phosphatase, PHPT1, was discovered [63,64]. PHPT1 is reported to act upon 3-pHis proteins like Gβ1, KCa3.1, TRPV5 and ACLY that are phosphorylated either by NME1 or NME2 [58,65–67]. The dephosphorylation of pHis proteins by PHPT1 follows a novel catalytic mechanism, where His53 acts as general base by extracting a proton from a water molecule thus activating it to carry out a nucleophilic attack on substrate proteins’ pHis releasing the phosphate (Figure 2C) [68]. While PHPT1 is localized in the cytoplasm, a recently described pHis phosphatase, PGAM5, a member of the histidine acid phosphatase superfamily, is localized to the mitochondria. (Note: Histidine acid phosphatases are a family of enzymes that use pHis as an enzyme intermediate, and they should not be confused with pHis phosphatases, which hydrolyze the pHis phosphoramidate bond; however, PGAM5 belongs to both categories). The catalytic mechanism involves a nucleophilic attack of the PGAM5 active site His105 on the pHis of a substrate protein to form 3-pHis-enzyme intermediate. The catalytic site uses Arg104, Arg152 and His230 to position the substrates, while Glu176 acts a proton donor to release the phosphate [69]. Prior work had shown that PGAM5 is as an atypical Ser/Thr phosphatase involved in oxidative stress, necroptosis and autophagy [69]. Recent studies by Panda et al. suggest that PGAM5 can even dephosphorylate His118 on NDPK-B/NME2 HK thus inhibiting KCa3.1 channel activation and subsequent inhibition of T-cell receptor stimulated Ca2+ influx and cytokine production in CD4+ T cells [70]. SixA is a bacterial pHis phosphatase that belongs to the histidine acid phosphatase superfamily, like PGAM5, and has a similar catalytic mechanism. Schulte et al. [71] showed that SixA dephosphorylates Npr, a phosphocarrier protein belonging to the nitrogen-related phosphotransferase system in E. coli. It is worth noting that PGAM5 and SixA selectively dephosphorylate 1-pHis [70,71].
Another phosphohistidine phosphatase, LHPP (phospholysine phosphohistidine inorganic pyrophosphate phosphatase) belongs to the haloacid dehalogenase (HAD) superfamily of hydrolases. In LHPP, a catalytic Asp acts as a nucleophile by extracting phosphate from a substrate to form a phosphoaspartyl intermediate in a magnesium-dependent manner, which is then hydrolyzed [72]. GWAS analysis of LHPP suggested a role in major depressive disorder (MDD) and alcohol dependence [73,74]. Hindupur et al. [75] reported that the LHPP acts a tumor suppressor gene in hepatocellular carcinoma (HCC), noting that its levels are down-regulated in human HCC, and, conversely, that phospho-NME1/2 and pHis protein levels are up-regulated, suggesting that elevated His phosphorylation might promote HCC. Subsequent studies have implicated LHPP’s role in various types of cancer [76–78]. However, LHPP pHis protein substrates have not yet been uncovered or validated biochemically. Even the role of LHPP as a protein phosphatase is in question, because the NagD clade of the HAD superfamily to which LHPP belongs is shared by proteins like pyridoxal phosphatase and phosphoglycolate phosphatase that are known to have smaller metabolite substrates [79]. Doubts about whether LHPP acts as a protein phosphatase are supported by the LHPP X-ray structure (PDB ID: 2X4D), which reveals a narrow opening to the catalytic site that may prevent access of a protein pHis residue to the catalytic site [79]. This leaves open the possibility that LHPP acts through an alternative mechanism to reduce pHis protein levels in cells. Meanwhile, a recent study by Chen et al. suggests that LHPP induces degradation of PKM2, a glycolytic enzyme, by a ubiquitin-mediated proteolytic pathway thus inhibiting the growth of glioblastoma [80].
Structural diversity of phosphohistidine proteins
To date, the PDB has 1544, 1181 and 739 protein or peptide structures that contain a phosphoserine (PDB chemical id: SEP), phosphothreonine (PDB chemical id: TPO) or phosphotyrosine (PDB chemical id: PTR) modification (Source: https://www.rcsb.org/). Due to its lability, currently there are only a few (<70) X-ray and NMR structures of proteins with pHis modification, including 7 (5 X-ray and 2 NMR) structures of proteins with 1-pHis (PDB chemical id: HIP), and 53 structures (52 X-ray and 1 NMR) with 3-pHis (PDB chemical id: NEP). Nevertheless, these relatively few structures still provide valuable insights into what kind of structural changes take place in a protein after pHis modification in comparison with the unphosphorylated protein, and how this is manifested in the protein’s function. They also provide insights into whether the amino acid environment around the pHis residue dictates the isomer specificity and stability of the pHis modification as well as providing valuable insights into structural motifs that favor the presence of pHis.
Structural changes in proteins dependent upon His phosphorylation vary widely. For example, proteins like the NDPKs exhibit essentially no structural changes upon phosphorylation, and HPr and enzymes from histidine acid phosphatase superfamily undergo only subtle changes. In contrast, enzymes in the acyl Co-A synthetase family can exhibit loop relocations of the order of tens of angstroms, and TCS HKs have prominent changes in the protein that have functional implications. NDPKs catalyze the transfer of γ-phosphate from ATP to a non-adenosine nucleoside diphosphate using a phosphoryl-His-enzyme intermediate complex. The structure of this phosphoenzyme complex was captured by the structures of slime mold and Drosophila NDPKs (PDB ids: 1NSP and 1NSQ, respectively) with 1-pHis modification [81]. Phosphorylation of the active site His was achieved by diffusing phosphoramidate into the crystals. The catalytic His (His122 in 1NSP) is located on a β-strand in a four-stranded antiparallel β-sheet and exposed to the solvent. Because the N3 position of His122 interacts with Glu (Glu133 in 1NSP), only the N1 position of His122 is available to undergo phosphorylation, which is then stabilized by a Tyr (Tyr56 in 1NSP) and water molecules. The 1-pHis modification did not result in any conformational changes in the protein (RMSD of 0.16 Å on Cα positions in 1NSP and 1KDN) (Figure 5A) [81].
Figure 5. Structural diversity of pHis proteins.

(A) NDPK from Dictyostelium is phosphorylated on His122. Cα overlaid structures of phosphorylated vs unphosphorylated forms reveals no detectable structural changes upon phosphorylation (inset) (NDPK- pale green and pHis-NDPK-light pink). The corresponding PDB IDs are provided in the figure. (B) MIPP in Bifidobacterium is phosphorylated on His45. Phosphorylation induces changes only in residues around the active site with respect to the substrate (IHS-D-myo-inositol hexaSulfate) binding (inset) (MIPP-pale green and pHis-MIPP-light pink) (figure adapted from Acquistapace et al. [85]). (C) The DesK HK from Bacillus subtilis undergoes phosphorylation on His188, which results in slight bending of the α1 helix, facilitating the phosphoryl group transfer onto Asp in the response regulator, DesR (DesK- pale green and pHis-DesK-light pink). (D) SCS is a heterodimer of α-(pale green and light pink) and β-subunits (gray and wheat). The catalytic His259 is present on the pHis segment or swinging loop of the α-subunit. The Cα-overlaid images porcine GTP-specific SCS when His259 is phosphorylated (light pink-α-subunit, wheat-β-subunit; PDB ID: 1EUD) vs nonphosphorylated (pale green-β-subunit, gray-β-subunit; PDB ID: 7JFP) show that the pHis segment moves 29.3 Å from the Coenzyme A and succinate-binding pocket (Site I; PDB ID: 1EUD, 1SCU and 6XRU) into the nucleotide-binding pocket (Site II; PDB ID: 7JFP) during a catalytic cycle.
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) (1TIP), phosphoglycerate mutase (PGAM1) (2HHJ), phytase (1QWO) and multiple inositol polyphosphate phosphatase (MIPP) (6RXG) are enzymes belonging to the histidine acid phosphatase family and act via an obligatory phosphohistidine intermediate [21,82–85]. The structures of these enzymes have a 3-pHis modification on their catalytic His in the RHG motif, where the His N1 interacts with the backbone amide of the Gly, and the Arg is involved in stabilizing the phosphoryl group on N3. In MIPP, phosphorylation of His causes large positional shifts in the atomic positions of the residues around the catalytic His with respect to the bound substrate (Figure 5B) [85]. The solution structures of the HPr pHis-containing proteins of the E. coli and B. subtilis (PDB ids:1PFH and 1JEM, respectively) PEP:sugar phosphotransferase systems show that the catalytic His is phosphorylated on the N1 position [86,87]. His15 is present at the N-terminus of an α-helix, with the N3 of the imidazole ring facing the core and being stabilized by Ser12, while the N1, which is phosphorylated, is exposed to the solvent. The negative charge on 1-pHis at the N-terminus of the helix is stabilized by the helix dipole. The phosphorylation does not cause any change in the overall structure (RMSD of 0.82 Å on Cα positions of PDB id: 1JEM and 2HID) in the HPr protein, but causes a strain in the dihedral angles of His which allows it to transfer the phosphoryl group to the EIIA [87]. Interestingly, while HPr undergoes 1-pHis modification, its upstream and downstream proteins, EI (PDB Id: 2HWG) and EIIA (PDB Id: 1VSQ) undergo 3-pHis modification [88,89].
Succinyl CoA synthetase (SCS or SUCLG1), ATP-citrate lyase (ACLY) and ADP-forming acetyl-CoA synthetase (ACD) have 3-pHis at the catalytic site and belong to the acyl-CoA synthetase family [90]. The hallmark of these enzymes is the presence of two catalytic sites — site I where substrate binds and site II for nucleotide binding (otherwise known as an ATP grasp fold). While ACD and SCS are proteins with two subunits — site I is located in α subunit and site II in β subunit, ACLY is a monomer with two sites embedded within itself. Succinate, citrate and acetate or their CoA counterparts are the substrates of SCS, ACLY and ACD, respectively, based on whether the anabolic or catabolic reaction is considered. The catalytic His, which is present on a segment known as the ‘phosphohistidine segment’ or ‘swinging loop’ (this loop is present on the α-subunit for SCS and ACD), undergoes phosphorylation at site I, and the loop then relocates to site II to transfer the phosphate moiety onto an NDP bound at site II.
