

Analysis of data from the Extended Analysis for Leukemia/Lymphoma Treatment (EXALT) trial revealed that guidance of treatment decisions based on single-cell functional drug testing is achievable and efficacious in relapsed and refractory hematologic cancers.
Abstract
Personalized medicine aims to match the right drug with the right patient by using specific features of the individual patient's tumor. However, current strategies of personalized therapy matching provide treatment opportunities for less than 10% of patients with cancer. A promising method may be drug profiling of patient biopsy specimens with single-cell resolution to directly quantify drug effects. We prospectively tested an image-based single-cell functional precision medicine (scFPM) approach to guide treatments in 143 patients with advanced aggressive hematologic cancers. Fifty-six patients (39%) were treated according to scFPM results. At a median follow-up of 23.9 months, 30 patients (54%) demonstrated a clinical benefit of more than 1.3-fold enhanced progression-free survival compared with their previous therapy. Twelve patients (40% of responders) experienced exceptional responses lasting three times longer than expected for their respective disease. We conclude that therapy matching by scFPM is clinically feasible and effective in advanced aggressive hematologic cancers.
Significance:
This is the first precision medicine trial using a functional assay to instruct n-of-one therapies in oncology. It illustrates that for patients lacking standard therapies, high-content assay-based scFPM can have a significant value in clinical therapy guidance based on functional dependencies of each patient's cancer.
See related commentary by Letai, p. 290.
This article is highlighted in the In This Issue feature, p. 275
INTRODUCTION
The overarching goal of precision oncology to match the right treatment with the right patient has prompted clinical trials for patients with refractory cancers to evaluate treatments targeting putative genetic tumor drivers (1–7). In most cancer cases, however, assessment of an individual's underlying genetic disease drivers has not been efficient at predicting therapy effectiveness, likely because of intratumor heterogeneity, dynamic changes, or our incomplete understanding of the genotype-to-phenotype relationship (8–10). Genomic tumor characterization and treatment matching have improved the management of some patients with hematologic malignancies, such as BRAF in hairy cell leukemia (11, 12), IDH1/2 in acute myeloid leukemia (AML; refs. 13–15), and Philadelphia chromosome in acute lymphoblastic leukemia (16, 17). However, most patients do not currently benefit from therapy selection based on molecular target identification. This is particularly true for patients with relapsed or refractory aggressive disease who still face a dire prognosis (18–20).
Genomically driven drug matching and targeted immunotherapies may be complemented by functional precision medicine strategies, such as high-throughput drug screening approaches, which are agnostic with respect to disease mechanisms and rely on direct measurements of cellular functions (21–24). Single-cell functional precision medicine (scFPM) integrates methods to assess differential cell fates in mixed cell populations derived from patients' real-time biopsy specimens after drug exposure (23). We conducted a prospective trial [Extended Analysis for Leukemia and Lymphoma Treatment (EXALT); NCT03096821] to determine clinical feasibility and the efficacy of scFPM to guide therapy choices for patients with aggressive hematologic cancers who exceeded all standard therapy lines (Fig. 1A). More specifically, we profiled the ex vivo efficacy of 139 drugs using high-content microscopy and image analysis on primary patient material to identify resistance-breaking treatments by using a progression-free survival (PFS) ratio of ≥1.3 as an outcome measure (Fig. 1B; Supplementary Table S1; Supplementary Fig. S1). An interim analysis reported results on the first 17 evaluable patients (23). Here, the final results of the completed EXALT study are presented.
Figure 1.

EXALT procedure and primary outcome measure. A, Viable cells from lymph node (LN), BM, or PB of patients with late-stage hematologic cancer were subjected to image-based scFPM. Target cells are identified by staining with fluorescent antibodies. Reports, automatically generated by the analysis pipeline, are discussed in a dedicated tumor board with patients treated accordingly. B, Our primary outcome measure was PFS ratio, defined as PFS(scFPM treatment)/PFS(previous treatment). A ratio of 1.3 is considered individually beneficial. DAPI, 4′,6-diamidino-2-phenylindole.
RESULTS
Patients
From 2015 to 2019, a total of 193 patients were screened, of whom 143 (74.1%) were eligible, enrolled, and could be tested by scFPM (study population). Seventy-six (53% of study population; Supplementary Table S2) patients could be evaluated according to study protocol, and 56 (39% of study population, primary analysis set) patients received treatment according to scFPM. Twenty patients received treatment based on physician's choice and thus served as an observational cohort (Fig. 2; Supplementary Table S2).
Figure 2.

CONSORT diagram of study patients.
Fifty-six patients represented the primary analysis data set after having received tumor board–recommended therapy guided by scFPM, and their clinical characteristics are detailed in Table 1. The median age of the patient cohort was 64 years (range, 23–86 years), and the median number of treatment lines before study entry was 3 (1–8). Seventeen patients (30%) had an Eastern Cooperative Oncology Group (ECOG) performance status of 2 or above. The median time was 5 (1–33) days from sampling to scFPM report, with longer times accounting for restaining due to updated immunophenotyping data, and 21 (4–77) days to treatment. The median follow-up was 718 days calculated by the reverse Kaplan–Meier method. Patients had diverse hematologic cancers, both common and rare, such as AML (14/56, 25%), aggressive B-cell non-Hodgkin lymphoma (B-NHL; 26/56, 46%), and T-cell non-Hodgkin lymphoma (T-NHL; 16/56, 28%; Table 1; Supplementary Table S2). Their unifying clinical feature was an aggressive disease, according to the World Health Organization (WHO) classification (25), that lacked standard treatment options.
Table 1.
Characteristics of scFPM-guided patients
| Characteristics | Count | % |
|---|---|---|
| All | 56 | 100 |
| Sex | ||
| Male | 35 | 63 |
| Female | 21 | 37 |
| Age, median (range), y | 64 (23–86) | |
| Disease group | ||
| Lymphoma | 38 | 68 |
| Leukemia | 18 | 32 |
| B-NHL | 22 | 39 |
| AML | 14 | 25 |
| T-NHL | 15 | 27 |
| ALL/LBL | 5 | 9 |
| Number of previous treatments, median (range) | 3 (1–8) | |
| Response—last treatment | ||
| CR | 15 | 27 |
| PR | 12 | 21 |
| SD | 6 | 11 |
| PD | 23 | 41 |
| Sampling—treatment in days (range) | 21 (4–77) | |
| ECOG at treatment start | ||
| ECOG 0 | 17 | 30 |
| ECOG 1 | 22 | 39 |
| ECOG 2 | 11 | 12 |
| ECOG 3 | 6 | 11 |
| Sample blast fraction | ||
| High (>50%) | 34 | 61 |
| Medium (10%–50%) | 13 | 23 |
| Low (<10%) | 8 | 14 |
| NA | 1 | 2 |
Abbreviation: ALL, acute lymphoblastic leukemia; LBL, lymphoblastic lymphoma; NA, information not available; PD, progressive disease; SD, stable disease.
