

Abstract
Background:
Stress responses are believed to involve corticotropin releasing factor (CRF), its two cognate receptors (CRF1 and CRF2), and the CRF-binding protein (CRFBP). Whereas decades of research has focused on CRF1, the role of CRF2 in the central nervous system (CNS) has not been thoroughly investigated. We have previously reported that CRF2, interacting with a C terminal fragment of CRFBP, CRFBP(10kD), may have a role in the modulation of neuronal activity. However, the mechanism by which CRF interacts with CRFBP(10kD) and CRF2 has not been fully elucidated due to the lack of useful chemical tools to probe CRFBP.
Methods:
We miniaturized a cell-based assay, where CRFBP(10kD) is fused as a chimera with CRF2, and performed a high-throughput screen (HTS) of 350,000 small molecules to find negative allosteric modulators (NAMs) of the CRFBP(10kD)-CRF2 complex. Hits were confirmed by evaluating activity toward parental HEK293 cells, toward CRF2 in the absence of CRFBP(10kD), and toward CRF1 in vitro. Hits were further characterized in ex vivo electrophysiology assays that target: 1) the CRF1+ neurons in the central nucleus of the amygdala (CeA) of CRF1:GFP mice that express GFP under the CRF1 promoter, and 2) the CRF-induced potentiation of N-methyl-D-aspartic acid receptor (NMDAR)-mediated synaptic transmission in dopamine neurons in the ventral tegmental area (VTA).
Results:
We found that CRFBP(10kD) potentiates CRF-intracellular Ca2+ release specifically via CRF2, indicating that CRFBP may possess excitatory roles in addition to the inhibitory role established by the N-terminal fragment of CRFBP, CRFBP(27kD). We identified novel small molecule CRFBP-CRF2 NAMs that do not alter the CRF1-mediated effects of exogenous CRF but blunt CRF-induced potentiation of NMDAR-mediated synaptic transmission in dopamine neurons in the VTA, an effect mediated by CRF2 and CRFBP.
Conclusion:
These results provide the first evidence of specific roles for CRF2 and CRFBP(10kD) in the modulation of neuronal activity and suggest that CRFBP(10kD)-CRF2 NAMs can be further developed for the treatment of stress-related disorders including alcohol and substance use disorders.
Keywords: Stress, CRF2, CRFBP, NAM, HTS, AUD
Introduction
Stress plays a critical role in the development of many psychiatric disorders including alcohol use disorder (AUD) and substance use disorder (SUD) [1]. Options for pharmacological intervention, whether FDA-approved for AUD or SUD, are limited and have only modest effects in a clinical setting [2]. The corticotropin releasing factor (CRF) system includes the binding of CRF to two G protein-coupled receptor (GPCR) subtypes, CRF1 and CRF2, and to a secreted binding protein (CRFBP). Dysregulation of CRF signaling in the central nervous system (CNS) has been proposed to underlie AUD and SUD [3, 4]. CRF binds to its receptors in a two-step mechanism. First, the C-terminus of CRF binds to the N-terminus of the receptor. Second, the N-terminus of CRF binds the extracellular loops of the receptor and activates signaling [5–7]. While the affinity of CRF for CRF1 and CRF2 is well established, the specific details of the role of CRF2 in the stress response are still unclear [8–11]. Furthermore, 20 years of dedicated research and development efforts to advance pharmacological agents that are specific antagonists of CRF1 [12, 13] have thus far not translated to humans, with CRF1 antagonists indicating a lack of efficacy in the treatment of AUD [14, 15], major depression [16], and anxiety [17]. At the cellular level, acute application of CRF increases vesicular γ-aminobutyric acid (GABA) release via activation of CRF1 [18–20] in the central nucleus of the amygdala (CeA) of rodents.
CRF binding protein (CRFBP) is a water-soluble, 37-kilodalton (kD) glycoprotein secreted through exocytosis [3], whose mRNA expression pattern in cortical and subcortical regions has suggested a role in stress responses [21]. It was reported that CRF modulates synaptic input by potentiating N-methyl-D-aspartate (NMDA)-mediated excitatory postsynaptic currents through CRFBP/CRF2 interactions in the ventral tegmental area (VTA) [22]. Later studies reported that CRFBP was co-expressed with CRF2 in rat VTA glutamatergic synaptosomes that originate from hypothalamic areas [23] and that CRFBP increases cell surface CRF2 expression [24].
In behavioral studies, CRFBP has been shown to play a key role via CRF2 in the modulation of cocaine seeking [25] and ethanol consumption [26]. We have further reported that while global knockout of the CRFBP gene CRHBP leads to increased ethanol consumption in mice, a selective downregulation of CRFBP in the center nucleus of the amygdala (CeA) by shRNA decreases ethanol consumption in ethanol-dependent rats [27]. Recently, the role of CRFBP has been evaluated in other psychiatric disorders and neurodegenerative diseases [28]. Together, these results support the hypothesis that CRFBP possesses additional functions beyond sequestering CRF and that modulation of its interaction with CRF2 may represent a novel pharmacological approach for the treatment of AUD and SUD.
In comparison to the CRF receptors, CRFBP has been far less investigated. This is due largely to the spontaneous proteolytic cleavage of the full-length 37 kD CRFBP to an N-terminal 27 kD fragment [CRFBP(27kD)] that can bind CRF and the C-terminal 10 kD fragment [CRFBP(10kD)] that does not bind CRF [29], and which has no known role. Recently, in a genetic study in individuals diagnosed with AUD, we discovered that there are some genetic variants in the CRFBP(10 kD) fragment that were associated with a greater risk for AUD and anxiety-related disorders, while other genetic variants were associated with reduced risk [27]. To further elucidate the functional role of these variants, we have developed novel chimeric cell-based assays stably expressing various CRFBP constructs tethered to both CRF1 and CRF2 [27], yielding chimeric CRFBP assays. Using these novel tools, we found that only the CRFBP(10kD) tethered to CRF2 can potentiate CRF-mediated Ca2+ mobilization [27], suggesting a dual role for CRFBP where CRFBP(27kD) acts to terminate CRF effects and where CRFBP(10kD) has a potential excitatory function [30, 27].
Currently, the only available pharmacological tool to probe the role of CRFBP is a truncated CRF peptide, CRF6–33, that prevents full-length CRF from binding to CRFBP [31]. Similarly, all available CRF2 antagonists [e.g., atressin 2B [32], antisauvagine 30 [33], and K 41498 [34] are peptides, and have limited utility due to their poor pharmacological properties. Furthermore, none of these reagents specifically target the CRFBP(10kD)-CRF2 interaction making it impossible to investigate this important interrelationship directly. Given our hypothesis that the interaction of these two proteins has a potential excitatory function on neurons [30, 27] and the lack of chemical probes for CRF2 and CRFBP, we sought to identify novel small molecule chemical probes that modulate CRF2-CRFBP utilizing our novel chimeric assay where CRF2 is tethered to the CRFBP(10kD) fragment and a fluorescent-based calcium sensor is utilized as a readout of signaling from this complex [27].
Here, we report the optimization, miniaturization, and application of this novel CRFBP-CRF2 assay for use in a high-throughput screen (HTS) to identify CRFBP-CRF2 modulators. From this HTS campaign, we identified two structurally distinct compounds that act as noncompetitive disruptors of the CRFBP-CRF2 complex, and have no effect on CRF2 function in the absence of CRFBP(10kD). These compounds will serve as chemical probes to further investigate the role of CRFBP(10kD) and CRF2 in vitro, ex vivo and in vivo and as starting points to optimize orally active CRFBP-CRF2 modulators suitable for advanced in vivo proof-of-concept studies for the treatment of AUD, SUD, and stress-related disorders.
Materials and methods
Materials
Materials and methods for the CRFBP-CRF receptor chimeras are extensively described in [27]. CRF, the CRF2 specific inhibitor, antisauvagine 30 (AS-30, # A4727) and the CRF1 selective antagonist, R121919, were purchased from Sigma-Aldrich (St. Louis, MO, US), FLIPR Calcium Assay Kits were purchased from Molecular Devices (Sunnyvale, CA, US), and 96-well plates (black wall, clear bottom, BIOCOAT, # 08774256) were purchased from Thermo Fisher Scientific (Waltham, MA, US). [35S]-Guanosine 5’-(γ-thio)triphosphate ([35S]-GTPγS) (250μCi; 9.25MBq) was supplied from Perkin-Elmer® (Boston, USA). Guanosine 5′-[γ-thio]triphosphate tetralithium salt (GTPγS), guanosine 5′-diphosphate sodium salt (GDP), 2-hydroxy-ethylpiperazine-N-2-ethane sulphonic acid (HEPES), DL-dithiothreitol, tricine, magnesium chloride (MgCl2) ethylenediaminetetraacetic acid (EDTA) and saponin were purchased from Sigma-Aldrich® (St. Louis, USA). Complete mini protease inhibitor cocktail tablets were purchased from Roche (Indianapolis, USA). Wheatgerm agglutinin SPA Beads were purchased from Amersham Biosciences (Little Chalfont, England). The cell culture, generation of stably transfected fusion plasmids, transfection and expression of human cells with the CRFBP-CRF receptor chimeras, cell selection and flow cytometry, Western blots, immunofluorescence, image analysis, quantification of receptor surface expression by ELISA and fluorescence-based calcium assay are extensively described in [27].
[35S]-GTPγS binding assay
HEK293 cells stably expressing the FLAG-CRFBP(10kD)-HA-CRF2 chimera and maintained at 37°C, 7% CO2 were suspended in a homogenization buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 3 mM MgCl2; 1 g brain tissue/20 mL buffer). Cell suspensions were centrifuged (14000 rpm, 15 min, 4°C), pellets were resuspended in the homogenization buffer, sonicated on ice, recentrifuged, and resuspended in HME assay buffer (100 mM HEPES pH 7.5, 10 mM NaCl, 5 mM MgCl2, 10 μg/mL saponin and one mammalian protease inhibitor tablet/25 mL). Binding assays were performed in 96-well plates in quadruplicate on ice with each reaction containing [35S]GTPγS (50 pM), cell membrane (10 μg protein), GDP (30 μM), and SPA beads (0.5 mg) with HME assay buffer and the CRF ligands. Non-specific binding was determined in the presence of unlabeled GTPγS (10 μM). Single drug dose-response curves of [35S]GTPγS stimulated binding were performed with CRF (10 nM - 10 μM) and inhibition of CRF2-mediated [35S]GTPγS stimulation was performed with CRF (1 μM; EC80) and AS-30 (10 nM - 10 μM). Membranes and GDP were incubated together for 20 min and then AS-30 was added 30 min prior to addition of CRF, before the [35S]-GTPγS and SPA beads were added. Assay plates were shaken for 45 min at 25°C, and centrifuged (1500 rpm, 5 min, 25°C) before [35S]GTPγS stimulated binding was assessed using the NXT TOPCOUNTERTM. [35S]GTPγS-stimulated binding is expressed as a percentage increase in basal [35S]GTPγS binding.