Site I: Citryl-CoA + Pi ⇔ E-Phosphate + CoA + Citrate (E is ACLY)
Succinyl-CoA + Pi ⇔ E-Phosphate + CoA + Succinate (E is SCS)
Acetyl-CoA + Pi ⇔ E-Phosphate + CoA + Acetate (E is ACD)
Site II: E-Phosphate + NDP ⇔ E + NTP (Common for all)
In the case of ACD, the phosphohistidine segment is 21 amino acids long (Gly242α–Val262α in PDB Id: 4YAK and 4YBZ) and shuttles the pHis ~37 Å from site I to II. To facilitate the relocation, the Gly245α to Thr255α α-helix partially unwinds and reforms into a longer helix stretching from the active site His254α to Ile258α [91]. On the other hand, the phosphohistidine segment of ACLY (Thr750–Ala768, which contains the catalytic His760 in PDB Id: 5TDZ, 5TDM, 5TDF and 5TES) relocates ~33 Å, but unlike ACD, there are no conformational changes in the secondary structure of its phosphohistidine segment [92]. The phosphohistidine segment of SCS (Gly248α–Gly270α with the catalytic His249 in PDB Id: 7JFP, 1EUD) bridges a gap of ~29 Å between sites I and II. Like ACD and unlike ACLY, SCS has secondary structural changes in the phosphohistidine segment to facilitate the relocation (Figure 5D) [22,93]. Interestingly, ACLY and SCS both have similar residues (GHAGA) around the catalytic His.
In DesK, a HK in the B. subtilis DesK/DesR TCS, phosphorylation on N3 of H188 located in a central helix α1 (PDB id: 3GIG, 5IUM), induces an asymmetric conformation with a kink of 33–35°, when compared with the 12–19° kink present on the unphosphorylated protein. This kink allows the protein to bend more towards the incoming response regulator DesR and poise the pHis for phosphotransfer to the acceptor Asp (Figure 5C) [94,95]. In the CheA/CheY TCS, compared with unphosphorylated CheA, 3-pHis phosphorylated CheA (PDB Id: 3KYI) undergoes a 2.1 Å rigid body translation towards the CheY, establishing linear geometry for the CheA catalytic His and the CheY acceptor Asp and a shorter distance for the phosphotransfer to happen compared with unphosphorylated HK [96].
Apart from the well-known proteins that undergo His phosphorylation, there are some unusual proteins in the PDB that exhibit a pHis modification. The structures of Dictyostelium discoideum discoidin II, a lectin domain containing protein located in the extracellular compartment of intracellular vesicles (PDB id: 2VME), and Pseudomonas sp. ω-transaminase (PDB Id: 4UHN) contain an N1 phosphorylated His residue that is surface exposed, but the functional significance of this modification, if any, is not known [97,98]. The structures of a Thermobifida fusca methyl transferase (PDB id: 2QE6), ERK2 (PDB id: 4QP1), an anti-PCSK9 monoclonal antibody (6E4Z), and ENPP, an ecto-nucleotide pyrophosphatase (PDB id: 6WFJ) have N3 phosphorylated His residues that are mostly localized on the surface. But, again, the functional significance of these modifications, if any, is not known. Notably, KSR1 (Kinase Suppressor of Ras), an ERK MAP kinase signaling pathway scaffolding protein, is phosphorylated by NME1 on Ser392 [31,33,99]. The reason for referring to these proteins as unusual is that they do not possess an annotated phosphohistidine residue. Some studies note that the phosphohistidine modification in these structures is merely a recombinant protein expression or crystallization artifact, but it may not be the case for all of them. Owing to the presence of these modifications, especially on the surface, we speculate that pHis modification in some of these proteins might have a role in transient protein–protein interactions and would be worth exploring.
Evolving perspective on the composition of phosphohistidine and phosphosites
The interest in uncovering the unexplored non-canonical phosphoproteome has peaked recently. However, one of the earliest attempts to determine the extent of phosphoramidation in cells was carried out by Chen et al. [15]. They injected rats undergoing liver regeneration with 32P inorganic phosphate to measure and compare the relative levels of P-N and P-O linkages in nuclear proteins, and found the P-N to P-O linkage ratio to be 0.5 to 4.5 (~10% P-N). In the slime mold Physarum polycephalum, 6% of the nuclear phosphoproteome was attributed to pHis, while pTyr was found to be <1% [100].
Early phosphoproteomic studies of this sort used in vivo radiolabeled 32P-proteins that were subjected to acid hydrolysis and quantified by thin layer chromatography (TLC) or electrophoresis (TLE). Such studies would not have detected any of the non-canonical phosphorylation events. Later on, the phosphoproteome analytical pipeline used 2D gels to separate the radiolabeled proteins and introduced MALDI-TOF mass spectrometry to identify the phosphoproteins [101]. Moving away from radioactive labeling, modern quantitative phosphoproteomic profiling employs label-free samples, novel enrichment and fractionation techniques coupled with advanced mass spectrometry tools to identify the phosphosites.
Though most phosphoproteomic studies follow this modern workflow, they are primarily useful for studying Ser/Thr/Tyr phosphorylation events. Here, we highlight studies published in the past three years that reported development of methods for studying non-canonical phosphorylation events, like pHis. IMAC (immobilized metal affinity chromatography) and titanium dioxide (TiO2) MOAC (metal oxide affinity chromatography), which use strongly acidic conditions (pH ≤2.0), are common phosphopeptide enrichment techniques for pSer, pThr and pTyr peptides. However, such strongly acid conditions mean that most pHis peptides are dephosphorylated before they can be recovered. In an attempt to get around this problem, Potel et al. developed a modified Fe+2-IMAC method at pH 2.3 that retained many pHis peptides intact [102]. Using this technique to analyze the E. coli phosphoproteome, they uncovered 246 pHis sites, which constitute 11.6% of the total phosphosites. They identified more pHis sites compared with a pan-pHis antibody based immunoprecipitation method [103]; however, both studies have 12 pHis sites in common.
The first analysis of the global eukaryotic phosphoproteome (i.e. canonical + noncanonical) was made in zebra fish larvae, using TiO2 enrichment followed by LC–MS/MS analyses, and estimated the pHis contribution in the total phosphoproteome to be 6.3% with 68 identified pHis sites [104]. Hardman et al. developed an enrichment method for non-canonical phosphopeptides, UPAX, which utilizes strong anion exchange chromatography at pH 6.8 that can retain the full spectrum of phosphosites, and combined UPAX with tandem mass spectrometry (MS/MS) to estimate the composition of the entire phosphoproteome in HeLa cells [105]. Consequently, they found 30% of the total phosphoproteome is contributed by non-canonical phosphorylations, uncovering 83 pHis true positive sites (ptmRS >0.90), accounting for ~2% of all identified sites. Interestingly, they found that depletion of the PHPT1 pHis phosphatase in HeLa cells did not result in a gross increase in number of pHis sites, either indicating the existence of other pHis phosphatases or that enzymatic dephosphorylation of pHis is not that important in HeLa cells. Another significant finding was that the pArg (4.8%) and pLys (10%) sites were identified more frequently than pHis sites in the HeLa cell phosphoproteome, but we know very little about the proteins with these modifications or their possible functions.
Building on the idea of using Zn-chelated Phos-tag beads to enrich P-N peptides [106], Hu et al. used bis (zinc(II)-dipicolylamine) containing sub-2 μM core-shell silica microspheres (SiO2@DpaZn) on tip to enrich N-phosphopeptides under neutral conditions [107]. As the nitrogen atom in the P-N bond is less electron withdrawing compared with the oxygen atom in a P-O bond, SiO2@DpaZn favors complexing with phosphoramidate bonds. As a result, this study had higher recovery rates for P-N peptides than P-O peptides, skewing the ratios (1 : 1 in E. coli and 3 : 1 in HeLa cell lysates for P-O vs P-N, respectively). In E. coli, out of 99 N-phosphosites uncovered, 19 were pHis while 38 and 42 were pLys and pArg respectively. In the case of HeLa cells, out of 3384 defined N-phosphosites, 611 contained pHis, 1618 pLys and 1155 pArg. The known E. coli pHis proteins that were identified in this study are phosphoenolpyruvate synthase and phosphoenolpyruvate-protein transferase (also known as EI of PTS system). However, these studies failed to identify pHis sites from well-characterized pHis proteins, such as NME1, PGAM1 and other NDPKs. Hydroxyapatite (HAP) chromatography in tandem with immunoaffinity purification (IAP) using a mixture of bead immobilized 1- and 3-pHis monoclonal antibodies followed by MS/MS was used by Adam et al. to enrich 77 pHis peptides, including the known PGAM1 3-pHis11 site [108].
As an alternative approach to using mass spectrometry for phosphoproteomic studies, recently Makwana et al. used 31P NMR to estimate the amount of pHis (1.8 fmol/cell) in human bronchial epithelial cells (16HBE14o-), which was one third of the amount of pSer/pThr combined (5.8 fmol/cell) and 20 times more than pTyr [109]. They also demonstrated that during the trypsinization step, pHis residue signals were reduced while the pSer/pThr residue signals spiked, which might be due to the transfer of phosphoryl group from pHis within a peptide or from orthophosphate diesters to pSer/pThr. Another recently developed approach for pHis peptide enrichment is usage of molecularly imprinted polymers (MIP) to bind pHis peptides in a sequence-independent manner. However, little was done with the MIP method beyond proof of concept experiments, and these polymers are yet to be adopted for phosphoproteomic studies [110]. Finally, an online database resource for pHis sites called HisPhosSite, which was launched in 2021 [111], curated 776 verified pHis sites reported in the literature along with 5378 predicted pHis sites based on various phosphoproteomic studies in prokaryotes and eukaryotes. This new database is a valuable addition to the histidine phosphorylation field.
