

Abstract
Purpose:
To explore relationships between biological gene expression signatures and pembrolizumab response.
Experimental Design:
RNA-sequencing data on baseline tumor tissue from 1,188 patients across seven tumor types treated with pembrolizumab monotherapy in nine clinical trials were used. A total of 11 prespecified gene expression signatures [18-gene T-cell–inflamed gene expression profile (TcellinfGEP), angiogenesis, hypoxia, glycolysis, proliferation, MYC, RAS, granulocytic myeloid-derived suppressor cell (gMDSC), monocytic myeloid-derived suppressor cell (mMDSC), stroma/epithelial-to-mesenchymal transition (EMT)/TGFβ, and WNT] were evaluated for their relationship to objective response rate (per RECIST, version 1.1). Logistic regression analysis of response for consensus signatures was adjusted for tumor type, Eastern Cooperative Oncology Group performance status, and TcellinfGEP, an approach equivalent to evaluating the association between response and the residuals of consensus signatures after detrending them for their relationship with the TcellinfGEP (previously identified as a determinant of pembrolizumab response) and tumor type. Testing of the 10 prespecified non-TcellinfGEP consensus signatures for negative association [except proliferation (hypothesized positive association)] with response was adjusted for multiplicity.
Results:
Covariance patterns of the 11 signatures (including TcellinfGEP) identified in Merck–Moffitt and The Cancer Genome Atlas datasets showed highly concordant coexpression patterns in the RNA-sequencing data from pembrolizumab trials. TcellinfGEP was positively associated with response; signatures for angiogenesis, mMDSC, and stroma/EMT/TGFβ were negatively associated with response to pembrolizumab monotherapy.
Conclusions:
These findings suggest that features beyond IFNγ-related T-cell inflammation may be relevant to anti–programmed death 1 monotherapy response and may define other axes of tumor biology as candidates for pembrolizumab combinations.
Translational Relevance.
To investigate axes of biology beyond T-cell inflammation for their relevance to response to anti–programmed death 1 therapy, the Merck–Moffitt and The Cancer Genome Atlas transcriptome datasets were used to define other key gene expression signatures independent of data from any pembrolizumab clinical trial. Testing of these other axes in a large cohort of multiple solid tumor types showed that angiogenesis, monocytic myeloid-derived suppressor cell, and stroma/epithelial-to-mesenchymal transition/TGFβ were negatively associated and thus may be predictors of resistance to pembrolizumab monotherapy and potential targets for combination therapy strategies.
Introduction
Immune checkpoint inhibitor (ICI) therapy has significantly improved clinical outcomes for patients with several tumor types (1). A number of tumor-intrinsic and tumor-extrinsic factors have been hypothesized to affect treatment responses (1), although the relative impact of these factors on response to ICI therapy remains poorly understood.
The degree of proinflammatory gene expression in the tumor microenvironment (TME) can be correlated with response to ICI therapy (2). For example, an IFNγ-related, 18-gene T-cell–inflamed gene expression profile (TcellinfGEP) has been associated with response to pembrolizumab across multiple tumor types (3). Programmed death ligand 1 (PD-L1) RNA expression is a component of the TcellinfGEP and evaluations of the correlation between PD-L1 combined positive score (by protein IHC) and the TcellinfGEP shows a moderate correlation of 0.55 to 0.60 in datasets examined, but the two are not deemed equivalent biomarkers (4). Furthermore, the antitumor immune response is likely also influenced by other elements of the TME, including immune and stromal cells, tumor vasculature, extracellular matrix, and modulatory cytokines, in addition to tumor-intrinsic pathways (5). Gene expression signatures associated with key cell types (e.g., myeloid-derived suppressor and stromal cells; ref. 6), oncogenic pathways [e.g., RAS and MYC (7)], and biological processes [e.g., proliferation (8), immunosuppression by WNT inhibition (9), hypoxia (10), glycolysis (11), and angiogenesis] may be relevant to cancer progression and response to ICI therapy. To our knowledge, no evaluation of transcriptomic correlates of response under a prespecified testing approach has been previously reported in a large dataset from treated patients enrolled in clinical trials across tumor types and characterized with rigorously assessed RECIST, version 1.1 (RECIST v.1.1).
This analysis employs a prospectively specified retrospective evaluation undergirded with a rigorous statistical approach to evaluate a short list of key gene expression signatures reflective of the TME. In addition to the 18-gene TcellinfGEP, we developed a selective but comprehensive consensus set of 10 RNA-sequencing (RNA-seq)–based gene expression signatures representing canonical biological axes of gene expression that are common across different tumor types and that may be associated with response or resistance to pembrolizumab monotherapy. Using data from baseline tumor tissue samples from 1,188 patients enrolled in pembrolizumab monotherapy studies across seven tumor types, we prospectively tested for associations between these biologically relevant RNA-seq–based gene signatures and objective response rate after accounting for the explanatory information provided in the TcellinfGEP using joint regression modeling. The hypothesis tested for each signature (except the proliferation signature, which was tested for a positive association with tumor response) was that the specified signature score was negatively associated with propensity for a tumor to respond to pembrolizumab after adjustment for the level of the TcellinfGEP.