Calcium assay in 384-well plates
The calcium assay described previously [27] was miniaturized for use in a 384-well plate format as follows: cells 15 μL, FLIPR dye 15 μL, AS-30 7.5 μL, CRF 7.5 μL, for a total of 45 μL as detailed below. HEK293 cells stably expressing the chimeras (CRFBP-CRF2α or CRFBP-CRF1) or individual receptors (CRF2α or CRF1) were rapidly thawed and centrifuged at 800 rpm for 12 min. Three vials (6e6 cells/mL each) were resuspended in 15 mL media containing 10% fetal bovine serum (FBS) in Dulbecco’s Modified Eagle Medium (DMEM), placed into a T75 flask, and incubated at 37°C, 5% CO2 for 3 days. When the cells reached ~70–80% confluency, the media was removed, and the cells were washed with 5 mL PBS without calcium or magnesium. After washing, an additional 5 mL PBS was added (no Ca2+, Mg2+), and the flask was allowed to incubate for 5 min. Cells were detached from the surface of the flask using repeated washing with a serological pipette, and upon complete resuspension were transferred to a 15 mL falcon tube, centrifuged at 800 rpm for 12 min, and finally resuspended in 5 mL 10% FBS/DMEM. A final cell concentration of 600,000 cells per mL was achieved by adding additional volumes of 10% FBS/DMEM, and 50 μL was dispensed into each individual well of 384-well plates (30,000 cells per well). The cells were returned to the incubator for 2 days. On the day of testing, the media was removed from each well and 50 μL PBS (no Ca2+, Mg2+) was added per well to wash the cells. The PBS was removed and 15 μL of 1% FBS/DMEM per well was added. Fresh assay buffer was prepared as follows for 100 mL buffer: 10X Hanks Balanced Salt Solution (HBSS), 10 mL; HEPES 1 M, 2 mL; distilled water, 87 mL; 100 × probenecid solution (250 mM), 1 mL (prepared by dissolving 71 mg probenecid in 1 mL 1 N NaOH); Bovine Serum Albumin (for 0.1% final) 100 mg; pH to 7.4. One vial of FLIPR Calcium 4 Dye (Molecular Devices) was diluted in 10 mL assay buffer and resuspended per assay plate. Next, 15 μL of dye was added to each well and the plate returned to the incubator for 1 h. For stimulation assays, compound source plates were prepared at a final concentration of 50 μM (5X for 7.5 μL injection onto 30 μL, i.e., 37.5 μL total volume per well after injection) to give a final concentration of 10 μM per well in the assay plate. After addition to the assay plate, the final concentration of DMSO was 1%. CRF (MW = 4757.45), purchased in 0.5 mg vials, was prepared as a 1 mM stock solution by addition of 105 μL distilled water. For inhibition assays, a new compound plate with agonist (CRF) was prepared at 6X. For the pre-determined IC80 concentration to inhibit of 1 μM of CRF, a 6 μM solution with a 40 μL volume per well (16 mL of 6 μM solution of CRF per plate) was prepared for a 7.5 μL injection onto 37.5 μL (45 μL total volume per well after injection) to give a final concentration of 1 μM CRF. Fluorescence was measured using a FLEXSTATION at 21°C (50 min for 384-well plate at 2 min for each column of 16 wells at a time).
Primary high-throughput and confirmation screening calcium assay in 1535-well plates
Additional assay optimization was performed to miniaturize the 384-well assay to 1536-well format as follows: cells 6 μL, FLIPR dye 2 μL, test compound via pin-tool 53 nL, 1 μL CRF, for a total of ~9 μL as detailed below. CRFBP-CRF2 HEK293 cells (1,000 cells / 6 μL per well) were plated in 1536-well black walled clear-bottom Poly-D-Lysine coated plates (Corning) in assay media (DMEM (1X), phenol red free, 1% defined FBS (Hyclone), 1X Penicillin/Streptomycin (P/S), and 1X L-Glutamine using a Multidrop Combi (Thermo). Assay plates containing cells were centrifuged at 500 rpm for 1 min, covered with Kalypsys lids, and incubated overnight at 37°C in the presence of 5% CO2. The following day, 2 μL per well of FLIPR Calcium 4 Dye (Molecular Devices), prepared in assay Buffer (1X Hanks’ balanced salt solution, 20 mM HEPES, 0.1% Bovine Serum Albumin (Sigma), and 2.5 mM probenecid (Sigma), pH 7.4) was added to each well of the assay plate at 0.5X according to the Manufacturer’s Instructions using the Multidrop Combi (Thermo) dispenser and the cells were incubated with dye for 1 h at 37°C, 5% CO2. Reagents are from Invitrogen unless otherwise stated.
Calcium flux and the resulting fluorescence was measured using a Functional Drug Screening System 7000 (FDSS7000; Hamamatsu, Tokyo, Japan). Baseline readings were taken (9 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and then test compounds (approximately 53 nL) were added at the 10 s time-point using the FDSS’s integrated 1536 well pin-tool from source library plates (Corning) containing 2 mM test compound stocks in 100% DMSO. At the 243 s time-point, 1 μL per well of either an EC100 CRF response (4.5 μM Final, prepared as a 9X stock in Assay Buffer), EC80 CRF (1 μM Final, prepared as a 9X stock in Assay Buffer), or vehicle response (Assay Buffer) was added to appropriate. Agonist hits were selected by comparing the amplitude of the responses at the time of test compound addition to the CRF maximal response on each plate. Compounds were tested at 13.25 μM for agonist responses and those with responses ≥ 50% of the CRF EC100 response were selected as hits for further study. Antagonist Hits were selected by comparing the amplitude of the responses at the time of EC80 CRF addition ± test compounds. Compounds were tested at 11.78 μM for antagonist responses and those compounds that inhibited > 50% of the CRF EC80 response were selected as hits for further study.
For confirmation screening, the CRFBP-CRF2 Ca2+ assay was performed in the identical manner as the primary screen with the following exceptions. For each test compound, eight assay plates were prepared, each of which received a different concentration of test compound by pin-tool in the FDSS7000 Ca2+ assay. For the agonist confirmation screening, compounds were tested at 53.0 μM, 26.5 μM, 13.3 μM, 6.63 μM, 3.31 μM, 1.66 μM, 0.828 μM, and 0.414 μM. For the antagonist confirmation screening, compounds were tested at 47.1 μM, 23.6 μM, 11.8 μM, 5.89 μM, 2.94 μM, 1.47 μM, 0.736 μM, and 0.368 μM. Agonist EC50 values were determined in the absence of CRF and antagonist IC50 values were determined in the presence of an EC80 concentration of CRF from each set of eight plates.
Z’ score calculation for quality control
The Z-factor is established from four parameters: the means (μ) and standard deviations (σ) of both the positive (p) and negative (n) controls (μp, σp, and μn, σn). Z-factor is computed as:
A Z’ factor between 0.5 and 1.0 has been shown to represent a robust and reliable assay [35].
Evaluation of the competitive or noncompetitive mechanism of action of novel CRFBP(10kD)-CRF2 modulators
A Schild analysis [36] was performed to determine whether the antagonist lead molecules act at the CRFBP-CRF2 complex by a competitive or noncompetitive mechanism relative to CRF. Dose-responses of CRF-induced increases in the Ca2+ assays were performed in the absence or presence of increasing concentrations (1.11 μM, 3.33 μM, 10 μM) of selected compounds in our primary CRFBP-CRF2 Ca2+ assay.
In vivo pharmacokinetic studies
Male C57BL/6J mice (n = 9 for each compound), weighing ~20 g, were purchased from The Jackson Laboratory (Bar Harbor, ME) and were acclimated to their surroundings for 1 week before the study. Mice were provided food and water ad libitum. To measure systemic plasma and brain exposure, MLS-0046818 was administered intraperitoneally (i.p.) in 5% DMSO, 10% Tween 80 in water, pH 7.0 and MLS-0219419 was administered i.p. in 5% DMSO, 10% Tween 80, 20% (2-hydroxypropyl)-beta-cyclodextrin (HPBCD) in water, pH 7.0. Both compounds were administered at 10 mg/kg. Blood samples were collected via the retro-orbital plexus at 0.25, 0.5, 1, 2, 4, 6, and 24 h. Whole blood was collected into EDTA tubes, centrifuged for 10 min at 14000 rpm, and the resulting plasma was collected and stored at −80°C until LC/MS/MS analysis. Whole brains were collected at 4, 6, and 26 h. Brain samples were rinsed in phosphate-buffered saline, snap-frozen, and stored at −80°C. Prior to LC/MS/MS analysis, brain samples were thawed to room temperature and subjected to mechanical homogenization using a Fisher PowerGen 125 (Fisher Scientific) on ice. Pharmacokinetic parameters were obtained from non-compartmental analysis (PkSolver [37]) of concentration-time profiles after compound administration.
Electrophysiology
In the central nucleus of the amygdala (CeA): Adult (minimum age of 10 weeks) male CRF1:GFP mice (n = 6) were bred in-house [38–41]. Mice were group-housed in a temperature and humidity-controlled vivarium on a 12-h reversed light/dark cycle (lights turn off at 8 AM) with food and water available ad libitum. All procedures involving the use of experimental animals in this study were approved by The Scripps Research Institute Institutional Animal Care and Use Committee and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were anesthetized with 3–5% isoflurane, decapitated, and the brains were quickly removed and placed in ice-cold oxygenated (95% O2 and 5% CO2) high-sucrose cutting solution containing: 5 mM HEPES pH 7.3, 206 mM sucrose, 2.5 mM KCl, 2.5 mM CaCl2, 7 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, 5 mM glucose. Coronal slices (300 μM) containing the CeA were sliced using a 1200S vibratome (Leica Microsystems, Buffalo Grove, IL) and incubated in artificial cerebrospinal fluid (ACSF) containing: 1.25 mM NaH2PO4 pH 7.3,130 mM NaCl, 3.5 mM KCl, 1.5 mM MgSO4, 2 mM CaCl2, 24 mM NaHCO3, 10 mM glucose; at 32°C for 30 min and then at room temperature for at least 30 min before use.
Whole-cell electrophysiological recordings were obtained from neurons in the medial subdivision of the CeA visualized with infrared differential interference contrast (IR-DIC) optics, and fluorescently labeled neurons were identified using Prior LED optics (Prior Scientific, Rockland, MA). Recordings were obtained using Multiclamp 700B amplifier, Digidata 1440A and pClamp 10 software (Molecular Devices, Sunnyvale, CA). Glass pipettes were pulled to a resistance of 3–6 MΩ and filled with an internal solution containing: 10 mM HEPES pH 7.2–7.3, 135 mM KCl, 5 mM EGTA, 2 mM MgCl2, 2 mM Mg-ATP, 0.2 mM Na-GTP; 290–300 mOsms; for whole-cell experiments. GABAA-mediated miniature inhibitory postsynaptic currents (mIPSCs) were pharmacologically isolated with glutamatergic transmission blockers [20 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 30 μM DL-2-amino-5-phosphonovalerate (AP-5); Tocris Bioscience (Ellisville, MI)] and a GABAB receptor blocker (1 μM CGP55845A; Tocris Bioscience), in the presence of tetrodotoxin (TTX). CeA cells were voltage-clamped at −60 mV. Series resistance was not compensated, and cells with a series resistance > 20 MΩ or with a > 20% change during the recording, as monitored by 10 mV pulses, or with a holding current ≥ 100 pA were excluded. Drugs were freshly dissolved in ACSF by adding a known concentration of the stock solutions and were bath, gravity perfused.
In the Ventral Tegmental Area (VTA), male and female mice (n = 44; 6–10 weeks) were anesthetized with Euthasol (Butler-Schein). Brains were rapidly removed and placed in ice-cold NMDG-based artificial Cerebrospinal Fluid (aCSF) cutting solution containing 20 mM HEPES, 92 mM NMDG, 25 mM glucose, 30 mM NaHCO3, 2.5 mM KCl, mM 1.2 NaPO4 saturated with 95% O2/5% CO2 with an osmolarity of 305–308 mOsm (Advanced Cell Diagnostics). The brains were cut in ice-cold NMDG solution to obtain horizontal VTA slices, 230 μm thick, using a Leica vibratome (Leica VT1200) and slices were incubated in warm NMDG solution (34°C) for < 5 min before being placed in a modified aCSF holding solution: 20 mM HEPES, 92 mM NaCl, 25 mM glucose, 30 mM NaHCO3, 2.5 mM KCl, 1.2 mM NaPO4 (305–308 mOsm). Slices were incubated at room temperature for at least 1 hour before recording.
Whole-cell patch clamp of the lateral VTA was performed under the guidance of IR-DIC optics on an Olympus BX51WI microscope. Cells were patched with 1.8–3 MΩ resistance glass microelectrodes containing the following internal solution: 20 mM HEPES,117 mM cesium methane-sulfonate, 0.4 mM EGTA, 2.8 mM NaCl, 5 mM TEA-Cl, 4 mM Mg-ATP, 0.4 mM Na-GTP, and 5 mM QX-314 (280 mOsm). Dopamine cells were identified by the following criteria: 1) large multipolar soma, 30–40 μm in diameter, 2) tonic firing of < 4Hz when the cell is attached, 3) large Ih current with a negative voltage step (identified immediately following break in). Slices were perfused at a rate of 2 mL per min with aCSF containing: 1.4 mM NaH2PO4, 126 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose (305–308 mOsm) in the presence of picrotoxin (100 μM) to block GABAA receptors and DNQX (10 μM) to block AMPA receptors. NMDA receptor currents were evoked with a bipolar stimulating electrode (150–400 μA; FHC) held at + 40 mV, filtered at 1 kHz with a Bessel filter, and were allowed to stabilize for 5–10 min before recording began. Drugs were washed on for 15–25 min prior to CRF application and for 2 min following application. CRF was washed on for 7 min. Series resistance was monitored online. If series changed more than 20%, the recording was discarded.