Optimized enrichment techniques to conserve pHis modification in biological samples should be complemented by compatible mass spectrometry tools. Unfortunately, MS also poses technical difficulties in conserving such labile modifications. The conventional usage of ESI-MS or MALDI in positive ion mode for sample ionization, and subsequent, collision induced dissociation (CID) fragmentation results in significant decay of pHis signal [16]. CID fragmentation generates a signature triplet for pHis peptides which corresponds to neutral losses of 98, 80 and 116 Da. This represents elimination of phosphoryl group from imidazole ring and two water molecules from the peptide backbone [103]. Introduction of negative ion mode and electron transfer induced dissociation (ETD) fragmentation methods, and more specifically, ETD fragmentation in negative ion mode (NETD) in basic conditions and UV-induced fragmentation are some of the recent promising developments to detect pHis as well as other N-phosphosites [112–114].
There are ~30 well known mammalian pHis proteins, but very few if any of the corresponding pHis peptides were recovered in the currently published pHis proteomic datasets. One reason could be that the pHis peptides obtained by trypsinization of proteins like ACLY (pHis760) and histone H4 (pHis18) are either too small or too long to be captured by MS analysis parameters, or they could be inherently less stable [108]. Moreover, there is little to no overlap in the pHis sites identified in most of the recent pHis proteome reports, which is in stark contrast with when different phosphoproteomic studies of pSer, pThr and pTyr are compared. To some extent this is to be expected, as different enrichment strategies were employed in each of study, which influences pHis selectivity and stability, and this combined with the use of different cell lines and acid-based column peptide fractionation methods, and the harshness of mass spectrometry will all have contributed to loss of the most labile pHis sites.
Another important hurdle in enrichment is stabilization of the pHis phosphosites. There are reports that suggest the phosphoryl group can be internally transferred from one amino acid to another in a peptide or a protein [115]. The internal rearrangement can be avoided and the time spent fractionating peptides under acidic conditions during HPLC separation could be reduced by using ultra-efficient chromatography involving slip flow nanofluidics [107,116]. The existence of the two isoforms of pHis is well known, but none of the MS-based phosphoproteomic methods discriminate the two isoforms, because their mass is the same. The literature suggests that proteins exclusively have either a 1-pHis or a 3-pHis modification. Hence, it becomes important to characterize new pHis N-phosphosites with regard to their isoform specificity. Here, the selective use of the 1-pHis and 3-pHis mAbs for affinity enrichment of pHis peptides and proteins can be an advantage [117].
Motif analysis of pHis sites in many of the reported pHis phosphoproteomic studies has shown that hydrophobic residues, i.e. Val. Leu and Ile, are enriched around the pHis. Hardman et al., found that out of 129 unique pHis sites they defined, 40 of them have hydrophobic residues around the pHis and commonly have either Leu/Val (sometimes Ile) at the +1 site relative to pHis [105]. Leu was also the hydrophobic residue that frequently populated the adjacent site of N-phosphopeptides in other studies [107,108]. The enrichment of peptides with a hydrophobic residue next to the pHis might be because histidine kinases prefer or the phosphohistidine phosphatases disfavor such a motif, or else the neighboring hydrophobic residues might reduce the rate of hydrolysis of pHis [107]. Another possibility is that pHis peptides with hydrophobic residues are preferentially enriched by the resins that were used.
In contrast with the phosphoproteomic results, residues that most frequently surround verified pHis sites curated from the literature are Ser, Arg, Asp, Lys in prokaryotes and Ser, Gly, Arg and Pro in eukaryotes [111]. Assuming that the activities of pHis phosphatases might lead to loss of pHis during cell lysis and enrichment, the addition of phosphohistidine phosphatase inhibitors during cell lysis, and tryptic digestion and pHis peptide enrichment might increase the number and variety of pHis sites recovered. Addition of chemical inhibitors or peptides that mimic the transition state of phosphatases, as demonstrated by Eerland et al. [118] using sulfonamide based transition state analogues with PHPT1, could be useful strategies to adopt. In silico methods are being developed using machine learning algorithms such as deep neural networks, hidden Markov models and random decision forests to make sense of complex patterns of phosphorylation sites from raw sequences and predict potential phosphorylation sites in proteins [119–121]. More recently, the PROSPECT and iPhosH-PseAAC webservers have been developed to predict the pHis sites in proteins with high accuracy [122,123]. If methods like these can be shown to be reliable, then they would complement the phosphoproteomic data and enrich our knowledge of the functions of pHis.
Non-hydrolyzable pHis analogues and the development of pHis antibodies
Antibodies are critical and versatile biochemical tools that are extensively used in research, diagnostics as well as therapeutics. Hence, having antibodies that selectively recognize the two different pHis modifications would be of great benefit to the pHis field, but the generation of useful anti-pHis antibodies took much longer than anticipated, especially compared with the time it took for first pTyr antibodies to be developed after its discovery, which was ~2 years [124,125]. Historically, many unsuccessful attempts were made to develop pHis antibodies [100]; the failure was presumably due to the rapid hydrolysis of phospho-epitope on the pHis-containing immunogen even before a host immune response was elicited in the immunized animal. The obvious solution to this problem was to use a non-hydrolyzable analogue of pHis for immunization. In this regard, the first pTyr antibodies were developed using a non-hydrolyzable pTyr analogue, p-azobenzyl phosphonate, as an immunogen [124]. Interestingly, one of the earliest reported anti-pTyr monoclonal antibodies generated using this pTyr analogue also recognized ACLY, which lacks pTyr but contains 3-pHis (structurally, the positions of the phosphoryl group on N3 in 3-pHis and 4-OH on pTyr are quite similar with respect to the peptide backbone). This crossreactivity gave credence to the idea that it would be possible to develop antibodies against pHis [126].
Two different approaches were considered for designing non-hydrolyzable pHis mimetics for 1-pHis and 3-pHis (Figure 6). The first is to replace an oxygen atom in the phosphoryl group with a less electronegative atom like sulfur, which reduces the rate of hydrolysis; thiophosphorylation of His can be done chemically or enzymatically using ATPγS as the kinase substrate. This approach was used to study pHis autophosphorylation and phosphotransfer to Asp in bacterial TCS [127,128], but there were no reported efforts to develop antibodies using this analogue. The second strategy of replacing P-N with a P-C phosphonate linkage in the imidazole ring gave rise to a bevy of analogues, and some of these were used in immunization. The use of phosphofurylalanine and phosphopyrrole pHis analogues as immunogens by Schenkel et al. [29] and Attwood et al. [10], respectively, yielded antibodies that have specificity only to the analogue but not the natural pHis. While the antibodies made against 4--phosphothiophen-2-yl alanine analogue by Lilley et al. [130] had significant crossreactivity with pTyr. Subsequently, Kee et al. and McAllister et al. independently reported synthesis of a pair of phosphoryltriazolylalanine (pTza) analogues, 1-phosphoryltriazolylalanine (1-pTza) and 3-phosphoryltriazoylalanine (3-pTza) [131,132] (see below), that are structurally very similar to the two isomers of phosphohistidine, 1-pHis and 3-pHis. As a proof of principle, Kee et al. incorporated the 3-pTza analogue into a synthetic peptide based on the sequence around pHis18 in histone H4, and coupled it to KLH for immunization. These antibodies were sequence specific [131].
Figure 6. Stable analogues of phosphohistidine.

Stable analogues of phosphohistidine were developed. Except for thiophosphohistidine, all the other analogues depicted here were used to raise antibodies against 1- and 3-pHis modifications.
To be useful for global pHis proteomic studies, one needs not only 1-pHis and 3-pHis specific antibodies, but, most importantly, sequence-independent pHis antibodies. In a subsequent study, with the goal of developing pan-3-pHis antibodies, Kee et al. used KLH-conjugated phosphoryl-triazolylethylamine (pTze) to immunize rabbits. Although these antibodies bound to a broader spectrum of substrates, they exhibited pTyr crossreactivity [133]. Moving away from the triazole-based analogues, efforts were put in to developing second generation pHis mimetics. Here, Kee et al., developed polyclonal antibodies against 3-pHis modification using phosphonopyrazolyl ethylamine (pPye), which is structurally and electronically similar to 3-pHis modification [134]. Although these antibodies fared well in recognizing pHis proteins like PGAM1, histone H4 and PtsI, they still were slightly crossreactive with pTyr. Lilley et al. [135] also developed polyclonal antibodies against 3-pHis using a second generation analogue, 4-phosphopyrazol-2-yl alanine (pPza). These antibodies did not show crossreactivity to pTyr, but they did recognize unphosphorylated His, which could be overcome by performing affinity depletion on the serum by using a His-KLH glutaraldehyde conjugated Sepharose column. However, these antibodies lacked isomer specificity, meaning, though these antibodies were raised against a 3-pHis analogue, they crossreacted with 1-pHis proteins like NDPK-A and NDPK-B.
Rather than using pHis analogues directly conjugated to KLH as haptens to generate sequence independent pHis antibodies, Fuhs et al. chose to use haptens in which the pTza analogues were embedded in a peptide backbone, generating a pair of degenerate nonapeptide libraries containing randomized Ala/Gly residues with the relevant pTza isomer in the central position that were coupled to KLH to immunize rabbits [117]. Enzymatic assays were used to generate 1-pHis-modified NME1 and 3-pHis-modified PGAM1, which were then used on immunoblots for polyclonal antibody validation, and subsequently, select rabbit hybridomas for monoclonal antibody (mAbs) production. From these efforts, three mAbs against 1-pHis (SC1-1, SC50-3, SC77-11) and four mAbs against 3-pHis (SC39-4, SC44-8, SC56-2 and SC60-2) emerged as promising tools [117]. Dot blots with pTza peptides derived from the known HK substrate proteins, His and pTyr peptides were used to prove that these mAbs are able to differentiate phosphorylated vs nonphosphorylated forms of His, and are also isomer specific, i.e. the 1-pHis mAbs only recognize the 1-pTza peptide, but not the 3-pTza peptide and vice versa (Supplementary Table S1). They also do not bind pTyr peptides, but do recognize any 1-/3-pTza embedded peptide in an isomer specific and largely sequence-independent manner. These antibodies were also able to detect intracellular pHis when tested by immunofluorescence staining. Interestingly, when used to stain proliferating HeLa cells, a 1-pHis mAb stained the outer membrane of phagosomes and actin filaments, while 3-pHis mAbs stained centrosomes, spindle poles and mitotic midbodies. Immunoaffinity purification using immobilized pHis mAbs followed by SILAC-based LC–MS/MS of HeLa cell lysates identified 280 and 156 1-pHis and 3-pHis proteins, respectively, although the pHis sites in these proteins were not identified [117]. In parallel to the pHis antibody development, techniques like Western blotting, immunohistochemistry and immunofluorescence were modified to conserve the pHis signals during the course of experiments [117,136–138].
