

Ischemic heart disease is one of the leading causes of morbidity and mortality in the USA. It is mainly caused by the narrowing or occlusion of coronary arteries by plaque buildup, leading to a limited supply of oxygen and nutrients to the cardiac muscle. This results in necrotic death of cardiomyocytes (CMs). CM necrosis leads to the production of cytokines, chemokines, and damage-associated molecular patterns (DAMPs), which recruit immune cells from the bone marrow (BM)[1]. Infiltrated immune cells secrete proteases and cytokines that mediate inflammatory responses and fibroblast activation[1]. Subsequently, the damaged cardiac muscle is replaced with extracellular matrix produced by activated fibroblasts, leading to myocardial remodeling and dysfunction. Attempts to restore blood vessels (a.k.a. therapeutic angiogenesis) reduced fibrosis and improved the performance of the infarcted heart[2]. A possible underlying mechanism is that the supply of oxygen and nutrients via new blood vessels would preserve CM survival and support the health and function of remaining cardiovascular cells, thereby preventing adverse cardiac remodeling. Thus, therapeutic angiogenesis has been considered one of the important therapeutic approaches for ischemic heart diseases.
Investigations in the late 1990s and early 2000s indicated that the BM contains stem/progenitor cells, which can differentiate into multiple cell types. These results instilled great hope and excitement for cell therapy with BM-derived cells. However, clinical trials showed that injection of unselected bone marrow mononuclear cells (BMMNCs) into the infarcted heart only modestly improved cardiac function[2]. Studies raised questions regarding the mechanism of BM-derived cells-mediated cardiac repair. The observation that BM-derived cells directly transdifferentiate into CMs was refuted by other subsequent studies. Instead, it was revealed that BM-derived cells promote cardiac repair via short-term paracrine or humoral effects[3]. Over the past decade, genetic lineage tracing and single-cell RNA-seq approaches helped us better understand the differential roles of subpopulations in BMMNCs during cardiac remodeling and repair. Among them, myeloid cells play important roles during the sterile inflammation of the heart. They infiltrate into the damaged heart, engulf cell debris, and secrete a variety of paracrine factors to communicate with other cell types[1]. Emerging evidence suggests that myeloid cells promote vascular repair by secreting paracrine factors. Thus, identification and functional characterization of myeloid cell-derived paracrine factors can be critical to better understanding the pathophysiology of cardiovascular diseases.
Reboll and colleagues identified paracrine crosstalk between myeloid and endothelial cells (ECs) for vascular repair during cardiac ischemic injury responses[4] [Figure 1]. They found that Meteorin-like (METRNL) secreted by infiltrated myeloid cells binds to KIT tyrosine kinase receptor expressed on a subset of ECs, promoting proliferation and migration of KIT+ ECs. METRNL was known to be produced by inflammatory macrophages, but its receptor had been unknown. By utilizing genetically engineered mice and cutting-edge techniques, Reboll et al. demonstrated that METRNL-KIT signaling is crucial for vascular regeneration and cardiac repair. Stem cell factor (SCF) is a previously known ligand for KIT[4]. Reboll et al. showed that both METRNL and SCF bind to KIT with high affinity[4]. In contrast to METRNL, however, SCF was not upregulated in the infarcted heart. Moreover, major source cells for SCF in the heart are ECs, fibroblasts, and CMs. This suggests that there is minimal competition between METRNL and SCF for binding to KIT in the injured heart. If so, we can ask what the function of METRNL in a steady state without injury would be. While mice lacking Scf or Kit are embryonically lethal, Metrnl knockout mice were viable without apparent defects. There are two possibilities for the divergent observations. First, the prenatal expression level of METRNL might be negligible. Second, SCF might play compensatory roles for METRNL signaling in the METRNL knockout mice. It would be interesting to examine the prenatal expression of METRNL, its molecular interplay with SCF, and its impact on embryogenesis and organogenesis.
Figure 1.
