

Abstract

Severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) is considered a global public health concern since it causes high morbidity and mortality. Recently, it has been reported that repurposed anti-COVID-19 drugs might interact with multidrug resistance ABC transporter, particularly ABCB1. In the current study, a series of thiourea derivatives were screened as potential inhibitors against SARS-CoV-2 by targeting the attachment of receptor binding domain (RBD) of spike protein with ACE2 and their interaction with human ABCB1 has also been explored. The results indicated strong impairment of RBD–ACE2 attachment by BB IV-46 with a percentage inhibition of 95.73 ± 1.79% relative to the positive control, while BB V-19 was proven inactive with a percentage inhibition of 50.90 ± 0.84%. The same compound (BB IV-46) interacted with ABCB1 and potentially inhibited cell proliferation of P-gp overexpressing cell line with an IC50 value of 4.651 ± 0.06 μM. BB V-19, which was inactive against SARS-CoV-2, was inactive against ABCB1 with a higher IC50 value of 35.72 ± 0.09 μM. Furthermore, molecular dynamics simulations followed by binding free-energy analysis explored the binding interaction of BB IV-46 and BB V-19 to RBD region of spike protein of SARS-CoV-2. The results confirmed that compound BB IV-46 interacted strongly with RBD with a significant binding energy (−127.0 kJ/mol), while BB V-19 interacted weakly (−29.30 kJ/mol). The key interacting residues of the RBD involved in binding included Leu441, Lys444, and Tyr449. This study highlights the importance of BB IV-46 against SARS-CoV-2; however, further pharmacokinetic and pharmacodynamics studies are needed to be done.
1. Introduction
Severe acute respiratory syndrome corona virus 2 [SARS-CoV-2; provisionally labeled 2019 novel coronavirus or 2019-nCoV (COVID-19)] is an RNA virus that infects humans. Early coronaviruses that infected humans include SARS and Middle-East respiratory syndrome.1−6 SARS-CoV-2 is closely linked to bat-SL-CoVZC45 and bat-SL-CoVZXC21.7−9 The outbreak of SARS-CoV-2 in China at the end of 2019 triggered a worldwide pandemic and is a major public health concern for today’s world.3 It has been designated as the most contagious agent of the century.10 Additionally, in developing countries like Pakistan, this caused real economic burden on the government. According to data published by the World Health Organization (WHO), more than 43,000 tests reported positive in 28 countries until February 11, 2020, with China accounting for more than 99% of the total cases.11 Recently, new variants including Omicron and Delta variants have emerged in December 2020 and were attributed to the emergence of new SARS-CoV-2 variants 501Y.V2 (B.1.351) in South Africa and variants 501Y.V1 (B.1.1.7) in the UK.12
Structurally, SARS-CoV-2 consists of four proteins, that is, envelope (E), membrane (M), nucleocapsid (N), and spike (S). The S protein is a transmembrane protein, consisting of two subunits S1 and S2.13 The S1 subunit (receptor binding domain, RBD) binds to the host receptor, angiotensin-converting enzyme 2 (ACE2), leading to the discharge of the RNA into the cells. However, the S2 subunit mediates the viral cell membrane fusion.14−16 These characteristics of S protein make it a vital therapeutic target.17,18 The published crystal structure of SARS-CoV-2 RBD–ACE2 complex revealed that the interface is composed of a total of 17 residues of the RBD in contact with 20 residues of ACE2. These residues of RBD include Lys417, Gly446, Tyr449, Tyr453, Leu455, Phe456, Ala475, Phe486, Asn487, Tyr489, Gln493, Gly496, Gln498, Tyr500, Asn501, Gly502, and Tyr505.19
During COVID-19 pandemic, several repurposed drugs were screened against SARS-COV-2 in clinics without prior in vitro studies. Later studies showed that a few of these drugs showed interactions with multidrug resistance ABC transporter and thus lead to severe complication in COVID-19 patients by affecting the absorption, distribution, metabolism, elimination, and toxicity properties of drugs.20 One of these repurposed drugs was ivermectin, which was known for its role in viral replication inhibition. Its absorption is mainly controlled by ABCB1 efflux transporter. However, at a higher dose, it also inhibits ABCB1 and thus penetrates in higher concentration and becomes neurotoxic.21 Moreover, altered expression (high/absent) of ABCB1 has also been reported in COVID-19 patients who received repurposed drugs directly in clinical trials due to overuse of drugs.22 Thus, any new drug candidates must be tested for interactions with these key transporters.