Lessons learnt from pHis mAbs
While many recent studies have used the pHis mAbs to track or quantify the pHis levels in various systems and understand the role of pHis in protein functions, we were inspired to study the mechanism of antigen recognition by these mAbs with an aim to improve their specificity and affinity [57,70,71,75,108,139–143]. To this end, we solved structures of five of the pHis mAbs developed by Fuhs et al. in complex with the cognate antigenic pTza peptides. The 1-pHis Fabs (SC1-1 and SC50-3) were complexed with a 1-pTza118-containing NME1 peptide, and 3-pHis Fabs (SC39-4, SC44-8 and SC56-2) were complexed with an ACLY 3-pTza760 analogue peptide [144]. The peptide-binding mode to the CDR regions of these structures was similar for both 1-pHis Fabs, and likewise for the three 3-pHis Fabs, but the modes differed between the 1-pHis Fabs and 3-pHis Fabs, providing a first hint at the isomer specificity of these antibodies. Nevertheless, one common feature of all the pHis mAbs is the presence of positive surface potential at the entrance to the CDR region, which directs the phosphate group placement. One distinguishing feature of the 1- and 3-pHis mAbs is the different modes of phosphate recognition in the phosphate-binding pocket in the CDR regions. However, these two modes of phosphate recognition are not new in nature. Surprisingly, these phosphate-binding modes are conserved among both natural and designed antibodies that recognize pTyr, pSer and pThr modifications. The 1-pHis mAbs interaction with the phosphate is heavily reliant on a salt bridge interaction between an Arg95 from heavy chain CDRH3 with two oxygen atoms of the phosphate group. 4G10, the pan-specific pTyr antibody, and its engineered counterpart also have a similar mode of interaction (Figure 5) [145]. Involvement of an Arg residue in phosphate binding is found in FMN-linked oxidoreductases and PRTase superfamily of proteins, along with SH2 and PTB domain-containing proteins that recognize pTyr-containing proteins.
Unlike the 1-pHis mAbs, the 3-pHis mAbs interact with phosphate through backbone amides of residues present in a GXXX motif, known as a ‘structural P-loop’, which is present in 13 superfamilies of proteins that recognize phosphate [146]. The SC44-8 3-pHis antibody has an even more specialized structure, known as a ‘nest’ motif with GXGX residues, such as those present in the ATPase and kinase superfamilies (Figure 7). In fact, Koerber et al. [147] designed phosphospecific antibodies against pTyr, pSer and pThr by installing this ‘nest’ motif into CDRH2 of a rat antibody scaffold.
Figure 7. The modes of phosphate recognition by phosphoantibodies.

(A) The 1-pHis mAbs (SC1-1 and SC50-3) interact with phosphate in the peptide (green) through bidentate Arg interactions as well as hydrogen bond interactions from the residues in heavy (blue) and light chain (light pink) CDRs. A similar interaction is observed in the 4G10 antibody, a pan-specific pTyr antibody (B). The second kind of interaction with phosphate is through the backbone interactions of SC44-8 antibody with the phosphate in the peptide. More than two consecutive residues form a ‘nest’ like structure and hydrogen bond with the phosphate. Most often, the residues in the ‘nest’ are dominated by Gly (C). This kind of interaction is observed in pTyr (D) and pThr (E) engineered antibodies, as well as in the phospho-state specific antibody against Tau protein, where Thr is phosphorylated (F).
The occurrence of similar phosphate recognition modes in various antibodies provides a critical piece of information for designing or selecting future phospho-specific antibodies from antibody libraries especially against other non-canonical phosphorylation modifications. The commonality in the phosphate recognition mode between the phosphate-recognizing proteins and the phosphoamino acid antibodies ensures that the antibodies follow an evolutionarily tested fool-proof mechanism for recognizing phosphoproteins, and, also, they are capable of competing or outcompeting phosphatases or other phosphate recognizing proteins in in vitro, in vivo and in situ settings. Though the mode of phosphate recognition is common among the pHis antibodies and other phosphoamino acid antibodies, there are still differences in how the interacting residues are distributed across the CDRs of heavy chain and light chain and the environment around the phosphate-binding pocket. Hence it is important to expand the structural repertoire of phosphoamino acid antibodies in order to facilitate structure-guided antibody engineering to generate more functional antibodies, and also, reduce cross-reactivity.
The other hallmark property of pHis mAbs is sequence independence. Sequence independent antibodies recognize the chemical moiety that they are raised against independent of the adjacent amino acids in a peptide or a protein. Development of sequence-independent antibodies becomes particularly important when not many target proteins are known, which is the case with the non-canonical phosphorylations, and also when the proteins lack a consensus phosphorylation sequence motif, which is the case with pHis proteins. Several studies previously used the pHis analogues directly conjugated to KLH for immunizations to develop sequence independent antibodies [10,129,130,134]. The resulting antibodies have had either no specificity for pHis or had cross reactivity with pTyr. However, usage of a degenerate library for Ala and Gly with pHis analogue as hapten gave rise to sequence independent antibodies (SC1-1, SC50-3, SC39-4 and SC56-2) that recognize pHis modified peptide or protein irrespective of the adjacent sequence [117]. Ala and Gly were chosen because they are not immunogenic chemical moieties, but the hydrogen bonding network between the main chain amide and carboxyl groups of the peptide with the antibody CDR residues contributed to the peptide recognition, and also, played an important role in dictating the depth of the phosphate-binding pocket by imposing steric restrictions [144]. The sequence independence of this panel of pHis mAbs becomes evident in the sequence breadth of pHis peptides enriched in the phosphoproteomic studies that used these antibodies [108,117]. Using a similar principle, Hauser et al., attempted development of antibodies against the pLys modification using 10-mer peptides with pLys analogues with phosphonate or phosphomonoester linkages [148,149]. Instead of Ala/Gly, the 10-mer peptides were randomized with all the amino acids except for Cys (Cys was used for KLH conjugation) to immunize rats. Though they obtained a B-cell clone that recognized pLys but not Lys, they were not successful in obtaining a corresponding hybridoma clone. Further development of these antibodies would light up the cryptic pLys field.
In contrast with the other pHis mAbs, SC44-8 is a 3-pHis mAb that is biased towards binding GpHAGA motif-containing proteins or peptides, and in this sense has a sequence dependence property [144]. The ordered CDRL3 loop of SC44-8 sterically constricts the maximal binding volume in the CDR binding pocket thus permitting only pHis peptides with small amino acids like Gly or Ala to bind. The crystal structure provided the molecular basis for SC44-8’s known bias towards proteins with a GpHAGA motif, which is present in proteins like ACLY and SCS. Hence, for studies of the global pHis proteome the SC44-8 mAb should be used with caution. Nevertheless, it is an appropriate antibody to identify unknown pHis proteins with this motif or a motif like XpHXXX, where X could be either Gly/Ala. This mAb could be further engineered to generate mAbs that distinguish pHis forms of ACLY and SCS proteins.
While the sequence-independent pHis mAbs have proved to be valuable tools for studying pHis proteins both in vivo and in vitro, as the number of identified pHis proteins and phosphosites increases, site-specific pHis antibodies will be needed to study His phosphorylation of individual proteins in greater detail, and understand the role of His phosphorylation in the protein’s functions. The conventional way of developing sequence-dependent antibodies is by using site-specific phosphopeptides for immunization. However, with the advent of antibody engineering, we can design and generate next generation antibody libraries using the available pHis mAbs and their structural information. Such libraries can be further subjected to iterative engineering in conjunction with phage display, yeast surface display or ribosome display library screening and selection to obtain superior antibodies with desirable properties [150].
Summary and future perspective
Phosphohistidine is a crucial post-translational modification ubiquitous to both prokaryotes and eukaryotes. During evolution, many distinct biological functions for pHis have emerged, ranging from enzyme intermediate to acting as a signaling molecule and regulating ion flux across membranes. The structures of pHis proteins provide insights into the role of pHis in their functions, and at the same time underscore the fact that no consensus sequence motif for pHis exists across families of pHis proteins. Moreover, the basis for the preference for either the 1- or 3-pHis isomer at a specific pHis site remains an enigma, although it seems to be dictated in part by local interactions with other residues in the pHis environment. The existence of unusual proteins where an unanticipated pHis modification was revealed by their X-ray crystal structures points to the fact that this labile modification may have a greater than expected role in physiology. Mutagenesis of these His residues would be one way to dissect the functional significance of such unexpected modifications.
Recent phosphoproteomic studies with optimized enrichment techniques to conserve the unstable modifications, have uncovered a large number of novel pHis proteins. Validation and functional analysis of these pHis sites is challenging, particularly because there is no natural analogue of His that can be used to make a non-phosphorylatable mutation. Likewise, neither Asp nor Glu can be used to mimic a pHis residue. Efforts to generate non-hydrolysable pHis analogues that can be incorporated into proteins at specific sites in vivo through the use of expanded genetic code technology should be undertaken [151–153]. If successful, pHis analogue-containing proteins could be used to uncover pHis-dependent interactomes and study their functions. The synthesis of stable pHis analogues was essential in raising the antibodies against the two pHis isoforms that have proved so useful in studying pHis proteins. The structures of pHis mAbs bound to their cognate pHis peptide antigens provided insights into the molecular basis for the phosphate recognition, which surprisingly revealed convergence with the phosphate-binding proteins and other phosphate-recognizing antibodies. The pHis antibody structural information has also aided in our understanding of the properties exhibited by these antibodies, and in the near future this information could be fed into an engineering pipeline to generate superior antibodies.