Materials and Methods
Datasets
Reference datasets: Merck–Moffitt (12) and The Cancer Genome Atlas (TCGA) databases (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) were used to develop consensus signatures. The Merck–Moffitt gene expression dataset contains 16,000 primary and 4,000 metastatic tumors and represents >25 different cancers (Supplementary Table S1; ref. 13). Gene expression data were obtained from primary and metastatic tumors of adult patients treated through the Total Cancer Care initiative, which was created by the H. Lee Moffitt Cancer Center (Tampa, FL; ref. 12). Tumor tissue was snap frozen. Macrodissection was performed in liquid nitrogen to maintain the frozen tissue and enrich the tumor content, followed by RNA extraction. The specimens were then arrayed on HuRSTA-2a520709 GeneChips (Affymetrix). These chips contain >52,000 probe sets, representing >22,000 unique genes. Additional details regarding the platform (e.g., probe set annotation, chip definition files) have been made public and can be accessed as platform GPL10379 at the NCBI Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL10379). TCGA data were obtained from the TCGA website (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Clinical trial (KEYNOTE) datasets: The current analysis included pretreatment tumor samples from patients with available RNA-seq data that passed quality control from nine single-arm or randomized clinical studies of pembrolizumab monotherapy (N = 1,188; Table 1): KEYNOTE-001/KEYNOTE-006–melanoma (NCT01295827/NCT01866319; n = 476; first-line and later setting; ipilimumab-treated and ipilimumab-naive); KEYNOTE-052–urothelial carcinoma (NCT02335424; n = 186; first-line setting); KEYNOTE-012/KEYNOTE-055–head and neck squamous cell carcinoma (NCT01848834/NCT02255097; n = 147; first-line and later setting; human papilloma virus–negative by whole exome sequencing); KEYNOTE-086–triple-negative breast cancer (NCT02447003; n = 132; first-line and later setting); KEYNOTE-059–gastric cancer (NCT02335411; n = 92; first-line and later setting); KEYNOTE-427–renal cell carcinoma (NCT02853344; n = 78; first-line setting); and KEYNOTE-100–ovarian cancer (NCT02674061; n = 77; recurrent setting). Detailed patient baseline and disease characteristics have been published for each respective study. The study protocols and all amendments were approved by the appropriate ethics committee at each center for each study. Each study was conducted in accordance with the protocol and its amendments, the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice, and local and national regulations. All participants provided written informed consent.
Table 1.
Study descriptions for RNA-seq dataset.
| Study | Clinicaltrials.gov registration | Cancer type | Treatment setting | Patient numbera | Patient characteristics | Treatment |
|---|---|---|---|---|---|---|
| KEYNOTE-001 | NCT01295827 | Melanoma | Second- or later-line treatment of ipilimumab-refractory advanced melanoma | 83 | Median (range) age: 62 (23–94) years; 59% male; ECOG 0/1: 70%/30%; PD-L1 positive: 73% | Pembrolizumab 2 mg/kg or 10 mg/kg Q2W, or 10 mg/kg Q3W |
| KEYNOTE-006 | NCT01866319 | Melanoma | First- or later-line treatment of unresectable stage III or metastatic melanoma; up to 1 previous systemic therapy | 393 | Median (range) age: 62 (18–88) years; 62% male; ECOG 0/1: 69%/31%; PD-L1 positive: 86% | Pembrolizumab 10 mg/kg Q3W or 10 mg/kg Q2W |
| KEYNOTE-052 | NCT02335424 | Urothelial carcinoma | First-line treatment for cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer | 186 | Median (range) age: 73 (34–91) years; 77% male; ECOG 0/1: 23%/76%; PD-L1 positive: 82% | Pembrolizumab 200 mg Q3W |
| KEYNOTE-012 | NCT01848834 | Head and neck squamous cell carcinoma | First- or later-line treatment of recurrent or metastatic squamous cell carcinoma of the head and neck | 88 | Median (range) age: 61 (25–83) years; 80% male; ECOG 0/1: 27%/73%; PD-L1 positive: 91%; Cohort B2: 72%, Cohort B: 28% | Pembrolizumab 10 mg/kg Q2W or 200 mg Q3W |
| KEYNOTE-055 | NCT02255097 | Head and neck squamous cell carcinoma | Second- or later-line treatment of platinum- and cetuximab-refractory head and neck cancer | 59 | Human papillomavirus–negative | Pembrolizumab 200 mg Q3W |
| Median (range) age: 61 (33–83) years; 75% male; ECOG 0/1: 14%/86%; PD-L1 positive: 86% | ||||||
| KEYNOTE-086 | NCT02447003 | Triple-negative breast cancer | First and later-line treatment for metastatic triple-negative breast cancer | 132 | All women | Pembrolizumab 200 mg Q3W |
| Median (range) age: 52 (26–82) years; ECOG 0/1: 58%/42%; Cohort A: 70%, Cohort B: 30%; PD-L1 positive: 75% | ||||||
| KEYNOTE-059 | NCT02335411 | Gastric or gastroesophageal junction cancer | First- or later-line treatment for patients with untreated recurrent or metastatic gastric or gastroesophageal junction adenocarcinoma | 92 | Median (range) age: 64 (24–89) years; 73% male; ECOG 0/1: 40%/60%; Cohort 1: 100%; PD-L1 positive: 62% | Pembrolizumab 200 mg Q3W |
| KEYNOTE-427 | NCT02853344 | Clear cell renal cell carcinoma | First-line treatment for patients with locally advanced or metastatic renal cell carcinoma | 78 | Cohort A | Pembrolizumab 200 mg Q3W |
| Median (range) age: 65 (39–87) years; 77% male; IMDC: Favorable, 37%; Intermediate, 51%; Poor, 12%; PD-L1 positive: 46% | ||||||
| KEYNOTE-100 | NCT02674061 | Ovarian cancer | First- or later-line treatment of recurrent ovarian cancer | 77 | Cohort A | Pembrolizumab 200 mg Q3W |
| Median (range) age: 62 (34–89) years; all women; ECOG 0/1: 69%/31% |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; IMDC, International Metastatic RCC Database Consortium; PD-L1, programmed death ligand 1; Q2W, every 2 weeks; Q3W, every 3 weeks.
aIncluding only patients receiving pembrolizumab monotherapy and for whom RNA-seq data were available.
RNA-seq
RNA-seq on formalin-fixed paraffin-embedded specimens from KEYNOTE trials was performed using the HiSeq 4000 platform (Illumina). The RNA-seq raw reads were processed using the customized RNA-seq data analysis pipeline in OmicSoft ArraySuite, version 9. Specifically, the raw reads were first filtered on the basis of quality control and then were aligned to the reference genome Human.B37.3 using OSA (14). After alignment, gene expression levels (raw read counts and fragments per kilobase of exon per million mapped fragments) were quantified using the RSEM algorithm (15) with the gene model Ensembl.R75. All samples and data were obtained with appropriate Institutional Review Board approvals.
Gene selection
A compilation of external resources, including the Merck–Moffitt and TCGA databases, was used to identify genes and signatures associated with the TcellinfGEP. Relatively good stability was observed across these two databases in terms of the pattern of all gene correlations with the GEP (Supplementary Fig. S1B). Beyond the 18 genes included in the GEP, other genes showed strong or moderate correlations with the GEP, and to account for FDR, we identified a cutoff for the correlation to determine membership of genes with a specified pattern. For example, all genes within the TcellinfGEP signature have a Spearman correlation of ≥0.6 with the signature itself. We also note that not all genes in the GEP signature are derived from the same cell type; however, this association at a correlation of approximately 0.6 would seem sufficient to define a pattern of gene expression that is highly replicable in terms of the member genes, ultimately showing reliable statistically significant associations with clinical outcome at the level of the individual genes within the signature.