Currently, there are vaccines available that may protect people from catching infection; however, an antiviral drug is a prerequisite to treat people once they have infection. Therefore, there is an urgent need for drug development against SARS-CoV-2 that is cheap and easily accessible to everybody.23 In this regard, thiourea derivatives can be considered the best source for anti-SARS-CoV-2 drugs. The thiourea moiety is an important synthon that has antibacterial, antiviral, antiproliferative, and cytotoxic properties.24 Organosulfur compounds such as thiourea and their derivatives are used in a variety of disciplines, including chemical synthesis and pharmaceutical industry. Due to this significance, a number of urea and thiourea derivatives have received approval as marketable drugs by FDA and EMA for the treatment of various human diseases, and as a result, pharmaceutical companies and academic researchers are currently working to develop promising drug candidates.25 Adedeji et al., 2013 screened a library of low-molecular-weight compounds as potential inhibitors for SARS-CoV and identified three compounds that inhibited SARS-CoV by different mechanisms. One of these was thiourea scaffold (SSAA09E1) that prevented entry of the virus into the cell by inhibiting viral membrane fusion to the host cell membrane.26 Van Tat et al., 2021 worked on selected anti-HIV-1 phenethyl-thiazole-thiourea compounds by building QSAR models to develop a lead compound that inhibits SARS-CoV-2 infection. Based on QSAR models, new derivatives were designed, followed by docking simulations into the RBD of SARS-COV-2 and HIV-1 receptor. The new derivatives were found potent against HIV-1 as well as SARS-CoV-2 as compared to the lead compound.27 This gives an indication that a wide range of molecules with excellent pharmacological properties and few adverse effects can be generated by strategically planning changes to thiourea derivative structures. In the current study, a series of new thiourea derivatives were screened as potential inhibitors of SARS-CoV-2 by targeting RBD–ACE2 attachment and their interaction with multidrug resistance ABCB1 transporter has also been explored. Furthermore, molecular dynamics (MD) simulations were performed, followed by binding free-energy analysis, to map the interacting residues of spike protein (receptor binding domain, RBD) that are involved in binding of thiourea derivatives and to compare the binding energetics of the most active and inactive ligands.
2. Materials and Methods
2.1. Synthetic Compounds
Thiourea derivatives were a kind gift from Prof. Dr. Khalid Mohammed Khan (H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi), which were synthesized by his group and characterized as urease inhibitors (Bano et al., 2018).
2.2. Inhibition of SARS-CoV-2
Inhibition assay was conducted using a SARS-CoV-2 inhibitor screening kit (AdipoGen, Life Sciences cat no. AG-48B-0001-KI01). This assay is based on a colorimetric ELISA kit, which measures the binding of the RBD of the spike S protein from SARS-CoV-2 to its human receptor ACE2. The assay is designed to highlight and characterize the impact of various inhibitory molecules on the inhibition of SARS-CoV-2 binding to any ACE2-expressing cells. The stepwise procedure for the assay is as follows: first of all, the 96-well ELISA microplate was coated by adding 100 μL/well of diluted spike (1 μg/mL). The plate was then covered with plastic film and left overnight at 4 °C. After incubation, wells were aspirated and any residual liquid was removed by inverting and blotting the plate against a clean absorbent paper. Next, 200 μL of blocking buffer was added and kept for 2 h at room temperature. Aspiration was repeated and 300 μL of 1× wash buffer was added to each well. Washing was done thrice. 100 μL of IMS-diluted thiourea derivatives was added to each well while 100 μL of IMS-diluted inhibitory control ACE2 (human), monoclonal antibody (mAb) (AC384) was added to one well as a positive control and incubated at 37 °C for 1 h. The wash procedure was repeated again. In the subsequent steps, 100 μL of the diluted HRP-labeled streptavidin (HRP) (1/200) was added and incubated for 1 h at room temperature, followed by aspiration and washing. For substrate development, 100 μL of ready-to-use TMB was added to each well and kept for 5 min at room temperature. Finally, 50 μL of the stop solution (2 M H2SO4) was added to stop the reaction. The measurements were taken at 450 nm on an ELISA plate reader (HT Company UK; HER 480).
2.3. Cytotoxicity Assay
Cytotoxicity assay was accomplished to test the response of thiourea derivatives against CCRF-CEM vcr1000 cell line, which was an ABCB1 overexpressing cell line. In a 96-well plate, these cells were carefully seeded at a density of 5000–10,000 cells/well; afterward, varying concentrations of thiourea derivatives were added to the wells and kept under overnight incubating conditions of 37 °C and 5% of CO2. The following day, MTT reagent [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was prepared in a concentration of 0.5 mg/mL and 20 μL of it was then added to each well and again incubated for 4 h. After incubation, MTT crystals were dissolved by adding 100 μL of DMSO and kept at room temperature for 20 min. Data from the cytotoxicity assay were generated by taking measurements at 450 nm/520 nm on ELISA (Biotic ELx800), and data analysis was performed using GraphPad Prism Version 7.