Data analysis
For the calcium assay, the data output contains a pre-assay scan of the plate and the maximum fluorescence observed over the 2-min read per well. The data are presented as the Relative Fluorescence Units (RFU) calculated as: fluorescence value/pre-assay value × 1,000,000. The Z-factor was determined from each plate using the mean ± SD RFU values of CRF (1 μM) alone and CRF (1 μM) with AS-30 (10 μM) (i.e., in presence of the CRF receptor inhibitor), as described. The stimulation assay was analyzed as a percentage of the mean RFU value of CRF (1 μM) alone (i.e., CRF = 100% stimulation). The inhibition assay was analyzed as the difference between the mean RFU value for CRF (1 μM) alone and the RFU of CRF (1 μM) in the presence of the inhibitor compound (AS-30 at 1 μM provides 100% inhibition). For inhibitor hits, > 70% inhibition of CRF (from the second assay run) was sought, but without any stimulation on its own (from the first assay run). Comparisons between groups were performed using an unpaired students t-test. Data are presented as mean (M) ± standard error (SEM). The [35S]-GTPγS bound (fmol/mg) was calculated based on the [35S]-GTPγS radioactivity, Bmax and counts per minute (CPM) of CRF treatment relative to untreated samples and presented as a percentage increase in basal activity. For this, the % increase over basal [35S]GTPγS activity was determined as follows:
For this, the stimulated signal is represented by the CPM from CRF treatments, the basal signal is determined from the vehicle treated CPMs, and non-specific binding is determined from wells containing unlabeled 10 μM GTPγS. [35S]-GTPγS stimulated binding by AS-30 either alone or in combination with CRF agonists, was assessed using each of CRF2 and CRF1-containing membranes. Differences are considered significant at * p < 0.05.
For electrophysiology measures of spontaneous inhibitory postsynaptic currents (sIPSCs), data were analyzed using Mini Analysis (Synaptosoft Inc., Fort Lee, NJ) and Clampfit 10.7 (Molecular Devices), and all events were visually confirmed. To obtain average baseline s/mEPSC, characteristics, events were binned into 3 min bins. Measurements for the effects of acute CRF (Tocris) application were made after 9 mins of CRF application and normalized to a pre-CRF application baseline.
Excitatory post-synaptic currents (EPSCs) were normalized to 7 min of averaged baseline responses, prior to CRF application. All points are averaged per minute. The M ± SEM analyses in bar graphs compare drug to vehicle and utilize a one-way ANOVA and post-hoc comparisons corrected using a Bonferroni multiple comparisons (** p < 0.01). For single comparisons across sex, two-tailed t-tests were used. All statistical tests were two-sided, and statistical significance was accepted if a * p-value < 0.05 unless noted by Bonferroni correction. Statistical Package for the Social Sciences (SPSS, v.22) (Armonk, NY, USA) was used to conduct the analysis and GraphPad Prism (v.7) was used to generate figures (La Jolla, CA, USA).
Results
Optimization and miniaturization of the CRFBP-CRF2 calcium assay in 384-well plate format
We previously reported that a CRFBP-CRF2 chimeric complex potentiates CRF-induced release of intracellular Ca2+ [27]. In order to further validate signaling from the CRFBP-CRF2 chimeric complex, we measured CRF-mediated coupling of the chimera in [35S]GTPγS binding assays to provide a convenient measure of CRF2 activity proximal to the receptor (Fig. 1). CRF produced a dose-dependent stimulation of [35S]GTPγS-binding in cell membranes expressing the CRFBP-CRF2 chimera (EC50 = 283 ± 35 nM) (Fig. 1 A). Furthermore, when CRF-stimulated (1 μM) [35S]GTPγS-binding was performed in the presence of AS-30 (10 nM – 10 μM), the binding was inhibited (IC50 = 298 ± 37 nM) (Fig. 1B), also indicating in this system that AS-30 may behave as an inverse agonist.
Fig. 1. Optimization of cell-based assay expressing FLAG-CRFBP(10kD)-HA-CRF2α using the [35S]GTPγS binding assay.
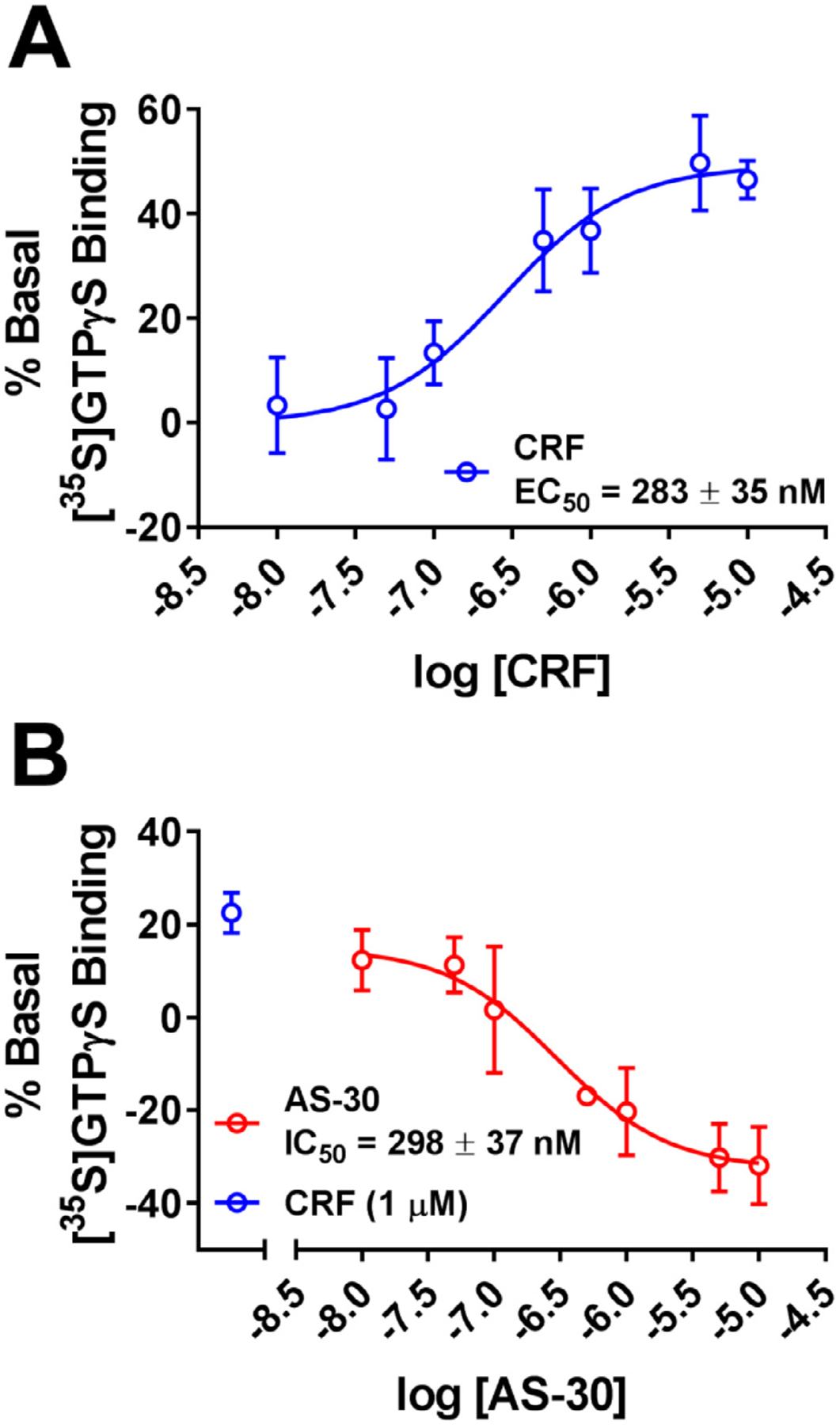
(A) CRF produced a dose dependent stimulation of [35S]GTPγS-binding FLAG-CRF-BP(10kD)-HA-CRF2α. (B) AS-30 inhibits CRF-stimulated (1 μM) [35S]GTPγS FLAG-CRF-BP(10kD)-HA-CRF2α binding. The values are expressed as M ± SEM percentage increase in basal [35S]GTPγS binding.
To improve the throughput and to increase HTS readiness for implementation, we rescaled the assay from the original 96-well to a 384-well format (Fig. 2). Starting from the optimized 96-well conditions that we previously described [27], we developed a total assay volume of 25–40 μL with 30,000 cells per well. The dye loading and pre-incubation times of antagonists and instrument settings for the FlexStation3 were identical to those for 96-well format. CRF-induced dose-dependent release of intracellular calcium (EC50 = 451 ± 3 nM, EC80 = 1 μM), as shown in Fig. 2A, and AS-30 dose-dependently inhibited CRF-(3 μM) induced intracellular Ca2+ release (IC50 = 27 ± 1 nM) as shown in Fig. 2B.
Fig. 2. Miniaturization of the calcium assay in 384-well format.
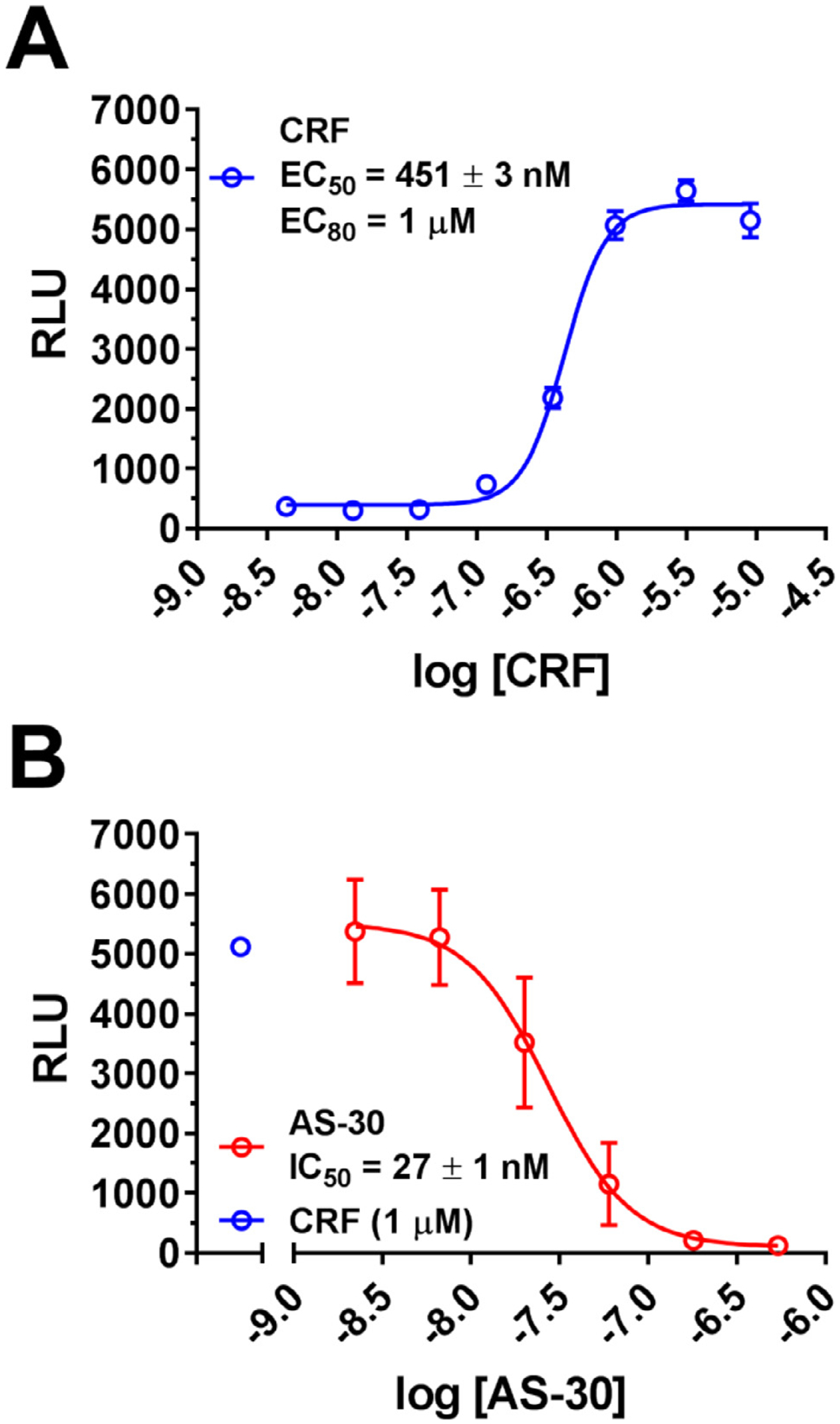
(A) Dose response curves for CRF-induced (1 pM-10 μM) intracellular calcium release in HEK293 cells expressing the FLAG-CRFBP(10kD)-HA-CRF2α (EC50 = 451 ± 1 nM). (B) Inhibition of CRF-induced (1 μM) intracellular calcium release in HEK293 cells expressing CRFBP(10kD)-CRF2α by CRF2 antagonist, AS-30 (2.2 nM - 0.54 μM) (IC50 = 27 ± 1 nM). Results are expressed as the M ± SEM relative fluorescence units (RFU), using 384-well assay format, calculated as agonist-induced maximum calcium peak/cell number × 1000.