Efficacy
Thirty of 56 patients (54%; 95% confidence interval, 40%–67%) from the primary analysis set reached a PFS ratio (PFS on scFPM-guided therapy compared with PFS on prior therapy) of ≥1.3 with a median PFS ratio of 3.4 [interquartile range (IQR), 2.2–5.7]. This indicates that their individual PFS on scFPM-guided treatment more than tripled compared with their most recent individualized response time. These findings led to the rejection of the null hypothesis of less than 15% of patients benefiting from scFPM-guided treatment (P < 0.0001, one-sided binomial test; Fig. 3A). The PFS on scFPM-guided treatment was significantly increased (HR, 0.58; P = 0.0093; Supplementary Fig. S2A). Notably, 13 of 56 patients (23%) were progression-free after 12 months on scFPM-guided therapy compared with 3 of 56 patients (5%) on their previous treatment. The objective response rate (ORR) was 55% for patients treated according to scFPM results, 60% for the lymphoid subgroup, and 41% for patients with myeloid neoplasms (Supplementary Fig. S2B). Eleven patients (∼20%) had an ongoing response at the censoring date (Fig. 3A) with a median PFS of 718 days. Moreover, 12 of 56 (21%) scFPM-guided patients experienced exceptional responses, defined as tripled PFS duration compared with expected response duration of the respective disease entity, based on criteria outlined by Wheeler and colleagues (Fig. 3B; ref. 26). Exceptional responders demonstrated better performance status (ECOG ≤1), response [complete response (CR) or partial response (PR)] to prior therapy, and an overrepresentation of a T-NHL diagnosis (7/12; Table 2). For the entire primary analysis cohort, the median PFS ratio was 1.47 (IQR, 0.5–3.51). Eight of 56 (14%) patients received an allogenous hematopoietic stem cell transplantation (HSCT) or donor lymphocyte infusion (DLI) as a consolidation after reaching CR on scFPM-guided treatment. This did not translate into a PFS benefit compared with patients in CR who did not receive consolidation with HSCT or DLI (Supplementary Fig. S2C).
Figure 3.

scFPM-guided treatment enhances PFS ratio in patients with advanced hematologic cancers and provides a survival benefit. A, Bar plot showing the PFS for all included, scFPM-guided patients: blue bars denote PFS in days for scFPM-guided treatment, red bars indicate last previous treatment, and asterisks denote ongoing response for scFPM treatment at the censoring date. PFS ratio is the following ratio: PFS(scFPM treatment)/PFS(previous treatment). Patient characteristics are color coded and stratified (leukemia vs. lymphoma, exceptional response vs. nonexceptional response, ECOG >1 vs. ECOG ≤1). B, Kaplan–Meier plot comparing PFS on scFPM-guided treatment with previous treatment in exceptional responders (n = 12). C, Bar plot showing PFS for all patients with an ECOG ≤1 (n = 39). Asterisks denote ongoing response for scFPM treatment at censoring date. D, Kaplan–Meier plot comparing PFS on scFPM treatment between patients with ECOG ≤1 (n = 39) versus ECOG>1 (n = 17). E, Bar plot showing PFS for all patients with OR on previous treatment. Asterisks denote ongoing response for scFPM treatment at censoring date. F, Kaplan–Meier plot comparing PFS on scFPM treatment stratified according to OR on last treatment (CR/PR: n = 27, SD/PD: n = 29). G, Scatter plot comparing PFS on last treatment to current treatment, for scFPM-guided versus physician's choice patients (paired Wilcoxon test). H, Kaplan–Meier plot comparing overall survival stratified according to scFPM-guided patients (n = 56) versus physician's choice patients (n = 20).
Table 2.
Characteristics of exceptional responders
| Characteristics | Count | % of all |
|---|---|---|
| Exceptional responders | 12 | 21 |
| Sex | ||
| Male | 6 | 50 |
| Female | 6 | 50 |
| Age, median (range), y | 60 (29–86) | |
| Disease group | ||
| Lymphoma | 9 | 75 |
| Leukemia | 3 | 25 |
| B-NHL | 2 | 17 |
| AML | 3 | 25 |
| T-NHL | 7 | 58 |
| ALL/LBL | 0 | 0 |
| Number of previous treatments, median (range) | 2 (2–9) | |
| Response—last treatment | ||
| CR | 7 | 58 |
| PR | 3 | 25 |
| SD | 1 | 8 |
| PD | 1 | 8 |
| Sampling—treatment in days (range) | 28 (4–56) | |
| ECOG at treatment start | ||
| ECOG 0 | 8 | 67 |
| ECOG 1 | 4 | 33 |
| ECOG 2 | 0 | 0 |
| ECOG 3 | 0 | 0 |
| Sample blast fraction | ||
| High (>50%) | 4 | 33 |
| Medium (10%–50%) | 6 | 50 |
| Low (<10%) | 1 | 8 |
| NA | 1 | 8 |
| Sample type | ||
| Bone marrow | 2 | 17 |
| Lymph node | 6 | 50 |
| Peripheral blood | 2 | 17 |
| Skin | 1 | 8 |
| Spleen | 1 | 8 |
Pretreatment performance status influenced benefit from scFPM-guided treatment, with a PFS ratio of ≥1.3 being reached by 62% of patients with ECOG ≤1 and by 35% of patients with ECOG >1 (Fig. 3C; Supplementary Fig. S2D). Median PFS was 207 days for patients with ECOG ≤1 compared with 29 days for patients with ECOG >1 (Fig. 3D). Patients who had an objective response (OR) consisting of CR or PR to their previous treatment had a longer PFS on scFPM-guided treatment (Fig. 3E and F). Furthermore, patients with T-NHL had an increased median PFS (235 days) on scFPM-optimized treatment in comparison to patients with B-NHL (60 days) and showed exceptional response in 44% of cases (Supplementary Fig. S2E and S2F; Supplementary Table S2). Somatic TP53 status was not included in the study protocol but was available for 28 patients. Patients whose cancer harbored a TP53 variant experienced significantly shorter PFS than those without TP53 variants (Supplementary Fig. S2G and S2H). The proportion of patients achieving a PFS ratio ≥1.3 was not significantly influenced by the ECOG performance status, OR to last treatment, lymphoid subgroup, or TP53 variant status (Supplementary Fig. S3A–S3D).
Age (≤60 vs. >60 years), sex, lineage (myeloid vs. lymphoid), number of previous treatment lines (≤2 vs. >2), disease subgroup (leukemia vs. lymphoma), and time from sampling to treatment start did not have an impact on PFS duration on scFPM-guided treatment (Supplementary Fig. S4A–S4H). We included a physician's choice cohort of 20 non–scFPM-treated patients who underwent scFPM analysis but, in consultation with their treating physician, decided on alternative treatment. Here, we observed that the PFS prolongation significantly improved in scFPM-treated patients but not in physician's choice patients (Fig. 3G; Supplementary Table S2). Patients treated based on scFPM had a significant overall survival benefit compared with the observational cohort (Fig. 3H, P = 0.035). Although this study was designed without a controlled comparator arm, the physician's choice cohort was comparable with regard to age, ECOG at treatment start, sample blast fraction, number of previous treatments, and response to previous treatment (Supplementary Fig. S5A–S5E).