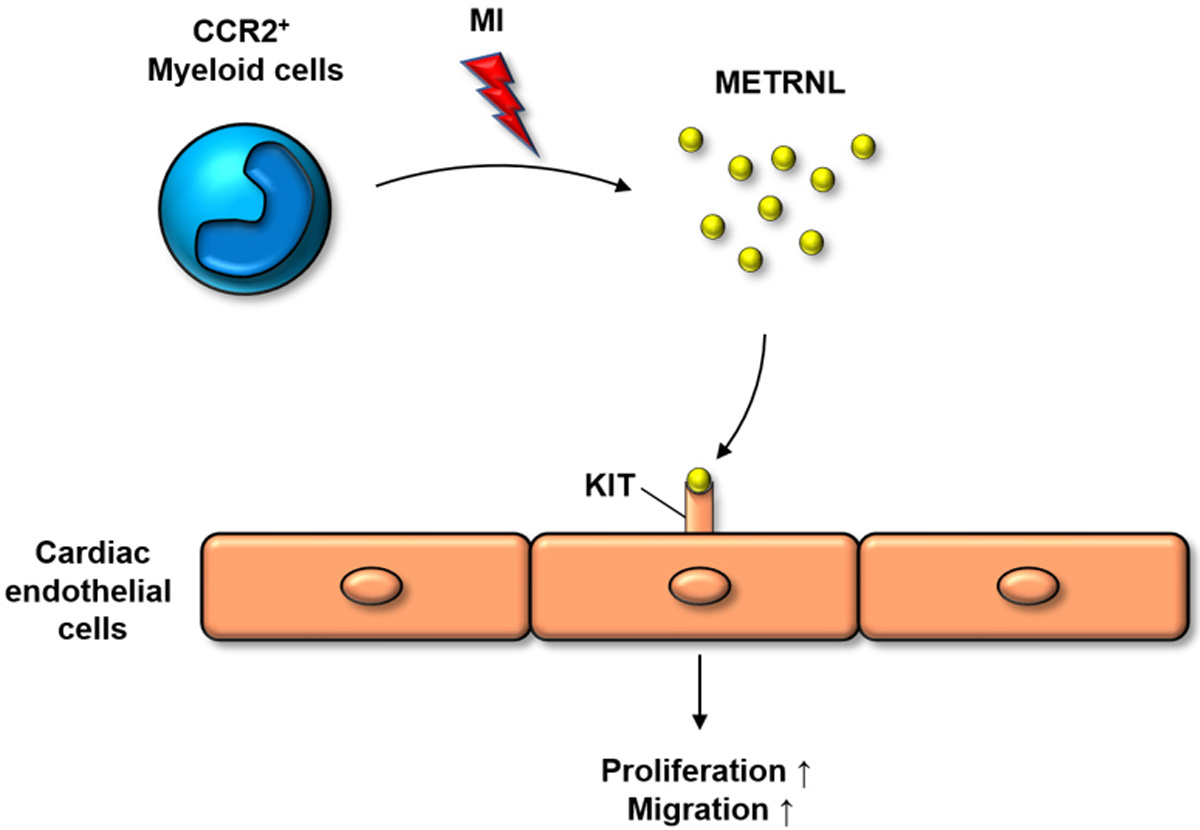
A schematic summary for the paracrine crosstalk between myeloid and endothelial cells (ECs) for vascular repair of the injured heart. In response to myocardial infarction (MI), CCR2+ myeloid cells infiltrate the heart and secrete METRNL which binds to KIT tyrosine receptor expressed on a subset of cardiac ECs. The KIT-METRNL signaling cascade induces the proliferation and migration of KIT+ ECs, contributing to tissue repair.
In addition, Reboll et al. made an interesting observation on the role of infiltrated macrophages (CCR2high) versus resident macrophages (CCR2low) in vascular repair[4]. Some reports claimed that BM-derived macrophages are angiogenic and reparative: infiltrated macrophages expressed higher angiogenic factors such as VEGF and MYDGF, whereas resident macrophages expressed pro-inflammatory factors such as TNFα and TGFβ1[5,6]. However, others reported that adult BM-derived macrophages are not helpful for angiogenesis and cardiac regeneration: inhibition of macrophage recruitment by a CCR2 inhibitor increased vascular density after cardiac injury[7]. In addition, resident macrophages that arise from the yolk sac were shown to promote angiogenesis[7] and prevent adverse cardiac remodeling[8]. In this study, Reboll et al. showed that METRNL is primarily expressed in infiltrated macrophages (CCR2high) rather than resident macrophages (CCR2low), claiming that BM-derived macrophages are essential for vascular repair[4]. Further studies are needed to address this controversy of whether angiogenic myeloid cells may exist in subpopulations of both recruited and resident macrophages.