To assess the quality of the screening data in the 96-well and 384-well formats, the Z’ factors were calculated for each plate and for the entire experiment as previously reported [35] (Fig. 3). Under the described 96-well experimental conditions (50,000 cell per well, 3 μM CRF ± 1 μM AS-30), the Z’ factor for the quality control assay in 96-well format was 0.62 (Fig. 3A). A value of Z’ = 0.54 (Fig. 3B) was achieved using the experimental conditions described above (30,000 cells and 1 μM CRF ± 1 μM AS-30 per well) in 384-well format, showing that the assay performance did not significantly degrade during the miniaturization process, and that the assay was amenable to HTS.
Fig. 3. Quality control of cell-based assay expressing FLAG-CRFBP(10kD)-HA-CRF2.
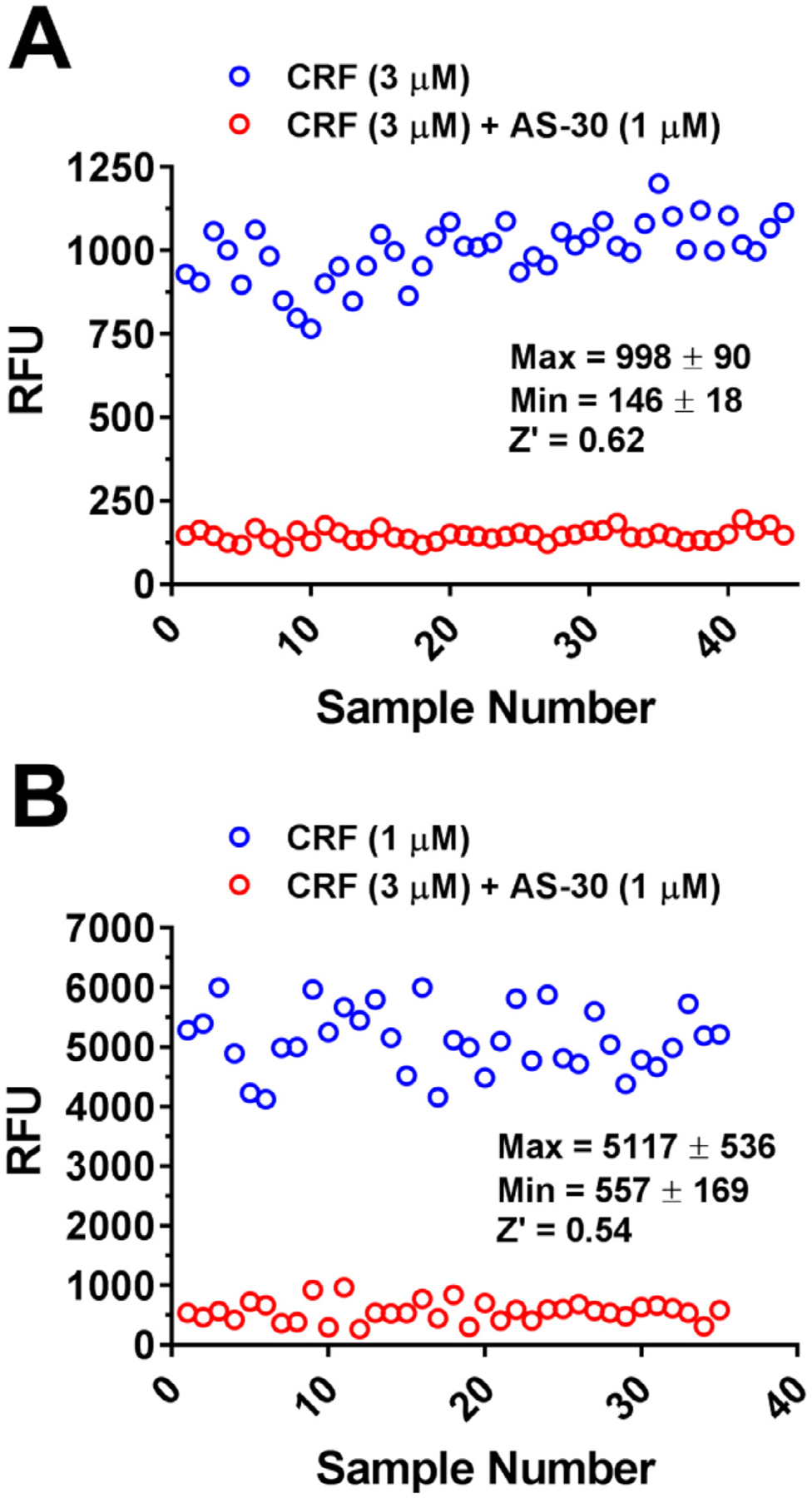
Assay quality control analysis from: (A) 96-well format (Z’ = 0.62) and (B) 384-well format (Z’ = 0.54) in HEK293 cells stably expressing the CRFBP(10kD)-CRF2. CRF (1 μM)-induced intracellular calcium (maximum RFU, EC80 concentration) was consistently inhibited by AS-30 (1 μM) (minimum RFU), n = 35. Results are expressed as the M ± SEM relative fluorescence units (RFU), calculated as agonist-induced maximum calcium peak/cell number × 1000.
We next configured a pilot screen of the Library of Pharmacologically Active Compounds (LOPAC, SIGMA) to test the performance of the HTS in 384-well format. LOPAC consists of 1280 small molecules of known structure and generally assigned function, but of unknown activity with respect to CRFBP-CRF2. Compounds that inhibited the assay by > 50% were considered “hits” (data not shown). For > 70% inhibition, the hit rate for 1280 compounds was 28%. The maximum signal for the assay was 5,921 ± 158 RFU while the minimum signal for the assay was −250 ± 28 RFU. As a control experiment, the inhibition of CRF (1 μM) was tested again prior to the library screening. Since one of our goals was to develop probes to investigate the interaction of CRFBP(10kD) with CRF2 to understand the physiological role of CRFBP in the central nervous system (CNS), we tested compounds in a primary screen for stimulation prior to the CRF inhibition assay. Compounds that produced > 10% stimulation compared to the RFU of CRF (1 μM) in the same assay plate in the primary screen were excluded. This was necessary to remove false positive compounds that produced interference with intracellular calcium homeostasis. The average Z’ factor across all the plates was determined to be 0.52 for the antagonist assay and 0.66 for the agonist assay.
Identification of CRFBP(10kD)-CRF2 modulators using a novel HTS of the Molecular Libraries Small Molecule Repository (MLSMR)
This assay was transferred to a Hamamatsu FDSS 7000 multimodal (fluorescence and luminescence) kinetic imaging system capable of measuring intracellular calcium flux in both 384 and 1534-well formats at the drug discovery facility at the Sanford Burnham Prebys Medical Discovery Institute (SBP). The photonic sensitivity of the FDSS is superior to the FlexStation; thus the assay quality control analysis for the 384-well format using the Hamamatsu FDSS 7000 calculated an improved Z’ both for the CRF activation (Z’ = 0.89) and inhibition (Z’ = 0.71). We further optimized the assay for a 1536-well format for screen implementation, resulting in an average plate Z’ of 0.60 and 0.59 for the agonist and antagonist screens, respectively. Given the robustness of the 1536-well assay, we then screened the National Institute of Health’s (NIH) Molecular Libraries Probes Center Network (MLPCN) collection of approximately 350,000 compounds to find modulators of the CRFBP(10kD)-CRF2 receptor complex. This library was screened simultaneously for agonist and antagonist hits. During the performance of the screening campaigns, 1,568 hits with activity > 50% of the CRF maximal response at a single concentration of 13.25 μM were identified in the agonist assay and 2,056 hits were identified in the antagonist assay with > 50% inhibition of the CRF EC80 at 11.78 μM. After cheminformatic filtering to remove pan-assay interference compounds (PAINS) and promiscuous compounds, as well as those no longer available from vendors, 708 agonist and 1728 antagonist liquid samples were re-ordered from the repository. The liquid samples were assayed in full dose-response curves (DRCs) in the respective primary assays to obtain EC50 (agonist) and IC50 (antagonist) potency values.
Chemistry and cheminformatics resources were further employed in the selection of both novel and chemically tractable molecules from the list of reconfirmed compounds. In total, 62 reconfirmed agonists and 37 reconfirmed antagonist structures were pursued through commercial vendors with additional related compounds acquired via analogue-by-catalogue. To eliminate compounds that may activate or inhibit the Ca2+ mobilization responses by a mechanism that is not specific to CRFBP(10kD)-CRF2 activation, hit compounds were screened against HEK293 cells not expressing the CRFBP(10kD)-CRF2 receptor complex. This secondary screen had a high attrition rate eliminating all of the reconfirmed agonist hits and all but four antagonist hits. These compounds fell into two distinct structural series (Fig. 4), those represented by the tetrazole-thiomethyl-oxadiazole, MLS-0046818 and those represented by the quinazolinone, MLS-0219419. These compounds were also evaluated for activity toward CRF2 in the absence of CRFBP(10kD) and for CRF1 activity and were found to be inactive in both secondary assays (Fig. 5).
Fig. 4. CRFBP(10kD)-CRF2 antagonist hits.
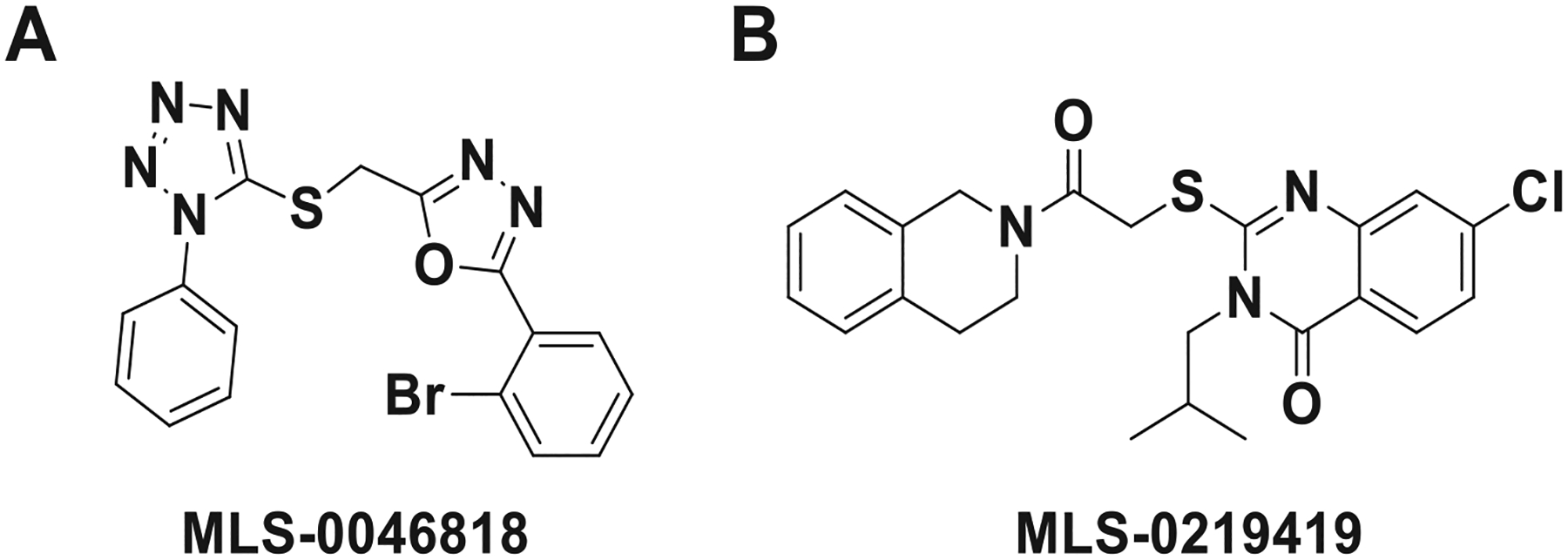
Compounds fell into two series: (A) represented by the tetrazole-thiomethyl-oxadiazole like MLS-0046818, and (B) represented by the quinazolinone like MLS-0219419.