Post Hoc Analysis
To investigate how well the actual received treatment matched the scFPM results by assessment of a matching score, we reanalyzed scFPM image data in a post hoc analysis. Updated image analysis pipelines and quality control criteria were used, resulting in the post hoc exclusion of 10 patients, with an analysis set of 66 patients consisting of 49 patients from the primary analysis cohort and 17 patients from the physician's choice set. The scFPM results were integrated across evaluated markers and given drugs per patient, leading to an integrated scFPM that was calculated in an automated fashion, blinded to patient outcome. Treatments with positive (>0) integrated scores, denoting overall post hoc support for the patient treatment, were considered as matching according to the individual drug-profiling report. Fifty-two patients (78%) obtained such positive scores and were therefore considered as having received scFPM-matched treatment (Supplementary Fig. S6A). Twenty-six of these 52 matched patients (50%) demonstrated an OR to treatment received, and 30 patients had a PFS improvement on scFPM-matched treatment (Supplementary Fig. S6B). Patients receiving matched treatment in the post hoc analysis exhibited an increase of PFS. After 12 months, 13 of 52 patients (28%) on matched treatment were progression-free compared with only 4 of 52 patients (8%) on previous treatment (Fig. 4A). Patients receiving non–scFPM-matched treatments did not demonstrate an improved PFS compared with their previous treatment (Fig. 4B). A positively scFPM-matched therapy resulted in a mean PFS of 276 days compared with 121 days on their previous treatment (P = 0.0039), whereas nonmatched therapy led to a mean PFS of 96 days with a mean previous PFS of 121 days (P = 0.51; Fig. 4C).
Figure 4.
![Figure 4. Post hoc analysis. A, Kaplan–Meier plot comparing scFPM-matched treatment with previous treatment. Dotted line denotes 1-year follow-up. B, Kaplan–Meier plot comparing non–scFPM-matched treatment with previous treatment. C, Paired scatter plot comparing nonmatched versus matched patients with regard to PFS ratio. Paired Wilcoxon test comparing PFS of previous treatment versus scFPM-matched/nonmatched treatment [H0: rank PFS(previous) = rank PFS(current)]. D, Kaplan–Meier plot of scFPM-matched treatment stratified according to ECOG <1 versus ECOG ≥1. E, Kaplan–Meier plot of scFPM-matched treatment stratified according to response on previous treatment. F, Kaplan–Meier plots comparing PFS for scFPM-matched patients stratified according to tumor cell content in the sample (high ≥50%, medium >10%, low ≥10%).](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9762339_372fig4.jpg)
Post hoc analysis. A, Kaplan–Meier plot comparing scFPM-matched treatment with previous treatment. Dotted line denotes 1-year follow-up. B, Kaplan–Meier plot comparing non–scFPM-matched treatment with previous treatment. C, Paired scatter plot comparing nonmatched versus matched patients with regard to PFS ratio. Paired Wilcoxon test comparing PFS of previous treatment versus scFPM-matched/nonmatched treatment [H0: rank PFS(previous) = rank PFS(current)]. D, Kaplan–Meier plot of scFPM-matched treatment stratified according to ECOG <1 versus ECOG ≥1. E, Kaplan–Meier plot of scFPM-matched treatment stratified according to response on previous treatment. F, Kaplan–Meier plots comparing PFS for scFPM-matched patients stratified according to tumor cell content in the sample (high ≥50%, medium >10%, low ≥10%).
Influencing factors for PFS were comparable for post hoc scFPM-matched treatment and scFPM-guided treatment: ECOG ≤1, OR to previous treatment, and lymphoid subtype positively influenced PFS on scFPM-matched treatment. In addition, matched patients with lymphomatous disease had a longer PFS than patients with leukemic disease (Fig. 4D and E; Supplementary Fig. S6C–S6F). We also observed that the relative cancer cell fraction in the sample influenced PFS on scFPM-matched treatment. In particular, patients with medium cancer cell percentages exhibited longer PFS on scFPM-matched treatment in comparison to patients with either low or high cancer cell percentages (Fig. 4F).
DISCUSSION
The EXALT trial aimed to offer individualized treatment for patients with aggressive hematologic cancers beyond curative options based on real-time ex vivo functional evaluation of drug responses. This single-arm open-label study demonstrates clinical feasibility of integrating an image-based scFPM approach into clinical routine. The primary endpoint of the study was clearly reached. In 54% of cases, scFPM-guided treatments led to a PFS prolongation of ≥1.3-fold of the patients' previous individual treatment response time as well as to an enhanced overall survival when comparing scFPM-guided patients with a physician's choice cohort. We found that 21% of patients showed disease-specific exceptional responses, defined by a threefold extension of absolute PFS duration compared with the expected median PFS duration. This definition was introduced in a study by Wheeler and colleagues (26), which investigated the underlying molecular mechanisms of exceptional responders and identified four broad categories accounting for favorable clinical outcomes: DNA damage response, intracellular signaling, immune engagement, and genetic characteristics of a favorable response. In our study, an ECOG score of 0 or 1 and response to previous treatment were the strongest predictors for a benefit of scFPM-guided treatment. Several clinical parameters such as age, sex, disease lineage, and number of prior treatments did not appear to predict response to scFPM-guided treatment.
Precision medicine trials that aim to match targeted therapies to molecular tumor profiles or gene variants have been emerging in recent years (Table 3; refs. 3, 27–31). Thus far, only few studies could indicate an improved outcome for patients receiving a genetically matched treatment (28, 30, 32). Functional precision medicine trials tailoring treatment strategies based on functional data, such as drug profiling, represent a complementary or alternative strategy and rely on the employed technological platform. Appreciation of the full potential of functional screening technologies, such as the image-based scFPM described here, for their capacity to improve clinical outcome is only now beginning (23). The results of the EXALT study presented here show that scFPM can be an effective tool for clinical decision-making and therapy optimization based on the functional characteristics of each patient's tumor.
Table 3.