Antibodies are valuable and versatile biochemical tools to study PTMs in vitro, but if pHis antibodies can be engineered to attain higher affinity and specificity, single chain scFv versions can be designed and expressed in vivo to probe pHis in the intracellular environment or to disrupt pHis-dependent processes. The use of antibody mimetics, like darpins, affimers, monobodies, nanobodies etc., to generate novel pHis specific reagents could also be valuable and cost-effective approach.
Systematic study and functional validation of the increasing number of pHis proteins is the need of the hour. In the future, components of the pHis regulome might prove to be valuable therapeutic targets. Lastly, pHis stands as a beacon for other non-canonical phosphorylations to follow.
Supplementary Material
Abbreviations
- ACD
acetyl-CoA synthetase
- ACLY
ATP citrate lyase
- CID
collision induced dissociation
- ETD
electron transfer induced dissociation
- HCC
hepatocellular carcinoma
- HK
histidine kinases
- IMAC
immobilized metal affinity chromatography
- MIP
molecularly imprinted polymers
- MIPP
multiple inositol polyphosphate phosphatase
- NAMPT
Nicotinamide phosphoribosyltransferase
- NDPK
nucleoside diphosphate kinases
- NMN
nicotinamide mononucleotide
- PEP
phosphoenolpyruvate
- PTMs
post-translational modifications
- RR
response regulator
- SCS
succinyl Co-A synthetase
- TCS
two-component signal
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Hunter T (2012) Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci 367, 2513–2516 10.1098/rstb.2012.0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adam K and Hunter T (2018) Histidine kinases and the missing phosphoproteome from prokaryotes to eukaryotes. Lab. Invest 98, 233–247 10.1038/labinvest.2017.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ni F, Fu C, Gao X, Liu Y, Xu PX, Liu L et al. (2015) N-phosphoryl amino acid models for P-N bonds in prebiotic chemical evolution. Sci. China Chem 58, 374–382 10.1007/s11426-015-5321-1 [DOI] [Google Scholar]
- 4.Boyer PD, Deluca M, Ebner KE, Hultquist DE and Peter JB (1962) Identification of phosphohistidine in digests from a probable intermediate of oxidative phosphorylation. J. Biol. Chem 237, PC3306–PC3308 10.1016/S0021-9258(18)50167-8 [DOI] [PubMed] [Google Scholar]
- 5.Hultquist DE, Moyer RW and Boyer PD (1966) The preparation and characterization of 1-phosphohistidine and 3-phosphohistidine. Biochemistry 5, 322–331 10.1021/bi00865a041 [DOI] [PubMed] [Google Scholar]
- 6.Hultquist DE (1968) The preparation and characterization of phosphorylated derivatives of histidine. Biochim. Biophys. Acta 153, 329–340 10.1016/0005-2728(68)90078-9 [DOI] [PubMed] [Google Scholar]
- 7.Klumpp S and Krieglstein J (2009) Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci. Signal 2, pe13 10.1126/scisignal.261pe13 [DOI] [PubMed] [Google Scholar]
- 8.Stock AM, Robinson VL and Goudreau PN (2000) Two-component signal transduction. Annu. Rev. Biochem 69, 183–215 10.1146/annurev.biochem.69.1.183 [DOI] [PubMed] [Google Scholar]
- 9.Bardy SL, Briegel A, Rainville S and Krell T (2017) Recent advances and future prospects in bacterial and archaeal locomotion and signal transduction. J. Bacteriol 199, e00203–e00217 10.1128/JB.00203-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attwood PV, Piggott MJ, Zu XL and Besant PG (2007) Focus on phosphohistidine. Amino Acids 32, 145–156 10.1007/s00726-006-0443-6 [DOI] [PubMed] [Google Scholar]
- 11.Kee JM and Muir TW (2012) Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem. Biol 7, 44–51 10.1021/cb200445w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuhs SR and Hunter T (2017) Phisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell Biol 45, 8–16 10.1016/j.ceb.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makwana MV, Muimo R and Jackson RF (2018) Advances in development of new tools for the study of phosphohistidine. Lab. Invest 98, 291–303 10.1038/labinvest.2017.126 [DOI] [PubMed] [Google Scholar]
- 14.Hauser A, Penkert M and Hackenberger CPR (2017) Chemical approaches to investigate labile peptide and protein phosphorylation. Acc. Chem. Res 50, 1883–1893 10.1021/acs.accounts.7b00170 [DOI] [PubMed] [Google Scholar]
- 15.Chen CC, Bruegger BB, Kern CW, Lin YC, Halpern RM and Smith RA (1977) Phosphorylation of nuclear proteins in rat regenerating liver. Biochemistiy 16, 4852–4855 10.1021/bi00641a016 [DOI] [PubMed] [Google Scholar]
- 16.Medzihradszky KF, Phillipps NJ, Senderowicz L, Wang P and Turck CW (1997) Synthesis and characterization of histidine-phosphorylated peptides. Protein Sci. 6,1405–1411 10.1002/pro.5560060704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Attwood PV, Ludwig K, Bergander K, Besant PG, Adina-Zada A, Krieglstein J et al. (2010) Chemical phosphorylation of histidine-containing peptides based on the sequence of histone H4 and their dephosphorylation by protein histidine phosphatase. Biochim. Biophys. Acta 1804, 199–205 10.1016/j.bbapap.2009.10.007 [DOI] [PubMed] [Google Scholar]
- 18.Stock JB, Stock AM and Mottonen JM (1990) Signal transduction in bacteria. Nature 344, 395–400 10.1038/344395a0 [DOI] [PubMed] [Google Scholar]
- 19.Hansen AL and Kay LE (2014) Measurement of histidine pKa values and tautomer populations in invisible protein states. Proc. Natl Acad. Sci. U.S.A 111, E1705–E1712 10.1073/pnas.1400577111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elzinga M, Collins JH, Kuehl WM and Adelstein RS (1973) Complete amino-acid sequence of actin of rabbit skeletal muscle. Proc. Natl Acad. Sci. U.S.A 70, 2687–2691 10.1073/pnas.70.9.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigden DJ (2008) The histidine phosphatase superfamily: structure and function. Biochem. J 409, 333–348 10.1042/BJ20071097 [DOI] [PubMed] [Google Scholar]
- 22.Fraser ME, James MN, Bridger WA and Wolodko WT (2000) Phosphorylated and dephosphorylated structures of pig heart, GTP-specific succinyl-CoA synthetase. J. Mol. Biol 299, 1325–1339 10.1006/jmbi.2000.3807 [DOI] [PubMed] [Google Scholar]
- 23.Gajewski S, Comeaux EQ, Jafari N, Bharatham N, Bashford D, White SW et al. (2012) Analysis of the active-site mechanism of tyrosyl-DNA phosphodiesterase I: a member of the phospholipase D superfamily. J. Mol. Biol 415, 741–758 10.1016/j.jmb.2011.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuckey JA and Dixon JE (1999) Crystal structure of a phospholipase D family member. Nat. Struct. Biol 6, 278–284 10.1038/6716 [DOI] [PubMed] [Google Scholar]
- 25.Ghosh A, Shieh JJ, Pan CJ, Sun MS and Chou JY (2002) The catalytic center of glucose-6-phosphatase. HIS176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J. Biol. Chem 277, 32837–32842 10.1074/jbc.M201853200 [DOI] [PubMed] [Google Scholar]
- 26.Burgos ES, Ho MC, Almo SC and Schramm VL (2009) A phosphoenzyme mimic, overlapping catalytic sites and reaction coordinate motion for human NAMPT. Proc. Natl Acad. Sci. U.S.A 106, 13748–13753 10.1073/pnas.0903898106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lascu I and Gonin P (2000) The catalytic mechanism of nucleoside diphosphate kinases. J. Bioenerg. Biomembr 32, 237–246 10.1023/A:1005532912212 [DOI] [PubMed] [Google Scholar]
- 28.Hutter MC and Helms V (2002) The mechanism of phosphorylation of natural nucleosides and anti-HIV analogues by nucleoside diphosphate kinase is independent of their sugar substituents. Chembiochem 3, 643–651 10.1002/1439-7633(20020703)3:7<643::AID-CBIC643>3.0.CO;2-L [DOI] [PubMed] [Google Scholar]
- 29.Boissan M, Schlattner U and Lacombe ML (2018) The NDPK/NME superfamily: state of the art. Lab. Invest 98, 164–174 10.1038/labinvest.2017.137 [DOI] [PubMed] [Google Scholar]
- 30.Lu Z and Hunter T (2018) Metabolic kinases moonlighting as protein kinases. Trends Biochem. Sci 43, 301–310 10.1016/j.tibs.2018.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartsough MT, Morrison DK, Salerno M, Palmieri D, Ouatas T, Mair M et al. (2002) Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of Ras via a histidine protein kinase pathway. J. Biol. Chem 277, 32389–32399 10.1074/jbc.M203115200 [DOI] [PubMed] [Google Scholar]
- 32.Wagner PD and Vu ND (2000) Histidine to aspartate phosphotransferase activity of nm23 proteins: phosphorylation of aldolase C on Asp-319. Biochem. J 346, 623–630 10.1042/bj3460623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adam K, Ning J, Reina J and Hunter T (2020) NME/NM23/NDPK and histidine phosphorylation. Int. J. Mol. Sci 21, 5848–5873 10.3390/ijms21165848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith DL, Bruegger BB, Halpern RM and Smith RA (1973) New histone kinases in nuclei of Rat tissues. Nature 246, 103–104 10.1038/246103a0 [DOI] [PubMed] [Google Scholar]
- 35.Chen CC, Smith DL, Bruegger BB, Halpern RM and Smith RA (1974) Occurrence and distribution of acid-labile histone phosphates in regenerating rat liver. Biochemistry 13, 3785–3789 10.1021/bi00715a026 [DOI] [PubMed] [Google Scholar]
- 36.Besant PG and Attwood PV (2000) Detection of a mammalian histone H4 kinase that has yeast histidine kinase-like enzymic activity. Int. J. Biochem. Cell Biol 32, 243–253 10.1016/S1357-2725(99)00119-3 [DOI] [PubMed] [Google Scholar]