2.4. Computational Methods
2.4.1. MD Simulation
All MD simulations were conducted using GROMACS28 Version 5.1.1 with a GROMOS united-atom force field (GROMOS96 53a6)29 for a time scale of 200 ns each at 300 K on a DELL Precision T5600 Workstation comprising 16 physical processor cores within two second-generation Intel. Xeon E5-2650 processors accelerated by NVIDIA Quadro K2000 graphic processor unit. For the MD simulations, the initial structure of the SARS-CoV-2 spike RBD S1 unit was obtained from the RCSB protein database (pdb id # 7CH5).30 The topology files of the thiourea derivatives were obtained from PRODRG. The electrostatic potential (ESP) charges were calculated using high-level quantum mechanical calculations for higher accuracy. Gaussian 09 rev. D.0131 software was used to compute the single-point energies of the downloaded compounds. For the calculations, a hybrid density functional (PBE0)32 with 25% Hartree Fock exchange33 was used with the triple ζ basis set Def2-TZVP34 and with empirical dispersion correction (D3) by Grimme.35−37 The solvent effects were included through the polarizable continuum model with the solvent model density parameter set by Truhlar.31,38−41 Water was used as a solvent in all the single-point energy calculations.42
Two systems were modeled; the first one contained RBD of spike protein and the active thiourea derivative (BB IV-46), while the second one consisted of RBD of spike protein and the inactive derivative (BB V-19), both positioned randomly in a cubic box and solvated using the SPCE water model. The simulations were performed in a replicate of three following the same protocol for both the systems. Each system was neutralized using counterions and then energy-minimized using the steepest descent for 50,000 steps. Next, the systems were equilibrated for 500 ps under periodic boundary conditions using NVT ensemble, followed by NPT. During equilibration, the temperature was kept constant at 300 K using a V-rescale thermostat,43 and the pressure was kept constant at 1 bar using a Parrinello–Rahman barostat with the time constant τ = 2.0 ps and a compressibility of 4.5 × 10–5 bar–1 in all three dimensions.44 Long-range electrostatic interactions were calculated using particle-mesh Ewald algorithm.45 A 1.4 nm cutoff was used for both the van der Waals interaction and the real space Ewald interaction. LINCS algorithm was used to constrain all the bonds during simulation.46 A 200 ns of MD was run for each system with a time step of 2 fs, and the frames were saved every 10,000 ps for subsequent analysis. The trajectories thus obtained were visualized using visual MD package47 and analyzed using tools available in GROMACS. The binding energy (BE) of both BB IV-46 and BB V-19 with the RBD of spike protein was calculated using the MM-PBSA method implemented in Gromacs.48
3. Results
3.1. Inhibition of SARS-CoV-2 Using Thiourea Derivatives
In the initial series of experiments, thiourea derivatives (n = 09) were screened for their inhibitory potential against the RBD of the S1 subunit of the SARS-CoV-2 spike protein using a SARS-CoV-2 inhibitor screening kit (as mentioned in method Section 2.1). mAb was used as a positive control. Percentage inhibition was plotted in the form of a bar graph as shown in Figure 1 and Table 1. The results revealed the highest percentage inhibition of 95.73 ± 1.79% for compound BB IV-46 (active), followed by compounds BB IV-52, BB IV-34, BB IV-60, BB V-21, and BB IV-45 with percentage inhibitions of 90.21 ± 3.69, 77.73 ± 1.08, 67.38 ± 4.92, 67.07 ± 5.39, and 65.52 ± 7.55%, respectively, relative to the positive control, while BB IV-8, BB IV-80, and BB V-19 show comparatively weaker activity with percentage inhibitions of 58.50 ± 0.70, 55.22 ± 7.02, and 50.90 ± 0.84%, respectively. However, compound BB V-19 was found to be the most inactive, having a percentage inhibition of 50.90 ± 0.84%.
Figure 1.

Percentage inhibition of spike protein (RBD) attachment to ACE2 receptor by thiourea derivatives in relation to the positive control.
Table 1. List of Percentage Inhibitions of Thiourea Derivatives.

3.2. Results of Cytotoxicity Assay
In another series of experiments, thiourea derivatives were screened for their cytotoxic properties using MTT in ABCB1 overexpressing CCRF-CEM vcr1000 cell line. The results revealed compounds BB IV-46, BB IV-52, BB IV-34, BB V-8, and BB IV-60 with potent cytotoxic activity by showing IC50 values of 4.651 ± 0.06, 23 ± 0.51, 13.45 ± 0.01, 6.17 ± 0.02, and 4.45 ± 0.08 μM, respectively, against P-gp (ABCB1) overexpressing cell lines. These three compounds were also found to be active against SARS-CoV-2, as shown in Section 3.1, while BB V-19, which was found to be inactive against SARS-CoV-2, also became inactive against ABCB1 by showing an IC50 value of 35.72 ± 0.10 μM. Overall, BB IV-46 indicates 9 folds higher activity as compared to BB V-19 (inactive). The results are shown in Table 2.
Table 2. Different IC50 Values of Thiourea Derivatives against ABCB1 Overexpressing Cell Line CCRF-CEM vcr1000.
| s. no. | code | IC50 ± SE |
|---|---|---|
| 1 | BB IV-80 | 7.511 ± 0.01 |
| 2 | BB IV-34 | 13.45 ± 0.01 |
| 3 | BB IV-45 | 11.93 ± 0.08 |
| 4 | BB IV-46 | 4.651 ± 0.06 |
| 5 | BB IV-60 | 4.450 ± 0.08 |
| 6 | BB IV-52 | 23 ± 0.51 |
| 7 | BB V-8 | 6.17 ± 0.02 |
| 8 | BB V-19 | 35.72 ± 0.09 |
| 9 | BB V-21 | 23.13 ± 0.02 |
4. Computational Methods
4.1. Results for MD Simulations
MD simulation is a powerful computational tool that explores the changes in the macroscopic properties of the systems as a result of inter- and intramolecular forces. In this study, two systems were chosen for modeling, including the active thiourea derivative (BB IV-46) and the inactive derivative (BB V-19) with RBD of spike protein for 200 ns. To investigate the conformational behavior of the system, tools such as root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), H-bond, secondary structure analysis, and MM-PBSA were used. The results show that the active compound, that is, BB IV-46, makes strong interactions with the RBD of the spike protein, which implies that it could potentially inhibit the binding of SARS-CoV-2 spike protein to ACE2 receptors.