Consequently, using this 0.6 correlation as a guide, consensus signatures beyond GEP were defined de novo from foundational datasets external to our study and independent of any pembrolizumab trial or exposure using an algorithm to select individual genes for membership in a consensus signature based on correlation, with reference signatures identified in the literature or from internal discovery outside of pembrolizumab trials (10, 11, 16, 17). For the current analysis, a reference signature was selected for a biological process (e.g., angiogenesis), and the correlation of all genes to the reference signature score in large datasets of gene expression profiles of human tumors was computed using data from both the Merck–Moffitt and TCGA databases. This approach allowed for derivation of coexpression patterns with expected high reproducibility across datasets while avoiding signature redundancy by mapping genes with similar behavior (with respect to the reference signature) into a single signature, thereby limiting the statistical adjustments to be applied for multiple hypotheses testing when evaluating these consensus signatures in patients treated with pembrolizumab.
If the correlation between an individual gene in the transcriptome and the reference signature score, as calculated using data from the two databases, differed by >0.2, the gene was removed from consideration for membership in that consensus signature. The two correlation values from the Merck–Moffitt and TGCA databases for the remaining genes were then averaged. Gene selection for the consensus sets then followed a hierarchical process, starting with all genes with an average correlation of ≥0.65 to the reference signature score being selected if ≥30 genes met this criterion. If not, the correlation threshold was lowered to ≥0.60. If <30 genes met these criteria, all genes with an average correlation of ≥0.55 were incorporated into the consensus signature. The algorithm aimed to avoid small sized signatures while staying consistent with our observation that genes in the 18-gene TcellinfGEP, which are validated predictors of response in moderate to large pan-tumor datasets (N > 500, but N < 1,000), tend to maintain a correlation of approximately 0.6 with the GEP itself.
TcellinfGEP score and development of consensus signatures
The TcellinfGEP score, as assessed using the RNA-seq platform, was calculated as the weighted sum of the predictor genes determined during the development of the TcellinfGEP on the NanoString platform (3). In addition to the TcellinfGEP, 10 consensus RNA expression signatures representative of canonical pathways associated with tumor biology and TME elements beyond the TcellinfGEP were defined using gene RNA coexpression patterns in the Merck–Moffitt and TCGA databases. These non-TcellinfGEP signature scores were calculated as the average of the genes (in log scale) in each signature gene set. The analytical framework for the development of these consensus signatures combined biological knowledge of tumor gene expression patterns in large foundational datasets with reference signatures from the literature (18, 19) to define a parsimonious set of signatures with a robust covariance structure related to a number of relevant biological patterns.
Our method generalizes the immune-centric approach taken by Thorsson and colleagues (20) by further synthesizing nonimmune elements of the TME into a small set of molecular signatures. We relied on observations related to the covariance of genes with TcellinfGEP in the Merck–Moffitt and TCGA databases as a template. The TcellinfGEP identifies a parsimonious gene expression pattern associated with response to programmed death 1 (PD-1) blockade using the NanoString platform as described in Ayers and colleagues (3). The resultant core set of genes, referred to as the 18-gene TcellinfGEP, captures a dominant predictive signature of response to PD-1 blockade in a pan-tumor setting (3). Several other reports have identified gene expression signatures significantly associated with an IFNγ-activated T-cell–inflamed microenvironment: immunoscore (21), chemokine signature (22), and cytolytic signature (23). All these signatures and GEPs (3, 21–23), as well as PD-L1/PD-L2 gene expression, exhibited similar covariance patterns in the Merck–Moffitt database (Supplementary Figs. S1 and S2).
Consensus RNA signatures reflective of TME and tumor biology, including signatures of proliferation, stroma/epithelial-to-mesenchymal transition (EMT)/TGFβ, RAS, MYC, WNT, hypoxia, glycolysis, angiogenesis, and granulocytic and monocytic myeloid-derived suppressor cells (gMDSC and mMDSC, respectively), were developed. A list of member genes for each consensus signature is provided in Supplementary Table S2 in the Supplementary Data and descriptions of each consensus signature are given in Supplementary Table S3 in the Supplementary Data; the overlap of gene membership between the signatures is minimal, reflecting the relative orthogonal nature of their biological substrate.
Evaluation of the pairwise correlation, indicating patterns for the 10 consensus signature scores plus the TcellinfGEP, led to similar results when using either the tissue-adjusted residual versions of the signature scores or the standard versions, suggesting these consensus signatures represent fairly robust patterns of coexpression across solid tumor types.
Tests for association between non-GEP consensus signature scores and confirmed objective response rate as per RECIST v.1.1 by independent central review (where objective response equals complete response plus partial response) were based on logistic regression analysis and adjusted for tumor type, Eastern Cooperative Oncology Group performance status, and the TcellinfGEP. Adjustment for the TcellinfGEP was performed to understand the additional explanatory value that any non-TcellinfGEP signatures had for objective response rate, an approach equivalent to evaluating the association between objective response rate and the residuals of consensus signatures after detrending for their relationship with the TcellinfGEP. Testing of the 10 prespecified consensus signatures for a postulated negative association (except proliferation, which had a hypothesized positive association) with objective response rate was adjusted for multiplicity using the Hochberg step-up procedure. Area under the receiver operating characteristic (AUROC) curve for the residual consensus signature score (after detrending the associations of tumor type and the GEP on the signature score) on objective response was used as a general measure of the discriminatory ability of the consensus signatures.
Importantly, the consensus signature gene sets were prespecified, and corresponding scores were calculated, prior to connecting RNA-seq data to clinical outcomes from the studies evaluated, thus ensuring blinding of the clinical data to the genomic analysts to avoid the risk of overfitting the data.
Data availability
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the United States and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
Access to data
Andrey Loboda had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Results
Details on consensus signatures and their correlation patterns are shown in Table 2 and visualized for the Merck–Moffitt dataset (Fig. 1A), TCGA dataset (Fig. 1B), and the independent pembrolizumab clinical trial patient analysis population (Fig. 1C), consisting of 1,188 patient samples collected from clinical trials with pembrolizumab monotherapy (which were not used to develop the consensus signatures). RNA-seq data from pembrolizumab clinical trials demonstrated highly concordant coexpression patterns for the signatures defined in the Merck–Moffitt and TCGA datasets.