- 37.Pfluger-Grau K and Gorke B (2010) Regulatory roles of the bacterial nitrogen-related phosphotransferase system. Trends Microbiol. 18, 205–214 10.1016/j.tim.2010.02.003 [DOI] [PubMed] [Google Scholar]
- 38.Galinier A and Deutscher J (2017) Sophisticated regulation of transcriptional factors by the bacterial phosphoenolpyruvate: Sugar phosphotransferase system. J. Mol. Biol 429, 773–789 10.1016/j.jmb.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 39.Herzberg O and Klevit R (1994) Unraveling a bacterial hexose transport pathway. Curr. Opin. Struct. Biol 4, 814–822 10.1016/0959-440X(94)90262-3 [DOI] [PubMed] [Google Scholar]
- 40.Falke JJ, Bass RB, Butler SL, Chervitz SA and Danielson MA (1997) The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu. Rev. Cell Dev. Biol 13, 457–512 10.1146/annurev.cellbio.13.1.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strauch MA and Hoch JA (1993) Signal transduction in Bacillus subtilis sporulation. Curr. Opin. Genet. Dev 3, 203–212 10.1016/0959-437X(93)90024-J [DOI] [PubMed] [Google Scholar]
- 42.Novick RP and Geisinger E (2008) Quorum sensing in staphylococci. Annu. Rev. Genet 42, 541–564 10.1146/annurev.genet.42.110807.091640 [DOI] [PubMed] [Google Scholar]
- 43.Hong HJ, Hutchings MI and Buttner MJ and Biotechnology and Biological Sciences Research Council, U. K. (2008) Vancomycin resistance vanS/VanR two-component systems. Adv. Exp. Med. Biol 631, 200–213 10.1007/978-0-387-78885-2_14 [DOI] [PubMed] [Google Scholar]
- 44.Xu Q, Porter SW and West AH (2003) The yeast YPD1/SLN1 complex: insights into molecular recognition in two-component signaling systems. Structure 11, 1569–1581 10.1016/j.str.2003.10.016 [DOI] [PubMed] [Google Scholar]
- 45.Maeda T, Wurgler-Murphy SM and Saito H (1994) A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369, 242–245 10.1038/369242a0 [DOI] [PubMed] [Google Scholar]
- 46.Singh KK (2000) The saccharomyces cerevisiae Sln1p-Ssk1p two-component system mediates response to oxidative stress and in an oxidant-specific fashion. Free Radic. Biol. Med 29, 1043–1050 10.1016/S0891-5849(00)00432-9 [DOI] [PubMed] [Google Scholar]
- 47.Boyce KJ and Andrianopoulos A (2013) Morphogenetic circuitry regulating growth and development in the dimorphic pathogen Penicillium marneffei. Eukaryot. Cell 12, 154–160 10.1128/EC.00234-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheen J (2002) Phosphorelay and transcription control in cytokinin signal transduction. Science 296, 1650–1652 10.1126/science.1071883 [DOI] [PubMed] [Google Scholar]
- 49.Cheng MC, Liao PM, Kuo WW and Lin TP (2013) The Arabidopsis ETHYLENE RESPONSE FACTOR1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 162, 1566–1582 10.1104/pp.113.221911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appleby JL, Parkinson JS and Bourret RB (1996) Signal transduction via the multi-step phosphorelay: not necessarily a road less traveled. Cell 86, 845–848 10.1016/S0092-8674(00)80158-0 [DOI] [PubMed] [Google Scholar]
- 51.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C and Thompson CB (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 10.1038/sj.onc.1208773 [DOI] [PubMed] [Google Scholar]
- 53.Wagner PD and Vu ND (1995) Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J. Biol. Chem 270, 21758–21764 10.1074/jbc.270.37.21758 [DOI] [PubMed] [Google Scholar]
- 54.Cuello F, Schulze RA, Heemeyer F, Meyer HE, Lutz S, Jakobs KH et al. (2003) Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 IN Gbeta. J. Biol. Chem 278, 7220–7226 10.1074/jbc.M210304200 [DOI] [PubMed] [Google Scholar]
- 55.Hippe HJ, Luedde M, Lutz S, Koehler H, Eschenhagen T, Frey N et al. (2007) Regulation of cardiac cAMP synthesis and contractility by nucleoside diphosphate kinase B/G protein beta gamma dimer complexes. Circ. Res 100, 1191–1199 10.1161/01.RES.0000264058.28808.cc [DOI] [PubMed] [Google Scholar]
- 56.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK et al. (2006) Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell 24, 665–675 10.1016/j.molcel.2006.11.012 [DOI] [PubMed] [Google Scholar]
- 57.Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele DJ et al. (2016) Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. eLife 5, e16093 10.7554/eLife.16093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai X, Srivastava S, Surindran S, Li Z and Skolnik EY (2014) Regulation of the epithelial Ca2+ channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol. Biol. Cell 25, 1244–1250 10.1091/mbc.e13-04-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muimo R, Hornickova Z, Riemen CE, Gerke V, Matthews H and Mehta A (2000) Histidine phosphorylation of annexin I in airway epithelia. J. Biol. Chem 275, 36632–36636 10.1074/jbc.M000829200 [DOI] [PubMed] [Google Scholar]
- 60.Fujitaki JM, Fung G, Oh EY and Smith RA (1981) Characterization of chemical and enzymatic acid-labile phosphorylation of histone H4 using phosphorus-31 nuclear magnetic resonance. Biochemistry 20, 3658–3664 10.1021/bi00515a055 [DOI] [PubMed] [Google Scholar]
- 61.Huebner VD and Matthews HR (1985) Phosphorylation of histidine in proteins by a nuclear extract of Physarum polycephalum plasmodia. J. Biol. Chem 260, 16106–16113 10.1016/S0021-9258(17)36207-5 [DOI] [PubMed] [Google Scholar]
- 62.Besant PG, Tan E and Attwood PV (2003) Mammalian protein histidine kinases. Int. J. Biochem. Cell Biol 35, 297–309 10.1016/S1357-2725(02)00257-1 [DOI] [PubMed] [Google Scholar]
- 63.Ek P, Pettersson G, Ek B, Gong F, Li JP and Zetterqvist O (2002) Identification and characterization of a mammalian 14-kDa phosphohistidine phosphatase. Eur. J. Biochem 269, 5016–5023 10.1046/j.1432-1033.2002.03206.x [DOI] [PubMed] [Google Scholar]
- 64.Klumpp S, Hermesmeier J, Selke D, Baumeister R, Kellner R and Krieglstein J (2002) Protein histidine phosphatase: a novel enzyme with potency for neuronal signaling. J. Cereb. Blood Flow Metab 22, 1420–1424 10.1097/01.wcb.0000045041.03034.99 [DOI] [PubMed] [Google Scholar]
- 65.Maurer A, Wieland T, Meissl F, Niroomand F, Mehringer R, Krieglstein J et al. (2005) The beta-subunit of G proteins is a substrate of protein histidine phosphatase. Biochem. Biophys. Res. Commun 334, 1115–1120 10.1016/j.bbrc.2005.06.200 [DOI] [PubMed] [Google Scholar]
- 66.Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H et al. (2008) Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc. Natl Acad. Sci. U.S.A 105, 14442–14446 10.1073/pnas.0803678105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klumpp S, Bechmann G, Maurer A, Selke D and Krieglstein J (2003) ATP-citrate lyase as a substrate of protein histidine phosphatase in vertebrates. Biochem. Biophys. Res. Commun 306, 110–115 10.1016/S0006-291X(03)00920-3 [DOI] [PubMed] [Google Scholar]
- 68.Gong W, Li Y, Cui G, Hu J, Fang H, Jin C et al. (2009) Solution structure and catalytic mechanism of human protein histidine phosphatase 1. Biochem. J 418, 337–344 10.1042/BJ20081571 [DOI] [PubMed] [Google Scholar]
- 69.Chaikuad A, Filippakopoulos P, Marcsisin SR, Picaud S, Schroder M, Sekine S et al. (2017) Structures of PGAM5 provide insight into active site plasticity and multimeric assembly. Structure 25,1089–1099 e1083 10.1016/j.str.2017.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panda S, Srivastava S, Li Z, Vaeth M, Fuhs SR, Hunter T et al. (2016) Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4(+) T cells. Mol. Cell 63, 457–469 10.1016/j.molcel.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schulte JE, Roggiani M, Shi H, Zhu J and Goulian M (2020) The phosphohistidine phosphatase SixA dephosphorylates the phosphocarrier NPr. J. Biol. Chem 296, 100090–100101 10.1074/jbc.RA120.015121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seifried A, Schultz J and Gohla A (2013) Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571 10.1111/j.1742-4658.2012.08633.x [DOI] [PubMed] [Google Scholar]
- 73.Polimanti R, Wang Q, Meda SA, Patel KT, Pearlson GD, Zhao H et al. (2017) The interplay between risky sexual behaviors and alcohol dependence: genome-Wide association and neuroimaging support for LHPP as a risk gene. Neuropsychopharmacology 42, 598–605 10.1038/npp.2016.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neff CD, Abkevich V, Packer JC, Chen Y, Potter J, Riley R et al. (2009) Evidence for HTR1A and LHPP as interacting genetic risk factors in major depression. Mol. Psychiatry 14, 621–630 10.1038/mp.2008.8 [DOI] [PubMed] [Google Scholar]
- 75.Hindupur SK, Colombi M, Fuhs SR, Matter MS, Guri Y, Adam K et al. (2018) The protein histidine phosphatase LHPP is a tumour suppressor. Nature 555, 678–682 10.1038/nature26140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu F, Chen Y and Zhu J (2020) LHPP suppresses proliferation, migration, and invasion and promotes apoptosis in pancreatic cancer. Biosci. Rep 40, BSR20194142 10.1042/BSR20194142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang X, Kang H, Xiao J, Shi B, Li X and Chen G (2020) LHPP inhibits the proliferation and metastasis of renal cell carcinoma. Biomed. Res. Int 2020, 7020924 10.1155/2020/7020924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun W, Qian K, Guo K, Chen L, Xiang J, Li D et al. (2020) LHPP inhibits cell growth and migration and triggers autophagy in papillary thyroid cancer by regulating the AKT/AMPK/mTOR signaling pathway. Acta Biochim. Biophys. Sin. (Shanghai) 52, 382–389 10.1093/abbs/gmaa015 [DOI] [PubMed] [Google Scholar]