4.1.1. Structural Dynamics of RBD
4.1.1.1. Root-Mean-Square Deviation
RMSD analysis is performed to follow the deviation of atoms of the system from their initial coordinates during the simulation. The results of this analysis give insights into the structural stability of the protein components and other domains involved in the systems such as ligands. We calculated RMSD of the protein to investigate the structural change in RBD of the spike protein in comparison to the original crystal structure as a response to BB IV-46 and BB V-19 ligand binding. There was a typical increase in RMSD observed at the beginning of simulation for both structures because of the initial kinetic force which plateaued after 100 ns in the case of BB IV-46 ligand binding to the RBD. However, in the presence of BB-V-19, there were still be some fluctuations at around 150 ns in which an increase in the RMSD was observed (Figure 2A). To inspect this divergence, we extracted the average structure coordinates from RBD–BB-V-19 complex; interestingly, we found that the negatively charged Asp428 comes in close contact with the counterpart BB-V-19 containing fluoride and iodide moieties at the terminal aromatic rings. As soon as the two negatively charged moieties come in closer contact, Asp428 moves away, and this is depicted in comparatively large fluctuations in the distance between Asp428 of RBD and negative groups on BB-V-19 ligand (see distance analysis in Figure 2B). This is also supported by a slight increase in the radius of gyration Rg value at the same time (Figure 2C). After some small initial fluctuations, RMSD values attain equilibrium and stay stable afterward. RMSD is frequently employed to evaluate the similarity of three-dimensional structures of two proteins. A similarity metric of this type is dependent on the protein’s dimension. For two exactly same structures, the RMSD is 0, and its value increases as the two structures become more dissimilar. In this study, an interesting observation was made, that is, a high average RMSD of RBD–BB IV-46 complex (see Figure 2D) as compared to RBD–BB V-19 complex, attributed to a small change in the secondary structure of RBD on binding with BB-IV-46. This change includes the conversion of residues 5–10 from α-helix in BB-IV-46 to turns and coils in BB-V-19, conversion of residues 23–26 in BB-IV-46 from 3–10 helix to turns and coils in BB-V-19, and conversion of residues 106–108 in BB-IV-46 from 3–10 helix to turns and coils in BB-V-19 as a result of ligand binding; these changes in secondary structures are shown figuratively in Figure S1. The small average RMSD of BB IV-46 and RBD–BB V-19 complex in both cases reflects a stable secondary structure of the RBD of the spike protein as reported in the literature earlier as well. We observed that no matter the small molecule was active or inactive, the protein showed minimal change in the conformation, meaning that even upon binding with the ligands, no substantial change in the secondary structure of RBD was observed, which means that the structure in itself is very stable. Studies show that RBD of spike protein is an independently foldable protein domain, resilient to conformational changes even upon mutations, and therefore an attractive target for vaccination (Figure 3).49
Figure 2.

(A) RMSD (RBD–BB-IV-46/BB-V-19) complexes; (B) RMSD (RBD alone in the presence of BB-IV-46/BB-V-19); (C) distance between key residues of RBD of spike protein and BB-V-19. The plot shows that out of four key residues that are involved in binding with BB-V-19, Asp428 shows maximum fluctuations; (D) RMSF of RBD in the presence of BB-IV-46/BB-V-19 showing flexibility of the key regions; (E) number of hydrogen bonds between RBD–BB-IV-46 and RBD–BB-V-19; (F) Rg of RBD in the presence of BB-IV-46 and BB-V-19.
Figure 3.

Plot of MM-PBSA binding free-energy contribution per residue of the complexes formed by (A) RBD–BB-IV-46 and (B) RBD–BB-V-19.
4.1.1.2. Radius of Gyration (Rg)
Rg analysis shows variation in overall compactness of the RBD as a result of ligand attachment. The average values for RBD in the presence of BB IV-46 and BB V-19 lie between 1.73 and 1.76 nm, respectively. These values point to only a subtle change in conformation of protein as a result of ligand binding. Rg of all complex systems were fairly consistent throughout the simulation, which suggests that the secondary structure elements have compact packing (see Figure 2C).
4.1.1.3. Root-Mean-Square Fluctuation
The residual fluctuation and flexibility of the backbone atoms of RBD from their average position on ligand binding were calculated using RMSF tool. The values of RMSF were plotted against the residues of the protein in order to reflect the flexibility of RBD’s residues as shown in Figure 2E. The figure shows that in both the systems, most residues have an RMSF value lower than 0.4 nm. As expected, terminal residues showed larger RMSF values, indicating greater flexibility. The RMSF of loops in the BB V-19 ligand-binding site was significantly higher than in the BB-IV-46 RBD structure. A small increase in the average RMSF value was observed for RBD bound to BB-IV-46 (0.24 nm) when compared to the RBD bound to BB-V-19 (0.21 nm). We reason that this increase in RMSF on binding with the active ligand might be indirectly caused by changing the conformational dynamics of the active site residues. The shaded areas in Figure 2E show the important regions (residues 439–449) with higher flexibility in RBD. An interesting observation is that these residues 439–449 form a cavity-like structure that serves as hot spots for binding with BB-IV-46 (see Figures 4 and 5). Once BB-IV-46 binds to this cavity, it does not unbind. The role and BE contribution of these residues is elaborated in the MM-PBSA Binding Free Energy section.