Table 2.
Overview of consensus gene sets.
| Signature | TcellinfGEP | Proliferation | Stroma EMT/TGFβ | RAS | MYC | WNT | Hypoxia | Glycolysis | Angiogenesis | gMDSC | mMDSC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of genes in consensus | 18 | 227 | 51 | 11 | 32 | 13 | 20 | 30 | 16 | 43 | 218 |
| Correlation with TcellinfGEP, TCGA | 1 | 0.07 | 0.23 | 0.11 | −0.09 | −0.20 | 0.20 | 0.21 | 0.10 | 0.47 | 0.81 |
| Correlation with TcellinfGEP, Merck–Moffitt | 1 | 0.10 | 0.22 | 0.10 | 0.04 | −0.11 | 0.20 | 0.25 | 0.02 | 0.46 | 0.80 |
Abbreviations: EMT, epithelial-to-mesenchymal transition; gMDSC, granulocytic myeloid-derived suppressor cell; mMDSC, monocytic myeloid-derived suppressor cell; TcellinfGEP, T-cell–inflamed gene expression profile; TCGA, The Cancer Genome Atlas; TGFβ, transforming growth factor β.
Figure 1.
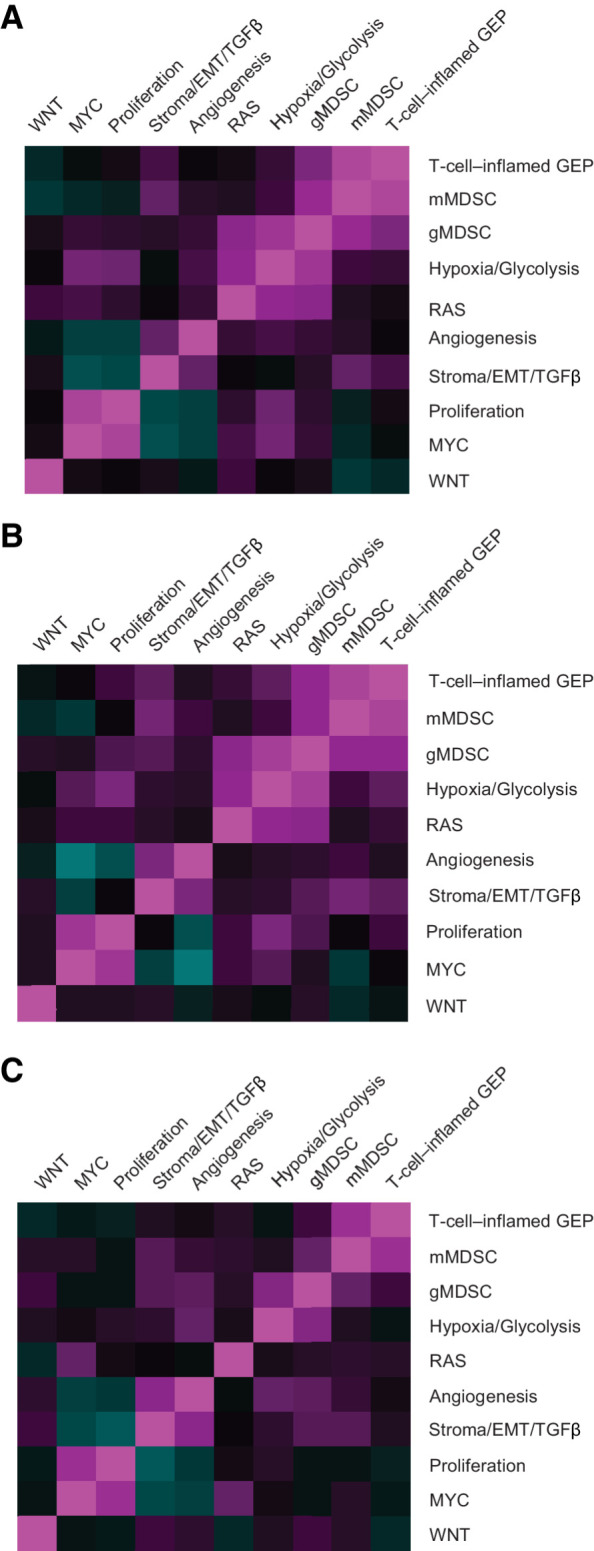
Pairwise Spearman correlation of consensus signatures. Merck–Moffitt dataset (A), TCGA dataset (B), and pooled pembrolizumab trial analysis population (C). EMT, epithelial-to-mesenchymal transition; GEP, gene expression profile; gMDSC, granulocytic myeloid-derived suppressor cell; mMDSC, monocytic myeloid-derived suppressor cell; TCGA, The Cancer Genome Atlas; TGFβ, transforming growth factor β.
The TcellinfGEP demonstrated the strongest association with response to pembrolizumab (Table 3), as previously described in a smaller sample size set (13). TcellinfGEP showed a moderate correlation with PD-L1 assay values, suggestive of their consistent readouts of an inflamed microenvironment, but nonredundant role in selecting patients most likely to benefit from pembrolizumab monotherapy.
Table 3.
Association of TcellinfGEP and consensus signatures with response to pembrolizumab.
| Signature | AUROC curvea (95% CI) | Nominal one-sided Pb | Multiplicity-adjusted Pc |
|---|---|---|---|
| TcellinfGEP | 0.63 (0.60–0.67) | 3.6 × 10–12 | N/A |
| Proliferation | 0.53 (0.49–0.56) | 0.09 | 0.45 |
| Stroma/EMT/TGFβ | 0.56 (0.52–0.60) | 0.0003 | 0.0023 |
| RAS | 0.52 (0.48–0.56) | 0.11 | 0.45 |
| MYC | 0.51 (0.47–0.55) | 0.41 | 0.82 |
| WNT | 0.52 (0.48–0.56) | 0.10 | 0.45 |
| Hypoxia | 0.51 (0.47–0.54) | 0.38 | 0.82 |
| Glycolysis | 0.48 (0.44–0.52) | 0.83 | 0.83 |
| Angiogenesis | 0.58 (0.54–0.61) | 0.0001 | 0.0009 |
| gMDSC | 0.53 (0.50–0.57) | 0.032 | 0.22 |
| mMDSC | 0.56 (0.53–0.60) | 0.0001 | 0.0009 |
Abbreviations: AUROC, area under the receiver operating characteristic; CI, confidence interval; EMT, epithelial-to-mesenchymal transition; gMDSC, granulocytic myeloid-derived suppressor cell; mMDSC, monocytic myeloid-derived suppressor cell; N/A, not applicable; TcellinfGEP, T-cell–inflamed gene expression profile; TGFβ, transforming growth factor β.