Fig. 5. MLS-0046818 and MLS-0219419 selectively antagonize CRFBP-CRF2 responses.
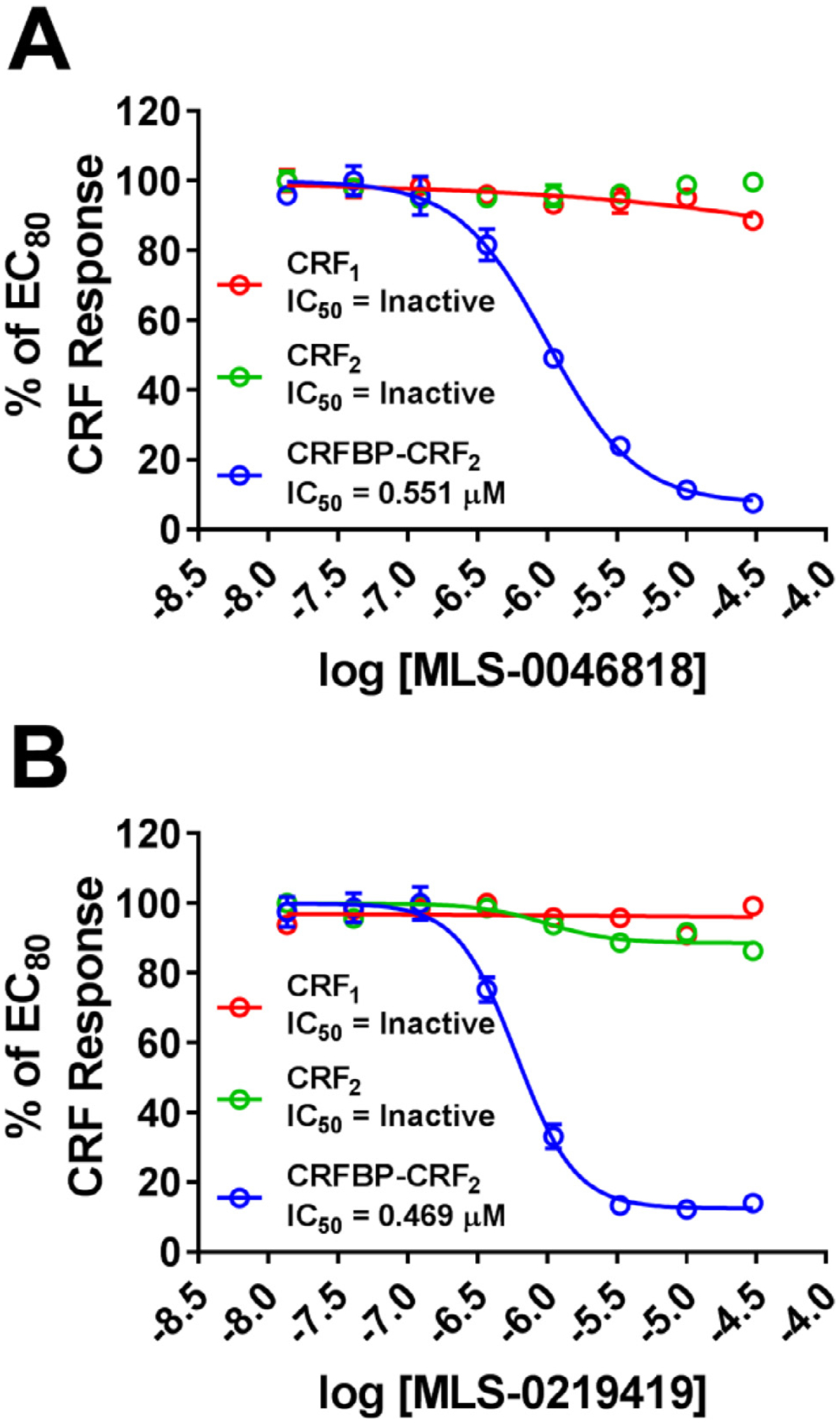
(A) MLS-0046818 and (B) MLS-0219419 dose-responses were performed in the presence of an EC80 concentration of CRF in CRF1 (Red), CRF2 (Green), and CRFBP-CRF2 (Blue) Ca2+ assays. MLS-0046818 and MLS-0219419 only inhibit the CRFBP-CRF2 response. Results are expressed as the M ± SEM of the % of the EC80 CRF response.
Evaluation of the competitive or noncompetitive mechanism of action of novel CRFBP(10kD)-CRF2 modulators
The maximum response to CRF was significantly decreased by increasing concentrations of both lead compounds (1.11 μM, 3.33 μM, 10 μM) with little to no change in the CRF EC50 (Fig. 6). These data are consistent with a noncompetitive interaction of MLS-0046181 and MLS-0219419 with the orthosteric (endogenous) CRF site. Together with the lack of antagonist activity of these compounds toward CRF2 in the absence of CRFBP (see Fig. 5), these compounds appear to act as negative allosteric modulators (NAMs) of the CRFBP-CRF2 complex.
Fig. 6. MLS-0046818 and MLS-0219419 noncompetitively antagonize CRF responses in CRFBP-CRF2 Ca2+ assays.
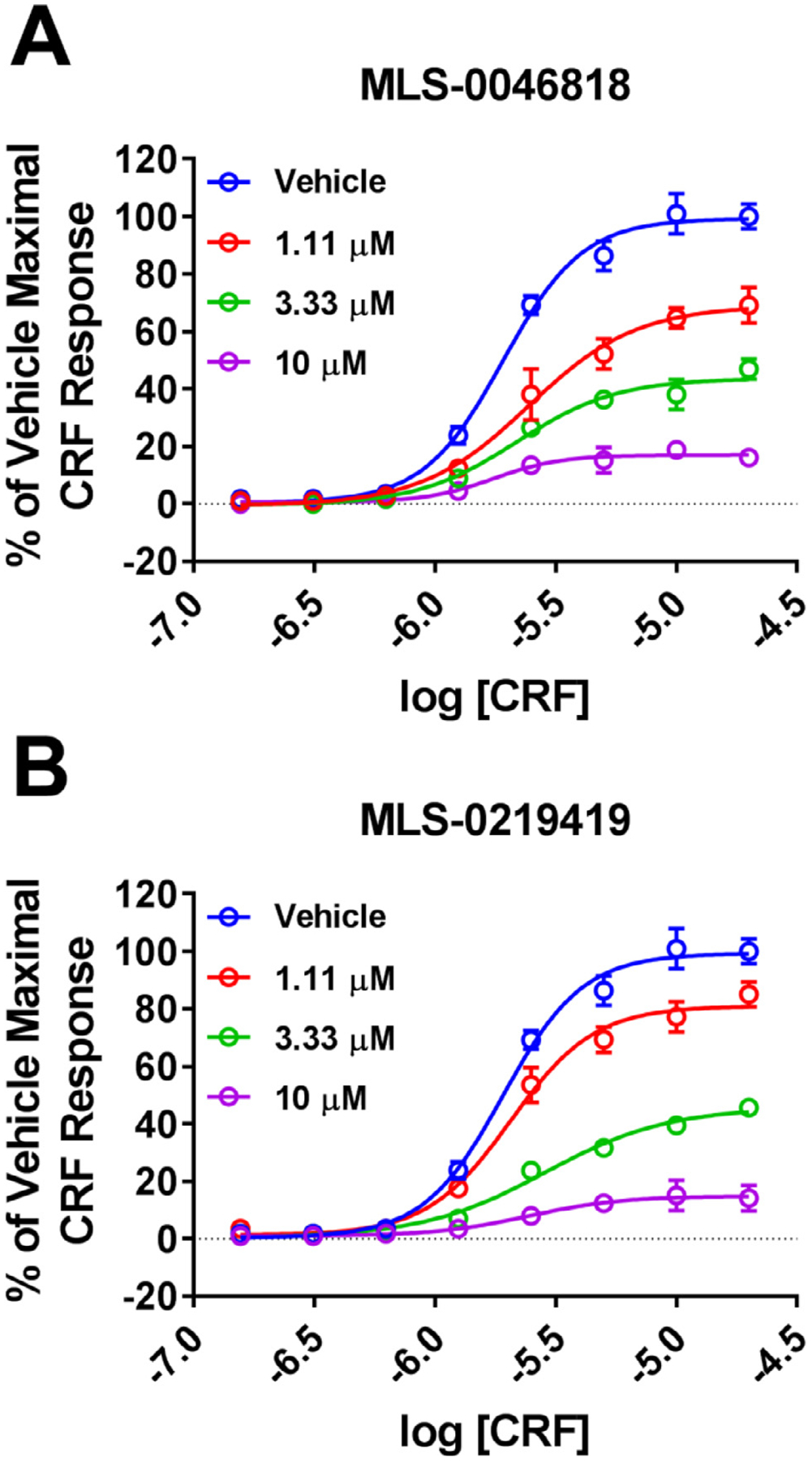
CRF responses were performed for (A) ± MLS-0046818 or (B) ± MLS-0219419 as indicated. CRF maximal responses are decreased with increasing concentrations of either MLS-0046818 or MLS-0219419. Results are expressed as the M ± SEM of the % of the vehicle treated maximal CRF response.
Initial in vitro absorption, distribution, metabolism, and excretion (ADME) and in vivo pharmacokinetic (PK) characterization of analogues
MLS-0046818 and MLS-0219419 were profiled in vitro to assess their drug-like properties and potential for systemic activity in rodent models of AUD (Table 1). Our initial plasma and microsomal stability data indicate that while MLS-0046818 and MLS-0219419 are stable in plasma (98.6 % and 71.5 % remaining at 1 h respectively), they are rapidly metabolized in both rat (0.5 % and 0.1 % remaining at 1 h respectively) or human (6.6 % and 0.1 % remaining at 1 h respectively) liver microsomes; these experiments were performed as we have previously described [42]. We then profiled both compounds for their plasma protein binding (PPB) and brain homogenate binding (BHB) to predict the unbound drug fraction in the plasma and brain respectively. Both PPB and BHB were measured by equilibrium dialysis by methods similar to those previously described utilizing rat plasma and rat brain homogenate [43]. While MLS-0219419 was highly bound to both plasma (0.1% free) and brain (0.0 % free), MLS-0046818 displayed a higher unbound drug fraction in both plasma (4.6% free) and brain (2.3% free), indicating that this series may be more favorable for achieving target exposure. Compounds were also evaluated for inhibition of cytochrome P450 (CYP450) enzymes using a fluorescence-based approach in insect microsomes to evaluate potential drug–drug interaction liabilities. When tested at 10 μM, MLS-0046818 and MLS-0219419 each displayed significant inhibition of CYP3A4 (91% and 76% inhibition respectively), CYP2C9 (86% and 102% inhibition respectively), and moderate inhibition of CYP1A2 (46% and 65% inhibition respectively). Alternatively, while MLS-0219419 showed no significant CYP2D6 inhibition (−21%), MLS-0046818 has the potential for CYP2D6 induction (−128% inhibition), which we will explore in more detail in future experiments. MLS-0219419 and MLS-0046818 were also tested for inhibition of the human Ether-à-go-go-Related Gene (hERG) potassium channel binding using a competition binding assay with a tracer at Reaction Biology (Malvern, PA). Both compounds also exhibited low to moderate hERG channel binding inhibition when tested at 10 μM (48% for MLS-0046818 and 86% for MLS-0219419).
Table 1.