Figure 4.
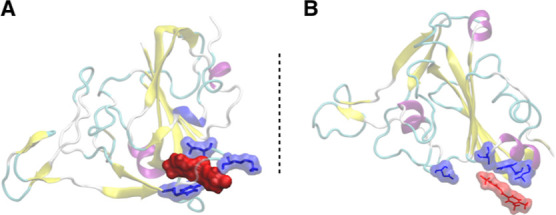
Binding of RBD with (A) BB-IV-46 and (B) BB-V-19. The figure suggests that BB-IV-46 fits well in RBD’s binding pocket and interacts with residues in close vicinity to a greater extent as compared to BB-V-19. The blue color indicates the interacting residues from RBD of spike protein, while the red color indicates the ligands. Both are represented as surfaces to give a better idea of the interaction.
Figure 5.
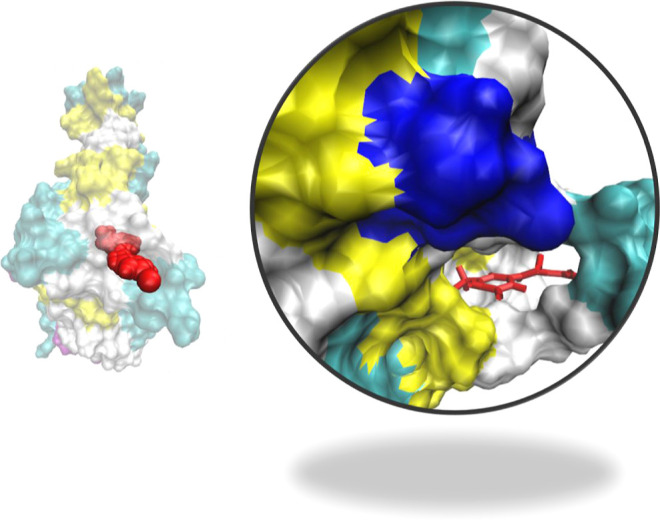
Binding of RBD with BB-IV-46. The figure suggests that BB-IV-46 fits well in RBD’s binding pocket. On the right side is the zoom image of the binding pocket. Red stick model represents the active ligand, that is, BB-IV-46.
4.1.1.4. Hydrogen Bond Analysis
The H-bond analysis shows the total number of hydrogen bonds between the RBD and both ligands, that is, BB IV-46 and BB V-19, throughout the simulation. Although a total of 18 hydrogen bonds formed between RBD and BB IV-46 and six hydrogen bonds formed between RBD and BB V-19, most of them are transient. On an average, only two hydrogen bonds persist for the RBD–BB IV-46 complex, while there was only one hydrogen bond for RBD–BB V-19. From the plot, we can see that the hydrogen bonds are very short-lived; they form and break throughout the simulation, and we can assume that they do not provide significant stability of the ligand–protein interaction (Figure 2F). They only serve to bring the ligand closer to the RBD, and once the ligand binds to RBD, the van der Waals and other electrostatic interactions come into play as confirmed by MM-PBSA analysis.
4.1.1.5. MM-PBSA Binding Free Energy
The average free BE is a critical parameter that suggests how effectively a ligand is bound to the protein. We used single trajectory approach in MM-PBSA study that does not consider any binding-induced structural changes. MM-PBSA method was used to compare the free energy of two different ligand-bound conformations of the same molecules, that is, the BE of the RBD with the BB-IV-46 and BB-V-19 ligands. The van der Waals forces and electrostatics were the main contributors toward the binding of RBD to the BB IV-46 and BB V-19 ligands as shown in Table 3, and the van der Waals energy contribution to the overall binding free energy was greater than that of electrostatics. The BEs of RBD with BB IV-46 and BB V-19 were −127.089 ± 12.00 and −29.30 ± 6.04 kJ/mol, respectively, indicating that BB IV-46 interacts strongly with RBD. The energy contribution for the RBD–BB IV-46 complex was from the residues Asn334, Arg346, Val401, Arg403, Arg408, Lys417, Leu441, Lys444, Gly446, Tyr449, Tyr451, Arg454, Arg457, Arg466, Phe497, Pro507, and Arg509, out of which Leu441, Lys444, and Tyr449 were the major contributors which are located in the already known binding site.19 Out of these, residues Asn439 to Tyr449 form a hydrophobic cavity that develops van der Waals interactions with BB IV-46. Residues Asn439, Gly446, Asn448, and Tyr449 are also reported in the literature because of their crucial role in binding with ACE2.50 This implies that the active compound, that is, BB IV-46, exploits the same region for binding as ACE2. Thus, it can be a potential inhibitor of RBD–ACE2 binding interactions. However, in the case of the RBD–BB V-19 complex, the highest contribution was obtained from Val382, Leu390, Phe392, and Asp428. Figure 3A shows the per residue contribution of various residues from RBD involved in binding with BB IV-46 resulting in maximum binding affinity with the lowest free energy, that is, −127.0 kJ/mol.