aFor the TcellinfGEP, predictor is residual score after adjusting for tumor type and for non-TcellinfGEP residual score after adjustment for tumor type and TcellinfGEP.
bFor the TcellinfGEP and for proliferation, statistical testing and AUROC curve estimation were based on hypothesized positive associations, whereas negative associations were posited for the remainder.
cConsensus signature tests were adjusted using Hochberg step-up procedure.
After adjusting for multiple testing, tumor type and other covariates, including the TcellinfGEP, the angiogenesis, mMDSC, and stroma/EMT/TGFβ signatures, exhibited significant negative associations with response (P < 0.05; Table 3), although the P values and the AUROC curve estimates indicated that the size of the effect was less than that observed for the TcellinfGEP and was modest (AUROC < 0.6) overall. The distributions of tumor type and TcellinfGEP-detrended versions of the signature scores are shown for responders and nonresponders (Fig. 2), demonstrating the consistency across cancer types in distribution differences between responders and nonresponders in the TcellinfGEP, angiogenesis, mMDSC, and stroma/EMT/TGFβ signatures.
Figure 2.
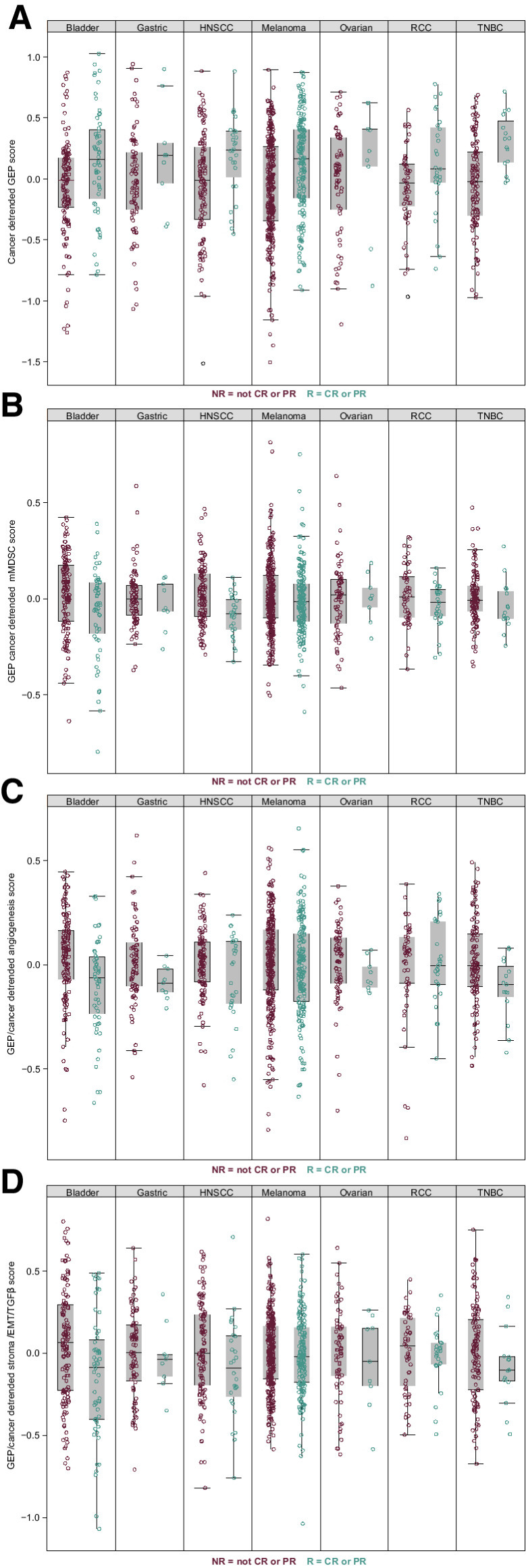
Associations of gene signatures with response to pembrolizumab across tumor types. T-cell–inflamed GEP (A), mMDSC (B), angiogenesis (C), and stroma/EMT/TGFβ signatures (D). CR, complete response; EMT, epithelial-to-mesenchymal transition; GEP, gene expression profile; HNSCC, head and neck squamous cell carcinoma; mMDSC, monocytic myeloid-derived suppressor cells; NR, nonresponder; PR, partial response; R, responder; RCC, renal cell carcinoma; TGFβ, transforming growth factor β; TNBC, triple-negative breast cancer.
When the AUROC was compared by tumor type, considerable variation was observed (Fig. 3). Consensus sets tended to be more accurate predictors of response in patients with triple-negative breast cancer or head and neck squamous cell carcinoma and less predictive for patients with melanoma, gastric cancer, or renal cell carcinoma, suggestive of particularities of individual cancer types that cannot be fully explained by the universal signatures we propose here.
Figure 3.

AUROC curve by signature and tumor type estimated in the direction of the hypothesized association. The symbols are sized to represent the population sizes of the different cohorts that influence the AUROC curve estimates shown in Table 2. Non-TcellinfGEP consensus signatures were evaluated after detrending for TcellinfGEP. AUROC, area under the receiver operating characteristic; EMT, epithelial-to-mesenchymal transition; GEP, gene expression profile; gMDSC, granulocytic myeloid-derived suppressor cells; HNSCC, head and neck squamous cell carcinoma; mMDSC, monocytic myeloid-derived suppressor cells; RCC, renal cell carcinoma; TcellinfGEP, T-cell–inflamed gene expression profile; TGFβ, transforming growth factor β; TNBC, triple-negative breast cancer.