Summary of precision medicine trials with comparator (randomized, PFS ratio, or observational control)
| Patients screened, n | Patients tested, n (%) | Patients with target availability, n (%) | Patients assigned to treatment, n (%) | Patients treated matched, n (%) | Patients evaluable for response, n (%) | Response rate | Biomarker type | Comparator | Study |
|---|---|---|---|---|---|---|---|---|---|
| 1,893 | 1,640 (87) | 341/827 (41) MALDI-TOF; 583/792 (74) TSACP; 14/21 (67) ASCP | 245/1,640 (15) | 84/1,640 (5) | ORR, 84/84 (100) | ORR per tested, 16/1,640 (<1) | Genetic; IHC; MALDI-TOF MS | Patients treated on genotype- matched trials compared with genotype-unmatched trials | IMPACT/COMPACT (2) |
| ORR per treatment assigned, 16/245 (6.5) | |||||||||
| ORR per treated and evaluable, 16/84 (19) | |||||||||
| 741 | 520 (70) sequenced; | 293/741 (40) | 195/741 (26) | 99/741 (13.4) | ORR, 98/99 (99) | ORR per tested, 4/293 (1.4) | Genetic; IHC | Patients randomized to molecularly targeted agent vs. treatment at physician's choice | SHIVA (3) |
| 638 (86) IHC | ORR per treatment assigned, 4/195 (2.1) | ||||||||
| 522 (70) gene copy number analysis | ORR per treated, 4/99 (4) | ||||||||
| 496 (67) complete profile | ORR per evaluable, 4/98 (4.1) | ||||||||
| 500 | 339 (68) | 317/339 (93.5) | 188/339 (55) | 122/339 (36); direct match, 45/339 (13.3) | ORR, 118/122 (97) | ORR per tested, 23/339 (7) | Genetic | Matched vs. nonmatched patients; comparison of TTF after enrollment to prior TTF | Genomic Profiling Assay in Phase I (4) |
| ORR per treatment assigned, 23/188 (12) | |||||||||
| ORR per treated, 23/122 (19) | |||||||||
| ORR per evaluable, 23/118 (19.5) | |||||||||
| TTF2/TTF1, NS | |||||||||
| 1,542 | 1,276 (83) | 534/1,542 (35) | 379/1,276 (30) | 143/1,276 (11.2) | ORR, 134/143 (94) | ORR per tested, 16/1,276 (1.3) | Genetic | Those who had received targeted therapy and those for whom no targeted therapy was available; previously published cohort | Tsimberidou et al. (6) |
| ORR per treatment assigned, 16/379 (4.2) | |||||||||
| ORR per treated, 16/143 (11.2) | |||||||||
| ORR per evaluable, 16/134 (12) | |||||||||
| 1,035 | 843 (81) | 411/1,035 (40) | 199/843 (24) | 199/843 (24) | PFS ratio, 193/199 (97); ORR, 194/199 (97.5) | PFS ratio ≥1.3 per evaluable, 63/193 (33) | Genetic | Intraindividual PFS ratio (PFS2/PFS1 >1.3) | MOSCATO 01 (7) |
| ORR per enrolled, 22/843 (3) | |||||||||
| ORR per treated and evaluable, 22/194 (11) | |||||||||
| 106 | 86 (81) | 84/86 (98) | 68/86 (79) | 66/86 (77) | PFS ratio and ORR, 66/66 (100) | PFS ratio ≥1.3 per evaluable, 18/66 (27) | Genetic; IHC/FISH | Intraindividual PFS ratio (PFS2/PFS1 >1.3) | Von Hoff et al. (27) |
| ORR per enrolled, 6/86 (7) | |||||||||
| ORR per treated and evaluable, 6/66 (9) | |||||||||
| 149 | 149 (100) | 73/149 (49) | 83/149 (56) | 73/149 (49) | PFS ratio, 53/73 (73); ORR, 69/73 (95) | PFS ratio ≥1.3 per evaluable, 24/53 (45) | Genetic; IHC | Intraindividual PFS ratio (PFS2/PFS1 >1.3) | I-PREDICT (28) |
| ORR per tested, 17/149 (11) | |||||||||
| ORR per treated, 17/73 (23) | |||||||||
| ORR per evaluable, 17/69 (25) | |||||||||
| 303 | 253 (84) | 158/253 (63) | 158/253 (62.4) | 124/253 (49) | PFS ratio and ORR, 107/124 (86) | PFS ratio >1.5 per evaluable, 24/107 (22.4); ORR per tested, 15/253 (6) | Genetic | Intraindividual PFS ratio (PFS2/PFS1 >1.5) | WINTHER (29) |
| ORR per assigned to treatment, 15/158 (9.5); ORR per treated, 15/124 (12) | |||||||||
| ORR per evaluable, 15/107 (14) | |||||||||
| 487 | 487 (100) | 317/487 (65) | 395/487 (81) | 224/487 (46) | OS, 395/487 (81) | 30-day mortality, 8/395 (2) | Genetic; IHC | Standard of care, palliative and investigational therapy vs. BEAT-AML substudy | BEAT-AML (30) |
| Median OS, 12.8 months | |||||||||
| OS estimate at 12 months, 54.7% | |||||||||
| No ORR reported | |||||||||
| 1,856 | 1,223 (66) | 282/1,223 (23) | 677/1,223 (55.4) | 46/1,223 (4) | OS, 677/1,223 (55.4) | Median OS matched therapy group vs. nonmatched group (2.58 vs. 1.51 years) | Genetic | Patients treated on matched therapy vs. treated with nonmatched therapy | Know Your Tumor Registry (32) |
| Median PFS, 10.93 vs. 4.53 months | |||||||||
| No ORR reported | |||||||||
| 2,340 | 1,484 (63) | 1,153/1,484 (78) | 1,138/1,484 (77) | 362/1,484 (24.4) | PFS ratio, 300 (13); best response comparison, 181 (8) | PFS ratio >1.3, 107/300 (36) | Genetic | Intraindividual PFS ratio (PFS2/PFS1 >1.3) | MASTER (38) |
| ORR, 43/181 (24) | |||||||||
| DCR, 100/181 (55) | |||||||||
| 193 | 143 (74) | 143/143 (100) | 76/143 (53) | 56/143 (39) | PFS ratio and ORR, 56/143 (39) | PFS ratio ≥1.3, 30/56 (54) | Functional | Intraindividual PFS ratio (PFS2/PFS1 >1.3) | EXALT-1 (this study) |
| ORR per tested, 31/143 (22) | |||||||||
| ORR per treated and evaluable, 31/56 (55.4) |
NOTE: Patients screened: number of patients who enrolled or signed the consent form. Patients tested: number of patients on whom the test was performed. Patients with target availability: number of patients in whom an actionable target was detected. Patients assigned to treatment: number of patients with a treatment recommendation. Patients treated matched: number of patients treated according to treatment recommendation. Patients evaluable for response: number of patients evaluable for outcome. Response rate: reporting on outcome per tested, treatment assigned, treated, and/or evaluable. Biomarker type: type of biomarker used in study for treatment matching. Comparator: what the outcome was compared with (control arm, intraindividual benefit, or observational cohort). Study: reference to original study.
Abbreviations: ASCP, AmpliSeq Cancer Panel; DCR, disease control rate; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight mass spectrometer; MS, mass spectrometry; NS, not significant; OS, overall survival; TSACP, TruSeq Amplicon Cancer Panel; TTF, time to treatment failure.