- 79.Gohla A (2019) Do metabolic HAD phosphatases moonlight as protein phosphatases? Biochim. Biophys. Acta Mol. Cell Res 1866, 153–166 10.1016/j.bbamcr.2018.07.007 [DOI] [PubMed] [Google Scholar]
- 80.Chen WJ, Chen LH, Wang J, Wang ZT, Wu CY, Sun K et al. (2021) LHPP impedes energy metabolism by inducing ubiquitin-mediated degradation of PKM2 in glioblastoma. Am. J. Cancer Res 11, 1369–1390 [PMC free article] [PubMed] [Google Scholar]
- 81.Morera S, Chiadmi M, LeBras G, Lascu I and Janin J (1995) Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 34, 11062–11070 10.1021/bi00035a011 [DOI] [PubMed] [Google Scholar]
- 82.Wang Y, Liu L, Wei Z, Cheng Z, Lin Y and Gong W (2006) Seeing the process of histidine phosphorylation in human bisphosphoglycerate mutase. J. Biol. Chem 281, 39642–39648 10.1074/jbc.M606421200 [DOI] [PubMed] [Google Scholar]
- 83.Lee YH, Olson TW, Ogata CM, Levitt DG, Banaszak LJ and Lange AJ (1997) Crystal structure of a trapped phosphoenzyme during a catalytic reaction. Nat. Struct. Biol 4, 615–618 10.1038/nsb0897-615 [DOI] [PubMed] [Google Scholar]
- 84.Xiang T, Liu Q, Deacon AM, Koshy M, Kriksunov IA, Lei XG et al. (2004) Crystal structure of a heat-resilient phytase from aspergillus fumigatus, carrying a phosphorylated histidine. J. Mol. Biol 339, 437–445 10.1016/j.jmb.2004.03.057 [DOI] [PubMed] [Google Scholar]
- 85.Acquistapace IM, Zi Etek MA, Li AWH, Salmon M, Kuhn I, Bedford MR et al. (2020) Snapshots during the catalytic cycle of a histidine acid phytase reveal an induced-fit structural mechanism. J. Biol. Chem 295, 17724–17737 10.1074/jbc.RA120.015925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Nuland NA, Boelens R, Scheek RM and Robillard GT (1995) High-resolution structure of the phosphorylated form of the histidine-containing phosphocarrier protein HPr from escherichia coli determined by restrained molecular dynamics from NMR-NOE data. J. Mol. Biol 246, 180–193 10.1006/jmbi.1994.0075 [DOI] [PubMed] [Google Scholar]
- 87.Jones BE, Rajagopal P and Klevit RE (1997) Phosphorylation on histidine is accompanied by localized structural changes in the phosphocarrier protein, HPr from bacillus subtilis. Protein Sci. 6, 2107–2119 10.1002/pro.5560061006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teplyakov A, Lim K, Zhu PP, Kapadia G, Chen CC, Schwartz J et al. (2006) Structure of phosphorylated enzyme I, the phosphoenolpyruvate: sugar phosphotransferase system sugar translocation signal protein. Proc. Natl Acad. Sci. U.S.A 103, 16218–16223 10.1073/pnas.0607587103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu J, Hu K, Williams DC Jr, Komlosh ME, Cai M and Clore GM (2008) Solution NMR structures of productive and non-productive complexes between the A and B domains of the cytoplasmic subunit of the mannose transporter of the Escherichia coli phosphotransferase system. J. Biol. Chem 283, 11024–11037 10.1074/jbc.M800312200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Galperin MY and Koonin EV (1997) A diverse superfamily of enzymes with ATP-dependent carboxylate-amine/thiol ligase activity. Protein Sci. 6, 2639–2643 10.1002/pro.5560061218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weisse RH, Faust A, Schmidt M, Schonheit P and Scheidig AJ (2016) Structure of NDP-forming acetyl-CoA synthetase ACD1 reveals a large rearrangement for phosphoryl transfer. Proc. Natl Acad. Sci. U.S.A 113, E519–E528 10.1073/pnas.1518614113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu J, Komakula A and Fraser ME (2017) Binding of hydroxycitrate to human ATP-citrate lyase. Acta Crystallogr. D Struct. Biol 73, 660–671 10.1107/S2059798317009871 [DOI] [PubMed] [Google Scholar]
- 93.Huang J and Fraser ME (2021) Second distinct conformation of the phosphohistidine loop in succinyl-CoA synthetase. Acta Crystallogr. D Struct. Biol 77, 357–368 10.1107/S2059798321000334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Albanesi D, Martin M, Trajtenberg F, Mansilla MC, Haouz A, Alzari PM et al. (2009) Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc. Natl Acad. Sci. U.S.A 106, 16185–16190 10.1073/pnas.0906699106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trajtenberg F, Imelio JA, Machado MR, Larrieux N, Marti MA, Obal G et al. (2016) Regulation of signaling directionality revealed by 3D snapshots of a kinase:regulator complex in action. eLife 5, e21422 10.7554/eLife.21422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bell CH, Porter SL, Strawson A, Stuart DI and Armitage JP (2010) Using structural information to change the phosphotransfer specificity of a two-component chemotaxis signalling complex. PLoS Biol. 8, e1000306 10.1371/journal.pbio.1000306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wilding M, Peat TS, Newman J and Scott C (2016) A beta-Alanine catabolism pathway containing a highly promiscuous omega-transaminase in the 12-aminododecanate-degrading Pseudomonas sp. strain AAC. Appl. Environ. Microbiol 82, 3846–3856 10.1128/AEM.00665-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aragao KS, Satre M, Imberty A and Varrot A (2008) Structure determination of discoidin II from dictyostelium discoideum and carbohydrate binding properties of the lectin domain. Proteins 73, 43–52 10.1002/prot.22038 [DOI] [PubMed] [Google Scholar]
- 99.Morrison DK (2001) KSR: a MAPK scaffold of the Ras pathway? J. Cell Sci 114, 1609–1612 10.1242/jcs.114.9.1609 [DOI] [PubMed] [Google Scholar]
- 100.Matthews HR (1995) Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade. Pharmacol. Ther 67, 323–350 10.1016/0163-7258(95)00020-8 [DOI] [PubMed] [Google Scholar]
- 101.Mijakovic I and Macek B (2012) Impact of phosphoproteomics on studies of bacterial physiology. FEMS Microbiol. Rev 36, 877–892 10.1111/j.1574-6976.2011.00314.x [DOI] [PubMed] [Google Scholar]
- 102.Potel CM, Lin MH, Heck AJR and Lemeer S (2018) Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat. Methods 15, 187–190 10.1038/nmeth.4580 [DOI] [PubMed] [Google Scholar]
- 103.Oslund RC, Kee JM, Couvillon AD, Bhatia VN, Perlman DH and Muir TW (2014) A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism. J. Am. Chem. Soc 136, 12899–12911 10.1021/ja507614f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao Y, Lee H, Kwon OK, Cheng Z, Tan M, Kim KT et al. (2019) Profiling of histidine phosphoproteome in danio rerio by TiO2 enrichment. Proteomics 19, e1800471 10.1002/pmic.201800471 [DOI] [PubMed] [Google Scholar]
- 105.Hardman G, Perkins S, Brownridge PJ, Clarke CJ, Byrne DP, Campbell AE et al. (2019) Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 38, e100847 10.15252/embj.2018100847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yuan ET, Ino Y, Kawaguchi M, Kimura Y, Hirano H, Kinoshita-Kikuta E et al. (2017) A phos-tag-based micropipette-tip method for rapid and selective enrichment of phosphopeptides. Electrophoresis 38, 2447–2455 10.1002/elps.201700175 [DOI] [PubMed] [Google Scholar]
- 107.Hu Y, Jiang B, Weng Y, Sui Z, Zhao B, Chen Y et al. (2020) Bis(zinc(II)-dipicolylamine)-functionalized sub-2 mum core-shell microspheres for the analysis of N-phosphoproteome. Nat. Commun 11, 6226 10.1038/s41467-020-20026-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adam K, Fuhs S, Meisenhelder J, Aslanian A, Diedrich J, Moresco J et al. (2019) A non-acidic method using hydroxyapatite and phosphohistidine monoclonal antibodies allows enrichment of phosphopeptides containing non-conventional phosphorylations for mass spectrometry analysis. bioRxiv 10.1101/691352 [DOI] [Google Scholar]
- 109.Makwana MV, van Meurs S, Hounslow AM, Williamson MP, Jackson RFW and Muimo R (2020) 31P NMR spectroscopy demonstrates large amounts of phosphohistidine in mammalian cells. bioRxiv 10.1101/2020.12.03.409540 [DOI] [Google Scholar]
- 110.Incel A, Arribas Diez I, Wierzbicka C, Gajoch K, Jensen ON and Sellergren B (2021) Selective enrichment of histidine phosphorylated peptides using molecularly imprinted polymers. Anal. Chem 93, 3857–3866 10.1021/acs.analchem.0c04474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao J, Zou L, Li Y, Liu X, Zeng C, Xu C et al. (2021) Hisphossite: a comprehensive database of histidine phosphorylated proteins and sites. J. Proteomics 243, 104262 10.1016/j.jprot.2021.104262 [DOI] [PubMed] [Google Scholar]
- 112.Kleinnijenhuis AJ, Kjeldsen F, Kallipolitis B, Haselmann KF and Jensen ON (2007) Analysis of histidine phosphorylation using tandem MS and ion-electron reactions. Anal. Chem 79, 7450–7456 10.1021/ac0707838 [DOI] [PubMed] [Google Scholar]
- 113.Riley NM, Rush MJ, Rose CM, Richards AL, Kwiecien NW, Bailey DJ et al. (2015) The negative mode proteome with activated Ion negative electron transfer dissociation (AI-NETD). Mol. Cell. Proteomics 14, 2644–2660 10.1074/mcp.M115.049726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Potel CM, Lemeer S and Heck AJR (2019) Phosphopeptide fragmentation and site localization by mass spectrometry: an update. Anal. Chem 91, 126–141 10.1021/acs.analchem.8b04746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wilk P, Jarmula A, Ruman T, Banaszak K, Rypniewski W, Ciesla J et al. (2014) Crystal structure of phosphoramide-phosphorylated thymidylate synthase reveals pSer127, reflecting probably pHis to pSer phosphotransfer. Bioorg. Chem 52, 44–49 10.1016/j.bioorg.2013.11.003 [DOI] [PubMed] [Google Scholar]