Table 3. Summary of BE Electrostatic Energy, van der Waals Energy, Polar Solvation Energy, and SASA Energy of BB-IV-46 and BB-V-19 in Complex with RBD.
| s. no. | RBD–ligand complex | binding energy (kJ/mol) | van der Waals energy (kJ/mol) | electrostatic energy (kJ/mol) | polar solvation energy (kJ/mol) | SASA energy (kJ/mol) |
|---|---|---|---|---|---|---|
| 1 | BB-IV-46 | –127.089 | –157.274 | –83.838 | 128.902 | –14.879 |
| 2 | BB-V-19 | –29.365 | –59.666 | –65.636 | 105.339 | –9.402 |
5. Discussion
Although several studies and direct clinical trials on repurposed drugs are available against SARS-CoV-2, findings related to their interactions with multi-drug resistance ABCB1 transporter are limited.51,52 There are currently no ways for accurately calculating the drug-transport interaction using an in silico approach; thus, only extensive in vitro research may aid in therapeutic applications in SARS-CoV-2 therapy. Therefore, there is an urgent need of in vitro screening of potential inhibitors against SARS-CoV-2 in association with ABCB1 transporter to develop a drug that might be safe to use in clinical trials and become cheap and easily accessible to everyone.53
The current project was conducted with an aim to develop a drug that may inhibit the invasion of SARS-CoV-2 by inhibiting the interaction of spike protein (RBD) with ACE2 and to explore the interaction of these drugs with MDR-ABCB1 transporter. The data indicated the potency of BB-IV-46 among all other studied thiourea derivatives against SARS-CoV-2 (95.73 ± 1.79%) as well as ABCB1 (4.651 ± 0.06 μM), while BB V-19 showed the least activity (50.90 ± 0.84% for SARS CoV-2; 35.72 ± 0.09 μM for ABCB1). The difference in activity was due to the strong binding of BB-IV-46 (−127.089 ± 12.00 kJ/mol) with RBD of spike protein that could probably inhibit RBD–ACE2 attachment as evident by MD simulations. The key interacting residues were found in already reported RBD–ACE2 interface. However, BB V-19 showed weak interactions (−29.30 ± 6.04 kJ/mol).
A number of groups have worked on inhibition of SARS-CoV-2 by targeting spike protein. Kim et al., 2021 reported geranin to effectively block the binding of spike protein with ACE2, and their MD simulations of docked complexes showed the interaction of geranin with amino acid residues Arg403, Tyr449, Tyr453, Gln493, Ser494, Gln498, Gly502, Tyr495, Gly496, Thr500, Asn501, and Tyr505 of the spike protein.54 Our data also demonstrated higher inhibitory potential of BB IV-46 [1,(4-iodo-phenyl)-3-(3-(tri-fluoromethyl)-phenyl thiourea)] against RBD–ACE2 attachment. Additionally, our study also highlights the interaction of BB IV-46 with ABCB1 by showing a very low IC50 value (4.651 ± 0.06 μM) when administered to ABCB1 overexpressing cell line (CCRF CEM vcr1000) as compared to BB V-19 (inactive derivative). This suggests that COVID-19 complications can be controlled by affecting the drug penetration. Thus, the result is found to be consistent with several earlier studies which proposed that the COVID-19 patients at a higher risk due to altered expression of ABCB1 and the interaction of repurposed drugs with this efflux transporter22 reported SSAA09E2 that blocked the interaction of spike protein of SARS-CoV-2 with ACE2 receptor. Again, our results are in line by showing impaired attachment of RBD–ACE2 by BB IV-46 in inhibition assays preventing the entry of the virus into the cell.
The difference in activity (seen in inhibition experiments) was supported by our MD simulation data which indicated the strong binding of BB IV-46 (−127.0 kJ/mol) into the reported RBD–ACE2 interface, therefore suggesting that spike protein might not get a chance to attach with ACE2; hence, the viral entry was prevented. The interacting residues were found to be Asn334, Arg346, Val401, Arg403, Arg408, Lys417, Leu441, Lys444, Gly446, Tyr449, Tyr451, Arg454, Arg457, Arg466, Phe497, Pro507, and Arg509, out of which Leu441, Lys444, and Tyr449 were the major contributors. In comparison, the inactive compound BB V-19 showed weak interactions in RBD (−29.30 ± 6.04 kJ/mol). In terms of electronic effects, BB IV-46 is the halogen-based compound, containing halogen moieties on both ends, and thus is active toward the aforementioned amino acids via halogen bonding (a noncovalent interaction between a positive region on a halogen atom and a negative site), which further depends on the halogen’s electron-deficient σ-hole. If the electron deficiency is significant enough, a region of positive ESP can form, which can interact (attractively) noncovalently with negative sites (such as a lone pair of a Lewis base) on other molecules. Furthermore, the positive character of the σ-hole increases in going from lighter to heavier atoms within a group and the remainder of the molecule becomes more electron-withdrawing.55 This also supports the difference in activity of BB IV-46 and BB V-19.