Several advantages in comparison to classic sequencing-based approaches can be considered. First, scFPM results were available within days for most patients. We could provide reports for 51 of 76 (67.1%) patients within 7 days. In contrast, a major setback uniformly described in other personalized medicine trials is patient deterioration or death during time of analysis (4, 33). Even in a proof-of-principle, nonoptimized setting, median turnover time for scFPM (5 days) surpassed the limit of current optimized protocols such as the BEAT AML, in which it was possible to deliver genomic analysis within 7 days for 95% of patients (30). In well-optimized settings, scFPM or similar functional approaches can offer reports between 36 and 96 hours postsampling. Second, no indirect inference from genomic data is needed to design treatment strategies. Matching treatment to genetic mutations may work for only a fraction of cancers in which strong, uniform driver mutations are clearly disease- initiating and disease-maintaining, such as in Philadelphia chromosome–dependent chronic myeloid leukemia. Most of the time, the molecular makeup is complex and intertwined with epigenetic and metabolic states that make correlation to therapeutic outcome impossible at our current state of knowledge (3). Progressive, personalized medicine trials approach this by more general matching scores incorporating common markers for immuno-oncology (e.g., tumor mutational burden, mismatch repair). This strategy could raise the limited matching numbers but has not yet been validated in a prospective fashion (28). In contrast, scFPM provides more accessible readouts, which also take nononcogenic vulnerabilities into account (34). Third, functional drug testing may offer treatment options for a higher number of patients, given that clinically validated, approved therapies matched to mutations are available for less than 10% of patients (32, 35, 36), whereas in personalized medicine trials, matched treatment can be allocated to between 4% and 77% (median 24%), dependent on the stringency of matching criteria (Table 3, column 5). Fourth, scFPM-guided therapy can serve as an effective strategy for rapid remission induction to bridge to stem cell transplantation, which still remains a valid and potentially curative treatment for many hematologic cancers. Fifth, the scFPM platform can systematically identify drug-repurposing opportunities with clinical relevance. In addition, although not done in this work, scFPM can be used for combinatorial drug testing. As most effective cancer therapies rely on a combination of agents, means to easily test combinatorial efficacy of drugs are urgently needed.
Despite the advantages, there are limitations to this approach. For instance, scFPM is based on the collection of viable cells, the procurement of which requires an intimate interplay between different hospital departments, such as surgery, pathology, and laboratory. However, as the concept of temporal tumor evolution is more and more recognized and thus real-time biopsy becomes common for personalized approaches, this hurdle can be expected to vanish gradually. Furthermore, we acknowledge the diversity of our patients with regard to disease histology, as a heterogeneous patient population was analyzed with many different neoplasms. To circumvent this, we used a PFS ratio as the primary endpoint, based on considerations of Von Hoff and colleagues (27) and Bailey and colleagues (37) and accepted by regulatory bodies (https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-evaluation-anticancer-medicinal-products-man-revision-6_en.pdf). The use of each patient as his or her own control allows us to evaluate the individual benefit, and it became common in precision medicine trials, such as the WINTHER and MASTER trials including heterogenous cancer entities (29, 38). Another limitation of the study is that most patients were heavily pretreated, which may have limited their ability to respond. Given the trial's nonrandomized design, we cannot exclude that prognostic or other aspects may have confounded the findings. We did not directly assess the activity of drug combinations ex vivo but could still successfully design drug combination treatment strategies that had a positive effect on the PFS in vivo. We focused on cell death as our assay readout and thus might have missed compounds that exhibit anticancer effects by other mechanisms. Moreover, the entire surface of each well in the plate was not imaged, and we tested a limited concentration range, but initial optimization experiments indicated that the obtained data were representative of the entire well. The wider applicability and scalability of the approach are currently being evaluated in additional studies. We envision a future deployment of the technology to centralized laboratories. Last, multiple-testing adjustments were not performed, and thus these data would need to be confirmed by future studies.
The EXALT trial shows that functional testing can be integrated into clinical workflows and provide individual benefit for patients with late-stage hematologic cancer. These platforms appear especially suited for hematologic malignancies, as primary patient material is more readily accessible as intact viable cells. Thus, it makes it possible to introduce a class of assays traditionally related to drug discovery and translational research to personalized medicine. Complementation of classic histology and molecular -omics data with functional assays such as scFPM could lead to a comprehensive way of cancer diagnosis and ultimately to improved patient care and outcome in an increasingly predictive manner. In particular, matching phenotypic characterization to molecular profiles may lead to a constantly improving molecular–mechanistic understanding. Perhaps an integrative approach combining molecular and functional profiles may represent an ideal precision medicine approach, and this remains to be explored in future studies and trials.
An important aspect for success of these approaches is a well-functioning multidisciplinary tumor board, which was implemented in this study and served as a key instrument to optimize individualized treatment strategies. Data integration of a post hoc analysis could confirm the matching of the guiding scFPM test results with the treatment actually received (Fig. 4A and B). This work is the basis for the recently initiated prospective randomized trial comparing scFPM with comprehensive genomic profiling and physicians' choice (EXALT-2, NCT04470947).
METHODS
Patients
In this open-label, one-arm study, we enrolled patients with confirmed aggressive hematologic cancers according to the WHO classification who had received at least two lines of treatment or had no standard therapy options. All patients provided written informed consent. Real-time biopsy specimens were obtained from every enrolled patient.
Study Oversight and Conduct
The study was approved by the independent ethics committee at the Medical University of Vienna (institutional review board votes: EK 1830/2015, 2008/2015, 1895/2015) and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. The study was designed by one author (P.B. Staber) and the sponsor (Medical University of Vienna, Vienna, Austria). The first and last authors wrote all manuscript drafts. All authors vouch for the completeness and accuracy of the data and the adherence of the study to the protocol.
Image-Based scFPM
Cancer cell–containing tissue was procured by biopsy, bone marrow aspirate, or peripheral blood draws (Fig. 1A). Single-cell suspensions of biopsy material containing tumor cells were suspended in RPMI containing 10% FBS and 1% penicillin–streptomycin, and 20,000 cells per well were plated on 384-well CellCarrier ultra-imaging plates (PerkinElmer) containing 136 to 139 drugs prespotted with an Echo (Labcyte; Supplementary Fig. S1; Supplementary Table S1) and incubated overnight (18–24 hours) at 37°C and 5% CO2. The drugs were tested in two different concentrations (1,000 nmol/L and 10,000 nmol/L) in duplicate or triplicate, respectively. After the incubation period, the cells were fixed with 0.5% formaldehyde and 1:1,000 Triton X in PBS, stained with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) for nuclear identification, and stained with antibodies to identify if the cell was malignant or healthy. The antibodies used to detect the target cancer cell population were based on established antigens for a given indication, as well as on pathology or laboratory medicine reports for each individual patient. Immunofluorescence staining, imaging by automated microscopy (Opera Phenix; Perkin Elmer), image analysis (CellProfiler; Broad Institute), data analysis (MATLAB), and quality control were done as described previously (23, 39). In short, each cell in the images was identified using single-cell image analysis and subjugated to machine learning–based single-cell quality control. Based on the marker expression levels, cells were scored as being either cancer (marker positive) or healthy. The fraction of cancer cells upon ex vivo drug treatment was subsequently compared with the cancer fraction in DMSO controls [leading to a relative cancer fraction (RCF)]. The RCFs were averaged across concentrations and replicates per drug and subsequently transformed to 1-RCF, such that positive scores denote on-target reduction of cancer cells induced by the ex vivo drug treatment.
Treatment Allocation
The results of the scFPM platform were presented at a formal multidisciplinary tumor board consisting of hematologists, pathologists, specialists from laboratory medicine, biologists, and pharmacists. The board then issued written treatment recommendations directly to the case manager and the treating physician (Fig. 1A). For the observational cohort, patients were treated according to physician's choice based on individual diagnosis and biomarkers (genetic, IHC, and FACS). Treatment was given according to European Union and Austrian legislation as an individual healing attempt within the named patient program and under the responsibility and supervision of the treating physician.