- 116.Wei BC, Rogers BJ and Wirth MJ (2012) Slip flow in colloidal crystals for ultraefficient chromatography. J. Am. Chem. Soc 134, 10780–10782 10.1021/ja304177m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M et al. (2015) Monoclonal 1- and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 162, 198–210 10.1016/j.cell.2015.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Eerland MF and Hedberg C (2012) Design and synthesis of an fmoc-SPPS-compatible amino acid building block mimicking the transition state of phosphohistidine phosphatase. J. Org. Chem 77, 2047–2052 10.1021/jo2025702 [DOI] [PubMed] [Google Scholar]
- 119.Song JN, Wang HL, Wang JW, Leier A, Marquez-Lago T, Yang BJ et al. (2017) Phosphopredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci. Rep 7, 6862–6880 10.1038/s41598-017-07199-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Luo FL, Wang MH, Liu Y, Zhao XM and Li A (2019) Deepphos: prediction of protein phosphorylation sites with deep learning. Bioinformatics 35, 2766–2773 10.1093/bioinformatics/bty1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang D, Zeng S, Xu C, Qiu W, Liang Y, Joshi T et al. (2017) Musitedeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 33, 3909–3916 10.1093/bioinformatics/btx496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Awais M, Hussain W, Khan YD, Rasool N, Khan SA and Chou KC (2021) iPhosH-PseAAC: identify phosphohistidine sites in proteins by blending statistical moments and position relative features according to the chou’s 5-step rule and general pseudo amino acid composition. IEEE/ACM Trans Comput Biol. Bioinform 18, 596–610 10.1109/TCBB.2019.2919025 [DOI] [PubMed] [Google Scholar]
- 123.Chen Z, Zhao P, Li FY, Leier A, Marquez-Lago TT, Webb GI et al. (2020) PROSPECT: a web server for predicting protein histidine phosphorylation sites. J. Bioinf. Comput. Biol 18, 2050018 10.1142/S0219720020500183 [DOI] [PubMed] [Google Scholar]
- 124.Ross AH, Baltimore D and Eisen HN (1981) Phosphotyrosine-containing proteins isolated by affinity chromatography with antibodies to a synthetic hapten. Nature 294, 654–656 10.1038/294654a0 [DOI] [PubMed] [Google Scholar]
- 125.Eckhart W, Hutchinson MA and Hunter T (1979) An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18, 925–933 10.1016/0092-8674(79)90205-8 [DOI] [PubMed] [Google Scholar]
- 126.Frackelton AR Jr, Ross AH and Eisen HN (1983) Characterization and use of monoclonal antibodies for isolation of phosphotyrosyl proteins from retrovirus-transformed cells and growth factor-stimulated cells. Mol. Cell. Biol 3, 1343–1352 10.1128/mcb.3.8.1343-1352.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carlson HK, Plate L, Price MS, Allen JJ, Shokat KM and Marletta MA (2010) Use of a semisynthetic epitope to probe histidine kinase activity and regulation. Anal. Biochem 397, 139–143 10.1016/j.ab.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lasker M, Bui CD, Besant PG, Sugawara K, Thai P, Medzihradszky G et al. (1999) Protein histidine phosphorylation: increased stability of thiophosphohistidine. Protein Sci. 8, 2177–2185 10.1110/ps.8.10.2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schenkels C, Erni B and Reymond JL (1999) Phosphofurylalanine, a stable analog of phosphohistidine. Bioorg. Med. Chem. Lett 9, 1443–1446 10.1016/S0960-894X(99)00209-7 [DOI] [PubMed] [Google Scholar]
- 130.Lilley M, Mambwe B, Jackson RF and Muimo R (2014) 4-Phosphothiophen-2-yl alanine: a new 5-membered analogue of phosphotyrosine. Chem. Commun. (Camb) 50, 9343–9345 10.1039/C4CC03393K [DOI] [PubMed] [Google Scholar]
- 131.Kee JM, Villani B, Carpenter LR and Muir TW (2010) Development of stable phosphohistidine analogues. J. Am. Chem. Soc 132, 14327–14329 10.1021/ja104393t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McAllister TE, Nix MG and Webb ME (2011) Fmoc-chemistry of a stable phosphohistidine analogue. Chem. Commun. (Camb) 47, 1297–1299 10.1039/C0CC04238B [DOI] [PubMed] [Google Scholar]
- 133.Kee JM, Oslund RC, Perlman DH and Muir TW (2013) A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat. Chem. Biol 9, 416–421 10.1038/nchembio.1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kee JM, Oslund RC, Couvillon AD and Muir TW (2015) A second-generation phosphohistidine analog for production of phosphohistidine antibodies. Org. Lett 17, 187–189 10.1021/ol503320p [DOI] [PubMed] [Google Scholar]
- 135.Lilley M, Mambwe B, Thompson MJ, Jackson RF and Muimo R (2015) 4-Phosphopyrazol-2-yl alanine: a non-hydrolysable analogue of phosphohistidine. Chem. Commun. (Camb) 51, 7305–7308 10.1039/C5CC01811K [DOI] [PubMed] [Google Scholar]
- 136.Kalagiri R, Adam K and Hunter T (2020) Empirical evidence of cellular histidine phosphorylation by immunoblotting using pHis mAbs. Methods Mol. Biol 2077, 181–191 10.1007/978-1-4939-9884-5_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Luhtala N and Hunter T (2020) Immunohistochemistry (IHC): Chromogenic detection of 3-Phosphohistidine proteins in formaldehyde-Fixed, frozen mouse liver tissue sections. Methods Mol. Biol 2077, 193–208 10.1007/978-1-4939-9884-5_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Adam K and Hunter T (2020) Subcellular localization of histidine phosphorylated proteins through indirect immunofluorescence. Methods Mol. Biol 2077, 209–224 10.1007/978-1-4939-9884-5_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang J, Gelman IH, Katsuta E, Liang Y, Wang X, Li J et al. (2019) Glucose drives growth factor-independent esophageal cancer proliferation via phosphohistidine-focal adhesion kinase signaling. Cell. Mol. Gastroenterol. Hepatol 8, 37–60 10.1016/j.jcmgh.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khan I and Steeg PS (2018) The relationship of NM23 (NME) metastasis suppressor histidine phosphorylation to its nucleoside diphosphate kinase, histidine protein kinase and motility suppression activities. Oncotarget 9, 10185–10202 10.18632/oncotarget.23796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Affandi T and McEvoy MM (2019) Mechanism of metal ion-induced activation of a two-component sensor kinase. Biochem. J 476, 115–135 10.1042/BCJ20180577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gardell SJ, Hopf M, Khan A, Dispagna M, Hampton Sessions E, Falter R et al. (2019) Boosting NAD+ with a small molecule that activates NAMPT. Nat. Commun 10, 3241–3252 10.1038/s41467-019-11078-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Adam K, Lesperance J, Hunter T and Zage PE (2020) The potential functional roles of NME1 histidine kinase activity in neuroblastoma pathogenesis. Int. J. Mol. Sci 21, 3319–3337 10.3390/ijms21093319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kalagiri R, Stanfield RL, Meisenhelder J, La Clair JJ, Fuhs SR, Wilson IA et al. (2021) Structural basis for differential recognition of phosphohistidine-containing peptides by 1-pHis and 3-pHis monoclonal antibodies. Proc. Natl Acad. Sci. U.S.A 118, e2010644118 10.1073/pnas.2010644118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mou Y, Zhou XX, Leung K, Martinko AJ, Yu JY, Chen W et al. (2018) Engineering improved antiphosphotyrosine antibodies based on an immunoconvergent binding motif. J. Am. Chem. Soc 140, 16615–16624 10.1021/jacs.8b08402 [DOI] [PubMed] [Google Scholar]
- 146.Kinoshita K, Sadanami K, Kidera A and Go N (1999) Structural motif of phosphate-binding site common to various protein superfamilies: all-against-all structural comparison of protein-mononucleotide complexes. Protein Eng. 12, 11–14 10.1093/protein/12.1.11 [DOI] [PubMed] [Google Scholar]
- 147.Koerber JT, Thomsen ND, Hannigan BT, Degrado WF and Wells JA (2013) Nature-inspired design of motif-specific antibody scaffolds. Nat. Biotechnol 31, 916–921 10.1038/nbt.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hauser A, Poulou E, Muller F, Schmieder P and Hackenberger CPR (2021) Synthesis and evaluation of non-hydrolyzable phospho-lysine peptide mimics. Chemistry. 27, 2326–2331 10.1002/chem.202003947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hauser A (2020) Chemical Approaches to Elucidate Lysine Phosphorylation, Humboldt university of Berlin, Berlin, Thesis Dessertation [Google Scholar]
- 150.Hattori T and Koide S (2018) Next-generation antibodies for post-translational modifications. Curr. Opin. Struct. Biol 51, 141–148 10.1016/j.sbi.2018.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Noren CJ, Anthony-Cahill SJ, Griffith MC and Schultz PG (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244, 182–188 10.1126/science.2649980 [DOI] [PubMed] [Google Scholar]
- 152.Liu CC and Schultz PG (2010) Adding new chemistries to the genetic code. Annu. Rev. Biochem 79, 413–444 10.1146/annurev.biochem.052308.105824 [DOI] [PubMed] [Google Scholar]
- 153.Hoppmann C, Wong A, Yang B, Li S, Hunter T, Shokat KM et al. (2017) Site-specific incorporation of phosphotyrosine using an expanded genetic code. Nat. Chem. Biol 13, 842–844 10.1038/nchembio.2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.