The following conclusions can be drawn from this study: first, the thiourea derivative, BB IV-46 [1,(4-iodo-phenyl)-3-(3-(tri-fluoromethyl)-phenyl thiourea)], is identified as a potential new inhibitor of SARS-CoV-2 that can prevent entry of the virus into the cell by blocking the interaction spike protein (RBD) to ACE2. Second, BB IV-46 also inhibited ABCB1 (potentially known for disease progression and complications) and thus can be considered as a better alternative toward the treatment of SARS-CoV-2. Last, BB IV-46 showed strong binding in RBD of spike protein, while the inactive compound showed weak interactions with RBD as shown by MD simulations, which provided strong evidence for the activity of BB IV-46. We believe that this study will be very helpful in the future for drug development against SARS-CoV-2. However, further experiments are needed to be done to check the effect of the same derivatives on viral replication.
Acknowledgments
The current research was funded by Higher Education Commission, Pakistan [award number(s): 20-3589].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04671.
Secondary structure analysis of BB-IV-46 and BB-V-19 on binding with RBD (PDF)
Author Contributions
M.A. and Z.P. performed the experiments. Z.P. and S.F. designed the study. S.F., B.K., and Z.P. prepared the initial draft of the manuscript. M.A., Z.P., and S.F. performed the statistical analysis and finalized the manuscript. Z.P., S.F., B.K., and A.K. helped in scientific discussion and revision of the manuscript. B.K. and M.A.H. performed MD simulations and the analysis. Compounds were generously provided by K.M.K. (H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi).
The authors declare no competing financial interest.
Supplementary Material
References
- de Wit E.; van Doremalen N.; Falzarano D.; Munster V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Madhavan M. V.; Sehgal K.; Nair N.; Mahajan S.; Sehrawat T. S.; Bikdeli B.; Ahluwalia N.; Ausiello J. C.; Wan E. Y. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. 10.1038/s41591-020-0968-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.-C.; Chen C.-S.; Chan Y.-J. The outbreak of COVID-19: An overview. J. Chin. Med. Assoc. 2020, 83, 217. 10.1097/jcma.0000000000000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Xu Y.; Gao R.; Lu R.; Han K.; Wu G.; Tan W. Detection of SARS-CoV-2 in different types of clinical specimens. J. Am. Med. Assoc. 2020, 323, 1843–1844. 10.1001/jama.2020.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; McGoogan J. Charakterystyka i ważne wnioski z epidemii choroby koronawirusowej 2019 (COVID-19) w Chinach: streszczenie raportu 72 314 przypadków z Chińskiego Centrum Kontroli i Zapobiegania Chorobom. J. Am. Med. Assoc. 2020, 323, 1239–1242. 10.1001/jama.2020.2648. [DOI] [Google Scholar]
- Yuan M.; Wu N. C.; Zhu X.; Lee C.-C. D.; So R. T. Y.; Lv H.; Mok C. K. P.; Wilson I. A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.-C.; Shih T.-P.; Ko W.-C.; Tang H.-J.; Hsueh P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coven J.; Gupta A.. Disparities in Mobility Responses to COVID-19; New York University, 2020; p 150. [Google Scholar]
- Kumar A.; Mishra D. C.; Angadi U. B.; Yadav R.; Rai A.; Kumar D. Inhibition Potencies of Phytochemicals Derived from Sesame Against SARS-CoV-2 Main Protease: A Molecular Docking and Simulation Study. Front. Chem. 2021, 9, 744376. 10.3389/fchem.2021.744376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V.; Beer J. C.; Sankaranarayanan N. V.; Swanson-Mungerson M.; Desai U. R. Discovering small-molecule therapeutics against SARS-CoV-2. Drug Discovery Today 2020, 25, 1535–1544. 10.1016/j.drudis.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson P.; Rolain J.-M.; Raoult D.. Chloroquine for the 2019 Novel Coronavirus SARS-CoV-2; Elsevier, 2020; Vol. 55, p 105923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARS-CoV-2 B. 1.1. 529 (Omicron) Variant—United States, December 1–8, 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1731. 10.15585/mmwr.mm7050e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; He Y.; Zhou Y.; Liu S.; Zheng B.-J.; Jiang S. The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Moore M. J.; Vasilieva N.; Sui J.; Wong S. K.; Berne M. A.; Somasundaran M.; Sullivan J. L.; Luzuriaga K.; Greenough T. C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Hu Y.; Wang Q.; Qi J.; Gao F.; Li Y.; Zhang Y.; Zhang W.; Yuan Y.; Bao J. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N.-H.; Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Yang Y.; Zhou Y.; Lu L.; Li F.; Jiang S. MERS-CoV spike protein: a key target for antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. 10.1080/14728222.2017.1271415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Li W.; Farzan M.; Harrison S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Telbisz Á.; Ambrus C.; Mózner O.; Szabó E.; Várady G.; Bakos É.; Sarkadi B.; Özvegy-Laczka C. Interactions of potential anti-COVID-19 compounds with multispecific ABC and OATP drug transporters. Pharmaceutics 2021, 13, 81. 10.3390/pharmaceutics13010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low Z. Y.; Yip A. J. W.; Lal S. K. Repositioning Ivermectin for Covid-19 treatment: Molecular mechanisms of action against SARS-CoV-2 replication. Biochim. Biophys. Acta, Mol. Basis Dis. 2022, 1868, 166294. 10.1016/j.bbadis.2021.166294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos C.; Olarte I.; Mendoza I.; Martinez A. Impact of the abcb1 drug resistance gene on complications associated with covid-19. HemaSphere 2021, 5, 381. [Google Scholar]