Furthermore, Reboll et al. add a new perspective to the endothelial heterogeneity in the heart[4]. They showed that there is a subset of cardiac ECs that express KIT and they proliferate in METRNL- or KIT-dependent manners under ischemic stress. On the other hand, KIT-negative ECs were expanded independently of METRNL-KIT signaling, suggesting that different subsets of ECs proliferate via distinct mechanisms in response to myocardial infarction. However, it is still unclear how only a subset of ECs express KIT but others do not. First, it is possible that KIT+ ECs have different developmental origins from other ECs. ECs can originate from angioblasts (mesoderm-derived endothelial precursors) and erythromyeloid progenitors. Thus, the origin of Kit+ ECs versus Kit− ECs remains to be determined. Using genetic lineage tracing approaches, van Berlo et al. showed that c-kit+ cells generate ECs in the heart in both physiological and pathological conditions[9]. The proportion of c-kit lineage ECs doubled after myocardial infarction[9], consistently with Reboll et al., where the population of Kit+ ECs increased in the infarcted heart[4]. Since c-kit is expressed in a variety of cell types, dissecting the origin of Kit+ ECs and manipulation of the precursor cells to promote the generation of KIT+ ECs could be an intriguing research subject for vascular repair and its resultant heart regeneration. Second, it is possible that KIT expression could be induced on ECs. A study showed that KIT expression was induced in mature human CD8+ T cells after primary activation[10]. It would be worthwhile to define the pathophysiologic conditions and molecular mechanisms that induce endothelial expression of KIT. Understanding the regulation of KIT expression on ECs could maximize the therapeutic potential of METRNL for vascular repair.
In summary, Reboll et al. revealed that a novel form of molecular crosstalk between infiltrated myeloid cells and a subset of cardiac endotshelial cells drives angiogenesis and tissue repair after myocardial infarction[4]. Furthermore, the authors identified METRNL as a key myeloid paracrine factor that induces the proliferation of KIT+ ECs. This study offers new perspectives on the controversy about the role of infiltrated macrophages in vascular regeneration and on the heterogeneity of cardiac ECs. Furthermore, this study lays the foundation for new research topics in the fields of cardioimmunology and vascular biology. Addressing the remaining questions will help us to better understand molecular and cellular mechanisms underlying ischemic heart disease and to develop innovative therapeutic strategies.
Financial support and sponsorship
The study was supported by grants from NHLBI (R01HL150887, R01HL156008, R61/33 HL154116) and AHA Transformational Project Award (20TPA35490282), and by National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (No. 2020R1A2C3003784, No. 2020M3A9I4038454).
Footnotes
Conflicts of interest
All authors declared that there are no conflicts of interest.
DECLARATIONS
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
REFERENCES
- 1.Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol 2018;18:733–44. [DOI] [PubMed] [Google Scholar]
- 2.Behfar A, Crespo-Diaz R, Terzic A, Gersh BJ. Cell therapy for cardiac repair-lessons from clinical trials. Nat Rev Cardiol 2014;11:232–46. [DOI] [PubMed] [Google Scholar]
- 3.Cho HJ, Lee N, Lee JY, et al. Role of host tissues for sustained humoral effects after endothelial progenitor cell transplantation into the ischemic heart. J Exp Med 2007;204:3257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reboll MR, Klede S, Taft MH, et al. Meteorin-like promotes heart repair through endothelial KIT receptor tyrosine kinase. Science 2022;376:1343–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sager HB, Hulsmans M, Lavine KJ, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 2016;119:853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korf-Klingebiel M, Reboll MR, Klede S, et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat Med 2015;21:140–9. [DOI] [PubMed] [Google Scholar]
- 7.Lavine KJ, Epelman S, Uchida K, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA 2014;111:16029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 2019;20:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Berlo JH, Kanisicak O, Maillet M, V et al. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014;509:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frumento G, Zuo J, Verma K, et al. CD117 (c-Kit) is expressed during CD8(+) T cell priming and stratifies sensitivity to apoptosis according to strength of TCR engagement. Front Immunol 2019;10:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.