ADME parameters for CRFBP-CRF2 NAMs.
| Parameter | MLS-0046818 | MLS-0219419 |
|---|---|---|
| Plasma Stability at 1 h (rat) | 98.6% | 71.5% |
| Microsomal Stability at 1 h | 0.5% (rat), 6.6% (human) | 0.1% (rat), 0.1% (human) |
| Plasma Protein Binding (rat) | 4.6% free | 0.1% free |
| Brain Homogenate Binding (rat) | 2.3% free | 0.0% free |
| CYP3A4 Inhibition at 10 μM | 91% | 76% |
| CYP2C9 Inhibition at 10 μM | 86% | 102% |
| CYP1A2 Inhibition at 10 μM | 46% | 65% |
| CYP2D6 Inhibition at 10 μM | −21% | −128% |
| hERG Binding Inhibition at 10 μM | 48% | 86% |
Finally, MLS-0046818 and MLS-0219419 were profiled in binding assays against a comprehensive panel of CNS receptors through the NIMH Psychoactive Drug Screening Program (PDSP; for experimental details please refer to the PDSP web site https://pdspdb.unc.edu/pdspWeb/). As shown in Supplemental Table 1, at a concentration of 10 μM, overall neither MLS-0046818 nor MLS-0219419 exhibited significant activity (>50% binding inhibition) against the majority of the targets. MLS-0046818 exhibited > 50% activity at 5HT2B (67%) and KOR (85%) receptors, which corresponded to inhibitory constant (Ki) values of 1496 nM and 2053 nM respectively.
Next, to evaluate plasma and brain exposure of MLS-0046818 and MLS-0219419, we determined plasma drug levels after intraperitoneal (i.p.) administration in mice (Fig. 7). Both compounds exhibit high nanomolar to low micromolar plasma levels and are nearly completely eliminated after 24 h (Table 2). Comparing the plasma Cmax exposure values to the in vitro IC50 values, MLS-0046818 achieved 4.8-fold above its in vitro CRFBP-CRF2 IC50 while MLS-0219419 achieved 1.7-fold above its in vitro CRFBP-CRF2 IC50. Samples from brains at 4 h, 6 h, and 24 h post-dose were also analyzed to provide an initial estimate of brain levels. MLS-0046818 exhibited a brain exposure of 56 nM (0.10 × IC50) at 4 h post-dose, 78 nM (0.14 × IC50) at 6 h post-dose, and 40 nM (0.07 × IC50) at 24 h post-dose. Alternatively, MLS-0219419 exhibited a brain exposure of 245 nM (0.52 × IC50) at 4 h post-dose, 764 nM (1.63 × IC50) at 6 h post-dose, and 78 nM (0.17 × IC50) at 24 h post-dose.
Fig. 7. In vivo PK properties in mouse plasma.
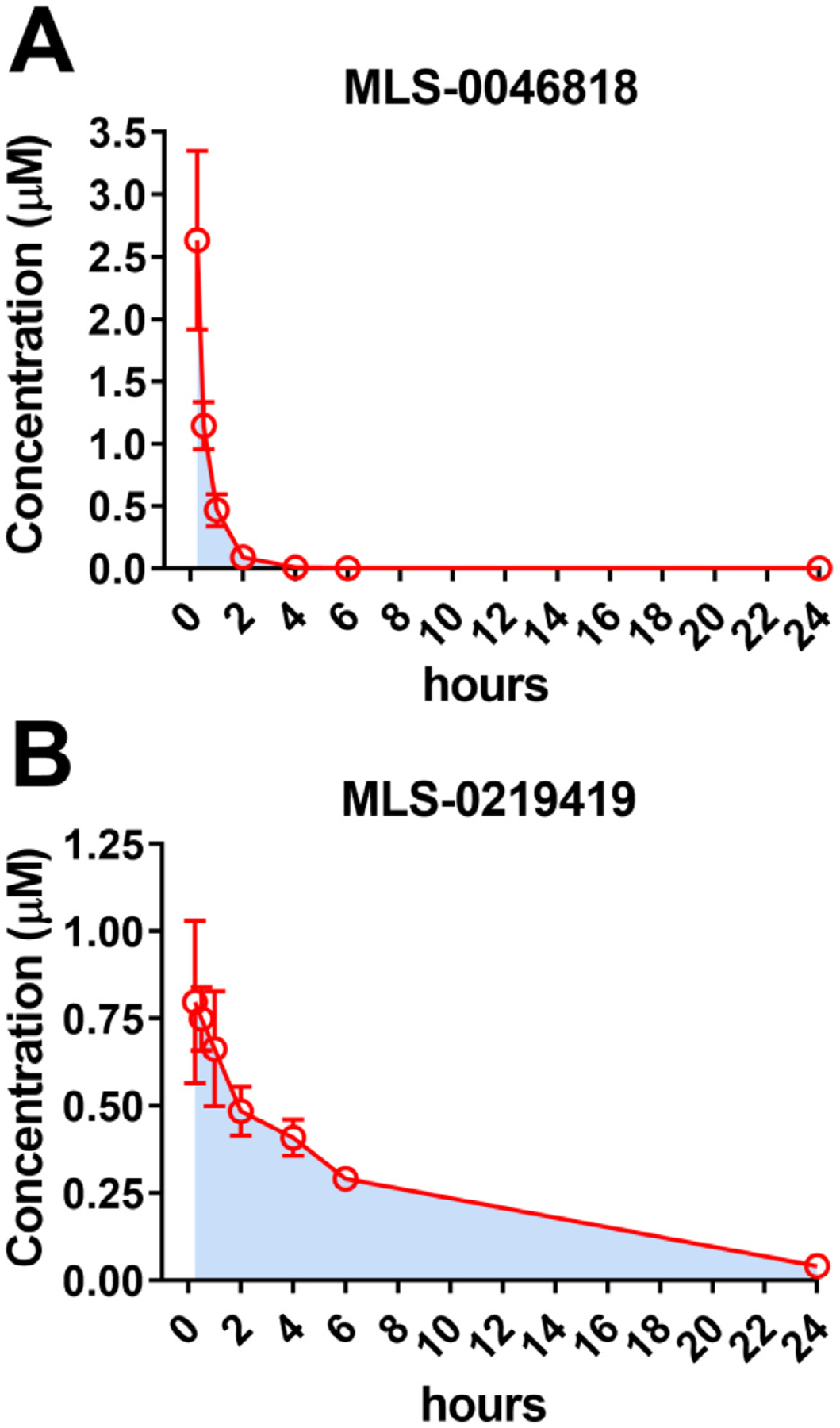
Mice were dosed 10 mg/kg i.p. with either (A) MLS-0046818 or (B) MLS-0219419 and drug levels were monitored over 24h in the mouse plasma. Data represented as M ± SEM of the blood plasma levels from three independent mice with AUC shown in light blue.
Table 2.
Pharmacokinetic parameters for CRFBP-CRF2 NAMs.
| PK Parameter | MLS-0046818 | MLS-0219419 |
|---|---|---|
| t1/2 | 0.49 h | 6.17 h |
| Tmax | 0.25 h | 0.25 h |
| Cmax | 2.633 μM | 0.797 μM |
| AUC0-t | 1.580 μmol/L*h | 5.798 μmol/L*h |
Evaluation of the effects of CRFBP-CRF2 modulators on CRF1+ neurons in the central nucleus of the amygdala (CeA)
The central nucleus of the amygdala (CeA) is comprised of GABAergic projection neurons and interneurons. Neurons that express CRF and CRF1 form a subset of a heterogenous neuronal population in the CeA [44]. It has been previously reported that CRF enhances GABAergic synaptic transmission in the CeA through CRF1-mediated increases in GABA release in both mice and rats [18–20]. Hence, to assess whether the identified NAMs are selective for CRF2, we utilized male CRF1-GFP reporter mice and recorded miniature inhibitory postsynaptic currents (mIPSCs) from CeA CRF1+ neurons (Fig. 8A). Acute application of MLS-0046818 (30 μM) in the presence of R121919 (1 μM), a CRF1 receptor antagonist, for 15 min showed no significant changes in mIPSC frequency, amplitude, rise time, or decay (Representative traces in Fig. 8B, data summary in Fig. 8C, n = 9) suggesting that this CRFBP-CRF2 NAM does not alter CRF1 activity. As CRF acts on presynaptic CRF1 receptors to increase GABA release [18–20], we then applied acute CRF (200 nM) for 9–12 minutes in 8 out of these 9 neurons treated with R121219 and MLS-0046818. CRF did not induce changes in mIPSC properties (Representative traces in Fig. 8B, data summary in Fig. 8D, n = 8) in these recordings, indicating that CRF1 receptors mediate the effect of CRF on action potential dependent GABA release in the CeA, which is blocked here with R121919.
Fig. 8. CRFBP-CRF2 modulators do not affect CRF1 activity on action potential dependent GABA transmission in the CeA.
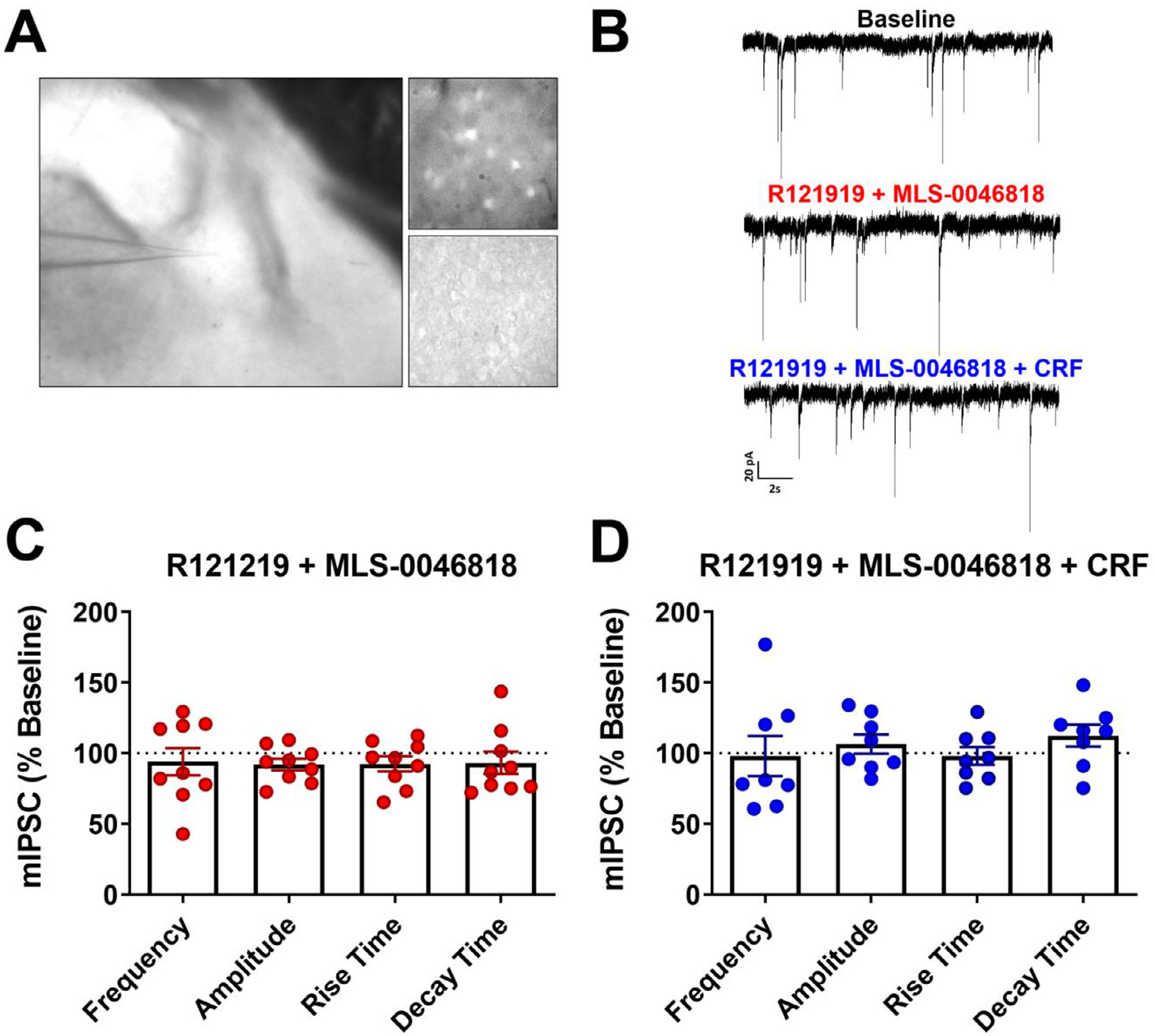
A) Electrophysiological recordings were performed from CRF1+ labeled neurons in CeA slices. B) Representative traces of mIPSCs at baseline, in the presence of R121219 (1 μM) and MLS-0046818 (30 μM), and R121219 (1 μM) + MLS-0046818 (30 μM) + CRF (200 nM). C) Application of R121219 (1 μM) and MLS-0046818 (30 μM) for 15 min does not induce significant changes: one sample t-test, p = 0.5575 (frequency), p = 0.0908 (amplitude), p = 0.2017 (rise time) and p = 0.4208 (decay) in mIPSC properties relative to baseline (one-sample t-test; n = 9 neurons/6 mice). D) Application of CRF (200 nM) following pre-treatment of brain slices with R121219 (1 μM) and MLS-0046818 (30 μM) for 15 min do not produce significant changes: one sample t-test, p = 0.8948 (frequency), p = 0.3619 (amplitude), p = 0.7754 (rise time) and p = 0.1513 (decay) in mIPSC properties relative to baseline (one-sample t-test, n = 8 neurons/4 mice).