The consensus signatures that were significant across solid tumor types can be mapped to corresponding parsimonious signatures analyzed by other groups in a clinical setting. A number of studies have explored signatures that are composed of overlapping but not identical sets of genes as ours but that generate scores highly concordant with corresponding scores from the consensus signatures developed in the current analysis (24, 25). For example, core biological pathways explored by Mariathasan and colleagues (25) in molecular profiles in tumors from patients with urothelial carcinoma receiving anti–PD-L1 therapy uniquely and strongly map to TcellinfGEP, stroma/EMT/TGFβ, proliferation, and angiogenesis consensus signatures (Supplementary Fig. S3A). Among these core biological pathways, CD8+ T-effector GEP and TGFβ (stroma/EMT/TGFβ) demonstrated significant associations with response and resistance, respectively. In another study with patients with renal cell carcinoma treated with anti–PD-L1 therapy, a different set of signatures was explored by McDermott and colleagues (24); these signatures again demonstrated significant correlations with TcellinfGEP, gMDSC, and angiogenesis signatures (Supplementary Fig. S3B). All genes illustrated in the study by McDermott and colleagues (24) showed a high correlation with the corresponding consensus signatures in our study.
Discussion
This analysis of prespecified RNA-seq–based consensus signatures of canonical biological pathways of the TME from a large dataset of patients treated with pembrolizumab confirms the importance of the IFNγ-related TcellinfGEP and demonstrated additional pathways that may be relevant to response and resistance to ICI therapy across multiple solid tumor types. Whereas the TcellinfGEP demonstrated a robust positive association with response to pembrolizumab, this analysis also indicates that signatures for angiogenesis, mMDSC, and stroma/EMT/TGFβ may be negative predictors of response to pembrolizumab monotherapy after adjusting for the levels of the TcellinfGEP, providing important directions of biological variation that currently are or can be exploited therapeutically.
The goal for the development and selection of consensus signatures was to choose a short but comprehensive list of coherent, reproducible, and nonredundant signatures consistent with literature that have a clear connection to canonical pathways, key cell types, and biological processes. These signatures were prespecified for prospective testing of RNA expression datasets across the set of MSD's pembrolizumab monotherapy studies. Our approach is novel for biomarker development and qualification and provides platform-independent evaluation of the proposed gene expression signatures across solid tumor types.
A review of key clinical publications that document response to anti–PD-1/PD-L1 therapy highlights unequivocally strong pairwise relationships between the biomarker scores generated using our consensus signatures and the signatures proposed in these other studies. Although gene expression signature definitions and naming will differ across different investigational groups, a high degree of concordance at the level of their signature scores is likely in many instances. Examining the relationships between the signature scores used across multiple studies published by different groups is necessary to understand where consistent findings have been observed. For example, our consensus profile for the stroma/EMT/TGFβ consensus signature not only demonstrated an association with resistance to anti–PD-1/PD-L1 treatment in the current analysis but also showed high concordance with anti–PD/PD-L1 resistance signatures defined in two other clinical studies (25, 26). Differently named signatures, but similar in terms of signature scores, mapping to our stroma/EMT/TGFβ signature were explored by Mariathasan and colleagues (25) and Wang and colleagues (26) and showed a significant association with a lack of response in PD-1 inhibitor–treated patients with urothelial carcinoma. Although these studies have assigned the signatures different names and have provided alternative explanations with regard to biological interpretation of the data, the proposed biomarkers are highly concordant and reflect the same convergent biology, likely related to the mesenchymal state of the tumors, infiltration of cancer-associated fibroblasts, and activation of the TGFβ pathway.
The selection of signatures in this report was somewhat similar to that in a previous report, which analyzed TCGA transcriptome data (N > 10,000) and a collection of previously published human immune signatures to determine a sparse, but comprehensive, set of five signatures to summarize the immune landscape of cancer (20). Those five signatures show high internal coherence (Supplementary Fig. S4A) and map to some of the consensus signatures we defined (Supplementary Fig. S5). Interestingly, the mMDSC consensus signature in our study and the monocytes/macrophages signature proposed by Thorsson and colleagues (20) demonstrate a 0.99 correlation coefficient in both the Merck–Moffitt and TCGA databases, consistent with our interpretation of the biology behind this consensus signature.
Liberzon and colleagues (27) also sought to reduce redundancy in large datasets by generating a hallmark gene set. However, they did not consistently use large cohorts of human tumor samples, and the genes within the signatures are typically less coherent (with an average correlation to signature score of <0.5; Supplementary Fig. S4B) compared with the consensus signatures we developed (Supplementary Fig. S4C).
The significant findings for the association with an outcome reported for the angiogenesis, mMDSC, and stroma/EMT/TGFβ signatures were consistent with the proposed role of these pathways as immune-suppressive axes, which may negatively impact the efficacy of single-agent immunotherapy (5). For example, MDSCs can promote tumor evasion and growth (5). Notably, the mMDSC signature score evaluated here demonstrates a strong positive association with the TcellinfGEP, which may reflect the biology of concurrent antitumor and compensatory immunosuppressive response at the tumor site. Complementary to mMDSCs, the production of angiogenic factors is known to suppress antigen-presenting cells and immune effector cells, augmenting the activity of regulatory T cells, MDSCs, and tumor-associated macrophages. Furthermore, the vascular phenotype associated with excessive expression of angiogenic factors may have an additional immunosuppressive effect, preventing the efficient trafficking of T cells within the TME (5). Excessive TGFβ may also have an immunosuppressive effect, acting as an additional mechanism of resistance to ICI therapy (5). Potential actionable targets behind biomarker-associated resistance in these signatures include multiple potential checkpoint and regulatory receptors, such as ILT3, ILT4, LAIR1, PILRA, SIRPA, SELPLG, and CSF1R, among those associated with the mMDSC signature. Similarly, angiogenesis modules contain multiple known targets, such as TIE1, TIE2, KDR, VEGFR, and additional potential targets critical for the biology of endothelial cells that contribute to this consensus signature. The stroma/EMT/TGFβ signature contains potential targets related to fibroblast biology (e.g., CD93 and PDGFRB), TGFβ signaling (e.g., sLRRC32), and EMT induction (e.g., AXL).