Assessments and Endpoints
Individual patient benefit was measured by the PFS ratio, defined as PFS achieved on scFPM-guided therapy to PFS observed on previous therapy 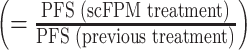 . A PFS ratio of ≥1.3 was considered beneficial based on considerations of Bailey and colleagues (37) and Von Hoff and colleagues (27), thereby using each patient as his or her individual control, which is a study endpoint for precision medicine studies as recommended by health agencies such as the European Medicines Agency and others. Our null hypothesis was that less than 15% of patients meet the primary endpoint, a common level of benefit for personalized medicine trials (1, 40). PFS was computed as the time from the first day of treatment to the date of first reported evidence of disease progression or relapse, initiation of new (unplanned) anticancer treatment, or death as a result of any cause. Commonly used and predefined sequential treatments (e.g., 3 + 7 followed by high-dose cytarabine followed by HSCT in AML) were considered as one line, and the PFS durations were summed. Patients not being able to reach the primary endpoint by the censoring date (January 30, 2020) were excluded regardless of response state. For the post hoc analysis, patients were assessed in terms of their actual received treatment and its matching level to the scFPM results. The scFPM imaging data from all evaluated patients were reanalyzed with upgraded image analysis pipelines and quality control criteria, which resulted in the exclusion of 10 patients. Therefore, the final post hoc set comprised 49 patients from the primary analysis cohort and 17 patients from the physician's choice set. The matching score of the post hoc analysis was calculated in an automated fashion, blinded to patient outcome. Sample blast fraction was determined histologically or from flow cytometry data using a three-tiered scale (≤10%, low; ≤50%, medium; >50%, high).
. A PFS ratio of ≥1.3 was considered beneficial based on considerations of Bailey and colleagues (37) and Von Hoff and colleagues (27), thereby using each patient as his or her individual control, which is a study endpoint for precision medicine studies as recommended by health agencies such as the European Medicines Agency and others. Our null hypothesis was that less than 15% of patients meet the primary endpoint, a common level of benefit for personalized medicine trials (1, 40). PFS was computed as the time from the first day of treatment to the date of first reported evidence of disease progression or relapse, initiation of new (unplanned) anticancer treatment, or death as a result of any cause. Commonly used and predefined sequential treatments (e.g., 3 + 7 followed by high-dose cytarabine followed by HSCT in AML) were considered as one line, and the PFS durations were summed. Patients not being able to reach the primary endpoint by the censoring date (January 30, 2020) were excluded regardless of response state. For the post hoc analysis, patients were assessed in terms of their actual received treatment and its matching level to the scFPM results. The scFPM imaging data from all evaluated patients were reanalyzed with upgraded image analysis pipelines and quality control criteria, which resulted in the exclusion of 10 patients. Therefore, the final post hoc set comprised 49 patients from the primary analysis cohort and 17 patients from the physician's choice set. The matching score of the post hoc analysis was calculated in an automated fashion, blinded to patient outcome. Sample blast fraction was determined histologically or from flow cytometry data using a three-tiered scale (≤10%, low; ≤50%, medium; >50%, high).
Response Evaluation
Response evaluation was based on response evaluation criteria in lymphoma (RECIL) and European LeukemiaNet (ELN) criteria for leukemia (41, 42). As patients with leukemia were not consistently followed up with bone marrow (BM) biopsy or aspirate, changes in peripheral blood (PB) were used equivalently: CR could not be stated with presence of blasts in PB. Analogous to ELN–morphologic leukemia-free state, absence of blasts in the peripheral blood without BM available was considered a hematologic leukemia-free state (HLFS) and classified as an OR. Comparable to morphologic criteria with ELN-PR, reduction of blasts in PB by at least 50% was regarded as PR [hematologic partial remission (HPR) = PR]. The white blood cell count had to be above 1 G/L for HLFS and HPR. ECOG performance status was extracted from patient charts and referral reports where available, or a median score of four independent reviewers blinded to outcome was used, based on chart notes, nursing reports, and discharge letters.
Exceptional response was defined as tripled PFS duration compared with median PFS for a given diagnosis. For T-NHL, 9 months of PFS was considered exceptional, and for aggressive B-NHL and myeloid disease, 18 months was considered exceptional based on the definition by Wheeler and colleagues (26) and others (18, 43, 44).
Response assessment was reviewed by an internal study committee consisting of three hematologists (P.B. Staber, K. Miura, U. Jaeger), a radiologist (M.E. Mayerhoefer), and a pathologist (C. Kornauth). Somatic mutation TP53 status was obtained from reports for routine diagnostics produced using certified tests in laboratories of either clinical or surgical pathology.
Statistical Analysis
The primary endpoint of this study was the percentage of patients reaching a PFS ratio of ≥1.3 with an H0 hypothesis of <15% patients meeting the primary endpoint. To test this hypothesis, a one-sided binomial test was applied with an α of 0.025. The null hypothesis could be rejected when at least 14 of 50 patients showed a PFS of ≥1.3. The predefined secondary endpoints were ORR, disease control rate, and PFS, including subgroup analysis (diagnostic group, performance status, age, sex, number of prior therapies, relative blast fraction). The analysis for exceptional responders was not preplanned. A detailed description of the analysis plan can be found in the supplementary materials. All statistical analyses were performed using the R statistical environment (R Core Team; R Foundation for Statistical Computing) and MATLAB (MathWorks). Survival times were compared using the Kaplan–Maier estimator, log-rank tests, and Cox proportional hazard models. Continuous variables were compared with Wilcoxon rank-sum tests. A P value of <0.05 was considered significant.