- Brown C. M.; Vostok J.; Johnson H.; Burns M.; Gharpure R.; Sami S.; Sabo R. T.; Hall N.; Foreman A.; Schubert P. L. Outbreak of SARS-CoV-2 infections, including COVID-19 vaccine breakthrough infections, associated with large public gatherings—Barnstable County, Massachusetts, July 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1059. 10.15585/mmwr.mm7031e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struga M.; Rosolowski S.; Kossakowski J.; Stefanska J. Synthesis and microbiological activity of thiourea derivatives of 4-azatricyclo [5.2. 2.02, 6] undec-8-ene-3, 5-dione. Arch. Pharmacal Res. 2010, 33, 47–54. 10.1007/s12272-010-2223-9. [DOI] [PubMed] [Google Scholar]
- Ronchetti R.; Moroni G.; Carotti A.; Gioiello A.; Camaioni E. Recent advances in urea- and thiourea-containing compounds: focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 2021, 12, 1046–1064. 10.1039/d1md00058f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A. O.; Severson W.; Jonsson C.; Singh K.; Weiss S. R.; Sarafianos S. G. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 2013, 87, 8017–8028. 10.1128/jvi.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tat P.; Hoa T. T.; Vo Ky A.; Nu Ngoc Han P. Novel SARS-CoV-2 Inhibitors from Phenethylthiazolethiourea Derivatives Using Hybrid QSAR Models and Docking Simulation. Smart Sci. 2021, 9, 165–185. 10.1080/23080477.2021.1914967. [DOI] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A.; Berendsen H. J. C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Soares T.; van der Vegt N. F. A.; van Gunsteren W. F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. 10.1007/s00249-004-0448-6. [DOI] [PubMed] [Google Scholar]
- Du S.; Cao Y.; Zhu Q.; Yu P.; Qi F.; Wang G.; Du X.; Bao L.; Deng W.; Zhu H. Structurally resolved SARS-CoV-2 antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy. Cell 2020, 183, 1013–1023.e13. 10.1016/j.cell.2020.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford CT, 2009; Vol. 121, pp 150–166.
- Blaha P.WIEN2K, an Augmented Plane Wave Local Orbitals Program for Calculating Crystal Properties Karlheinz Schwarz; Technische Universität Wien: Austria, 2001. [Google Scholar]
- Robb M.; Cheeseman J.; Scalmani G.. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009.; b Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Barone V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J. Chem. Phys. 2000, 112, 2427–2435. 10.1063/1.480808. [DOI] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Mennucci B.; Tomasi J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. 10.1063/1.473558. [DOI] [Google Scholar]
- Crescenzi O.; Tomaselli S.; Guerrini R.; Salvadori S.; D’Ursi A. M.; Temussi P. A.; Picone D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment: Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Brand W.; Oosterhuis B.; Krajcsi P.; Barron D.; Dionisi F.; Bladeren P. J.; Rietjens I. M.; Williamson G. Interaction of hesperetin glucuronide conjugates with human BCRP, MRP2 and MRP3 as detected in membrane vesicles of overexpressing baculovirus-infected Sf9 cells. Biopharm. Drug Dispos. 2011, 32, 530–535. 10.1002/bdd.780. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H.; Fraaije J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A.; g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Basu S.; Chakravarty D.; Bhattacharyya D.; Saha P.; Patra H. K. Plausible blockers of Spike RBD in SARS-CoV2-molecular design and underlying interaction dynamics from high-level structural descriptors. J. Mol. Model. 2021, 27, 191. 10.1007/s00894-021-04779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe C.; Okiyama Y.; Tanaka S.; Fukuzawa K.; Honma T. Molecular recognition of SARS-CoV-2 spike glycoprotein: quantum chemical hot spot and epitope analyses. Chem. Sci. 2021, 12, 4722–4739. 10.1039/d0sc06528e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y.-F.; Chien C.-S.; Yarmishyn A. A.; Lin Y.-Y.; Luo Y.-H.; Lin Y.-T.; Lai W.-Y.; Yang D.-M.; Chou S.-J.; Yang Y.-P.; Wang M.-L.; Chiou S.-H. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. 10.3390/ijms21072657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Trivedi N. Disease-drug and drug-drug interaction in COVID-19: Risk and assessment. Biomed. Pharmacother. 2021, 139, 111642. 10.1016/j.biopha.2021.111642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrmann J. M. Intracellular ABCB1 as a Possible Mechanism to Explain the Synergistic Effect of Hydroxychloroquine-Azithromycin Combination in COVID-19 Therapy. AAPS J. 2020, 22, 86. 10.1208/s12248-020-00465-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. S.; Chung H.-S.; Noh S. G.; Lee B.; Chung H. Y.; Choi J.-G. Geraniin Inhibits the Entry of SARS-CoV-2 by Blocking the Interaction between Spike Protein RBD and Human ACE2 Receptor. Int. J. Mol. Sci. 2021, 22, 8604. 10.3390/ijms22168604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. 10.1039/c3cp00054k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.