Identifying factors that may limit the efficacy of ICI therapy may also identify patients who could benefit from combination therapy with antiangiogenic, anti-TGFβ, or other targeted therapies (5, 28–30). These data provide opportunities for bedside-to-bench exploration of relevant biology, and a rational basis for combination strategies. For example, multiple combinations of ICIs with anti-angiogenesis agents have been approved by the FDA, affirming their viability as an effective combination strategy (31, 32) and underscoring the importance of the relationship between angiogenesis and response to ICI. These results are consistent with our identification of angiogenesis as a signature negatively correlated to response and suggest that our described analytic approach may have mechanistic relevance to clinical development strategies. For instance, TGFβ-modulating agents (33) are being tested in combination with ICIs, with promising results (30). Finally, agents that modulate the mMDSC axis are also being evaluated in the clinic (34). Future evaluation of signatures in combination studies may provide insight into the biological context in which combination therapy has the greatest incremental impact.
It is also important to note that we could not replicate some previously reported negative associations for the WNT, MYC, and gMDSC signatures and clinical outcome. WNT- and MYC-related pathways are hypothesized to be involved in immune exclusion processes (35, 36). However, the overall influence of these signatures on response was not observed, and no noteworthy negative correlation between these signatures and GEP was observed in this analysis of specimens from pembrolizumab studies across solid tumor types. We anticipated that the gMDSC signature would be associated with resistance to pembrolizumab, as has been reported by McDermott and colleagues (24) for atezolizumab in advanced renal cell carcinoma; however, we did not identify such a relationship. Several differences in our analysis may account for this. First, we used RECIST-defined response as the primary clinical endpoint, given the noncontrolled single-arm treatment setting. Our analysis was also inherently aimed at an approach across multiple solid tumor types. In contrast, McDermott and colleagues (24) used overall survival as the primary endpoint and was focused on renal cell carcinoma.
Our study, which investigated outcomes in patients treated with pembrolizumab, is inherently limited by its retrospective, uncontrolled, and exploratory nature. The tumor types represent a convenient sample of single-arm trials with data available at the time the analyses were conducted. The tumor types represented in our combined RNA-seq dataset are not equally distributed (albeit all modeling was adjusted for tumor type as a covariate). By convention, for the evaluation of tumor response, patients whose best overall response was stable disease were treated as nonresponders; however, in the evaluation of “resistance” or “suppressive” TME axes, we acknowledge that this stable disease group represents a mixed population in terms of long-term prognosis and that treating this entire group as nonresponders may not be the most powerful approach to defining drug activity.
Our focus was an approach across multiple solid tumor types aimed at evaluating some well-recognized TME dimensions using prespecified set features; no de novo discovery effort to find tumor-specific resistance patterns was pursued. The strengths of our results are in the prespecified testing of key consensus signatures and in the large sample size (N = 1,188) of patients treated with pembrolizumab. To our knowledge, this is the largest dataset of pembrolizumab monotherapy evaluated with transcriptome analysis and our approach may serve as an important benchmark for evaluation of RNA-seq–based biomarkers in future studies and analyses. Further evaluation of these molecular signatures in these and other tumor types, and in randomized trials, may provide additional insight into their prognostic or predictive character, or both.
Conclusions
A set of prespecified consensus gene expression signatures using RNA-seq data derived from multiple tumor types external to studies of pembrolizumab were confirmed as resistance patterns of pembrolizumab studies across multiple solid tumor types. Our findings represent important additions in the exploration of RNA-seq–based biomarkers in the anti–PD-1 monotherapy setting due to power of the dataset, prospective testing of a parsimonious list of predefined signatures, and introduction of novel signatures into a clinical setting that can continue to be followed for further validation in more homogeneous, large, late-phase randomized trials. In addition, TME elements beyond IFNγ-related T-cell inflammation show promise for understanding the biology of ICI therapy response and highlight the potential to develop rational combination strategies.
Supplementary Material
Acknowledgments
This study was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. The authors thank the participants and their families and all investigators and site personnel. The authors also thank Andrea Webber, Petar Jelinic, Raluca Predoiu, and Mohini Rajasagi of Merck & Co., Inc., Kenilworth, NJ, USA for their contributions. Medical writing and/or editorial assistance was provided by Holly C. Cappelli, PhD, CMPP, and Dana Francis, PhD, of ApotheCom (Yardley, PA, USA). This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
R. Cristescu reports other support from Merck & Co., Inc., Kenilworth, NJ, USA during the conduct of the study; in addition, R. Cristescu has a patent pending related to the application of stroma signature (WO 2020/167619), is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and is a stockholder of Merck & Co., Inc., Kenilworth, NJ, USA. M. Nebozhyn reports personal fees, nonfinancial support, and other support from Merck & Co., Inc., Kenilworth, NJ, USA during the conduct of the study as well as nonfinancial support and other support from Merck & Co., Inc., Kenilworth, NJ, USA outside the submitted work. C. Zhang reports personal fees and other support from Merck & Co., Inc., Kenilworth, NJ, USA outside the submitted work; in addition, C. Zhang has a patent application pending, reports personal fees and other support from Merck & Co., Inc., Kenilworth, NJ, USA outside the submitted work, has a patent pending related to the application of stroma signature (WO 2020/167619), and is currently an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and is a stockholder of Merck & Co., Inc., Kenilworth, NJ, USA. A. Albright is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. L. Huang reports personal fees from Merck & Co., Inc., Kenilworth, NJ, USA and other support from Merck & Co., Inc., Kenilworth, NJ, USA outside the submitted work. A. Wang reports other support from Merck & Co., Inc., Kenilworth, NJ, USA outside the submitted work. H. Ma reports a patent for Gene expression based biomarker of tumor response to PD-1 antagonists issued to European Patent Office. Z.A. Cao reports employment at Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and is a stockholder of Merck & Co., Inc., Kenilworth, NJ, USA. M. Morrissey reports other support from Merck & Co., Inc., Kenilworth, NJ, USA during the conduct of the study. A. Ribas reports personal fees from Amgen, Chugai, Genentech, Merck, Novartis, Roche, Sanofi and Vedanta, 4C