Authors' Disclosures
G.I. Vladimer reports other support from Allcyte GmbH and other support from Exscientia GmbH during the conduct of the study; other support from Allcyte GmbH; and other support from Exscientia GmbH outside the submitted work; in addition, G.I. Vladimer has a patent for EP 3 704 484 A1 pending and licensed to Exscientia GmbH and a patent for EP 3 198 276 A1 pending and licensed to Exscientia GmbH; much of the screening work was performed while a postdoc at CeMM; manuscript drafting/data interpretation during employment, shareholder, and founder of Allcyte, which used the techniques described in this manuscript for translational research (not involved in clinical data collection). After final submission but before acceptance, Allcyte GmbH was acquired by Exscientia. M. Bergmann reports personal fees from Boehringer Ingelheim; grants from Boehringer Ingelheim, and grants from Bristol Myers Squibb outside the submitted work. K. Geissler reports personal fees from Novartis, personal fees from Celgene, and personal fees from Jazz outside the submitted work. B.L. Hartmann reports personal fees and nonfinancial support from Janssen, personal fees and nonfinancial support from Amgen, personal fees and nonfinancial support from Takeda, and personal fees and nonfinancial support from Abbvie during the conduct of the study. T. Heinemann reports grants from the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation—project number 389640585) and grants from the Swiss National Science Foundation (PP00P3_163961) during the conduct of the study. N. Krall reports grants from WWTF, grants from Marie Curie Fellowship, and grants from SNF Postdoc Fellowship during the conduct of the study; and reports being an employee of Allcyte GmbH and an employee of Exscientia GmbH, outside the submitted work; in addition, N. Krall has a patent for EP 3 704 484 A1 pending, assigned to Exscientia GmbH, and a patent for EP 3 198 276 A1 pending, assigned to Exscientia GmbH; and is a founder and shareholder of Allcyte GmbH. Allcyte GmbH was bought by Exscientia Ltd. and is now registered under Exscientia GmbH as a fully owned subsidiary of Exscientia Ltd. Nikolaus Krall holds shares in Exscientia Ltd. M.E. Mayerhoefer reports grants from NIH National Cancer Institute (NCI) during the conduct of the study; personal fees from Siemens, personal fees from General Electric, and personal fees from Bristol Myers Squibb outside the submitted work. K. Miura reports personal fees from AstraZeneca, personal fees from Chugai, personal fees from Kyowa Kirin, personal fees from Takeda, personal fees from Bristol-Myers Squibb, personal fees from Nippon Shinyaku, personal fees from SymBio, and personal fees from Ono Parma outside the submitted work. G.W. Prager reports personal fees from Merck, personal fees from Roche, personal fees from Amgen, personal fees from BMS, personal fees from MSD, personal fees from Servier, personal fees from Sanofi, personal fees from Lilly, personal fees from Bayer, and personal fees from Ipsen outside the submitted work. M. Raderer reports personal fees from Ipsen, personal fees from Eli Lilly, personal fees from Ipsen, personal fees from Gilead, and personal fees from Novartis outside the submitted work. W.R. Sperr reports personal fees from AbbVie, personal fees from BMS-Celgene, personal fees and nonfinancial support from Daiichi Sankyo, personal fees from Deciphera, personal fees from Jazz, personal fees from Novartis, grants and personal fees from Pfizer, personal fees from StemLine, and grants and personal fees from Thermo Fisher outside the submitted work. P. Valent reports grants from Allcyte during the conduct of the study; grants and personal fees from Celgene-BMS, personal fees from Pfizer, personal fees from Novartis, personal fees from AOP Orphan, and personal fees from Blueprint outside the submitted work. C.C. Zielinski reports other support from MSD, other support from AstraZeneca, grants from Pfizer, personal fees from Athenex, other support from Roche, other support from BMS, other support from Servier, other support from Eli Lilly, other support from Takeda, other support from Daiichi Sankyo, and other support from Haolzyme outside the submitted work; and a patent with Imugene. N. Zojer reports personal fees and nonfinancial support from Amgen, personal fees and nonfinancial support from Celgene/BMS, personal fees and nonfinancial support from Janssen, personal fees and nonfinancial support from Sanofi, and personal fees and nonfinancial support from Takeda outside the submitted work. G. Superti-Furga reports grants from Vienna Science and Technology Fund, grants from Austrian Science Fund, and grants from ERC—European Research Council during the conduct of the study; in addition, G. Superti-Furga has a patent for EP 3 704 484 A1 pending and licensed to Exscientia GmbH, previously Allcyte GmbH, and a patent for EP 3 198 276 A1 pending and licensed to Exscientia GmbH, previously Allcyte GmbH; and was shareholder and founder of Allcyte GmbH. After final submission but before acceptance, Allcyte GmbH was acquired by Exscientia Ltd. and became Exscientia GmbH. B. Snijder reports grants from Swiss National Science Foundation during the conduct of the study; grants from Roche and personal fees from AbbVie outside the submitted work; in addition, B. Snijder has a patent for EP3198276A1 pending to Austrian Academy of Sciences; Dr. Snijder was scientific cofounder and shareholder of Allcyte GmbH and currently is shareholder of Exscientia Ltd. P.B. Staber reports grants from Vienna Science and Technology Fund, grants from Austrian Science Fund; grants and personal fees from F. Hoffmann-La Roche AG; personal fees from Amgen, Takeda, AbbVie, Janssen Cilag, Medical University Vienna, Incyte, Celgene, BMS; personal fees from General Hospital Vienna during the conduct of the study, and personal fees from AstraZeneca outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
C. Kornauth: Resources, data curation, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. T. Pemovska: Data curation, formal analysis, validation, investigation, writing–original draft, project administration, writing–review and editing. G.I. Vladimer: Formal analysis, validation, investigation, methodology, writing–original draft. G. Bayer: Resources, investigation. M. Bergmann: Resources, investigation. S. Eder: Resources, investigation. R. Eichner: Resources, investigation. M. Erl: Resources, investigation. H. Esterbauer: Resources, investigation. R. Exner: Resources, investigation. V. Felsleitner-Hauer: Resources, investigation. M. Forte: Resources, investigation. A. Gaiger: Resources, investigation. K. Geissler: Resources, investigation. H.T. Greinix: Resources, investigation. W. Gstöttner: Resources, investigation. M. Hacker: Resources, investigation. B.L. Hartmann: Resources, investigation. A.W. Hauswirth: Resources, investigation. T. Heinemann: Resources, investigation. D. Heintel: Resources, investigation. M.A. Hoda: Resources, investigation. G. Hopfinger: Resources, investigation. U. Jaeger: Conceptualization, resources, investigation. L. Kazianka: Resources, data curation, investigation, project administration. L. Kenner: Resources, investigation. B. Kiesewetter: Resources, investigation. N. Krall: Conceptualization, resources, data curation, software, formal analysis, validation, investigation, methodology, project administration. G. Krajnik: Resources, investigation. S. Kubicek: Resources, investigation, methodology. T. Le: Resources, investigation. S. Lubowitzki: Data curation, investigation. M.E. Mayerhoefer: Resources, investigation, visualization. E. Menschel: Resources, investigation. O. Merkel: Resources, investigation. K. Miura: Resources, data curation, investigation. L. Müllauer: Resources, investigation. P. Neumeister: Resources, investigation. T. Noesslinger: Resources, validation. K. Ocko: Resources, investigation. L. Öhler: Resources, investigation. M. Panny: Resources, investigation. A. Pichler: Resources, investigation, project administration. E. Porpaczy: Resources, investigation. G.W. Prager: Resources, investigation. M. Raderer: Resources, investigation. R. Ristl: Conceptualization, data curation, formal analysis, visualization, methodology. R. Ruckser: Resources, investigation. J. Salamon: Resources, investigation. A.-I. Schiefer: Resources, investigation. A.-S. Schmolke: Resources, data curation, investigation, project administration. I. Schwarzinger: Resources, investigation. E. Selzer: Resources, investigation. C. Sillaber: Resources, investigation. C. Skrabs: Resources, investigation. W.R. Sperr: Resources, investigation. I. Srndic: Resources, data curation, formal analysis, validation, investigation. R. Thalhammer: Resources, investigation. P. Valent: Resources, investigation. E. van der Kouwe: Resources, data curation, investigation, project administration. K. Vanura: Resources, investigation. S. Vogt: Resources, investigation. C. Waldstein: Resources, investigation. D. Wolf: Resources, investigation. C.C. Zielinski: Resources, investigation. N. Zojer: Resources, investigation. I. Simonitsch-Klupp: Resources, data curation, investigation, project administration, writing–review and editing. G. Superti-Furga: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing. B. Snijder: Conceptualization, resources, data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. P.B. Staber: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