Evaluation of the effects of CRFBP-CRF2 modulators on CRF-mediated NMDAR potentiation in ventral tegmental area (VTA)-dopamine (DA) neurons
We recorded NMDAR excitatory postsynaptic currents (EPSCs) from VTA dopamine (DA) neurons in acute brain slices from male and female mice. A CRF dose (1 μM) known to maximally potentiate NMDAR responses [26, 22] was washed onto the slices. As we have observed previously [26, 22], CRF potentiated NMDA receptor EPSCs recorded from VTA-DA neurons (Fig.s 9A) and there was no difference in NMDAR EPSCs between male and female mice (Supplemental Fig. 1S). We next evaluated the ability of each of the two CFRBP-CRF2 NAMs to block this CRF potentiation of NMDAR EPSCs. We found that at a concentration of 30 μM, MLS-0046818 or MLS-0219419 blocked the CRF effect (Fig. 9B and 9C), again with no difference in response between male and female mice (Supplemental Fig. 1S). The summary of these data are shown in Fig. 9D.
Fig. 9. NMDAR potentiation by CRF and blockade by CRFBP-CRF2 NAMs.
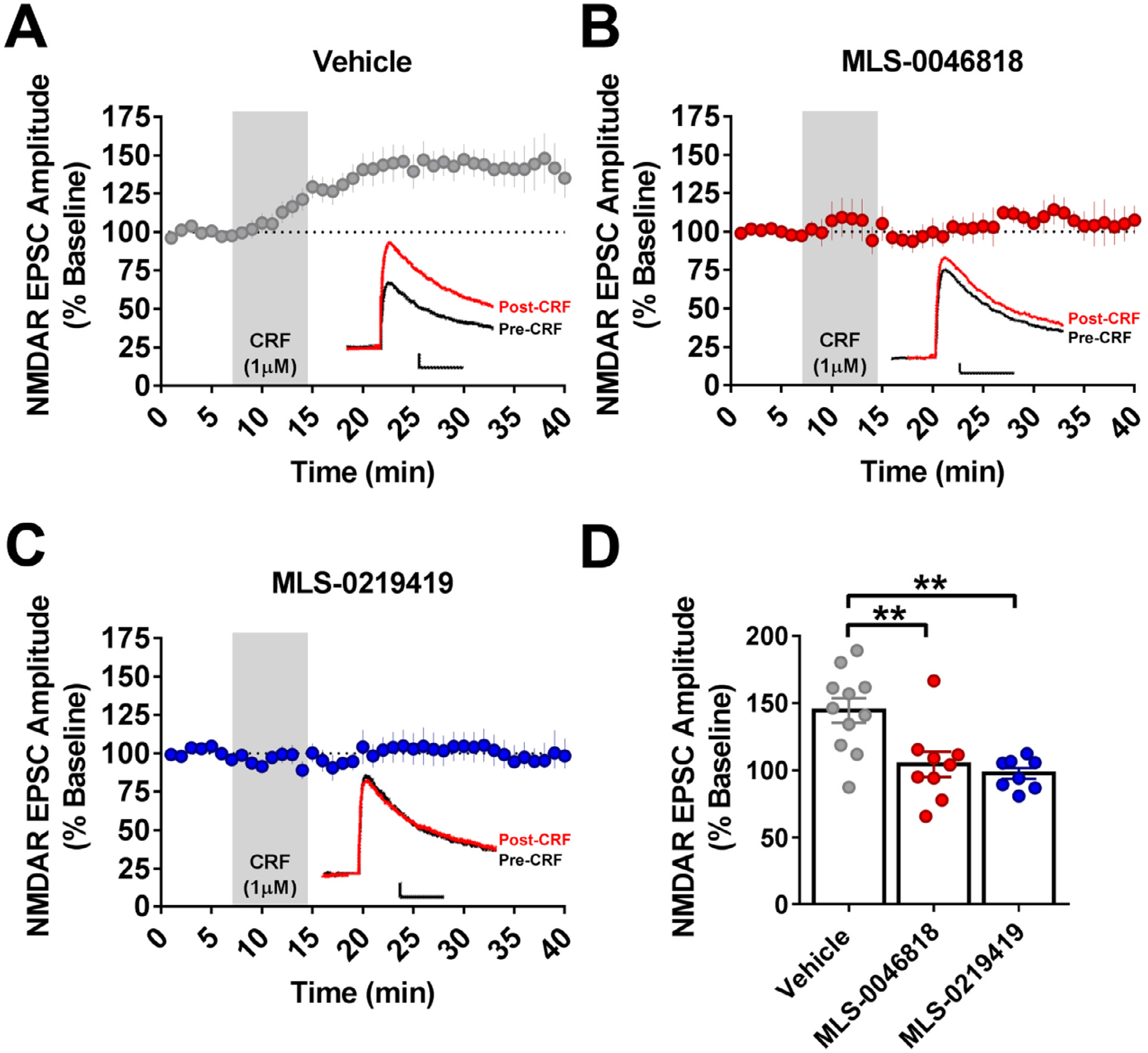
(A) CRF potentiated NMDA receptor EPSCs recorded from VTA-DA neurons (n = 8, 11). 30 μM of (B) MLS-0046818 (n = 4, 9) and (C) MLS-0219419 (n = 4, 8) block CRF potentiation of NMDAR EPSCs. (D) Summary of the M EPSC ± SEM. The asterisk indicates significant differences between treatment and vehicle samples (** p < 0.01), (n = mice, cells). Traces: Black, pre-CRF; Red, Post-CRF; Scale bar 25pA/ 25msec.
Discussion
We have identified and validated the first small molecule chemical probes for use as research tools to evaluate the roles of CRFBP in the CNS with the potential to pharmacologically treat AUD and other psychiatric disorders [28].
We previously investigated the role of the full length CRFBP(37kD) and its two cleavage fragments CRFBP(27kD) and CRFBP(10kD) on the signal transduction of CRF1 and CRF2 receptors in vitro by establishing cell lines in which these various CRFBP constructs are tethered to either CRF1 or CRF2 [27]. In these studies, we established that the CRFBP(10kD) fragment tethered to CRF2 results in increased Ca2+ signaling from the CRF2 receptor relative to the non-tethered CRF2. However, the equivalent CRFBP(10kD)-CRF1 receptor complex and CRF1 or CRF2 complexes with either the full length CRFBP(37kD) or the CRFBP(27kD) fragment do not show increases in Ca2+ signaling relative to their non-tethered counterparts [27].
While the CRFBP(10kD)-CRF2 chimera did not represent the natural biological action in vivo, it provided the first in vitro step to investigate the association of CRF with the complex, CRFBP(10kD)/CRF2, and its role in modulating endocrine activation [30, 45]. We previously reported that the entire construct, FLAG-CRFBP(10kD)-HA-CRF2, was capable of internalization after CRF stimulation [27, 46], thus, this in vitro chimeric tool appears to behave in a similar manner to the non-chimeric CRF2. To further characterize the chimeric receptor complex, we showed that CRF produced a dose-dependent stimulation of [35S]GTPγS-binding in cell membranes expressing the FLAG-CRFBP(10kD)-HA-CRF2 chimera and this stimulation was inhibited by a CRF2 antagonist. We miniaturized the cell-based assay to a 384-well format, validated the assay utilizing the LOPAC1280 library in a pilot screen, and transferred the assay to a Hamamatsu FDSS 7000 system at the SBP drug discovery facility where we found an improved signal response. Together, these studies support the robustness of the assay and its suitability for screening the MLPCN library to identify small molecule modulators. Importantly, this system removed the need to make and purify CRFBP and facilitated the development of a simple fluorescence calcium assay amenable to HTS.
Cell-based HTS assays have been primarily based on ligand-receptor interactions [47]. This design has been successful in the drug discovery process due to the specificity and high affinity of natural ligands to their corresponding receptor in the same system. However, the in vivo biological system interactions at the receptor level are not limited to a one-to-one interaction (ligand-receptor), but often are the consequence of multiple component interactions including those with soluble binding proteins [47]. Most proteins function in a physiological environment under crowded conditions. Our approach allowed us to create a more realistic in vitro environment in which proteins are expressed in a cell-like, dense state [48]. This innovation forms the basis of the development of the HTS assay that we employed to identify novel small molecule allosteric modulators of the CRFBP-CRF2 receptor complex.
Finally, the effect of our novel small molecule allosteric modulators on electrophysiological recordings in brain slices provides validation of their neuroactive properties in a physiologically relevant system. Previous studies indicate that VTA NMDAR EPSCs are potentiated by CRF through an interaction with CRFBP and CRF2 [22]. The current study demonstrates that this effect of CRF on NMDAR-mediated EPSCs in the VTA can be blocked through allosteric inhibition of the CRFBP complex with CRF2. This is supported by the observation that a selective concentration of each of two NAMs (30 μM) inhibited the strong response to CRF (1 mM) in the VTA. In addition, CRF-CRF1 induced increases in GABA release in the CeA are not affected by a CRFBP-CRF2 NAM. Further investigation will be required to elucidate the mechanism through which the binding between CRF and CRFBP facilitates the actions of CRF at CRF2. Our results, however, support a permissive role for CRFBP where it facilitates the binding of CRF to the CRF2. When CRF binds to CRFBP, it forms a stable dimer, and undergoes conformational modifications [45]. It is possible that our compounds may interfere with conformational changes that allow CRF to signal when bound to CRF2.
It is often desirable for pharmacotherapies to modulate CNS receptor signaling rather than completely inhibiting it, as this more closely mirrors native temporal neuronal signaling [49]. As a therapeutic approach for the treatment of AUD and SUD, our aim in this study was to discover and develop allosteric modulators that act specifically on the stress system which may reduce craving [50]. Altogether, the efforts of this study have resulted in validated chemical probes for use as research tools to delineate the roles of CRFBP in the CNS. However, suboptimal in vitro potencies, in vitro ADME/T, and in vivo PK properties such as poor systemic exposure of current CRFBP-CRF2 probes would impede our efforts to obtain proof-of-concept in vivo efficacy data in rodent models of AUD and SUD. Towards a therapeutic objective, we have initiated lead optimization efforts to further develop these scaffolds with regard to their potency, ADME/T and PK properties. We aim to identify candidates with the requisite properties, including brain penetration, suitable for comprehensive in vivo evaluation. These next generation probes will allow us to better investigate the role of CRFBP in the consumption of addictive substances and facilitate the development of effective treatments targeting CRFBP for AUD and SUD.
Supplementary Material
Acknowledgements
The authors would like to thank the following for funding: the National Institute on Alcohol Abuse and Alcoholism (NIAAA): R01 AA026589 (DJS); K01 AA023867, R01 AA027760, R21 AA027614 (CLH-K), R01 AA021491 and R01 AA029841 (MR), T32 AA007456 (RP); the National Institute of General Medical Sciences (NIGMS), Center of Biomedical Research Excellence (COBRE, P20 GM130414) (CLH-K); the National Institute of Mental Health (NIMH) K99 MH123673 and NIGMS FI2 GM128622 (TCF); the National Institute Drug Abuse (NIDA) R21 DA209966 and R33 DA029966 (NDPC; SEB); NIDA Intramural Research Program (TCF); NIH Fast Track Award (SEB); and the National Cancer Institute (NCI) Cancer Center Support Grant P30 CA030199. Ki determinations and receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. The high-throughput screen was funded by the MLPCN (HG005033). LHS acknowledges support for the Conrad Prebys Center for Chemical Genomics at Lake Nona from the Florida Translational Research Program (COHK8). SEB acknowledges the State of California for Medical Research on Alcohol and Substance Abuse through the University of California, San Francisco. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of Competing Interests
LHS is currently an employee of Fate Therapeutics, San Diego, CA, United States. SV and BSH are currently employees of Alchem Laboratories Corp, Alachua, FL, United States. ES is currently an employee of Chugai Pharmaceutical Co. Ltd., Tokyo, Japan. MPH is currently an employee of Boundless Bio, La Jolla, CA, United States. MM is an employee of Kite Pharmaceuticals, CA, United States. AB is currently an employee of GIA (Global Institutes on Addiction), Miami, FL.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.slasd.2022.09.005.