Biomed, Appia, Apricity, Arcus, Highlight, Compugen, ImaginAb, Kalthera-Immpact Bio, MapKure, Merus, Rgenix, Lutris, PACT Pharma, Synthekine, Tango, Advaxis, CytomX, Five Prime, RAPT, Isoplexis, and Kite-Gilead, and grants from Agilent and Bristol Myers Squibb outside the submitted work. P. Grivas reports nonfinancial support from Merck during the conduct of the study as well as personal fees and nonfinancial support from AstraZeneca; personal fees from Astellas Pharma, Bayer, Dyania Health, Exelixis, Foundation Medicine, Genentech/Roche, Genzyme, Guardant Health, Heron Therapeutics, Infinity Pharmaceuticals, Janssen, Regeneron Pharmaceuticals, Seattle Genetics, 4D Pharma PLC, and Urogen; grants and personal fees from Bristol Myers Squibb, EMD Serono, GlaxoSmithKline, Immunomedics/Gilead, Merck, Mirati Therapeutics, QED Therapeutics, and Pfizer; grants and nonfinancial support from Clovis Oncology; and grants from Bavarian Nordic and Debiopharm outside the submitted work. D.W. Cescon reports nonfinancial support and other support from Merck during the conduct of the study as well as personal fees from AstraZeneca, Exact Sciences, Gilead, Novartis, and Roche; personal fees and other support from GlaxoSmithKline Merck; grants and personal fees from Pfizer; and other support from Inivata outside the submitted work; in addition, D.W. Cescon has a patent (US62/675,228) for methods of treating cancers characterized by a high expression level of spindle and kinetochore associated complex subunit 3 (ska3) gene issued, and is a steering committee member for clinical trials: Merck and GlaxoSmithKline. T.K. McClanahan reports a patent pending for Angiogenesis and mMDSC gene expression based biomarker of tumor response to PD-1 antagonists; in addition, T.K. McClanahan is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. A. Snyder reports other support from Merck & Co., Inc. and Two River, Inc outside the submitted work. M. Ayers reports other support from Merck & Co., Inc., Kenilworth, NJ, USA during the conduct of the study; in addition, M. Ayers has a patent for GEP biomarker issued. J. Lunceford reports a patent pending for Angiogenesis and mMDSC gene expression based biomarker of tumor response to PD-1 antagonists. A. Loboda reports other support from Merck & Co., Inc., Kenilworth, NJ, USA during the conduct of the study; in addition, A. Loboda has a patent pending related to the application of stroma signature (WO 2020/167619). No disclosures were reported by the other authors.
Authors' Contributions
R. Cristescu: Conceptualization, formal analysis, investigation, writing–original draft. M. Nebozhyn: Formal analysis, investigation, visualization, writing–original draft. C. Zhang: Formal analysis. A. Albright: Conceptualization, investigation, methodology. J. Kobie: Formal analysis, investigation. L. Huang: Formal analysis, writing–review and editing. Q. Zhao: Formal analysis, investigation, writing–review and editing. A. Wang: Formal analysis. H. Ma: Formal analysis. Z.A. Cao: Formal analysis, investigation. M. Morrissey: Formal analysis. A. Ribas: Formal analysis, investigation, writing–review and editing. P. Grivas: Investigation, writing–review and editing. D.W. Cescon: Conceptualization, formal analysis, writing–review and editing. T.K. McClanahan: Conceptualization, formal analysis, writing–review and editing. A. Snyder: Formal analysis, writing–original draft. M. Ayers: Conceptualization, formal analysis, supervision. J. Lunceford: Conceptualization, formal analysis, writing–original draft. A. Loboda: Conceptualization, formal analysis, investigation, writing–original draft.
References
- 1. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jamieson NB, Maker AV. Gene-expression profiling to predict responsiveness to immunotherapy. Cancer Gene Ther 2017;24:134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ott PA, Bang Y-J, Piha-Paul SA, Razak ARA, Bennouna J, Soria J-C, et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 2019;37:318–27. [DOI] [PubMed] [Google Scholar]
- 5. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book 2019;39:147–64. [DOI] [PubMed] [Google Scholar]
- 6. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352:227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pabla S, Conroy JM, Nesline MK, Glenn ST, Papanicolau-Sengos A, Burgher B, et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J Immunother Cancer 2019;7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 10. Chi J-T, Wang Z, Nuyten DSA, Rodriguez EH, Schaner ME, Salim A, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med 2006;3:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loboda A, Nebozhyn M, Cheng C, Vessey R, Huang P, Dai H, et al. Biomarker discovery: identification of a growth factor gene signature. Clin Pharmacol Ther 2009;86:92–96. [DOI] [PubMed] [Google Scholar]
- 12. Fenstermacher DA, Wenham RM, Rollison DE, Dalton WS. Implementing personalized medicine in a cancer center. Cancer J 2011;17:528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ayers M, Nebozhyn M, Cristescu R, McClanahan TK, Perini R, Rubin E, et al. Molecular profiling of cohorts of tumor samples to guide clinical development of pembrolizumab as monotherapy. Clin Cancer Res 2019;25:1564–73. [DOI] [PubMed] [Google Scholar]
- 14. Hu J, Ge H, Newman M, Liu K. OSA: a fast and accurate alignment tool for RNA-seq. Bioinformatics 2012;28:1933–4. [DOI] [PubMed] [Google Scholar]
- 15. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res 2010;70:2264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics 2010;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res 2019;25:2116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, et al. The immune landscape of cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 2013;39:11–26. [DOI] [PubMed] [Google Scholar]
- 22. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol 2011;179:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L, Saci A, Szabo PM, Chasalow SD, Castillo-Martin M, Domingo-Domenech J, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun 2018;9:3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018;118:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res 2019;25:5449–57. [DOI] [PubMed] [Google Scholar]
- 30. Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 2019;20:711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. KEYTRUDA® (pembrolizumab) injection, for intravenous use. 10/2020. Merck Sharp & Dohme Corp.: Whitehouse Station, NJ, USA; 2020:92. [Google Scholar]
- 32. Genentech Inc. TECENTRIQ® (atezolizumab) injection, for intravenous use. South San Francicso, CA; 4/2017. [Google Scholar]
- 33. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JL, et al. Dual targeting of TGF-beta and PD-L1 via a bifunctional anti-PD-L1/TGF-betaRII agent: status of preclinical and clinical advances. J Immunother Cancer 2020;8:e000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Siu LL, Wang D, Hilton J, Geva R, Rasco D, Abraham AK, et al. Initial results of a phase I study of MK-4830, a first-in-class anti–immunoglobulin-like transcript 4 (ILT4) myeloid-specific antibody in patients (pts) with advanced solid tumours. Ann Oncol 2020;31:S462. [Google Scholar]
- 35. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 2018;175:984–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 2019;25:3074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the United States and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.