Supplementary Material
Supplementary Data containing the statistical analysis plan, Supplemental Figures 1-6 and Supplemental Tables 1 and 2
Acknowledgments
We thank the patients and their families for their trust in taking part in this study. The study was academically funded and supported by the Medical University Vienna, the General Hospital Vienna, and the Research Center for Molecular Medicine (CeMM) of the Austrian Academy of Sciences. We gratefully acknowledge funding from the Vienna Science and Technology Fund; (LS16-034, to G. Superti-Furga and U. Jaeger), the Austrian Science Fund (F4704-B20, to P. Valent; F4711-B20, to G. Superti-Furga; and P27132-B20, to P.B. Staber), and the European Molecular Biology Organization Long Term Fellowship (1543-2012, to G.I. Vladimer; 733-2016 to T. Pemovska). B. Snijder acknowledges funding from the ETH Zurich, the Swiss National Science Foundation (PP00P3_163961, PP00P3_194809, and CRSII5_193832), and the European Research Council (SCIPER; 803063).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
References
- 1. Flaherty KT, Gray R, Chen A, Li S, Patton D, Hamilton SRet al. The molecular analysis for therapy choice (NCI-MATCH) trial: lessons for genomic trial design. J Natl Cancer Inst 2020;112:1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FAet al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med 2016;8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tourneau CL, Delord J-P, Gonçalves A, Gavoille C, Dubot C, Isambert Net al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncology 2015;16:1324–34. [DOI] [PubMed] [Google Scholar]
- 4. Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner Ret al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res 2016;76:3690–701. [DOI] [PubMed] [Google Scholar]
- 5. Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn Jet al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol 2015;33:3817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsimberidou A-M, Wen S, Hong DS, Wheler JJ, Falchook GS, Fu Set al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res 2014;20:4827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Massard C, Michiels S, Ferté C, Deley M-CL, Lacroix L, Hollebecque Aet al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov 2017;7:586–95. [DOI] [PubMed] [Google Scholar]
- 8. Johnson DB, Zhao F, Noel M, Riely GJ, Mitchell EP, Wright JJet al. Trametinib activity in patients with solid tumors and lymphomas harboring BRAF non-V600 mutations or fusions: results from NCI-MATCH (EAY131). Clin Cancer Res 2020;26:1812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EPet al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH Trial (EAY131) subprotocol W. J Clin Oncol 2020;38:2407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jhaveri KL, Wang XV, Makker V, Luoh S-W, Mitchell EP, Zwiebel JAet al. Ado-trastuzumab emtansine (T-DM1) in patients with HER2-amplified tumors excluding breast and gastric/gastroesophageal junction (GEJ) adenocarcinomas: results from the NCI-MATCH trial (EAY131) subprotocol Q. Ann Oncol 2019;30:1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MPet al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tiacci E, Park JH, De Carolis L, Chung SS, Broccoli A, Scott Set al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen Ket al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yen K, Travins J, Wang F, David MD, Artin E, Straley Ket al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov 2017;7:478–93. [DOI] [PubMed] [Google Scholar]
- 15. Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins Jet al. Discovery of AG-120 (ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett 2018;9:300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Secker-Walker LM, Craig JM, Hawkins JM, Hoffbrand AV. Philadelphia positive acute lymphoblastic leukemia in adults: age distribution, BCR breakpoint and prognostic significance. Leukemia 1991;5:196–9. [PubMed] [Google Scholar]
- 17. Towatari M, Yanada M, Usui N, Takeuchi J, Sugiura I, Takeuchi Met al. Combination of intensive chemotherapy and imatinib can rapidly induce high-quality complete remission for a majority of patients with newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia. Blood 2004;104:3507–12. [DOI] [PubMed] [Google Scholar]
- 18. Crump M, Neelapu SS, Farooq U, Neste EVD, Kuruvilla J, Westin Jet al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 2017;130:1800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrara F, Lessi F, Vitagliano O, Birkenghi E, Rossi G. Current therapeutic results and treatment options for older patients with relapsed acute myeloid leukemia. Cancers 2019;11:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kantarjian HM, DiNardo CD, Nogueras-Gonzalez GM, Kadia TM, Jabbour E, Bueso-Ramos CEet al. Results of second salvage therapy in 673 adults with acute myelogenous leukemia treated at a single institution since 2000. Cancer 2018;124:2534–40. [DOI] [PubMed] [Google Scholar]
- 21. Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan Det al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015;160:977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda Aet al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 2013;3:1416–29. [DOI] [PubMed] [Google Scholar]
- 23. Snijder B, Vladimer GI, Krall N, Miura K, Schmolke A-S, Kornauth Cet al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol 2017;4:e595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krall N, Superti-Furga G, Vladimer GI. Patient-derived model systems and the development of next-generation anticancer therapeutics. Curr Opin Chem Biol 2020;56:72–8. [DOI] [PubMed] [Google Scholar]
- 25. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein Het al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Rev. 4th ed. Lyon, France: IARC;2017. [Google Scholar]
- 26. Wheeler DA, Takebe N, Hinoue T, Hoadley KA, Cardenas MF, Hamilton AMet al. Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell 2021;39:38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Von Hoff DD, Stephenson JJ, Rosen P, Loesch DM, Borad MJ, Anthony Set al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–83. [DOI] [PubMed] [Google Scholar]
- 28. Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CBet al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med 2019;25:744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodon J, Soria J-C, Berger R, Miller WH, Rubin E, Kugel Aet al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med 2019;25:751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burd A, Levine RL, Ruppert AS, Mims AS, Borate U, Stein EMet al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med 2020;26:1852–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SLet al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar Aet al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21:508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev 2020;86:102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136:823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dienstmann R, Jang IS, Bot B, Friend S, Guinney J. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov 2015;5:118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prasad V. Perspective: the precision-oncology illusion. Nature 2016;537:S63. [DOI] [PubMed] [Google Scholar]
- 37. Bailey CH, Jameson G, Sima C, Fleck S, White E, Von Hoff DDet al. Progression-free survival decreases with each subsequent therapy in patients presenting for phase I clinical trials. J Cancer 2012;3:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hullein Jet al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov 2021;11:2780–95. [DOI] [PubMed] [Google Scholar]
- 39. Vladimer GI, Snijder B, Krall N, Bigenzahn JW, Huber KVM, Lardeau C-Het al. Global survey of the immunomodulatory potential of common drugs. Nat Chem Biol 2017;13:681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney Cet al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from mypathway, an open-label, phase IIa multiple basket study. J Clin Oncol 2018;36:536–42. [DOI] [PubMed] [Google Scholar]
- 41. Younes A, Hilden P, Coiffier B, Hagenbeek A, Salles G, Wilson Wet al. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol 2017;28:1436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner Tet al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Foster C, Kuruvilla J. Treatment approaches in relapsed or refractory peripheral T-cell lymphomas. F1000Res 2020;9:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Medeiros BC. Is there a standard of care for relapsed AML? Best Pract Res Clin Haematol 2018;31:384–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data containing the statistical analysis plan, Supplemental Figures 1-6 and Supplemental Tables 1 and 2