References
- [1].KOOB GF, SCHULKIN J. Addiction and stress: An allostatic view. Neurosci Biobehav Rev 2019;106:245–62. [DOI] [PubMed] [Google Scholar]
- [2].Burnette EM, Nieto SJ, Grodin EN, Meredith LR, Hurley B, Miotto K, Gillis AJ, Ray LA. Novel agents for the pharmacological treatment of alcohol use disorder. Drugs 2022;82:251–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 2004;44:525–57. [DOI] [PubMed] [Google Scholar]
- [4].HAASS-KOFFLER CL, BARTLETT SE. Stress and addiction: contribution of the corticotropin releasing factor (CRF) system in neuroplasticity. Front Mol Neurosci 2012;5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].GRACE CR, PERRIN MH, DIGRUCCIO MR, MILLER CL, RIVIER JE, VALE WW, RIEK R. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci U S A 2004;101:12836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].HOARE SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today 2005;10:417–27. [DOI] [PubMed] [Google Scholar]
- [7].NIELSEN SM, NIELSEN LZ, HJORTH SA, PERRIN MH, VALE WW. Constitutive activation of tethered-peptide/corticotropin-releasing factor receptor chimeras. Proc Natl Acad Sci U S A 2000;97:10277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet 2000;24:410–14. [DOI] [PubMed] [Google Scholar]
- [9].Bale TL, Lee KF, Vale WW. The role of corticotropin-releasing factor receptors in stress and anxiety. Integr Comp Biol 2002;42:552–5. [DOI] [PubMed] [Google Scholar]
- [10].COSTE SC, KESTERSON RA, HELDWEIN KA, STEVENS SL, HEARD AD, HOLLIS JH, MURRAY SE, HILL JK, PANTELY GA, HOHIMER AR, HATTON DC, PHILLIPS TJ, FINN DA, LOW MJ, RITTENBERG MB, STENZEL P, STENZEL-POORE MP. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet 2000;24:403–9. [DOI] [PubMed] [Google Scholar]
- [11].KISHIMOTO T, RADULOVIC J, RADULOVIC M, LIN CR, SCHRICK C, HOOSHMAND F, HERMANSON O, ROSENFELD MG, SPIESS J. Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet 2000;24:415–19. [DOI] [PubMed] [Google Scholar]
- [12].ROBERTO M, SPIERLING SR, KIRSON D, ZORRILLA EP. Corticotropin-releasing factor (CRF) and addictive behaviors. Int Rev Neurobiol 2017;136:5–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].SPIERLING SR, ZORRILLA EP. Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology (Berl) 2017;234:1467–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].KWAKO LE, SPAGNOLO PA, SCHWANDT ML, THORSELL A, GEORGE DT, MOMENAN R, RIO DE, HUESTIS M, ANIZAN S, CONCHEIRO M, SINHA R, HEILIG M. The corticotropin releasing hormone-1 (CRH1) receptor antagonist pexacerfont in alcohol dependence: a randomized controlled experimental medicine study. Neuropsychopharmacology 2015;40:1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].SCHWANDT ML, CORTES CR, KWAKO LE, GEORGE DT, MOMENAN R, SINHA R, GRIGORIADIS DE, PICH EM, LEGGIO L, HEILIG M. The CRF1 antagonist verucer-font in anxious alcohol-dependent women: translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology 2016;41:2818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry 2008;165:617–20. [DOI] [PubMed] [Google Scholar]
- [17].Coric V, Feldman HH, Oren DA, Shekhar A, Pultz J, Dockens RC, Wu X, Gentile KA, Huang SP, Emison E, Delmonte T, D’souza BB, Zimbroff DL, Grebb JA, Goddard AW, Stock EG. Multicenter, randomized, double-blind, active comparator and placebo–controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety 2010;27:417–25. [DOI] [PubMed] [Google Scholar]
- [18].NIE Z, SCHWEITZER P, ROBERTS AJ, MADAMBA SG, MOORE SD, SIGGINS GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 2004;303:1512–14. [DOI] [PubMed] [Google Scholar]
- [19].NIE Z, ZORRILLA EP, MADAMBA SG, RICE KC, ROBERTO M, SIGGINS GR. Presynaptic CRF1 receptors mediate the ethanol enhancement of GABAergic transmission in the mouse central amygdala. ScientificWorldJournal 2009;9:68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].ROBERTO M, CRUZ MT, GILPIN NW, SABINO V, SCHWEITZER P, BAJO M, COTTONE P, MADAMBA SG, STOUFFER DG, ZORRILLA EP. Corticotropin releasing factor–induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biological psychiatry 2010;67:831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].POTTER E, BEHAN DP, LINTON EA, LOWRY PJ, SAWCHENKO PE, VALE WW. The central distribution of a corticotropin-releasing factor (CRF)-binding protein predicts multiple sites and modes of interaction with CRF. Proc Natl Acad Sci U S A 1992;89:4192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].UNGLESS MA, SINGH V, CROWDER TL, YAKA R, RON D, BONCI A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron 2003;39:401–7. [DOI] [PubMed] [Google Scholar]
- [23].SLATER PG, NOCHES V, GYSLING K. Corticotropin-releasing factor type-2 receptor and corticotropin-releasing factor-binding protein coexist in rat ventral tegmental area nerve terminals originated in the lateral hypothalamic area. Eur J Neurosci 2016;43:220–9. [DOI] [PubMed] [Google Scholar]
- [24].SLATER PG, CERDA CA, PEREIRA LA, ANDRES ME, GYSLING K. CRF binding protein facilitates the presence of CRF type 2alpha receptor on the cell surface. Proc Natl Acad Sci U S A 2016;113:4075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].WANG B, YOU ZB, RICE KC, WISE RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacology (Berl) 2007;193:283–94. [DOI] [PubMed] [Google Scholar]
- [26].Albrechet-Souza L, Hwa LS, Han X, Zhang EY, Debold JF, Miczek KA. Corticotropin releasing factor binding protein and crf2 receptors in the ventral tegmental area: modulation of ethanol binge drinking in C57BL/6J Mice. Alcohol Clin Exp Res 2015;39:1609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].HAASS-KOFFLER CL, HENRY AT, MELKUS G, SIMMS JA, NAEMMUDDIN M, NIELSEN CK, LASEK AW, MAGILL M, SCHWANDT ML, MOMENAN R, HODGKINSON CA, BARTLETT SE, SWIFT RM, BONCI A, LEGGIO L. Defining the role of corticotropin releasing factor binding protein in alcohol consumption. Transl Psychiatry 2016;6:e953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].CURLEY DE, WEBB AE, SHEFFLER DJ, HAASS-KOFFLER CL. Corticotropin releasing factor binding protein as a novel target to restore brain homeostasis: lessons learned from alcohol use disorder research. Front Behav Neurosci 2021;15:786855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].WOODS RJ, KEMP CF, DAVID J, SUMNER IG, LOWRY PJ. Cleavage of recombinant human corticotropin-releasing factor (CRF)-binding protein produces a 27-kilodalton fragment capable of binding CRF. J Clin Endocrinol Metab 1999;84:2788–94. [DOI] [PubMed] [Google Scholar]
- [30].HAASS-KOFFLER CL. The corticotropin releasing factor binding protein: a strange case of Dr. Jekyll and Mr. Hyde in the stress system? Alcohol 2018;72:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Behan DP EB, Souza DE, Lowry PJ, Potter E, Sawchenko P, Vale WW. Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front Neuroendocrinol 1995;16:362–82. [DOI] [PubMed] [Google Scholar]
- [32].RIVIER J, GULYAS J, KIRBY D, LOW W, PERRIN MH, KUNITAKE K, DIGRUCCIO M, VAUGHAN J, REUBI JC, WASER B, KOERBER SC, MARTINEZ V, WANG L, TACHE Y, VALE W. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J Med Chem 2002;45:4737–47. [DOI] [PubMed] [Google Scholar]
- [33].RUHMANN A, BONK I, LIN CR, ROSENFELD MG, SPIESS J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2beta-selective antisauvagine-30. Proc Natl Acad Sci U S A 1998;95:15264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].LAWRENCE AJ, KRSTEW EV, DAUTZENBERG FM, RUHMANN A. The highly selective CRF(2) receptor antagonist K41498 binds to presynaptic CRF(2) receptors in rat brain. Br J Pharmacol 2002;136:896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].ZHANG JH, CHUNG TD, OLDENBURG KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73. [DOI] [PubMed] [Google Scholar]
- [36].Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 1959;14:48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].ZHANG Y, HUO M, ZHOU J, XIE S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 2010;99:306–14. [DOI] [PubMed] [Google Scholar]
- [38].HERMAN MA, CONTET C, JUSTICE NJ, VALE W, ROBERTO M. Novel subunit-specific tonic GABA currents and differential effects of ethanol in the central amygdala of CRF receptor-1 reporter mice. Journal of Neuroscience 2013;33:3284–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].HERMAN MA, CONTET C, ROBERTO M. A functional switch in tonic GABA currents alters the output of central amygdala corticotropin releasing factor receptor-1 neurons following chronic ethanol exposure. Journal of Neuroscience 2016;36:10729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].JUSTICE NJ, YUAN ZF, SAWCHENKO PE, VALE W. Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. Journal of Comparative Neurology 2008;511:479–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].WOLFE S, SIDHU H, PATEL R, KREIFELDT M, D’AMBROSIO S, CONTET C, ROBERTO M. Molecular, morphological, and functional characterization of corticotropin-releasing factor receptor 1-expressing neurons in the central nucleus of the amygdala. Eneuro 2019:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DHANYA RP, SHEFFLER DJ, DAHL R, DAVIS M, LEE PS, YANG L, NICKOLS HH, CHO HP, SMITH LH, D’SOUZA MS, CONN PJ, DER-AVAKIAN A, MARKOU A, COSFORD ND. Design and synthesis of systemically active metabotropic glutamate subtype-2 and -3 (mGlu2/3) receptor positive allosteric modulators (PAMs): pharmacological characterization and assessment in a rat model of cocaine dependence. J Med Chem 2014;57:4154–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].KALVASS JC, MAURER TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 2002;23:327–38. [DOI] [PubMed] [Google Scholar]
- [44].GILPIN NW, HERMAN MA, ROBERTO M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biological psychiatry 2015;77:859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].LOWRY PJ, KOERBER SC, WOODS RJ, BAIGENT S, SUTTON S, BEHAN DP, VALE W, RIVIER J. Nature of ligand affinity and dimerization of corticotrophin-releasing factor-binding protein may be detected by circular dichroism. J Mol Endocrinol 1996;16:39–44. [DOI] [PubMed] [Google Scholar]
- [46].SIMMS JA, HAASS-KOFFLER CL, BITO-ONON J, LI R, BARTLETT SE. Mifepristone in the central nucleus of the amygdala reduces yohimbine stress-induced reinstatement of ethanol-seeking. Neuropsychopharmacology 2012;37:906–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].TIAN YE, WU LH, MUELLER WT, CHUNG FZ. A screening strategy based on differential binding of ligand to receptor and to binding proteins: screening for compounds interacting with corticotrophin-releasing factor-binding protein. J Biomol Screen 1999;4:319–26. [DOI] [PubMed] [Google Scholar]
- [48].HAASS-KOFFLER CL, NAEEMUDDIN M, BARTLETT SE. An analytical tool that quantifies cellular morphology changes from three-dimensional fluorescence images. J Vis Exp 2012:e4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].LEWIS JA, LEBOIS EP, LINDSLEY CW. Allosteric modulation of kinases and GPCRs: design principles and structural diversity. Curr Opin Chem Biol 2008;12:269–80. [DOI] [PubMed] [Google Scholar]
- [50].HAASS-KOFFLER CL, LEGGIO L, KENNA GA. Pharmacological approaches to reducing craving in patients with alcohol use disorders. CNS Drugs 2014;28:343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.